

Abstract
The title compound is a new aminocoumarin derivative, C17H13NO4, and was synthesized by the condensation of 2-aminophenol and 3-acetyl-4-hydroxycoumarin. An intramolecular N—H⋯O hydrogen bond generates an S(6) ring motif. In the crystal, the molecules are linked into chains extending in the [010] direction by O—H⋯O hydrogen bonds. There is also a π–π stacking interaction between the bicyclic coumarin fragment and the phenol ring [centroid–centroid distance = 3.7510 (14) Å], and these ring systems form between them a dihedral angle of 53.3 (2)°. Intermolecular hydrogen bond C—H⋯O hydrogen bonding is also observed in the interconnection of the crystal packing.
Related literature
For related structures, see: Traven et al. (2000 ▶); Malecka et al. (2004 ▶); Mechi et al. (2009 ▶); Ghouili et al. (2011 ▶); Ketata et al. (2012 ▶). For the properties of coumarin derivatives, see: Bordin et al. (1995 ▶); Hamdi et al. (2010 ▶); Mahidol et al. (2004 ▶).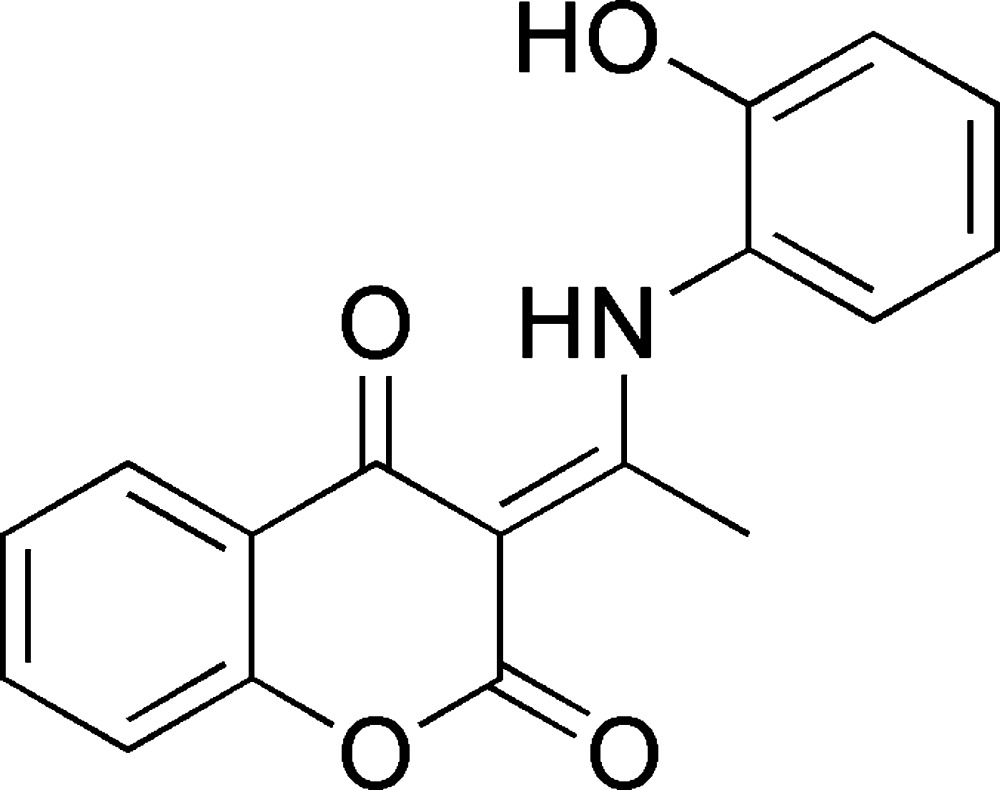
Experimental
Crystal data
C17H13NO4
M r = 295.29
Monoclinic,
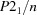
a = 12.5596 (4) Å
b = 7.5870 (3) Å
c = 14.3433 (6) Å
β = 94.660 (2)°
V = 1362.25 (9) Å3
Z = 4
Mo Kα radiation
μ = 0.10 mm−1
T = 293 K
0.16 × 0.13 × 0.10 mm
Data collection
Bruker SMART CCD area-detector diffractometer
11784 measured reflections
3891 independent reflections
1721 reflections with I > 2σ(I)
R int = 0.053
Refinement
R[F 2 > 2σ(F 2)] = 0.058
wR(F 2) = 0.194
S = 0.91
3891 reflections
251 parameters
H atoms treated by a mixture of independent and constrained refinement
Δρmax = 0.23 e Å−3
Δρmin = −0.20 e Å−3
Data collection: SMART (Bruker, 2001 ▶); cell refinement: SMART; data reduction: SAINT (Bruker, 2001 ▶); program(s) used to solve structure: SHELXS97 (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008 ▶); molecular graphics: ORTEP-3 for Windows (Farrugia, 2012 ▶); software used to prepare material for publication: WinGX (Farrugia, 2012 ▶).
Supplementary Material
Crystal structure: contains datablock(s) I, global. DOI: 10.1107/S160053681301934X/ff2111sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S160053681301934X/ff2111Isup2.hkl
Supplementary material file. DOI: 10.1107/S160053681301934X/ff2111Isup3.cml
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| O4—H12⋯O2i | 0.82 | 1.92 | 2.742 (2) | 175 |
| N1—H13⋯O3 | 0.88 (3) | 1.75 (3) | 2.537 (3) | 148 (3) |
| C8—H4⋯O2i | 0.93 | 2.59 | 3.274 (3) | 131 |
Symmetry code: (i) 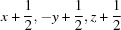 .
.
Acknowledgments
Professor A. Ben Salah is acknowledged for his contribution to the X-ray diffraction data collection at the Laboratory of Materials Science and the Environment, University of Sfax, Tunisia.
supplementary crystallographic information
Comment
Coumarin derivatives have a wide range of biological properties. They possess pharmacological activities, mainly anticoagulant. They also have anti-tumor, anti-oxidants and anti-inflammatory properties (Bordin et al., 1995; Mahidol et al., 2004). In continuation of our structural and biological studies of coumarin derivatives (Mechi et al., 2009; Hamdi et al., 2010; Ghouili et al., 2011; Ketata et al., 2012), we present the crystal structure of the title compound, a new derivative of amino coumarins.
In the crystal, the title compounds adopts a conformation where the dihedral angle between the plane of the bicyclic coumarin fragment and the phenol ring is 53.3 (2)°. The structure of our solid compound is stable thanks to the intermolecular hydrogen bonds O4—H12···O2. The crystal structure of C17H13N1O4 shows a π-π stacking interaction alternating between two inverted molecules. The stacking is observed along the b axis with distance 3.361 (4) Å between the layers of coumarin and phenol rings [centroid-centroid distance 3.7510 (14) Å].
The structure exhibits intramolecular hydrogen bonding N1—H13···O3, similar to that observed in amino coumarin analogue (Malecka et al. 2004), and we observe also a strong p-electron delocalization effect when comparing with O—H···O in 3-acetyl-4-hydroxycoumarin (Traven et al. 20000). In fact, the distances C1—C2 = 1.437 (3) Å in the title compound is longer then that one of the 3-acetyl-4-hydroxycoumarin (1.399 (1) Å) and C1—C5 = 1.428 (3) Å is shorter then 1.454 (1) Å. The elongation of N1—C5; C1—C2; C1—C11 and C11—O2 distances (see the table of bond lengths) and the shortening of O4—C6, C6—C4, C4—N1, C5—C1 and C2—O3 bond lengths, in comparison with those of the compound C13H13N1O4 (Malecka et al. 2004) could be related to the electron-donor effect of the phenolic group in the title compound.
The linkage between the coumarin system and aminophenol ring exhibits bond lengths O3—C2 = 1.251 (3) Å, C2—C1 = 1.437 (3) Å, C1—C5 = 1.428 (3) Å, C5—N1 = 1.316 (3) Å and N1—C4 = 1.422 (3) Å, suggesting that all non-hydrogen atoms between the electron-donors and acceptors are highly conjugated, leading to a π-bridge for the charge transfer from aminophenol ring to coumarin system.
Experimental
The amino coumarin was synthesized by the condensation of an equimolar amount of 2-aminophenol and 3-acetyl-4-hydroxycoumarin in absolute ethanol. After four hours of reflux, the reaction mixture was left crystallizing at room temperature. The compound obtained is presented as transparent crystals of light yellow color with shape and size suitable for the structural study of X-ray single-crystal. Yield:(90%). mp= 446 K. IR: ν 3155 (NH), 2973 (OH), 1666 (>C=O), 1609 (C=C), 1102 (C—O); 1HNMR: δp.p.m.: 2.54 (s, 3H, Hmethyl), 6.51–7.14 (m, 4H, Ar—H), 7.23–8.12 (m, 4H, Ph—H), 10.45 (s, 1H, OH), 15.20 (s, 1H, NH); 13CNMR: (p.p.m.): 20.30 (C methyl), 97.11 (C3), 116.27–134.34 (C arom), 151.40 (C—OH), 161.66 (C=O lactone), 175.90 (C—N), 180.19 (C=O ketone),
Refinement
The hydrogen atoms are fixed geometrically and refined as riding with the exception of the H13, which was located from electron density difference map and is refined isotropically. Uiso(H) values of the H atoms were set at 1.2Ueq or 1.5Ueq of the parent atom.
Figures
Fig. 1.

The molecular structure of the title compound showing 50% probability displacement ellipsoids and the atomic numbering. Dashed line denotes hydrogen bond.
Fig. 2.
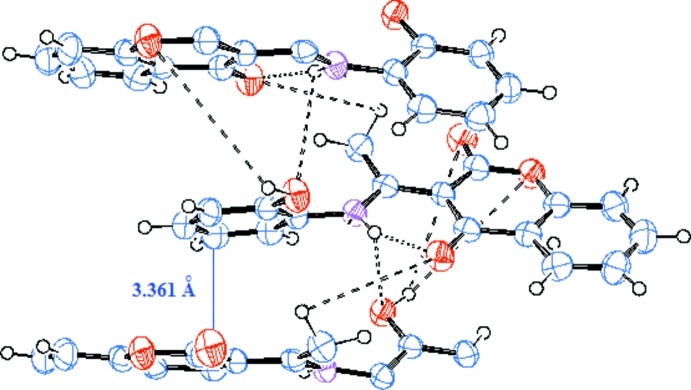
Hydrogen bonds between molecules of the title compound.
Crystal data
| C17H13NO4 | F(000) = 616 |
| Mr = 295.29 | Dx = 1.440 Mg m−3 |
| Monoclinic, P21/n | Melting point: 446 K |
| Hall symbol: -P2yn | Mo Kα radiation, λ = 0.71073 Å |
| a = 12.5596 (4) Å | Cell parameters from 1721 reflections |
| b = 7.5870 (3) Å | µ = 0.10 mm−1 |
| c = 14.3433 (6) Å | T = 293 K |
| β = 94.660 (2)° | Needle, yellow |
| V = 1362.25 (9) Å3 | 0.16 × 0.13 × 0.10 mm |
| Z = 4 |
Data collection
| Bruker SMART CCD area-detector diffractometer | 1721 reflections with I > 2σ(I) |
| Radiation source: fine-focus sealed tube | Rint = 0.053 |
| Graphite monochromator | θmax = 29.8°, θmin = 2.1° |
| φ and ω scans | h = −14→17 |
| 11784 measured reflections | k = −9→10 |
| 3891 independent reflections | l = −19→20 |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.058 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.194 | H atoms treated by a mixture of independent and constrained refinement |
| S = 0.91 | w = 1/[σ2(Fo2) + (0.0922P)2 + 0.2201P] where P = (Fo2 + 2Fc2)/3 |
| 3891 reflections | (Δ/σ)max < 0.001 |
| 251 parameters | Δρmax = 0.23 e Å−3 |
| 0 restraints | Δρmin = −0.20 e Å−3 |
Special details
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| O1 | 0.12600 (13) | 0.1466 (2) | 0.90032 (12) | 0.0480 (5) | |
| N1 | 0.22554 (16) | 0.0473 (3) | 1.22516 (14) | 0.0375 (5) | |
| C1 | 0.17836 (17) | 0.0955 (3) | 1.06478 (16) | 0.0350 (5) | |
| O3 | 0.34691 (12) | −0.0400 (2) | 1.09988 (12) | 0.0484 (5) | |
| C2 | 0.27686 (18) | 0.0180 (3) | 1.04068 (17) | 0.0378 (6) | |
| C3 | 0.29714 (18) | 0.0128 (3) | 0.94130 (17) | 0.0389 (6) | |
| C4 | 0.21353 (18) | 0.0298 (3) | 1.32227 (17) | 0.0368 (6) | |
| O4 | 0.38154 (13) | 0.1659 (2) | 1.35092 (12) | 0.0473 (5) | |
| H12 | 0.4244 | 0.1965 | 1.3941 | 0.071* | |
| C5 | 0.15554 (17) | 0.1107 (3) | 1.16048 (17) | 0.0363 (6) | |
| O2 | 0.01364 (14) | 0.2201 (3) | 1.00033 (13) | 0.0556 (5) | |
| C6 | 0.29532 (19) | 0.0900 (3) | 1.38614 (18) | 0.0376 (6) | |
| C7 | 0.22130 (19) | 0.0795 (3) | 0.87552 (18) | 0.0417 (6) | |
| C8 | 0.2850 (2) | 0.0704 (3) | 1.48055 (18) | 0.0439 (6) | |
| H4 | 0.3384 | 0.1118 | 1.5238 | 0.053* | |
| C9 | 0.05868 (19) | 0.2032 (4) | 1.19047 (19) | 0.0475 (7) | |
| H5 | 0.0586 | 0.1977 | 1.2573 | 0.071* | |
| H11 | −0.0045 | 0.1470 | 1.1623 | 0.071* | |
| H7 | 0.0598 | 0.3242 | 1.1710 | 0.071* | |
| C10 | 0.2392 (2) | 0.0838 (4) | 0.78132 (19) | 0.0546 (7) | |
| H10 | 0.1871 | 0.1274 | 0.7375 | 0.065* | |
| C11 | 0.10187 (19) | 0.1569 (3) | 0.99164 (18) | 0.0416 (6) | |
| C12 | 0.3922 (2) | −0.0510 (3) | 0.9114 (2) | 0.0481 (7) | |
| H1 | 0.4431 | −0.0990 | 0.9548 | 0.058* | |
| C13 | 0.12623 (19) | −0.0559 (3) | 1.35386 (19) | 0.0451 (7) | |
| H2 | 0.0735 | −0.1007 | 1.3111 | 0.054* | |
| C14 | 0.1168 (2) | −0.0752 (4) | 1.4483 (2) | 0.0503 (7) | |
| H3 | 0.0575 | −0.1314 | 1.4695 | 0.060* | |
| C15 | 0.4121 (2) | −0.0446 (4) | 0.8187 (2) | 0.0545 (7) | |
| H8 | 0.4768 | −0.0848 | 0.7996 | 0.065* | |
| C16 | 0.1960 (2) | −0.0104 (4) | 1.51091 (19) | 0.0497 (7) | |
| H6 | 0.1893 | −0.0214 | 1.5747 | 0.060* | |
| C17 | 0.3343 (2) | 0.0230 (4) | 0.7537 (2) | 0.0583 (8) | |
| H9 | 0.3471 | 0.0268 | 0.6908 | 0.070* | |
| H13 | 0.283 (2) | 0.007 (4) | 1.201 (2) | 0.061 (9)* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| O1 | 0.0451 (10) | 0.0600 (12) | 0.0366 (10) | 0.0120 (9) | −0.0099 (8) | 0.0012 (9) |
| N1 | 0.0327 (11) | 0.0452 (12) | 0.0330 (11) | 0.0007 (9) | −0.0072 (9) | −0.0010 (9) |
| C1 | 0.0327 (11) | 0.0370 (13) | 0.0335 (13) | 0.0004 (10) | −0.0089 (10) | 0.0011 (10) |
| O3 | 0.0378 (9) | 0.0621 (12) | 0.0433 (10) | 0.0091 (8) | −0.0085 (8) | 0.0041 (9) |
| C2 | 0.0347 (12) | 0.0368 (13) | 0.0398 (13) | −0.0011 (10) | −0.0104 (10) | 0.0017 (11) |
| C3 | 0.0387 (13) | 0.0355 (13) | 0.0413 (14) | −0.0040 (11) | −0.0045 (11) | −0.0031 (11) |
| C4 | 0.0355 (12) | 0.0391 (13) | 0.0342 (13) | 0.0009 (10) | −0.0057 (10) | −0.0007 (11) |
| O4 | 0.0390 (9) | 0.0621 (12) | 0.0390 (10) | −0.0135 (8) | −0.0080 (8) | 0.0009 (9) |
| C5 | 0.0339 (12) | 0.0330 (13) | 0.0403 (14) | −0.0055 (10) | −0.0082 (10) | −0.0012 (10) |
| O2 | 0.0406 (10) | 0.0748 (14) | 0.0484 (11) | 0.0151 (9) | −0.0141 (8) | 0.0016 (10) |
| C6 | 0.0403 (13) | 0.0340 (13) | 0.0375 (13) | −0.0002 (10) | −0.0032 (10) | −0.0001 (11) |
| C7 | 0.0436 (14) | 0.0406 (14) | 0.0393 (14) | −0.0002 (11) | −0.0069 (11) | −0.0026 (11) |
| C8 | 0.0474 (14) | 0.0477 (16) | 0.0351 (14) | −0.0028 (12) | −0.0063 (11) | −0.0026 (12) |
| C9 | 0.0409 (13) | 0.0541 (17) | 0.0462 (16) | 0.0054 (12) | −0.0047 (12) | −0.0015 (13) |
| C10 | 0.0630 (18) | 0.0593 (18) | 0.0395 (16) | 0.0063 (15) | −0.0078 (13) | 0.0033 (13) |
| C11 | 0.0399 (13) | 0.0428 (15) | 0.0400 (15) | −0.0012 (12) | −0.0094 (11) | 0.0003 (12) |
| C12 | 0.0413 (13) | 0.0493 (16) | 0.0519 (17) | 0.0012 (12) | −0.0073 (12) | −0.0047 (13) |
| C13 | 0.0377 (13) | 0.0486 (16) | 0.0475 (16) | −0.0029 (11) | −0.0056 (12) | −0.0019 (12) |
| C14 | 0.0471 (15) | 0.0493 (16) | 0.0554 (18) | −0.0022 (12) | 0.0104 (13) | 0.0016 (14) |
| C15 | 0.0489 (15) | 0.0655 (19) | 0.0492 (17) | −0.0035 (14) | 0.0040 (13) | −0.0079 (15) |
| C16 | 0.0601 (17) | 0.0510 (16) | 0.0384 (14) | 0.0040 (14) | 0.0057 (13) | 0.0006 (13) |
| C17 | 0.0656 (18) | 0.067 (2) | 0.0427 (16) | −0.0056 (16) | 0.0035 (14) | −0.0031 (15) |
Geometric parameters (Å, º)
| O1—C7 | 1.374 (3) | C7—C10 | 1.388 (4) |
| O1—C11 | 1.370 (3) | C8—C16 | 1.376 (4) |
| N1—C5 | 1.316 (3) | C8—H4 | 0.9300 |
| N1—C4 | 1.419 (3) | C9—H5 | 0.9600 |
| N1—H13 | 0.88 (3) | C9—H11 | 0.9600 |
| C1—C5 | 1.429 (3) | C9—H7 | 0.9600 |
| C1—C2 | 1.437 (3) | C10—C17 | 1.369 (4) |
| C1—C11 | 1.441 (3) | C10—H10 | 0.9300 |
| O3—C2 | 1.252 (3) | C12—C15 | 1.373 (4) |
| C2—C3 | 1.469 (3) | C12—H1 | 0.9300 |
| C3—C7 | 1.381 (3) | C13—C14 | 1.376 (4) |
| C3—C12 | 1.388 (3) | C13—H2 | 0.9300 |
| C4—C13 | 1.382 (3) | C14—C16 | 1.376 (4) |
| C4—C6 | 1.396 (3) | C14—H3 | 0.9300 |
| O4—C6 | 1.359 (3) | C15—C17 | 1.393 (4) |
| O4—H12 | 0.8200 | C15—H8 | 0.9300 |
| C5—C9 | 1.498 (3) | C16—H6 | 0.9300 |
| O2—C11 | 1.223 (3) | C17—H9 | 0.9300 |
| C6—C8 | 1.379 (3) | ||
| C7—O1—C11 | 122.26 (19) | C5—C9—H11 | 109.5 |
| C5—N1—C4 | 127.4 (2) | H5—C9—H11 | 109.5 |
| C5—N1—H13 | 112 (2) | C5—C9—H7 | 109.5 |
| C4—N1—H13 | 121 (2) | H5—C9—H7 | 109.5 |
| C5—C1—C2 | 120.5 (2) | H11—C9—H7 | 109.5 |
| C5—C1—C11 | 119.9 (2) | C17—C10—C7 | 119.2 (3) |
| C2—C1—C11 | 119.6 (2) | C17—C10—H10 | 120.4 |
| O3—C2—C1 | 123.5 (2) | C7—C10—H10 | 120.4 |
| O3—C2—C3 | 118.8 (2) | O2—C11—O1 | 113.1 (2) |
| C1—C2—C3 | 117.7 (2) | O2—C11—C1 | 127.5 (2) |
| C7—C3—C12 | 118.6 (2) | O1—C11—C1 | 119.4 (2) |
| C7—C3—C2 | 119.3 (2) | C15—C12—C3 | 121.0 (3) |
| C12—C3—C2 | 122.0 (2) | C15—C12—H1 | 119.5 |
| C13—C4—C6 | 120.0 (2) | C3—C12—H1 | 119.5 |
| C13—C4—N1 | 121.1 (2) | C4—C13—C14 | 120.4 (2) |
| C6—C4—N1 | 118.8 (2) | C4—C13—H2 | 119.8 |
| C6—O4—H12 | 109.5 | C14—C13—H2 | 119.8 |
| N1—C5—C1 | 118.2 (2) | C13—C14—C16 | 119.3 (2) |
| N1—C5—C9 | 118.7 (2) | C13—C14—H3 | 120.4 |
| C1—C5—C9 | 123.0 (2) | C16—C14—H3 | 120.4 |
| O4—C6—C8 | 123.5 (2) | C12—C15—C17 | 119.3 (3) |
| O4—C6—C4 | 117.4 (2) | C12—C15—H8 | 120.4 |
| C8—C6—C4 | 119.1 (2) | C17—C15—H8 | 120.4 |
| O1—C7—C3 | 121.7 (2) | C8—C16—C14 | 121.0 (3) |
| O1—C7—C10 | 117.2 (2) | C8—C16—H6 | 119.5 |
| C3—C7—C10 | 121.1 (2) | C14—C16—H6 | 119.5 |
| C6—C8—C16 | 120.1 (2) | C10—C17—C15 | 120.7 (3) |
| C6—C8—H4 | 119.9 | C10—C17—H9 | 119.6 |
| C16—C8—H4 | 119.9 | C15—C17—H9 | 119.6 |
| C5—C9—H5 | 109.5 |
Hydrogen-bond geometry (Å, º)
| D—H···A | D—H | H···A | D···A | D—H···A |
| O4—H12···O2i | 0.82 | 1.92 | 2.742 (2) | 175 |
| N1—H13···O3 | 0.88 (3) | 1.75 (3) | 2.537 (3) | 148 (3) |
| C8—H4···O2i | 0.93 | 2.59 | 3.274 (3) | 131 |
Symmetry code: (i) x+1/2, −y+1/2, z+1/2.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: FF2111).
References
- Bordin, F., Chilin, A., Dall Acqua, F., Guiotto, A. & Manzini, P. (1995). Rod Il Farm. 50, 479–488. [PubMed]
- Bruker (2001). SMART and SAINT Bruker AXS Inc., Madison, Wisconsin, USA.
- Farrugia, L. J. (2012). J. Appl. Cryst. 45, 849–854.
- Ghouili, A. & Ben Hassen, R. (2011). Acta Cryst. E67, o2209. [DOI] [PMC free article] [PubMed]
- Hamdi, N., Bouabdallah, F., Romerosa, A. & Ben Hassen, R. (2010). C. R. Chim. 13, 1261–1268.
- Ketata, I., Mechi, L., Ben Ayed, T., Dusek, M., Petricek, V. & Ben Hassen, R. (2012). Open J. Inorg. Chem. 2, 33–39.
- Mahidol, C., Ploypradith, P., Sahakitpichan, P. & Wongbundit, S. (2004). Angew. Chem. Int. Ed. 43, 866–868. [DOI] [PubMed]
- Malecka, M., Grabowski, S. J. & El_zbieta, B. (2004). Chem. Phys. 297, 235–244.
- Mechi, L., Chtiba, S., Hamdi, N. & Ben Hassen, R. (2009). Acta Cryst. E65, o1652–o1653. [DOI] [PMC free article] [PubMed]
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Traven, V. F., Manaev, A. V., Safronova, O. B., Chibisova, T. A., Lyssenko, K. A. & Antipin, M. Yu. (2000). Russ. J. Gen. Chem. 70, 798–808.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) I, global. DOI: 10.1107/S160053681301934X/ff2111sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S160053681301934X/ff2111Isup2.hkl
Supplementary material file. DOI: 10.1107/S160053681301934X/ff2111Isup3.cml
Additional supplementary materials: crystallographic information; 3D view; checkCIF report