

Abstract
Channelrhodopsins are used to optogenetically depolarize neurons. We engineered a variant of channelrhodopsin, denoted Red-activatable Channelrhodopsin (ReaChR), that is optimally excited with orange to red light (λ ~ 590 to 630 nm) and offers improved membrane trafficking, higher photocurrents, and faster kinetics compared with existing red-shifted channelrhodopsins. Red light is more weakly scattered by tissue and absorbed less by blood than the blue to green wavelengths required by other channelrhodopsin variants. ReaChR expressed in vibrissa motor cortex was used to drive spiking and vibrissa motion in awake mice when excited with red light through intact skull. Precise vibrissa movements were evoked by expressing ReaChR in the facial motor nucleus in the brainstem and illuminating with red light through the external auditory canal. Thus, ReaChR enables transcranial optical activation of neurons in deep brain structures without the need to surgically thin the skull, form a transcranial window, or implant optical fibers.
Keywords: brainstem, cell culture, light-activated, motor cortex, vibrissae
Introduction
Channelrhodopsins (ChRs) are light-gated, non-specific cation channels that allow the selective depolarization of genetically targeted cells1-6. Currently available ChRs, however, are limited by action spectra that typically peak at 450 to 545 nm7-11. In mammalian systems, such blue-green wavelengths have limited penetration depths into neural tissue12 since they are strongly absorbed by endogenous chromophores such as flavins, hemoglobin, and melanin, and are scattered more strongly than yellow-red wavelengths. A common mean to circumvent this attenuation and achieve deep ChR excitation is to insert a thin optical fiber into the target neural tissue13. However, this invasive procedure requires precise stereotaxic positioning that may be difficult to perform when ChR is expressed in subcortical nuclei and the fiber may damage neuronal and/or vascular structures en route to the target. The practical issue of efficient light delivery can be solved by creating a red-shifted ChR with spectral peaks near or above 600 nm, where light absorption by hemes and scattering cross-sections drop off steeply12. Such ChRs could facilitate the stimulation of deep structures with a light source placed outside the tissue or region of interests and thus mitigate the need for invasive stimulation.
In this study, we engineered a new variant of channelrhodopsin named ReaChR for Red-activatable Channelrhodopsin. Compared to the previously described ChRs with significant red spectral shift, i.e., VChR114 and C1V1(E122T)11, ReaChR has improved membrane trafficking and expression in mammalian cells, more robust spectral response above 600 nm, and enhanced steady-state response to light with wavelengths longer than 600 nm. When ReaChR was expressed in mouse vibrissae motor cortex (vM1), we could trigger temporally precise neuronal responses by illumination of the cortical surface with 617 nm light through a trans-cranial window, or evoke vibrissae movements through the intact skin and bone at wavelengths ranging from 470 to 655 nm. Expression of ReaChR in the facial motor nucleus (FN) of the brainstem allowed us to reliably activate vibrissa motoneurons with wavelengths up to 655 nm through a non-invasive placement of a light emitting diode (LED) at the opening of the external auditory canal. Thus, ReaChR enables effective and temporally precise non-invasive stimulation of deep brain structures with ChR technology.
Results
Development of a red-activatable channelrhodopsin
The red-shifted channelrhodopsin VChR114, which has minimal trafficking to the membrane as well as poor expression in mammalian cells11, 15, served as a template to engineer an efficient red-light activated channelrhodopsin. To improve the membrane trafficking, we examine the superior membrane trafficking of the variant ChIEF7, which gives almost exclusive plasma membrane expression with minimal cytosolic aggregation in mammalian cells without the need for additional trafficking signals, unlike many other ChR variants (Supplementary Fig. 1)15. We thus replaced the N-terminus of VChR1 prior to the first transmembrane domain with the corresponding ChIEF sequence, (Fig. 1a). The new construct, denoted as C-VChR1, has considerably improved membrane trafficking15 (Fig. 1c; P < 0.001 for VChR1 vs. C-VChR1). To increase the expression level, we replaced transmembrane domain F of VChR1 with the corresponding VChR2 helix, a strategy previously shown to increase the expression level of ChR in ChR1/ChR2 chimeras16. This new variant, named VCOMET for VChR Optimized for Membrane Expression and Trafficking, has a red-shifted response spectra similar to C-VChR1, with spectral peaks at 590 and 530 nm for maximum and steady-state/plateau response, respectively (Supplementary Fig. 2b). VCOMET expresses strongly in mammalian cells and retains robust membrane trafficking (Supplementary Fig. 1). It yields greater photocurrent in HEK293 cells (39 ± 8 pA/pF, mean ± S.E.M, n = 13) compared to C-VChR1 (6.9 ± 1.0 pA/pF, n = 13; P < 0.01; Fig. 1d) and comparable photocurrents to mammalian-codon optimized ChEF (oChEF; 48 ± 8 pA/pF, n = 12; Fig. 1d & Supplementary Fig. 2a) and mammalian-codon optimized ChIEF (oChIEF; 52 ± 7 pA/pF, n = 7). VCOMET has a reversal potential (12 ± 2 mV, n = 8) that is significantly higher from oChIEF (4 ± 1 mV, n = 5; P < 0.05) but not the Calcium-translocating channelrhodopsins CatCh17 (5 ± 1 mV, n = 6) in physiological saline (H = 10.39, k = 4, P = 0.0155 for comparisons between VCOMET, ChIEF, CatCh and ReaChR), and reduced inward rectification similar to oChIEF (Supplementary Fig. 2c)7.
Figure 1. Basic properties of ReaChR compared to C-VChR1 (VChR1 with ChIEF N-terminal) and C1V1(E122T).
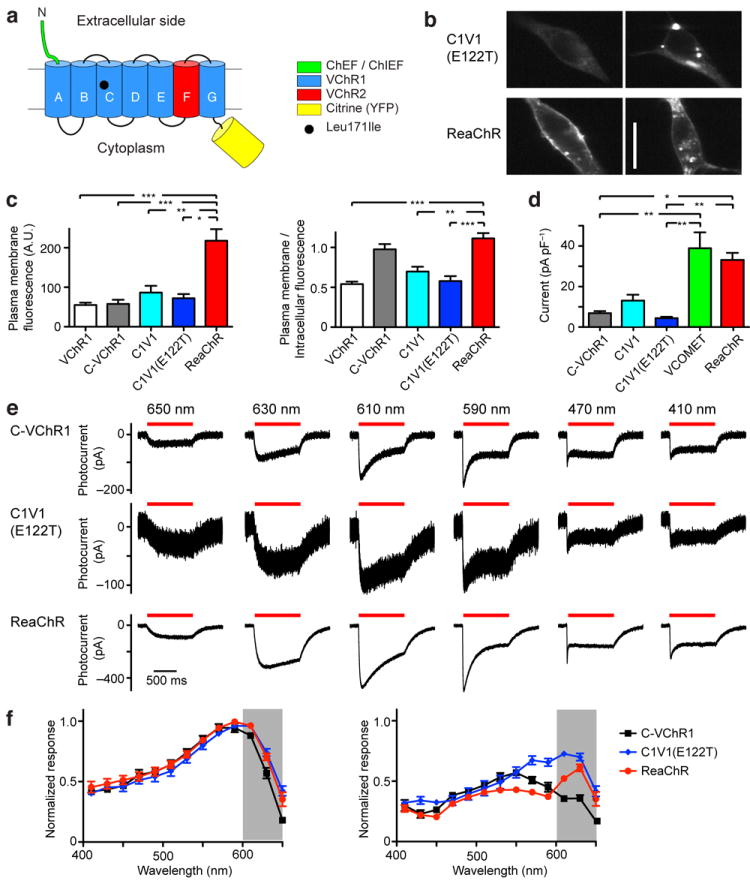
(a) Schematic design of ReaChR. ReaChR consists of the N-terminal of the ChEF/ChIEF variant, transmembrane domain A-E and G of VChR1, transmembrane domain F of VChR2, and a Leu171Ile point mutation. (b) Examples of C1V1(E122T) and ReaChR expression in HEK293 cells as visualized by fluorescence of fused Citrine. Much of ReaChR expression was at the plasma membrane, whereas C1V1(E122T) expression was more intracellular, with strong aggregation. (c) The relative plasma membrane expression level (left) and plasma membrane/intracellular fluorescence ratio (right) of VChR1, C-VChR1, C1V1, C1V1(E122T) and ReaChR as measured with Citrine fluorescence. (d) The mean photocurrent amplitudes of C-VChR1, C1V1, C1V1(E122T), VCOMET and ReaChR recorded from HEK293 cells. The current amplitudes were measured at the wavelengths that evoked the greatest response of each variant and normalized to cell capacitance. (e) The response of C-VChR1 (top row), C1V1(E122T) (middle row), and ReaChR (bottom row) to 650, 630, 610, 590, 470, and 410 nm light of same photon flux (5.1 × 1016 photon/mm2/s). (f) The spectra of the maximum response (left) and steady-state/plateau response (right) of C-VChR1 (n =7), C1V1(E122T) (n = 8) and ReaChR (n = 7). The responses were normalized to the maximum response of each cell. For statistical comparisons, Kruskal-Wallis tests were used with post-hoc Dunn’s tests on all pairs of variants. H = 31.63, k = 5, P < 0.0001 for the left panel of (c), H = 41.28, k = 5, P < 0.0001 for the right panel of (c), H = 48.02, k = 7, P < 0.0001 for (d). The statistical tests for (d) also include comparison to oChEF and oChIEF. Only significant differences detected with Dunn’s tests with VCOMET or ReaChR are shown on the graphs (*, **, *** indicate significance levels of < 0.05, P < 0.01 and P < 0.001, respectively). Scale bar in (a): 20 μm. The graphs in (c), (d) and (f) are presented as mean ± S.E.M.
While VCOMET responded strongly to light above 600 nm (Supplementary Fig. 2b), the responses at these longer wavelengths were desensitizing and failed to recover completely in the dark without reconditioning with 410 nm light (Supplementary Fig. 3). We thus sought to reduce the desensitization of VCOMET to light above 600 nm through known point mutations of ChRs. The ChETA mutation17 did not red-shift nor reduce the desensitization of VCOMET. The corresponding H134R mutation of ChR218 slowed the channel kinetics and thus degraded the temporal fidelity of the ChR. One mutation, L171I, which corresponds to the same position of the ChIEF mutation7, increased the amplitude of the photo-response at both 610 and 630 nm light by a reduced level of desensitization (Figs. 1e & 2, Supplementary Fig. 4) and thus led to an increase in the steady-state/plateau spectral peak at 630 nm (Fig. 1f). This formed ReaChR, the new channelrhodopsin. ReaChR retained the reversal potential (7 ± 4 mV, n = 6), reduced inward rectification, photocurrent amplitude (34 ± 4 pA/pF; n = 9), membrane trafficking, and expression of VCOMET (Fig. 1b-d). The one limitation of ReaChR relative to C-VChR1 is the slower channel closure rate after the termination of the light pulse; τ1/e = 137 ± 7 ms (n = 11) for ReaChR compared to τ1/e = 85 ± 4 ms (n = 9) for C-VChR1 (P < 0.05) (Fig. 2d & Table 1).
Figure 2. Kinetics of C-VChR1, C1V1(E122T) and ReaChR.
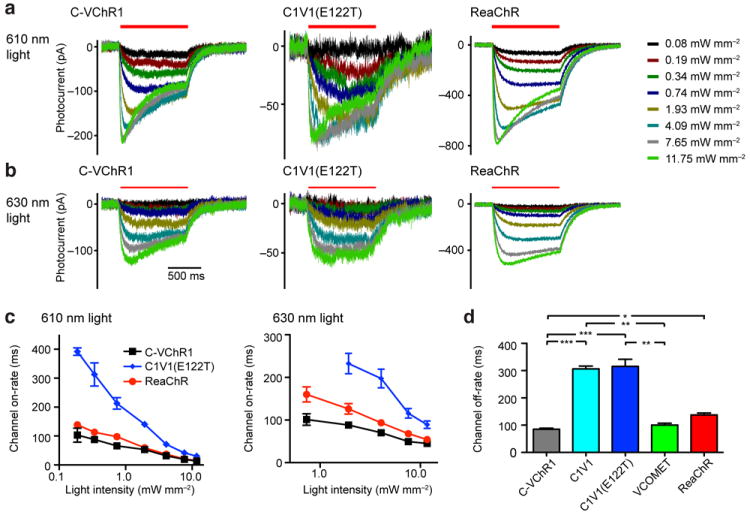
(a) Representative responses of a C-VChR1- (left), C1V1(E122T)- (middle) and ReaChR (right)-expressing HEK293 cell to 0.08, 0.19, 0.34, 0.74, 1.93, 4.09, 7.65, and 11.75 mW/mm2 of 610 nm light. (b) The same cells in (a) responding to 630 nm light of varying intensities. (c) Channel onset time constants of C-VChR1 (n = 10), C1V1(E122T) (n = 6) and ReaChR (n = 10) to 610 nm (left) and 630 nm (right) light of different intensities. (d) Channel closure time constants of C-VChR1 (n = 11), C1V1 (n = 6), C1V1(E122T) (n = 8), VCOMET (n = 9) and ReaChR (n = 11). For (d), Kruskal-Wallis test was used with post-hoc Dunn’s multiple comparison tests on all pairs of variants (H = 36.76, k =5, P < 0.0001). *, ** and *** indicate P < 0.05, P < 0.01 and P < 0.001 levels of significance as detected by Dunn’s tests, respectively. The graphs in (c) and (d) are presented as mean ± S.E.M.
Table 1.
Comparisons of the channel kinetics and properties of C-VChR1, VCOMET, ReaChR, C1V1, and C1V1(E122T).
| Variant | Response spectra (nm) | Mean membrane fluorescence (A.U.) | Mean ratio of membrane to cytosol fluorescence | Mean photocurrent (pA/pF) | Channel on-rate τ1/e (ms) 610 nm at 7.6 mW/mm2 | Channel on-rate τ1/e (ms) 630 nm at 7.6 mW/mm2 | Channel off-rate τ1/e (ms) | |
|---|---|---|---|---|---|---|---|---|
| Maximum | Steady-state | |||||||
| C-VChR1 | ~570 | ~550 | 58 ± 11 (n = 29) |
0.98 ± 0.07 (n = 29) |
6.9 ± 1.1 (n = 13) |
19.4 ± 0.8 (n = 11) |
49.4 ± 2.0 (n = 10) |
84.9 ± 3.9 (n = 11) |
| VCOMET | ~590 | ~530 | N/D | 1.10 ± 0.07 (n = 26) ++ |
38.9 ± 7.8 (n = 13) |
N/D | N/D | 100.4 ± 6.6 (n = 9) |
| ReaChR | ~590 | ~630 | 218 ± 29 (n = 21) |
1.12 ± 0.07 (n = 21) |
33.5 ± 3.6 (n = 9) |
20.7 ± 0.6 (n = 11) |
68.1 ± 4.2 (n = 10) |
137.2 ± 7.1 (n = 11) |
| C1V1 | N/D | N/D | 87 ± 17 (n = 26) |
0.70 ± 0.06 (n = 26) |
13.0 ± 3.0 (n = 10) |
N/D | N/D | 306.3 ± 10.3 (n = 6) |
| C1V1 (E122T) | ~600 | ~610 | 72 ± 11 (n = 16) |
0.58 ± 0.06 (n = 16) |
4.4 ± 0.6 (n = 10) |
41.5 ± 4.6 (n = 7) |
115.8 ± 11.1 (n = 6) |
315.4 ± 26.0 (n = 8) |
Electrophysiological characterizations were made under voltage-clamp recordings in HEK293 cells where the membrane potential can be accurately clamped at -60 mV. Plasma membrane and intracellular fluorescence were measured in HEK293 cells transiently expressing the channelrhodopsin fused to Citrine imaged with a confocal laser-scanning microscope. N/D: not determined.
indicates the value of VCOMET was acquired in a separate experiment with different pixel dwelling time settings.
Comparisons of red-shifted ChR variants in cell culture
The ChR variant C1V1(E122T) has been shown to excite neuronal membrane potential with 630 nm light that is pulsed for 50 ms11. To gauge the potential benefit of ReaChR, over C1V1 and related constructs, we characterized the attributes of ReaChR in comparison with C1V1, C1V1(E122T), and VCOMET (Table 1). C1V1 and C1V1(E122T) both expressed strongly in HEK293 cells as visualized with Citrine19 fluorescence of the C-terminal fusion protein. However, both C1V1 and C1V1(E122T) did not traffic to the membrane as well as ReaChR or VCOMET in these cells and showed high levels of intracellular aggregation and strong cytosolic fluorescence (Fig. 1b & c). The mean photocurrent of C1V1 expressed in HEK293 cells (13 ± 3 pA/pF, n = 10) was smaller than that for VCOMET or ReaChR (Fig. 1d), possibly caused by reduced efficiency in trafficking to the plasma membrane (Fig. 1c). Introduction of the E122T mutation, to form C1V1(E122T), reduced the C1V1 photocurrent two- to three-fold (4 ± 1 pA/pF, n = 10; Fig. 1d), although its action spectrum is red-shifted, with peaks at 590 and 610 nm for the maximum and steady-state/plateau responses, respectively (Fig. 1e,f). The channel kinetics of C1V1(E122T) are significantly slower than that of VCOMET and C-VChR1, with channel closure time constants of τ1/e = 315 ± 26 ms (n = 8), although similar to that of the parent construct C1V1 (τ1/e = 306 ± 10 ms, n = 6; Fig. 2d). The activation kinetics of C1V1(E122T) is also 1-1/2- to 3-fold slower than ReaChR and C-VChR1 at the same light intensities for 610 and 630 nm light (Fig. 2c). However, the photocurrent amplitudes of C-VChR1, ReaChR and C1V1(E122T) display a similar functional form in response to incident light intensity (Supplementary Fig. 4). An interesting observation is that with C-VChR1, C1V1(E122T) and ReaChR, increasing the stimulation intensity of 610 nm light leads to the reduction of steady-state/plateau response amplitudes at higher light intensities (Fig. 2a & Supplementary Fig. 4). As ReaChR has improved membrane trafficking, greater photocurrents, and faster kinetics compared to C1V1(E122T), we propose that ReaChR is a robust means to stimulate neurons with red-orange to red light.
Comparison of ChR variants in cultured neurons
We compared the properties of ReaChR with other ChR variants in cultured neurons20. ReaChR traffics to the membrane well and expresses strongly in neurons (n = 22), as was observed in HEK293 cells (Fig. 3a). The variants C1V1(E122T) (n = 13) and C1V1(E122T) with an additional trafficking sequence, denoted C1V1(E122T)-TS (n = 17), trafficked to the membrane as well as ReaChR in cultured neurons (Fig. 3a,b), unlike the greater trafficking observed for ReaChR in HEK293 cells. However, the levels of C1V1(E122T) or C1V1(E122T)-TS expression are approximately five-fold lower than ReaChR under identical conditions (Fig. 3a,b). Occasionally, cells with high expression levels of C1V1(E122T) and C1V1(E122T)-TS were observed, although these cells occurred too infrequently to be of general utility.
Figure 3. Characterization and comparison of different channelrhodopsin variants in primary cultured hippocampal neurons.
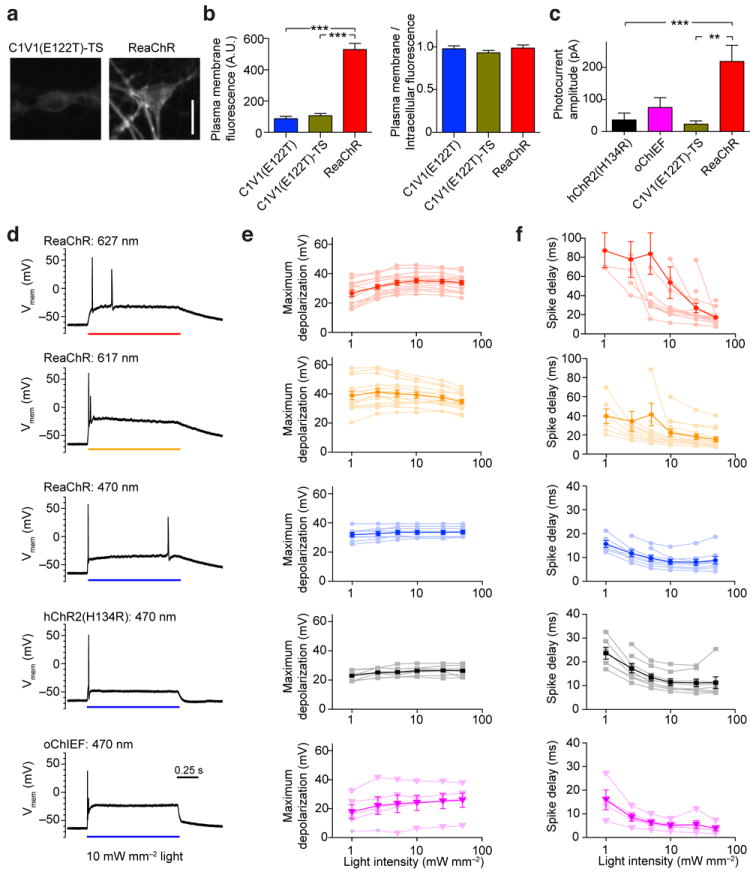
(a) Representative confocal images of neurons expressing C1V1(E122T)-TS-Citrine and ReaChR-Citrine. (b) The quantification of plasma membrane fluorescence (left) and plasma membrane / cytosolic fluorescence ratio (right) of C1V1(E122T)-Citrine, C1V1(E122T)-TS-Citrine and ReaChR-Citrine. (c) Photocurrent amplitudes of primary cultured neurons expressing hChR2(H134R), oChIEF, C1V1(E122T)-TS and ReaChR as measured with voltage-clamping recordings. The hChR2(H134R) and oChIEF-expressing neurons were stimulated with 10 mW/mm2 of 470 nm light and C1V1(E122T)-TS and ReaChR-expressing neurons were stimulated with 10 mW/mm2 light of 590 nm. (d) Representative recordings of neurons expressing different channelrhodopsin variants to 1 s of 10 mW/mm2 of LED light of the indicated wavelength under current-clamp recordings. (e) Quantification of the level of light-induced depolarization in neurons expressing the different variants to increasing intensity of light. (f) Quantification of the action potential latency from the onset of the light pulse to increasing intensity of light. For statistical comparisons, Kruskal-Wallis tests were used with post-hoc Dunn’s multiple comparison tests on all pairs of variants. H = 37.66, k = 3, P < 0.0001 for the left panel of (b), H = 1.09, k = 3, P = 0.58 for the right panel of (b), H = 18.32, k = 4, P = 0.0004 for (c). Scale bar in (a): 20 μm. The graphs in (b) and (c) are presented as mean ± S.E.M. Lighter color traces in (e) and (f) are responses of individual cells and darker color traces are mean ± S.E.M. *, ** and *** indicate 0.05, 0.01 and 0.001 level of significance as detected by Dunn’s test.
To compare the photocurrent amplitudes of different ChR variants, titer-matched lentivirus with a truncated human synapsin 1 promoter (hSyn) were used to express hChR2(H134R)-EYFP, oChIEF-Citrine, C1V1(E122T)-TS-Citrine, and ReaChR-Citrine in cultured neurons. Cells were selected for recording based on their morphology under bright-field visualization. We did not use fluorescence signal-based selection, nor the selective α-CamKIIα promoter20, to avoid bias in the selection process. In all cases, ChRs were activated by 10 mW/mm2 of light at the respective maximum response wavelengths. Photocurrents were recorded under voltage-clamp. We observed that currents from ReaChR-Citrine expressing cells (219 ± 50 pA, n = 16) were many-fold greater than from hChR2(H134R) (37 ± 21 pA, n = 16) and C1V1(E122T)-TS (23 ± 11 pA, n = 14)-expressing neurons but non-significantly greater than oChIEF-expressing neurons (75 ± 31 pA, n = 17) (Fig. 3c). None of the ReaChR-expressing cells tested were unresponsive to light, whereas many hChR2(H134R) (n = 9) and C1V1(E122T)-TS (n = 5) expressing neurons did not have a detectable response to light.
To test the utility of ReaChR to induce suprathreshold depolarization in neurons, we recorded from cells under current-clamp without blockers of voltage-gated channels. Current was injected through the patch-pipette to maintain the membrane potentials of all cells at -65 mV; junction potentials were not corrected. In ReaChR-expressing neurons, we were able to achieve suprathreshold depolarization with blue (470 nm), red-orange (617 nm), and red light (627 nm), in response to 1 s light pulses (Fig. 3d). The level of depolarization and the spike-delayed time were dependent on the wavelength, light intensity, and expression level, in addition to the membrane properties of the neurons, i.e., cell type, membrane resistance, and capacitance. Cells expressing ReaChR retained normal morphological appearances and physiological membrane properties (Supplemental Fig. 5), suggesting minimal toxic effects with expression. With 617 nm light, we were able to achieve 25 to 45 mV depolarization (mean = 35 mV) in the expressing cells (Fig. 3e) and trigger action potentials with a 7 to 40 ms delay time (mean = 15 ms; Fig. 3f) when stimulated at high light intensity (n = 14). With 627 nm light, we were able to achieve 24 to 44 mV depolarization (mean = 34 mV; Fig. 3e) and spike delays of 7 to 35 ms (mean = 17 ms; Fig. 3f)(n = 14). Blue light at 470 nm was also effective to achieve suprathreshold depolarization of ReaChR-expressing cells, with depolarization of 30 to 40 mV (mean = 34 mV; Fig. 3e) and a spike delay of 4 to 19 ms mean = 9 ms; Fig. 3f)(n = 8). In comparison, the maximum depolarization achieved in neurons expressing hChR2(H134R) was 22 to 32 mV (mean = 26 mV; Fig. 3e) and the spike delay ranged from 7 to 25 ms (mean = 11 ms) (Fig 3f)(n = 8). In oChIEF-expressing neurons, the level of depolarization was 8 to 38 mV (mean = 26 mV; Fig. 3e) and the spike delay ranged from 1.6 to 7 ms (mean = 8 ms; Fig. 3f) when stimulated with 470 nm light (n = 5).
Pulsed illumination at 10 Hz was an effective trigger for spiking in ReaChR-expressing neurons at 470, 617, and 627 nm (Fig. 4a). Light pulses of weak intensity and short duration often lead to the loss of temporal fidelity from insufficient depolarization (Fig. 4b). Light pulses of too high an intensity lead to extra action potentials and depolarization block as a result of insufficient repolarization between light pulses were the main reasons for a loss in temporal fidelity (Fig. 4c). In addition to stimulation intensity and pulse duration, cell membrane properties and the expression level of ReaChR affect the ability to trigger action potentials in response to pulsed light. Thus, one specific stimulation protocol, e.g., 0.5 ms of 5 mW/mm2 of 617 nm light or 1 ms of 10 mW/mm2 of 627 nm light, that achieved complete fidelity in one ReaChR-expressing cell could fail to trigger action potentials in another cell with different membrane properties and expression level. The same phenomenon was also observed with stimulation at 5 Hz (Supplementary Fig. 6).
Figure 4. The responses of neurons expressing the different channelrhodopsin variants to 10 Hz pulsed light stimulation train.
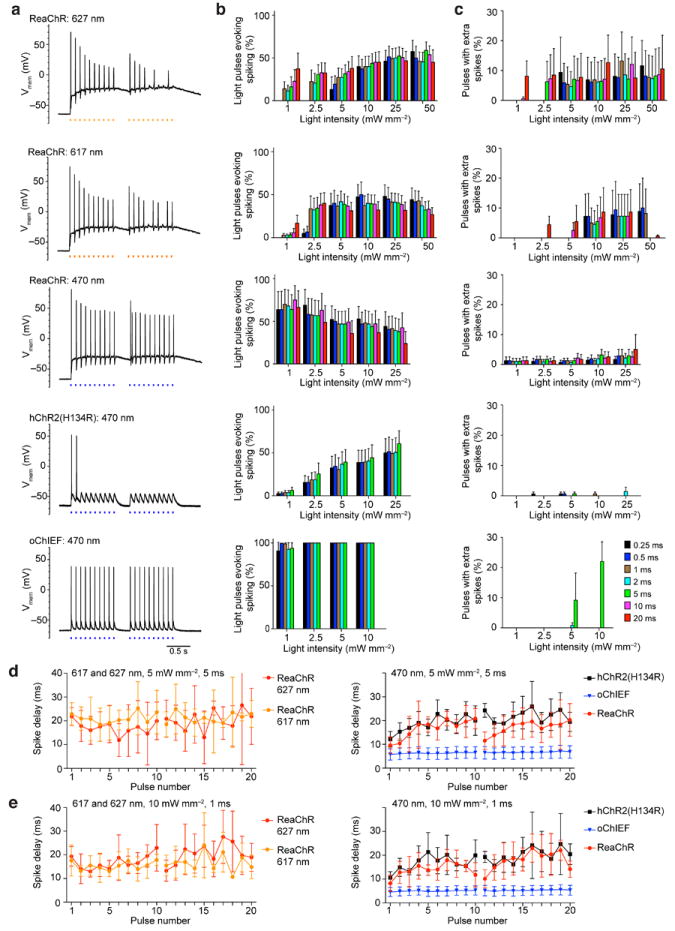
(a) Example recordings of neurons expressing the indicated channelrhodopsin variants to 10 Hz light stimulation at 10 mW/mm2 of 1 ms duration. Two 10 pulses trains 250 ms apart were used to simulate two bursting episodes. Current were injected to sustain the resting membrane potential at -65 mV. (b) The percentage of pulses resulting in action potentials of varying light intensity and pulse duration recorded from neurons expressing the different variants. (c) Percentage of pulses resulting in extra action potentials of varying light intensity and pulse duration of the different variants. 5 - 11 cells were tested in each conditions shown in (b) and (c). (d) and (e) Summaries of the mean latency and the standard deviation of light-triggered action potential of each pulse in ReaChR, hChR2(H134R) and oChIEF-expressing neurons to the indicated wavelength, intensity and duration. Pulses failed to trigger action potential were not included in the analysis, each trace is the average of 5-8 cells and each data points is the average of 2 – 8 values from the cells. (b) and (c) are mean ± S.E.M. (d) and (e) are mean ± S.D.
With regard to the fidelity of spike generation in neurons with ChR variants other than ReaChR, lower firing reliabilities were observed with hChR2(H134R)-expressing cells in response to 470 nm of light. This variant introduced a high level of channel desensitization that led to subthreshold depolarization with light pulses toward the end of a pulse train (Fig. 4a). As the overall level of depolarization observed with hChR2(H134R) was lower, extra spiking was rare. Cells expressing oChIEF had the highest temporal precision, as measured by the percentage of pulses achieving suprathreshold depolarization, shortest spike delay and variability in spike latency (Fig. 4b-e) and this higher fidelity is sustained at higher stimulation frequency (Supplementary Fig. 6). Yet even with oChIEF, extra action potentials after strong depolarization can also be observed in high expressers. The spike latency and the variability were similar between ReaChR and hChR2(H134R), with higher variability in response to stimulation pulses later in the pulse train.
Excitation of ReaChR expressing neurons in vivo
As red light can penetrate mammalian tissues in vivo with less attenuation compared to blue or green light12, we used ReaChR to stimulate deep brain structures in vivo. Two areas were targeted for in vivo stimulation: First, the vibrissa primary motor cortex (vM1), which is involved in the control and execution of vibrissa motion21. Second, the facial motor nucleus of the 7th cranial nerve (FN), whose motoneurons innervate the muscles responsible for vibrissa movements. ReaChR-Citrine was incorporated into a recombinant adeno-associated virus (rAAV) with a hSynapsin promoter, and injected into either vM1 cortex or FN of individual mice.
ReaChR expression in vM1 cortex was confirmed three weeks after injection. The mice were head-fixed and vibrissae movements monitored with high-speed video, either during light anesthesia with 1.0 - 1.5 % (v/v) isoflurane or while the mice were awake (Fig. 5). For electrophysiological recordings, the bone over vM1 was removed, the zone of infection mapped with epifluorescence imaging of the cortical surface, and a 617 nm LED positioned 10 mm above the surface of the brain (Fig. 5a,b). Short pulses of light, 1 to 20 ms in duration, were used to activate ReaChR expressing neurons (Fig. 5c) and evoke spikes, as recorded extracellularly (Fig. 5d-f). ReaChR expressing vM1 cortical neurons were reliably photo-activated at short latencies and with low temporal jitter (Fig. 5d). The evoked activity of a representative cortical single unit is shown in Figure 5d-f. This unit, which most likely expressed ReaChR, fired one or more spikes in response to 2 ms pulses with sub-millisecond jitter and a short, 2.4 ms spike latency. The unit was reliable activated with trains of light pulses that ranged from 5 to 30 Hz. Increasing the rate of optical activation to greater than 20 Hz resulted in a larger number of extra spikes per pulse (Fig. 5f). We observed a monotonic increase in the number of evoked spikes and of the spiking probability as a function of stimulus duration (n = 8 single units; Fig. 5g,h). Spiking probability was above 0.95 when stimulus duration was 5 ms or longer (Fig. 5h). When stimulus rate exceeded 10 Hz the population response increased (P = 0.002, one-sided KS-test; Fig. 5i) and the latency to the first spike decreased (P = 0.01, one-sided KS-test; Fig. 5j); these effects most likely result from the slow closing kinetics of ReaChR.
Figure 5. In vivo expression and utilization of ReaChR for cortical control.
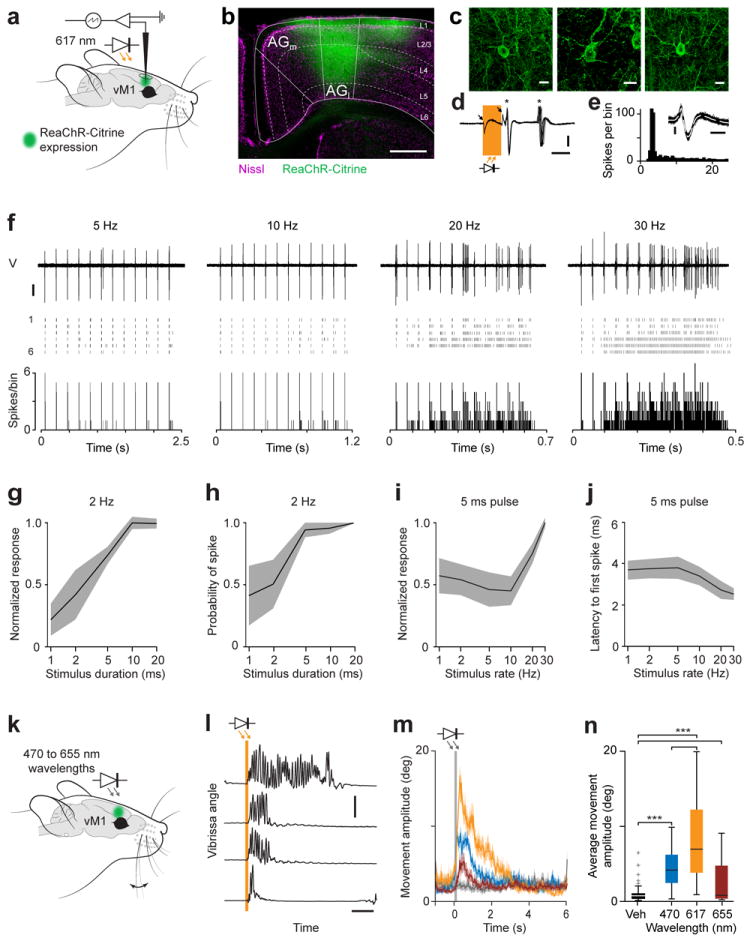
(a) Schematic of vM1 stimulation and recording. ReaChR was expressed in vM1 and visualized by Citrine fluorescence (green). vM1 was exposed for electrophysiology and ReaChR-expressing neurons activated with 617 nm light. (b) Coronal section through medial (AGm) and lateral (AGl) agranular vM1 of an infected mouse show ReaChR (green) expression. Neurons counter-labelled with a fluorescent marker (NeuroTrace; magenta). Scale bar: 500 μm. (c) ReaChR-expressing neurons in vM1. Scale bar: 10 μm. (d) Photo-activated spikes in a ReaChR-expressing neuron recorded in vivo during anaesthesia (recording depth = 410 μm). Single 2 ms pulses of 617 nm light (orange bar) evoked one or more spikes (indicated by *; n = 12 pulses at 2 Hz). Arrows indicate low amplitude artefacts associated with on- and off of light. Scale bars: 100 μV and 2 ms. (e) Spike interval histogram and waveforms (inset) of same unit, demonstrating the unit as a single neuron. Scale bars: 100 μV and 0.5 ms. (f) ReaChR-expressing neuron activated with 2 ms pulses of 617 nm light at 5, 10, 20 and 30 Hz (12 pulses). Top panel: Voltage trace during a train of 12 pulses (100 μV scale bar). Middle panel: Rasters of spikes during 6 trains. Bottom panel: Peristimulus time histogram (PSTH) of spikes across all trains. (g) Spikes per pulse (normalized to max) and (h) probability of one or more evoked spikes across a population of ReaChR-expressing neurons (n = 8) as a function of pulse duration (2 – 20 ms; pulse rate 2 Hz). (i) Number of spikes per pulse (normalized to max) and (j) the latency to the first spike neurons as a function of stimulus rate (1 – 30 Hz; pulse duration 5 ms). (k) Schematic of in vivo activation of vM1 with ReaChR (green) through intact skin and bone of awake mice. Vibrissae movements were measured with high-speed video. (l) Traces of vibrissae movements in response to single, 100 ms pulses (orange arrows and bar) of 617 nm light emitted by an LED placed 10 mm above the skin. Increasing values denote vibrissae protraction. Scale bars: 40 deg and 1 s. (m) Absolute movement amplitudes evoked by 100 ms pulses of 470, 617, and 655 nm light through intact skin in ReaChR-expressing mice (n = 3 mice, 10 stimulus repetitions per condition/mouse). Additionally, 3 mice injected with viral vehicle solution (mock control) were stimulated through-skin with 617 nm light (black). (n) Boxplots of movements during the first second after vM1 photo-activation through intact skull of mock transduced (black, n = 3 mice; 617 nm illumination) and ReaChR-expressing mice (n = 3 mice; 470 to 655 nm wavelengths). Vertical lines indicate data range, boxes the 25th to 75th percentile ranges and central mark the median. Light intensity in all panels was 100 mW light output.
Consistent with in vivo neural activation of ReaChR expressing neurons, we found that vibrissae movements could be elicited by LED illumination in awake mice (Fig. 5k-n). Removal of skin and bone was not required to evoke these movements, and placing the stimulating LEDs 10 mm above the fur line was sufficient in all cases (Fig. 5k). Movements were elicited with both blue (470 nm) and red (617 and 655 nm) illumination. Evoked movements were characteristic of those observed with electrical stimulation of vM1 cortex, displaying both a rapid component22, 23 and a sustained period, albeit with unpredictable duration, of rhythmic whisking that outlasted the stimulus24(Fig. 5l,m).
Our in vivo cortical experiment demonstrated stronger evoked movement responses to 617 nm compared to 470 nm light (P < 0.0001, one-sided KS-test; Fig. 5m,n), consistent with larger responses with orange-red light in cultured HEK293 cells (Fig. 1f and Supplementary Fig. 7) and the greater attenuation of blue versus red light in mammalian tissue12. We confirmed that these movements were caused by photo-activation of ReaChR by comparison with movements measured in a cohort of mock-transduced mice injected with the viral vehicle (black, Fig. 5m,n). Since we were able to evoke movements with 470 nm light in ReaChR-expressing mice, we tested whether movement could be evoked non-invasively with a standard blue-light activated ChR variant. We thus expressed hChR2(H134R)-EYFP in vM1 cortex with identical rAAV vector and viral titer. Vibrissa movements could be evoked in mice expressing hChR2(H134R) in vM1 cortex with 470 nm light illumination (n = 3), but these movements were infrequent, significantly smaller, and short-lasting compared to evoked movements with 617 nm light in ReaChR expressing mice (Supplementary Fig. 7c-f).
Durations and amplitudes of movements evoked by vM1 cortical stimulation in ReaChR expressing mice were unpredictable (Fig. 5l), mirroring results obtained with electrical stimulation in awake animals24 but nonetheless complicating comparisons of different stimulation parameters. As a more stringent test of the sensitivity of ReaChR to different wavelengths in vivo, we targeted ReaChR to mystacial motoneurons in FN of the brainstem that directly control vibrissa movements (Fig. 6a,b). It has previously been shown that in deeply anaesthetized animals, direct electrical stimulation of these neurons can reliably drive vibrissa movements, producing a relatively noise-free and controlled behavioural output25. In mice, the FN is located at a depth of up to 6 mm in the ventral part of the brainstem. We delivered light at 470, 530, 591, 617, 627 and 655 nm wavelengths in a non-invasive manner by placing an LED at the opening of the external auditory canal, thus illuminating the brainstem at a distance of 8 to 10 mm from the midline through intact tissue and bone (Fig. 6c). The external ear canal provides a convenient anchor point for non-invasive LED placement, bypasses fur and provides an opportunity to limit photo-activation to one side of the brainstem. LEDs were typically flashed for periods of 100 ms at 1 Hz with a 1 - 100 mW light output; we found no visual indications of any heat related damage to outer ear tissue. Activation of ReaChR expressing neurons elicited highly reproducible and temporally precise vibrissae movements, consistent with direct activation of motoneurons (Fig. 6d,e; Supplementary Video 1). Although a topographic organization of the facial muscle representation within FN has been documented, the exact distribution of all motoneurons innervating muscles involved in vibrissa control is not known26. Both protraction and retraction of the vibrissae are actively controlled by multiple extrinsic and intrinsic muscles22, 27. We found no consistent pattern between the intra-FN locations of infected motoneurons and evoked movement trajectories, and in several mice both net protraction and retraction could be elicited by adjusting light intensities appropriately (Fig. 6d,e). As example data, figure 6d shows evoked movements in a mouse where light stimulation led to the retraction of all vibrissae, independent of light intensity (10 to 100 mW light output, 100 ms pulses; Supplementary Video 2). Evoked retractions were three-times larger when ReaChR was activated at 617 nm as compared to 470 nm (40 versus 13 degrees, respectively, at maximum amplitude. Similarly, light stimulation of a different mouse (Fig. 6e) produced 6-fold larger movements at 617 nm compared to 470 nm (19 degrees versus 3 degrees, respectively, at maximum amplitude). This pattern of stronger activation at 617 nm compared to 470 nm was observed in all ReaChR expressing mice (Fig. 6f).
Figure 6. In vivo expression and utilization of ReaChR for brainstem activation.
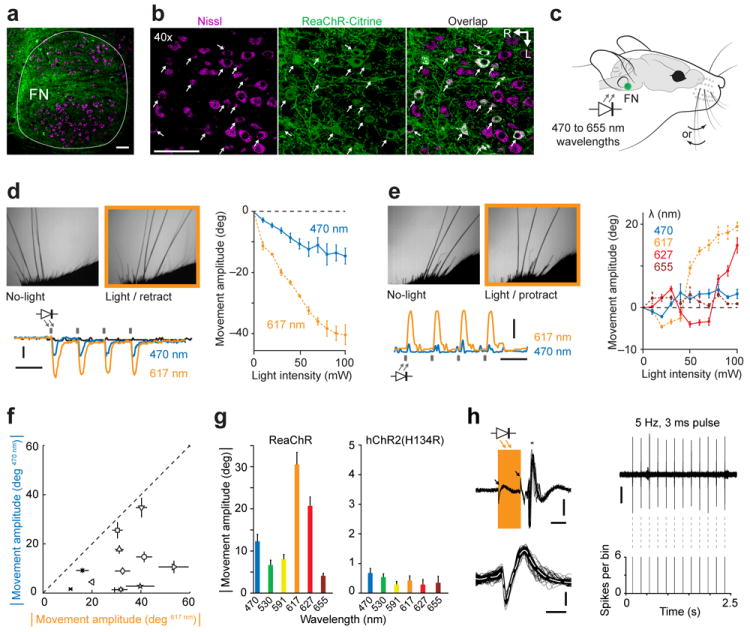
(a) Horizontal section through FN showing ReaChR-expressing neurons and associated processes (green). Cells were labelled with a fluorescent marker (NeuroTrace; magenta). (b) Confocal images of ReaChR-expressing motoneurons (arrows). Scale bar: 100 μm. R: rostral, L: lateral. (c) Schematic of awake mice expressing ReaChR in FN photo-activated by LEDs placed at the opening of the external auditory canal. Vibrissae movements were recorded with high-speed video. (d) Vibrissae movements evoked by stimulating FN in a ReaChR-expressing mouse. Left: Video frames (top) show vibrissae in the reference protracted position (No-light) and at peak retraction during photo-activation (Light), and traces of movements (bottom) in response to 470, and 617 nm light. Black trace indicates movement of contralateral C2 vibrissa. Right: Movement amplitude as a function of light output power. (e) Vibrissa movements evoked by stimulating FN in another ReaChR-expressing mouse. Left: Frames (top) show vibrissae in the reference retracted position (No-light) and at peak protraction during stimulation (Light), and traces of movements (bottom) in response to 470 and 617 nm light. Right: Movement amplitude as a function of light output power. Vertical gray bars in d-e indicate 100 ms light pulses (100 mW light output), and scale bars 10 degrees and 1 s. (f) Population data of absolute movement amplitudes evoked by 470 and 617 nm through-ear illumination (100 ms pulses at 1 Hz; n = 12 mice). (g) Comparison of movement amplitudes evoked with light between 470 and 655 nm in ReaChR (left, n = 12 mice) and hChR2(H134R) (right, n = 3 mice) expressing mice (100 mW light output). Movement amplitudes at same wavelengths were larger in ReaChR compared to hChR2(H134R) expressing mice (P < 0.001, KS-test). (h) In vivo recording of a single-unit in the FN region (based on stereotaxic coordinates) during photo-activation with 617 nm light (100 mW light output). Left, top: Voltage trace aligned to a 3 ms light pulse (orange bar, n = 12 pulses). Small artefacts are associated with on and off of light pulses (arrows). A single spike was evoked after each pulse (*) with low temporal jitter and short latency (4 ms) suggesting the unit expressed ReaChR and was directly activated. Scale bars: 100 μV and 2 ms. Left, bottom: Overlapped spikes from same unit demonstrating this as a single neuron. The unit was not spontaneously active and fired a single spike in response to the light pulse. Scale bars: 100 μV and 0.5 ms. Right, top: Voltage trace recorded during a single train of 12 light pulses (100 μV scale bar). Right, middle: Rasters of spikes during 6 trains. Right, bottom: PSTH of spikes across all trains.
Vibrissa movements in ReaChR expressing mice could be evoked across a broad range of wavelengths, consistent with our in vitro results. The largest movements were evoked at 617 nm, followed by 627 nm and 470 nm. Smaller vibrissa movements were also evoked with 530, 591 and 655 nm illumination (Fig. 6g). We confirmed that these movements resulted from photo-activation of ReaChR, and not LED related heating of neural tissue or visually evoked responses, by failing to observe any movement in a cohort of mice expressing the blue-light activated hChR2(H134R) channel in FN (n = 15; Fig. 6g and Supplementary Fig. 7g-i) and by recording single-units in the brainstem of ReaChR expressing mice that reliably responded with short latencies and with sub-millisecond temporal jitter to light stimulation (Fig. 6h).
The variability in whisking between individual mice in response to illumination of the FN is great (Fig. 6f). This was likely caused by variations in the exact location of ReaChR expressing motoneurons within the FN, which is ~1 mm in width, and/or by high expression levels in individual motoneurons that resulted in very low spiking thresholds. In principle, variability could also be caused by direct stimulation of the facial motor nerve. We confirmed that motoneuron axons were ReaChR-Citrine labelled and that vibrissa movements could also be evoked by direct illumination of the peripheral motor nerve through the skin of the lower cheek. These movements, however, were smaller and of shorter duration than through-ear evoked movements (Supplementary Fig. 7a). Lastly, movements were not evoked when stimulating through the ear contra-lateral to the side of ReaChR expression (617 nm, 100 mW light output), which shows that the lateralization of optogenetically evoked brainstem activity can be controlled with through-skull stimulation (Supplementary Fig. 7b).
Discussion
The optimal wavelength range for optical imaging into mammalian tissue is near infrared, where light scattering decreases with increasing wavelength and absorption by enodogenous chromophores is reduced, yet absorption by water is still negligible. This paves the way for deeper penetration of light into tissue with reduced attenuation12. Optogenetic tools to manipulate neuronal activity should, ideally, be excited by near infrared light. Current development of ChRs has produced many new variants, such as ChR2/H134R, ChETA, TC, SFO/D156A, ChD, oChEF, oChIEF, CatCh, and ChRGR, that are maximally activated by blue and green lights7,8,10,17,18,28,29. Many of these variants have improved properties regarding the kinetics, expression and level of desensitization compared with the native ChR2. However, there has been little progress in the development of red-shifted ChRs.
In this study, we engineered two red-shifted ChR variants, VCOMET and ReaChR, that had strong membrane expression in mammalian cells. The ReaChR variant responded strongly to red light, with a secondary steady-state spectral peak at 630 nm, in addition to a primary steady-state spectral peak at ~ 540 nm. The mechanism for this shift involves the L171I mutation in VCOMET. Past work attributed this position to the red-shift of the response spectrum of VChR1 (L126) relative to ChR2 (I131)14, and implies this L171I modification should lead to a blue-shift in the response spectrum. However, this is based on charge distribution of all-trans retinal in the binding pocket of bacteriorhodopsin in the dark state. In bacteriorhodopsin, some transient intermediate photocycle states have absorption spectra above 600 nm whereas the initial activation of the protein has λmax of ~ 570 nm30. It appears the main effect of the L171I mutation in ReaChR is to extend the life-time of the red-shifted ‘transient’ states and reduce the desensitization of the transient state, leading to greater photocurrent to red light. In support of this hypothesis, we found differential spectral peaks with the ‘transient’ and ‘steady-state’ responses of C-VChR1 and VCOMET with the ‘transient’ states having λmax of ~570 to 590 nm and the steady-state having λmax of ~530 nm (Fig. 1f & Supplementary Fig. 2b).
The initially described red-shifted channelrhodopsin, VChR1, does not express well in mammalian cells and traffics poorly to the membrane, limiting its utility for in vivo applications14, 15. The recently published variant C1V1 and its derivatives were reported to have improved expression, membrane trafficking and kinetic properties compared to VChR111. However, in our testing, C1V1 and its variants are still limited in their membrane trafficking and expression compared to VCOMET and ReaChR, in addition to having slower channel kinetics than VCOMET and ReaChR (Figs. 1, 2). The most red-shifted C1V1 derivative, i.e., C1V1(E122T), has a spectral peak at ~600 nm and slower channel kinetics and limited expression compared with ReaChR (Figs. 1-3). The E122T mutation variant also has a reduced photocurrent, as previously reported11, that may result from the position of this residue within the putative channel pore of ChR as predicted from the crystal structure of ChEF31. Thus, the reduction of C1V1(E122T) photocurrent and slow kinetics negate the spectral advantage that the E122T mutation provides. In contrast, ReaChR is strongly expressed, consistently traffics to the membrane, and can be used to reliably trigger temporally-precise spiking.
Although ReaChR has an improved spectral response to light at wavelengths greater than 600 nm, it still suffers slow kinetics in its channel closure rate compared to variants such as ChR2, ChIEF, ChD and ChR2/ChETA7, 8, 10, 17, 18, 28, 29. In the blue light-activated channelrhodopsin variants, single point mutations that improved the channel kinetics also reduced the channel’s light sensitivity7, 15. The other known ChR mutation that increased the channel kinetics, E123T of ChR2 / E162T of C1V1 / E163T of VCOMET, does so at the expense of reduced light sensitivity and reduced photocurrent17. In the context of ReaChR, this E to T mutation increased the channel kinetics (off-rate < 30 ms) and surpassed C1V1(E162T) in terms of kinetics11, but blue-shifted the spectral peak to 550 nm (Supplementary Fig. 8) and increased the desensitization of the channel. We did not find incorporation of this mutation useful in the current development of ReaChR. Important future goals in developing red-shifted ChR should include improving light sensitivities and faster kinetics simultaneously, and reducing the activation of these variants by light below 500 nm to allow for the independent manipulation of two populations of neurons with two different wavelengths of light.
We were able to exploit the spectral advantages of ReaChR to achieve efficient activation of expressing neurons through fur, skin and intact bone of adult mice with red-orange (617 nm) and red (627 nm) light, and even far-red (655 nm) light (Figs. 5 and 6). This enables novel in vivo applications of transcranial and deep brain stimulation with optogenetics. Transcranial stimulation is essential for many chronic studies, as cranial windows can lead to activated microglia and astrocytes as part of an inflammatory response32 that in turn alters neuronal physiology33 and plasticity34 as well as pial vasculature35, 36. Alternatives to our approach for non-invasive transcranial stimulation include clinically approved treatments for several neurological disorders and stroke, such as electroconvulsive therapy, repetitive transcranial magnetic stimulation and transcranial direct-current stimulation. These techniques do not, however, have the spatial and cellular specificity that can be achieved through optogenetics. Thus, optogenetic approaches utilizing red-shifted ChRs, such as ReaChR, have the potential to improve the efficiency of transcranial stimulation to treat neurological disorders.
In the present study, the channelrhodopsin ReaChR was introduced through injections of an engineered virus directly into the region of interest. In future work, it may be possible to label specific populations of neurons via retrograde transport of ReaChR from a known target37, 38. Thus, for example, pools of motoneurons in the brainstem or the spinal could be labelled via injection into specific muscles, rather than into motor nuclei themselves. Such an approach would significantly reduce the invasiveness of an optogenetic strategy for future therapeutic use. Further, one could label opposing muscle groups with short- versus long-wavelength ChRs to permit differential optical control of opposing muscle groups. The weak absorption of short-wavelengths by ReaChR will not compromise this strategy if the anatomically deeper motor pools are labelled with ReaChR. Lastly, ReaChR may facilitate neurological treatment and neuroprosthetic control through transcranial and possibly transvertebral stimulation.
Methods
Molecular cloning and transgene expression
Channelrhodopsin chimeras and point mutants were generated by over-lapping polymerase-chain reaction with Phusion polymerase-chain reaction mix (New England Biolabs, Ipswich, MA, USA). Genes coding mammalian codon optimized VChR1, VChR2 and ChIEF were synthesized separately according the published peptide sequence (GenScript, Piscataway, New Jersey). The gene encoding hChR2(H134R)-EYFP was a generous gift from Dr. Karl Deisseroth (Stanford University). The gene and peptide sequences of ReaChR and C1V1(E122T) are shown in Supplementary Figures 9 and 10. Standard digestion and ligation techniques were used to insert the mutant into the expression vector. For experiments with HEK293 cells, the channelrhodopsin variants were expressed with pcDNA3.1 vector under the CMV promoter and fused to the YFP variant Citrine19 with an in-frame XhoI restriction site (Leu-Glu). Cells were transfected with Fugene HD (Roche, Basel, Switzerland). In the experiments measuring membrane expression of hChR2, ChIEF, VChR1, C-VChR1 and VCOMET in primary cultured neurons, ChR-Citrine fusions (with an in-frame ClaI restriction site) were expressed under the CAG promoter with a WPRE sequence inserted after the stop codon of the ChR-Citrine coding sequence (plasmid courtesy of Dr. Karel Svoboda, Janelia Farm Research Campus). In the experiments measuring membrane expression of C1V1(E122T), C1V1(E122T)-TS and ReaChR, ChR-Citrine fusions (with an in-frame XhoI restriction site) were expressed under the hSyn promoter with a WPRE sequence inserted after the stop codon of the ChR-Citrine coding sequence in an AAV2 vector (plasmid courtesy of Dr. Lin Tian, Howard Hughes Medical Institute Janelia Farm Research Campus). The resulting constructs (at the same DNA concentration) were electroporated into the neurons prior to plating (Lonza, Walkersville, MD). For the stimulation of cultured primary hippocampal neurons with red light, the ReaChR-Citrine was introduced with recombinant lentivirus.
Lentivirus and recombinant adeno-associated virus (rAAV) production
The gene encoding hChR2(H134R)-EYFP, C1V1(E122T)-TS-Citrine, oChIEF-Citrine and ReaChR-Citrine were subcloned into a generation 2 lentiviral construct with the hSyn promoter. The lentivirus was made according to the protocols at http://vectorcore.salk.edu/protocols.php with minor modifications. In brief, 293A cells (Life Technologies, Carlsbad, CA) were grown to 85 % confluency and transfer vectors containing ChR-FP, psPAX2 and pMD2.G (gifts from Professor Didier Trono) were transfected with calcium phosphate (Clontech, Mountainview, CA). Virus particles were harvested from serum-free medium and concentrated with 20 % sucrose cushion with ultracentrifugation. The titre of lentivirus was estimated with Lentivirus Rapid Quantitation Kit (Cell Biolabs Inc. San Diego CA, USA) in parallel just before each infection. The lentiviral vector was a gift from Dr. Ed Boyden (MIT). Recombinant AAV with serotype 8 containing hChR2(E134R)-EYFP and ReaChR-Citrine was produced and purified according to the protocols at http://vectorcore.salk.edu/protocols.php by transfecting AAV2 ITR vector (gift from Dr. Lin Tian, Howard Hughes Medical Institute Janelia Farm Research Campus) containing ReaChR-Citrine, and the helper plasmids XX6-80 and XR8 (National Vector Biorepository) into 293A cells. rAAV2/8 were released from the cells by freeze-thawing and purified with iodixanol gradient purification. The virus was further concentrated using an Amicon Ultra centrifugal filter (Millipore, Billerica, MA) with 50kDa cut-off. The rAAV titre was measured by the Salk Vectorcore service with qPCR and was estimated to be 3 × 1013 GC/mL.
Measurement of relative efficiencies of channelrhodopsin membrane expression
Relative levels of plasma membrane expression of ChR-Citrine fusions in HEK293 and cultured cortical neurons were measured by imaging on a Zeiss Live 5 Confocal microscope with Zen software (Thornwood, NY). Transfections and measurements of expression were performed with the same concentrations of DNA electroporated into the neurons. The transfected cells were imaged with the same setting for comparison. The measurements of expression were performed on all cells with detectable visible fluorescence and normal cell morphology non-discriminatively in the culture dish to ensure fair comparisons, typically starting from one end of the dish to the other. Membrane expressions were measured by taking the mean fluorescence intensities of the membrane and the cytosol of a single in-focus imaging plane with ImageJ software. In the experiments where channelrhodopsin photocurrents were normalized to membrane fluorescence, images were acquired with an EMCCD camera (Photometric, Tucson, AZ) at 512 × 512 resolution with Slidebook (Intelligent Imaging Innovations, Inc., Denver, CO) in epi-fluorescence mode prior to electrophysiological recording.
Cell culture, electrophysiological recordings and stimulation of cultured cells
Characterization of spectral response, reversal potential, kinetics and membrane trafficking of C-VChR1, C1V1, C1V1(E122T), VCOMET, ReaChR, CatCh and oChEF were done with whole-cell patch clamping on 293A cells 2 days after transfection. HEK293 cells, which have smaller size, high membrane resistance and minimal expression of voltage-gated ion channels, were used for these characterizations to reduce the known voltage-clamping errors in neurons39. All recordings were performed with an extracellular solution containing 118 mM NaCl, 3 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 10 mM HEPES, 20 mM glucose (pH 7.35, 310 mOsm) and an intracellular solution containing 110 mM Cs-methanesulfonate, 30 mM tetraethylammonium chloride, 10 mM EGTA, 10 mM HEPES, 1 mM CaCl2, 1 mM MgCl2, 2 mM Mg-ATP, 0.15 mM Na3-GTP (pH 7.25, 285 mOsm), except for the reversal potential measurement where the intracellular solution contained 125 mM K-gluconate, 10 mM K4-BAPTA, 5 mM NaCl, 1 mM CaCl2, 10 mM HEPES, 2 mM MgATP and 0.3 mM Tris-GTP (pH 7.25, 285 mOsm). Series resistance compensation (at 70 - 75 %) was used in kinetics and reversal potential measurements in HEK293 cells. HEK293 cells with low membrane resistance (< 200 MΩ) were often discarded from analysis and testing due to inaccuracy of voltage-clamping in these conditions. These cells are typically found in clusters. All chemicals were acquired from Sigma-Aldrich (St. Louis, MO). Electrophysiological characterization of the channelrhodopsin in neurons were performed on 14 to 25 day in vitro neurons extracted from P2 neonatal Sprague-Dawley rat pups after at least 9 days of infection with lentivirus. For the measurement of photocurrent amplitude in neurons, the recording was performed with the Cs-methanesulfonate based intracellular solution to block potassium channel, and 1 μM TTX and 100 μM Cd2+ were included in the extracellular solution to block voltage-gated sodium and calcium channels, respectively. 10 μM NBQX, 10 μM bicuculline and 20 μM APV were also included in the extracellular solution to block fast synaptic transmission (Tocris, Ellisville, MO). To ensure the accuracy of voltage-clamping in neurons, light illumination was limited to a circular area of 150 μm diameter around the soma to reduce space-clamp error, and series resistance was compensated at 65 %. 470 nm light was used to activate hChR2(H134R) and oChIEF for photocurrent measurement, and 590 nm light for C1V1(E122T)-TS and ReaChR. Although the experimenters were not blinded to the groups, cells tested were selected based on the healthy appearance under bright field illumination without visualization of the fluorescence to ensure unbiased selection. All cells tested were included in the analysis regardless of detectable responses to light. For testing the effectiveness of light on evoking action potentials, the recordings were performed with K-gluconate based intracellular solution in the presence of extracellular 10 μM NBQX, 10 μM bicuculline and 20 μM APV. Recordings were performed in current-clamp mode with current injected to sustain -65 mV membrane potential at rest. In pulsed stimulation experiments, cells failed to reach suprathreshold depolarization with 20 ms of 50 mW/mm2 light pulses were excluded from the analysis. For experiments of neurons with 10 Hz stimulation under current-clamp, ReaChR-Citrine and oChIEF-Citrine expressing cells were selected based on bright field morphology without visualization with fluorescence. High membrane EYFP fluorescence was used to select hChR2(H134R) cells for recordings, as most cells with medium to low level of fluorescence failed to reach threshold when stimulated with light. Recordings were performed with the Axopatch 200B patch clamp amplifier (Molecular Devices, Union City, CA) and acquired through Digidata 1320 (Molecular Devices) to a PC (Dell, Austin, TX) running pClamp 9.2 or pClamp 10 software (Molecular Devices). Analysis was conducted with AxographX (Sydney, New South Wales, Australia). Junction potentials were not corrected.
Most light stimulation was provided from a monochromator xenon light source (Polychrome IV, TILL Photonic, Victor, NY) with 15 nm half-width. Shuttering and intensity of light were controlled with a custom-fitted 25 mm mechanical shutter (Vincent Associates, Rochester, NY) and a neutral density wheel (Thorlab, Newton, NJ). For the spectral response measurements and experiments where light intensity was not described, cells were stimulated with ~ 5.1 × 1016 photon/mm2/s across the light spectra. The photon flux of the monochromator at each wavelength was measured with a power meter and integrating sphere (Newport Corporation, Irvine, CA) placed on the objective and the photon flux was corrected with the neutral density wheel to give the same flux. The response spectra of ChRs were measured with 1 s light pulse from 650 to 410 nm of the same photon flux at 20 nm interval, with maximum response as the maximal photocurrent obtained within the 1 s pulse at the described wavelength and the steady-state response measured between 0.95 – 1.00 s after the onset of the stimulation light pulse. For most experiments, 410 nm light was delivered 20 s after the initial stimulation to re-condition the protein prior to the delivery of the next testing pulse 20 s later. Stimulation of neurons under current-clamp was conducted with 470 nm (SR-05-B0040), 617 nm (SR-05-H2070) or 627 nm (SR-05-D2050) LED illumination (Luxeonstar, Brantford, Ontario, Canada). The LEDs were connected to a computer-controlled current source (Mightex; Pleasanton, CA) coupled to a quartz fiber into the microscope.
Expression and stimulation of channelrhodopsin-expressing neurons in vivo
Recombinant AAV2/8-hSyn-ReaChR-Citrine or AAV2/8-hSyn-hChR2(H134R)-EYFP virus was injected stereotaxically into the vibrissa motor (vM1) cortex or the facial motor nucleus (FN) in the brainstem of C57BL/6 female mice using a nano-liter injector (Nanoject II, Drummond, Broomall, PA) under isoflurane anaesthesia (2 % v/v). Injection coordinates for vM1 cortex was 1 mm posterior, 1 mm lateral and 0.8 mm ventral, and for FN 4.8 – 5.6 mm posterior, 1.2 – 1.4 mm medial and 5.3 – 6.0 mm ventral to bregma. The skin was retracted to expose the injection coordinates, and a single 0.3 mm diameter hole was drilled for each injection site. Suspension of virus was injected in 10 × 50 nl aliquots every 5 minutes (total volume 500 μl) in vM1, and in 5 × 12 μl aliquots (total volume 60 μl) every 5 minutes in the FN. The injection hole was filled with sterile saline and topped with a thin layer of antibiotic ointment after the injection pipette was withdrawn. The skin was sutured back in place, and the animal left to recover for the viral incubation period. After a waiting period of 4 weeks, the mice were prepared for in vivo experiments. The injections of virus were done on 3 month-old mice and the testings were performed 3-6 weeks later. All animals were housed on a normal 12 hour light/dark cycle in groups between 3-5 per cage and all animals were tested during their light cycle.
One day prior to experiments, a single 10 mm incision was made perpendicular to the midline and caudal to lambda, the skull exposed and a titanium alloy head-bar attached to the skull using cyanoacrylate gel and a layer of dental cement (C&B-Metabond, Parkell Inc., Edgewood, NY) during isoflurane anesthesia. The skin rostral to lambda was not damaged, and the skull under it was neither exposed nor covered by any amount of cement, except for electrophysiological recordings in vM1 where a new craniotomy was opened where the virus had previously been injected. The following days, mice were head-fixed either when awake or during isoflurane (0.5 – 2 %), as indicated. ReaChR was activated in vivo by LEDs with wavelengths of 470 (LXML-PB01-0030), 530 (LXML-PM01-0100), 591 (LXM2-PL01-0000), 617 (LXM2-PH01-0070), 627 (LXM2-PD01-0050) and 655 nm (LXM3-PD01-0260; Luxeonstar, Brantford, Ontario, Canada) connected to a computer-controlled current source (Mightex; Pleasanton, CA).
Single units in vM1 and FN were recorded extracellularly with 9 – 12 MOhm tungsten electrodes (UEWLCESMLN1G; FHC Inc., Bowdoin, ME), buffered and pre-amplified (Alpha Omega Co., Alpharetta, GA), and acquired onto a PC (PCIe-6361, National Instruments, Austin, TX) using custom-written software for Matlab (MathWorks, Natick, MA). Spikes were sorted using the Chronux library in Matlab (http://chronux.org).
Vibrissa movements were monitored with high speed video36 and tracked offline using the WhiskerTracker software40 (code available on https://github.com/pmknutsen/whiskertracker). Following in vivo experiments, the mice were perfused with 4% paraformaldehyde, brains extracted and ReaChR expression confirmed with epifluoresence imaging and localized histologically using a fluorescent Nissl counterstain (NeuroTrace Cat# N21479, Life Technologies, Carlsbad, CA). The bone overlying the injections sites was inspected following perfusions, and was invariably found to have regrown entirely. In most mice, the bone that had regrown after viral injections appeared thickened. Only animals in which the channelrhodopsin expression in the correct regions with post-mortem histochemical analysis were included in the analysis of spiking and vibrissae movement. Although the experimenter was not blinded to group status, the measurement was automated so blinding status would not affect the outcome of the experiment. All animal procedures, including the extraction of primary neurons, were approved by the UCSD Institutional Animal Care and Use Committee.
Statistical analysis
Comparisons of the properties of channelrhodopsin variants in culture were done with the non-parametric Krustal-Wallis test with post-hoc Dunn’s multiple comparison tests between all possible pairs. Only the P-value for Krustal-Wallis tests were shown. The statistical analysis was done with Graphpad Prism 5.0 (San Diego, CA). The comparisons in vivo were conducted with non-parametric one-sided Kolmogorov-Smirov tests (KS test) in Matlab. All values are presented as mean ± S.E.M. All data in graphs are presented as mean ± S.E.M with the exception of the 10 Hz stimulation experiments and spike latency analysis where standard deviation was shown to illustrate the variability. No statistical methods were used to pre-determine sample sizes but our sample sizes were similar to those reported in previous publications7, 20. Most analysis in cultured cells was summarized from multiple transfection or transduction experiments. The results of the statistical analysis are available in the online Excel file.
Supplementary Material
Acknowledgments
J.Y.L. was funded by Foundation of Research, Science and Technology New Zealand, and P.M.K was supported by a Long-Term Fellowship from the Human Frontier Science Program (HFSP). The project was supported by grants to R.Y.T. from the National Institutes of Health (NS027177) and Howard Hughes Medical Institute and grants to D.K. from the National Institutes of Health (DA029706, OD006831, and NS058668). We thank Dr. Philbert Tsai for suggesting the name ReaChR. AAV2-ReaChR-Citrine and pLenti-ReaChR-Citrine constructs can be requested from http://www.tsienlab.ucsd.edu.
Abbreviations
- CatCh
Calcium Translocating Channelrhodopsin
- ChR
Channelrhodopsin
- EYFP
enhanced yellow fluorescent protein
- FN
Facial motor nucleus
- LED
light emitting diode
- rAAV
recombinant adeno-associated virus
- ReaChR
Red-activatable Channelrhodopsin
- VCOMET
VChR Optimized for Membrane Expression and Trafficking
- vM1
vibrissae motor cortex
Footnotes
Author contributions
J.Y.L. designed and developed the ReaChR constructs. J.Y.L. conducted and analyzed the experiments in HEK293 and neuron cultures. P.M.K and A.M. conducted and analyzed the in vivo experiments. D.K. and R.Y.T. contributed to the design and analysis of the experiments. All authors contributed to the writing and discussions of the manuscripts.
References
- 1.Arrenberg AB, Stainier DY, Baier H, Huisken J. Optogenetic control of cardiac function. Science. 2010;330:971–974. doi: 10.1126/science.1195929. [DOI] [PubMed] [Google Scholar]
- 2.Boyden ES, Zhang F, Bamberg E, Nagel G, Deisseroth K. Millisecond-timescale, genetically targeted optical control of neural activity. Nat Neurosci. 2005;8:1263–1268. doi: 10.1038/nn1525. [DOI] [PubMed] [Google Scholar]
- 3.Bruegmann T, et al. Optogenetic control of heart muscle in vitro and in vivo. Nat Methods. 2010;7:897–900. doi: 10.1038/nmeth.1512. [DOI] [PubMed] [Google Scholar]
- 4.Nagel G, et al. Channelrhodopsin-1: a light-gated proton channel in green algae. Science. 2002;296:2395–2398. doi: 10.1126/science.1072068. [DOI] [PubMed] [Google Scholar]
- 5.Nagel G, et al. Channelrhodopsin-2, a directly light-gated cation-selective membrane channel. Proc Natl Acad Sci U S A. 2003;100:13940–13945. doi: 10.1073/pnas.1936192100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Adamantidis AR, Zhang F, Aravanis AM, Deisseroth K, de Lecea L. Neural substrates of awakening probed with optogenetic control of hypocretin neurons. Nature. 2007;450:420–424. doi: 10.1038/nature06310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lin JY, Lin MZ, Steinbach P, Tsien RY. Characterization of engineered channelrhodopsin variants with improved properties and kinetics. Biophys J. 2009;96:1803–1814. doi: 10.1016/j.bpj.2008.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wen L, et al. Opto-current-clamp actuation of cortical neurons using a strategically designed channelrhodopsin. PLoS One. 2010;5:e12893. doi: 10.1371/journal.pone.0012893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Govorunova EG, Spudich EN, Lane CE, Sineshchekov OA, Spudich JL. New Channelrhodopsin with a Red-Shifted Spectrum and Rapid Kinetics from Mesostigma viride. MBio. 2011;2 doi: 10.1128/mBio.00115-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kleinlogel S, et al. Ultra light-sensitive and fast neuronal activation with the Ca(2)+-permeable channelrhodopsin CatCh. Nat Neurosci. 2011;14:513–518. doi: 10.1038/nn.2776. [DOI] [PubMed] [Google Scholar]
- 11.Yizhar O, et al. Neocortical excitation/inhibition balance in information processing and social dysfunction. Nature. 2011;477:171–178. doi: 10.1038/nature10360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tromberg BJ, et al. Non-invasive in vivo characterization of breast tumors using photon migration spectroscopy. Neoplasia. 2000;2:26–40. doi: 10.1038/sj.neo.7900082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aravanis AM, et al. An optical neural interface: in vivo control of rodent motor cortex with integrated fiberoptic and optogenetic technology. J Neural Eng. 2007;4:S143–156. doi: 10.1088/1741-2560/4/3/S02. [DOI] [PubMed] [Google Scholar]
- 14.Zhang F, et al. Red-shifted optogenetic excitation: a tool for fast neural control derived from Volvox carteri. Nat Neurosci. 2008;11:631–633. doi: 10.1038/nn.2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lin JY. A user’s guide to channelrhodopsin variants: features, limitations and future developments. Exp Physiol. 2011;96:19–25. doi: 10.1113/expphysiol.2009.051961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang H, et al. Molecular determinants differentiating photocurrent properties of two channelrhodopsins from Chlamydomonas. J Biol Chem. 2008 doi: 10.1074/jbc.M807632200. [DOI] [PubMed] [Google Scholar]
- 17.Gunaydin LA, et al. Ultrafast optogenetic control. Nat Neurosci. 2010;13:387–392. doi: 10.1038/nn.2495. [DOI] [PubMed] [Google Scholar]
- 18.Nagel G, et al. Light activation of channelrhodopsin-2 in excitable cells of Caenorhabditis elegans triggers rapid behavioral responses. Curr Biol. 2005;15:2279–2284. doi: 10.1016/j.cub.2005.11.032. [DOI] [PubMed] [Google Scholar]
- 19.Griesbeck O, Baird GS, Campbell RE, Zacharias DA, Tsien RY. Reducing the environmental sensitivity of yellow fluorescent protein. Mechanism and applications. J Biol Chem. 2001;276:29188–29194. doi: 10.1074/jbc.M102815200. [DOI] [PubMed] [Google Scholar]
- 20.Mattis J, et al. Principles for applying optogenetic tools derived from direct comparative analysis of microbial opsins. Nat Methods. 2012;9:159–172. doi: 10.1038/nmeth.1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hill DN, Curtis JC, Moore JD, Kleinfeld D. Primary motor cortex reports efferent control of vibrissa motion on multiple timescales. Neuron. 2011;72:344–356. doi: 10.1016/j.neuron.2011.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Berg RW, Kleinfeld D. Vibrissa movement elicited by rhythmic electrical microstimulation to motor cortex in the aroused rat mimics exploratory whisking. J Neurophysiol. 2003;90:2950–2963. doi: 10.1152/jn.00511.2003. [DOI] [PubMed] [Google Scholar]
- 23.Brecht M, Schneider M, Sakmann B, Margrie TW. Whisker movements evoked by stimulation of single pyramidal cells in rat motor cortex. Nature. 2004;427:704–710. doi: 10.1038/nature02266. [DOI] [PubMed] [Google Scholar]
- 24.Haiss F, Schwarz C. Spatial segregation of different modes of movement control in the whisker representation of rat primary motor cortex. J Neurosci. 2005;25:1579–1587. doi: 10.1523/JNEUROSCI.3760-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Herfst LJ, Brecht M. Whisker movements evoked by stimulation of single motor neurons in the facial nucleus of the rat. J Neurophysiol. 2008;99:2821–2832. doi: 10.1152/jn.01014.2007. [DOI] [PubMed] [Google Scholar]
- 26.Martin MR, Lodge D. Morphology of the facial nucleus of the rat. Brain Res. 1977;123:1–12. doi: 10.1016/0006-8993(77)90639-4. [DOI] [PubMed] [Google Scholar]
- 27.Dorfl J. The musculature of the mystacial vibrissae of the white mouse. J Anat. 1982;135:147–154. [PMC free article] [PubMed] [Google Scholar]
- 28.Berndt A, Yizhar O, Gunaydin LA, Hegemann P, Deisseroth K. Bi-stable neural state switches. Nat Neurosci. 2009;12:229–234. doi: 10.1038/nn.2247. [DOI] [PubMed] [Google Scholar]
- 29.Berndt A, et al. High-efficiency channelrhodopsins for fast neuronal stimulation at low light levels. Proc Natl Acad Sci U S A. 2011;108:7595–7600. doi: 10.1073/pnas.1017210108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shichida T, Matuoka S, Hidaka Y, Yoshizawa T. Absorption spectra of intermediates of bacteriorhodopsin measured by laser photolysis at room temperatures. Biochimica et Biophysica Acta. 1983;723:240–246. [Google Scholar]
- 31.Kato HE, et al. Crystal structure of the channelrhodopsin light-gated cation channel. Nature. 2012 doi: 10.1038/nature10870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu HT, Pan F, Yang G, Gan WB. Choice of cranial window type for in vivo imaging affects dendritic spine turnover in the cortex. Nat Neurosci. 2007;10:549–551. doi: 10.1038/nn1883. [DOI] [PubMed] [Google Scholar]
- 33.Hauss-Wegrzyniak B, Lynch MA, Vraniak PD, Wenk GL. Chronic brain inflammation results in cell loss in the entorhinal cortex and impaired LTP in perforant path-granule cell synapses. Exp Neurol. 2002;176:336–341. doi: 10.1006/exnr.2002.7966. [DOI] [PubMed] [Google Scholar]
- 34.Grutzendler J, Kasthuri N, Gan WB. Long-term dendritic spine stability in the adult cortex. Nature. 2002;420:812–816. doi: 10.1038/nature01276. [DOI] [PubMed] [Google Scholar]
- 35.Sohler TP, Lothrop GN, Forbes HS. The poial circulation of normal, non-anesthetized animals. Part 1. Description of a method of observation. Journal of Pharmacology and Experimental Therapy. 1941;71:325–330. [Google Scholar]
- 36.Drew PJ, et al. Chronic optical access through a polished and reinforced thinned skull. Nat Methods. 2010;7:981–984. doi: 10.1038/nmeth.1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gradinaru V, et al. Targeting and readout strategies for fast optical neural control in vitro and in vivo. J Neurosci. 2007;27:14231–14238. doi: 10.1523/JNEUROSCI.3578-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Osakada F, et al. New rabies virus variants for monitoring and manipulating activity and gene expression in defined neural circuits. Neuron. 2011;71:617–631. doi: 10.1016/j.neuron.2011.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Armstrong CM, Gilly WF. Access resistance and space clamp problems associated with whole-cell patch clamping. Methods Enzymol. 1992;207:100–122. doi: 10.1016/0076-6879(92)07007-b. [DOI] [PubMed] [Google Scholar]
- 40.Knutsen PM, Derdikman D, Ahissar E. Tracking whisker and head movements in unrestrained behaving rodents. J Neurophysiol. 2005;93:2294–2301. doi: 10.1152/jn.00718.2004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.