

Abstract
Background
Aromatic essential oils extracted from fresh fruits of Litsea cubeba (Lour.) Pers., have diverse medical and economic values. The dominant components in these essential oils are monoterpenes and sesquiterpenes. Understanding the molecular mechanisms of terpenoid biosynthesis is essential for improving the yield and quality of terpenes. However, the 40 available L. cubeba nucleotide sequences in the public databases are insufficient for studying the molecular mechanisms. Thus, high-throughput transcriptome sequencing of L. cubeba is necessary to generate large quantities of transcript sequences for the purpose of gene discovery, especially terpenoid biosynthesis related genes.
Results
Using Illumina paired-end sequencing, approximately 23.5 million high-quality reads were generated. De novo assembly yielded 68,648 unigenes with an average length of 834 bp. A total of 38,439 (56%) unigenes were annotated for their functions, and 35,732 and 25,806 unigenes could be aligned to the GO and COG database, respectively. By searching against the Kyoto Encyclopedia of Genes and Genomes Pathway database (KEGG), 16,130 unigenes were assigned to 297 KEGG pathways, and 61 unigenes, which contained the mevalonate and 2-C-methyl-D-erythritol 4-phosphate pathways, could be related to terpenoid backbone biosynthesis. Of the 12,963 unigenes, 285 were annotated to the terpenoid pathways using the PlantCyc database. Additionally, 14 terpene synthase genes were identified from the transcriptome. The expression patterns of the 16 genes related to terpenoid biosynthesis were analyzed by RT-qPCR to explore their putative functions.
Conclusion
RNA sequencing was effective in identifying a large quantity of sequence information. To our knowledge, this study is the first exploration of the L. cubeba transcriptome, and the substantial amount of transcripts obtained will accelerate the understanding of the molecular mechanisms of essential oils biosynthesis. The results may help improve future genetic and genomics studies on the molecular mechanisms behind the chemical composition of essential oils in L. cubeba fruits.
Introduction
Litsea cubeba (Lour.) Pers (mountain pepper). an evergreen or deciduous dioecious tree or shrub, belongs to a species of the genus Litsea in the Lauraceae family. It is a tropical and subtropical plant distributed in southeastern Asia, southern China, Japan, and Taiwan. The fruit and bark of L. cubeba are commonly used as traditional herbal medicines for the treatment of stomachaches, cold, hiccups, inflammation, gastric cavity crymodynia, headaches, coronary heart disease and atopic eczema [1,2]. Aromatic essential oils extracted from the fresh fruits are flavor enhancers in foods, cosmetics, and cigarettes [3]. At the same time, the essential oils of L. cubeba exhibit a range of bioactivities such as antioxidant [4], antitermite [5], larvicidal [6], cytotoxic [7], neuropharmacological [8] and antimicrobial [9] activities. L. cubeba is a genetically diploid (2n=2x=24) plant [10]. Despite the medicinal and economic importance of L. cubeba, little genomic information is available for this non-model genus, and only 40 nucleotide sequences have been deposited in the NCBI GenBank database (as of the 22nd of May 2013). To date, three genes involved in monoterpene synthases have been functionally characterized [11], showing the potential for genomic research on this plant. However, a lack of sequence data has limited extensive and intensive research. Therefore, it is necessary to explore genomic data sources of L. cubeba for gene discovery and further functional studies.
In the essential oils of L. cubeba, the dominant components are monoterpenes represented mainly by neral and geranial, which are cis-trans isomers of citral [12]. In addition, it also contains sesquiterpene and other non-terpenes [7,12]. The universal precursors for terpenoids are two branched unsaturated diphosphate isoprene units, isopentenyl pyrophosphate (IPP) and dimethylallyl pyrophosphate (DMAPP), which come from the mevalonate (MVA) and 2-C-methyl-Derythritol 4-phosphate (MEP) pathways, respectively [13,14]. IPP and DMAPP can be converted into each other by IPP isomerase (IPPI) [15]. IPP and DMAPP are catalyzed to form geranyl diphosphate (GPP) by GPP synthase, while IPP is converted into farnesyl diphosphate (FPP) by FPP synthase [16]. Subsequently, monoterpenes and sesquiterpenes are generated from the precursors GPP and FPP through the action of the monoterpene synthase (mono-TPS) and sesquiterpene synthase (sesqui-TPS), respectively [17]. However, it is difficult to differentiate the induction patterns of the individual TPSs due to the high level of sequence identity among TPS family members, which indicates the rapid evolution of a species-specific paralogous gene cluster [18]. The discovery and identification of key genes responsible for the terpenoid biosynthesis in L. cubeba could help regulate the composition of essential oils.
RNA sequencing (RNA-seq) has become a powerful technology to profile transcriptomes due to its high-throughput, accuracy, and reproducibility [19]. In plants, RNA-seq has accelerated the investigation of the complexity of gene transcription patterns, functional analyses and gene regulation networks [20]. Due to the limited genomic sources for L. cubeba, it is important to identify whole transcripts for complete gene expression profiling by RNA-seq. In the present work, an RNA-seq project for L. cubeba was initiated. Eleven RNA samples, including various tissues and fruits of different development and ripening stages, were sequenced using the high-throughput Illumina deep sequencing technique. In addition, we estimated the expression profiles of key genes responsible for terpenoid biosynthesis. The transcriptome sequencing from L. cubeba may help improve future genetic and genomics studies on the molecular mechanisms behind the chemical composition of the fruit’s essential oils.
Results
RNA-Seq and de novo transcriptome assembly
In previous works, the de novo assembly of short reads without a reference genome was a challenge despite the development of many bioinformatics software tools for data assembly and analysis [21,22]. To maximize the range of transcript diversity, a mixed RNA sample from four tissues and seven different developmental stages of fruits was prepared for RNA-seq using the Illumina HiSeqTM 2000. After a stringent quality check, we obtained by sequencing 3.66 million raw reads and 6.66 gigabase pairs (Gbp) with an average GC content of 47.66% (File S1). We defined the reads with Q≥20 and no ambiguous “N” as high-quality reads. After the removal of adaptor sequences and exclusion of contaminated or short reads, 23,460,490 high-quality reads were assembled into 1,973,896 contigs (Table 1) using SOAPdenovo [23]. Using the Trinity de novo assembly program, next-generation short-read sequences were assembled into 99,060 transcripts with a mean length of 680.34 bp. The transcripts were subjected to cluster and assembly analyses. Finally, we harvested a total of 68,648 unigenes with an average length of 834 bp, which included 10,270 unigenes (14.96%) with lengths greater than 1 kb. These results showed that the throughput and sequencing quality was high enough for the following analyses.
Table 1. Length distribution of assembled contigs, transcripts, and unigenes.
| Nucleotide length (bp) | Contigs | Transcripts | Unigenes |
|---|---|---|---|
| 0-100 | 1,758,303 | 0 | 0 |
| 100-200 | 134,073 | 0 | 0 |
| 200-300 | 34,458 | 33,002 | 28,329 |
| 300-400 | 14,452 | 15,481 | 12,137 |
| 400-500 | 7,512 | 8,861 | 6,134 |
| 500-600 | 4,722 | 6,157 | 3,822 |
| 600-700 | 3,256 | 4,621 | 2,678 |
| 700-800 | 2,578 | 3,923 | 2,100 |
| 800-900 | 2,047 | 3,319 | 1,732 |
| 900-1000 | 1,697 | 2,921 | 1,446 |
| 1000-1100 | 1,475 | 2,648 | 1,284 |
| 1100-1200 | 1,296 | 2,305 | 1,158 |
| 1200-1300 | 1,111 | 2,057 | 1,019 |
| 1300-1400 | 844 | 1,773 | 839 |
| 1400-1500 | 824 | 1,579 | 799 |
| 1500-1600 | 731 | 1,364 | 663 |
| 1600-1700 | 650 | 1,199 | 615 |
| 1700-1800 | 577 | 1,057 | 553 |
| 1800-1900 | 465 | 978 | 455 |
| 1900-2000 | 407 | 804 | 424 |
| 2000-2100 | 390 | 719 | 379 |
| 2100-2200 | 322 | 638 | 316 |
| 2200-2300 | 267 | 524 | 261 |
| 2300-2400 | 194 | 424 | 202 |
| 2400-2500 | 202 | 420 | 193 |
| 2500-2600 | 176 | 363 | 183 |
| 2600-2700 | 121 | 262 | 126 |
| 2700-2800 | 91 | 230 | 115 |
| 2800-2900 | 87 | 191 | 86 |
| 2900-3000 | 72 | 174 | 82 |
| 3000-3100 | 71 | 146 | 71 |
| >3000 | 425 | 920 | 447 |
| Total number | 1,973,896 | 99,060 | 68,648 |
| Total length | 132,955,711 | 67,394,111 | 39,398,812 |
| N50 length | 87 | 1053 | 834 |
| Mean length | 67.35699905 | 680.3362709 | 573.9251253 |
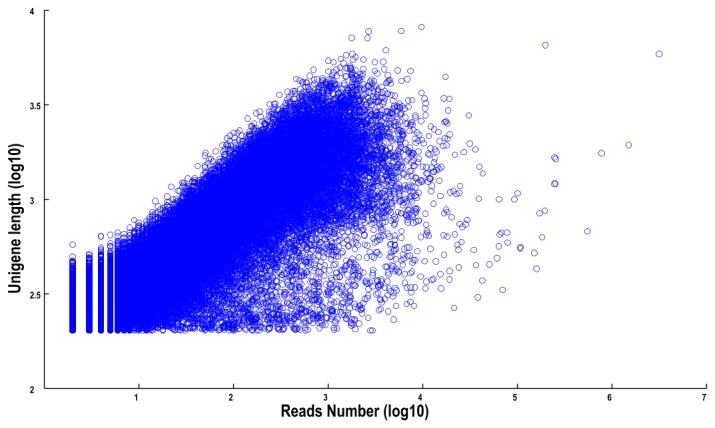
The length distributions of contigs, transcripts and unigenes are shown in Table 1, revealing that the distribution of transcripts showed a similar tendency to that of the unigenes. The N50 values of transcripts and unigenes were 1053 bp and 834 bp, respectively. As expected for a randomly fragmented transcriptome, there was a positive relationship between the length of a given unigene and the number of reads (Figure 1). In addition, we made an Open Reading Frame (ORF) prediction analysis. ORFs were predicted using getORF from the EMBOSS package and most unigenes (99.5%) were identified as having ORFs starting at an ‘ATG’ codon. To facilitate the access and use of the L. cubeba transcriptome sequencing data, the raw paired-end sequence data in the FASTQ format was deposited in the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) database with accession number SRA080286.
Figure 1. The dependence of unigene lengths on the number of reads assembled into each unigene.
Using the number of reads per kilobase of exon model per million mapped reads (RPKM) approach, the expression of each unigene was estimated by reading the depth of deep sequencing. According to Figure 2, the expression of each unigene varied similarly with sequencing depth. The expression of unigenes ranged from 0 to 22,608 RPKM with an average of 18 RPKM. Of 65,535 unigenes, 54,734 (83.5%) had a very low expression level of less than 10 RPKM. In addition, we found that most of the unigenes with high RPKM values were seed storage proteins, such as albumin, globulin and vicilin-like embryo storage protein. A higher proportion of fruits (seven different ripening stages) in the 11 samples may be an explanation for this result.
Figure 2. Assessment of assembly quality.
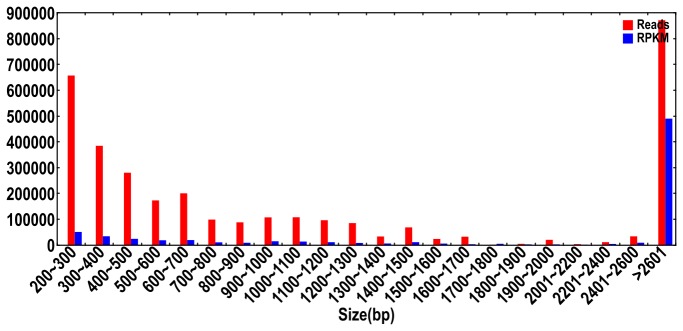
Distribution of unique-mapped reads and RPKM (reads per kb per million reads) of the assembled unigenes.
Functional annotation and classification
Only 56% of unigenes (38,439) were annotated with a threshold of 10-5 by performing a BLASTX search against diverse protein databases, including the NCBI nonredundant protein (Nr) database, NCBI non-redundant nucleotide sequence (Nt) database, UniProt/Swiss-Prot, Kyoto Encyclopedia of Genes and Genomes (KEGG), Cluster of Orthologous Groups of proteins (COG), UniProt/TrEMBL and PlantCyc database. The overall functional annotation for L. cubeba is listed in Table 2. According to the BLASTX results, 36,041 (52.5%) unigenes have homologous proteins in the Nr protein database. Interestingly, 17,272 (48%) unigenes showed significant homology with sequences of Vitis vinifera, and 13% and 12% of the mapped sequences have a high similarity with sequences of Populus trichocarpa and Ricinus communis, respectively (Figure 3). Furthermore, 25,806 (37.6%) unigenes had significant matches in the Nt database, and 25,606 (37.3%) unigenes had similarity to proteins in the Swiss-Prot database.
Table 2. Functional annotation of the L. cubeba.
| Annotated databases | Unigenes | Percentage of unigenes |
|---|---|---|
| Nr-annotation | 36,041 | 52.5% |
| Nt-annotation | 25,806 | 37.6% |
| SwissProt-annotation | 25,606 | 37.3% |
| TrEMBL-annotation | 35,732 | 52.1% |
| GO-annotation | 25,340 | 36.9% |
| KEGG-annotation | 7,702 | 11.2% |
| COG-annotation | 9,803 | 14.3% |
| PlantCyc-annotation | 12,963 | 18.9% |
| Total | 38,439 | 56.0% |
Figure 3. Species distribution of the top BLAST hits in Nr dababase.

36,041 BLASTX-hit unigenes were calculated. Species with proportions of more than 1% are shown.
Generally, GO was used to classify the functions of the assembled transcripts and describe gene products in terms of their associated biological processes, cellular components, and molecular functions. A total of 25,340 unigenes were distributed into the three categories with 46.28% in biological processes, 15.93% in molecular functions, and 37.79% in cellular components (Figure 4). To better review GO classification, each GO term was further clustered to its parent term. The results showed that the three largest biological processes were cellular processes, metabolic processes, and response to stimulus (Figure 4). Meanwhile, most of the genes were classified into the molecular functions of binding, catalytic activity, and transporter activity. In cellular components, the major classifications for these gene products were cell, cell part, and organelle. The results indicated that most of the sequenced genes were responsible for fundamental biological regulation and metabolism.
Figure 4. Functional annotation of assembled sequences based on gene ontology (GO) categorization.
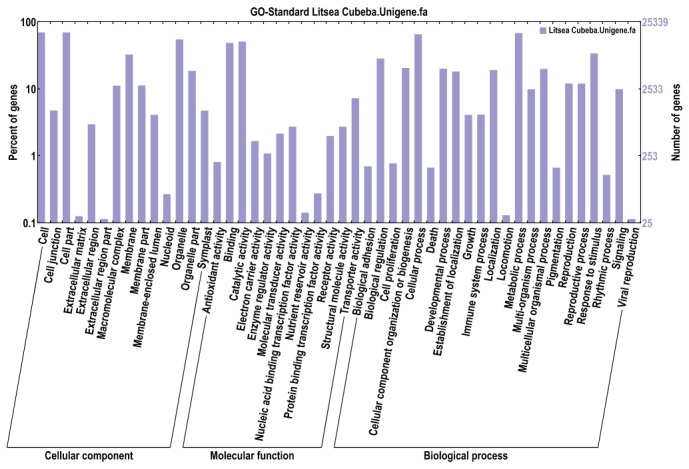
The unigenes are summarized into three main categories: cellular component, molecular function and biological process.
Based on the Nr annotation, only 9,803 of 68,648 (14.3%) unigenes could be aligned to the COG database to predict and classify possible functions. According to the annotation of COG, these genes were classified into 25 different functional classes, including RNA processing and modification, energy production and conversion, carbohydrate transport and metabolism, signal transduction mechanisms, lipid transport and metabolism, coenzyme transport and metabolism (Figure 5). The cluster for general function prediction (2,629; 18.76%) represented the largest group, followed by replication, recombination and repair (1,453; 10.37%), transcription (1,310; 9.35%), signal transduction mechanisms (1,092; 7.79%), translation, ribosomal structure and biogenesis (1,017; 7.26%), posttranslational modification, protein turnover and chaperones (949; 6.77%), carbohydrate transport and metabolism (732; 5.22%), amino acid transport and metabolism (657; 4.69%), and energy production and conversion (583; 4.16%). However, only a few unigenes were assigned to cell motility and nuclear structure (18 and 2 unigenes, respectively). In addition, no unigene was assigned to extracellular structures. Furthermore, 417 (2.98%) unigenes were assigned to lipid transport and metabolism and 272 (1.94%) unigenes were assigned to coenzyme transport and metabolism (Figure 5). However, unigenes involved in terpenoid biosynthesis were found in the categories of lipid transport and metabolism and coenzyme transport and metabolism.
Figure 5. Clusters of orthologous groups (COG) classification.
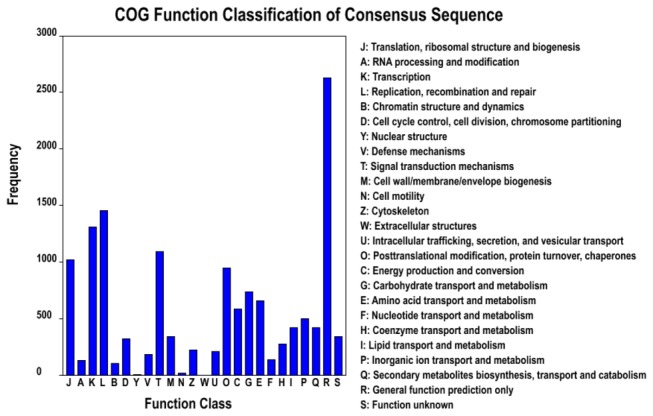
In total, 25,806 of the 68,648 sequences with Nr hits were grouped into 25 classifications.
The KEGG database can be used to categorize gene functions with an emphasis on biochemical pathways. To better understand biological pathways in L. cubeba, a BLASTX search against the KEGG protein database was made on the assembled unigenes. Only 7,702 (11.2%) unigenes were assigned to 297 KEGG pathways (File S2). The predicted pathways represented the majority of plant biochemical pathways including metabolism, genetic information processing, cellular processes, and organism systems. The pathways with highest unigene representation were Chromosome (ko03036, 480 unigenes, 2.98%), followed by Ribosome (ko03011, 423 unigenes, 2.62%) and Spliceosome (ko03041, 409 unigenes, 2.54%). In our sequence dataset, 61 unigenes containing the MEP and MVA pathways were found to be potentially related to terpenoid backbone biosynthesis. At least one unigene from the transcriptome database of L. cubeba corresponded to the enzymes participating in the two pathways except 4-diphosphocytidyl-2C-methyl-D-erythritol synthase (MCT). The database contained more than one unigene each for 3-hydroxy-3-methylglutaryl-CoA reductase (HMGR), phosphomevalonate kinase (PMVK), farnesyl diphosphate synthase (FPPS), 1-deoxy-D-xylulose-5-phosphate synthase (DXS), 1-hydroxy-2-methyl-2-(E)-butenyl-4-diphosphate synthase (HDS), 1-hydroxy-2-methyl-2-(E)-butenyl- 4-diphosphate reductase (HDR) and geranyl diphosphate synthase (GPPS) (Figure 6). Thus, the large amount of transcriptomic information may accelerate the study of terpenoid biosynthesis in L. cubeba.
Figure 6. Monoterpenes and sesquiterpenes biosynthetic pathway in L.cubeba (adapted from Gahlan et al and Ma et al [57, 58]).

Each enzyme name is followed in parentheses by the number of unigenes homologous to gene families encoding this enzyme. AACT: acetoacetyl-CoA thiolase; HMGS: 3-hydroxy-3-methylglutaryl-CoA synthase; HMGR: 3-hydroxy-3-methylglutaryl-CoA reductase; MVK: mevalonate kinase; PMVK: phosphomevalonate kinase; MVD: mevalonate diphosphate decarboxylase; DXS: 1-deoxy-dxylulose-5-phosphate synthase; DXR: 1-deoxy-D-xylulose-5-phosphate reductoisomerase; MCT: 4-diphosphocytidyl-2C-methyl-D- erythritol synthase; CMK: 4-diphosphocytidyl-2C-methyl-D-erythritol kinase; MECPS: 2C-methyl-D-erythritol 4-phosphate cytidylyltransferase; HDS: 1-hydroxy-2-methyl-2-(E)-butenyl-4-diphosphate synthase; HDR: 1-hydroxy-2-methyl-2-(E)-butenyl-4-diphosphate reductase; IPPI: isopentenyl-diphosphate isomerase; FPPS: farnesyl diphosphate synthase; GPPS: geranyl diphosphate synthase; Sesqui-TPS: Sesquiterpene synthase; Mono-TPS: Monoterpene synthase; CAS: (+)-3-carene synthase; CUS: beta-cubebene synthase; OS: Trans-ocimene synthase; GES: Geraniol synthase; KS: Ent-kaurene synthase; LS: limonene synthase; THS: Alpha-thujene synthase; TERS: Alpha-terpineol synthase; LIS: Linalool synthase; PINS: pinene synthase; EAS: 5-epi-aristolochene synthase; CAD: delta-cadinene synthase; GAS: Germacrene A synthase; NES/LIS: nerolidol/linalool synthase.
The PlantCyc database is a plant-specific metabolic pathway database, which includes experimentally supported, computationally predicted, and hypothetical pathways and enzymes. In the total unigenes (12,963) annotated using the PlantCyc database, we focused on terpenoid pathways (285 unigenes) to identify clues related to the medicinal properties of L. cubeba (Figure 7). A major share of the unigenes related to the terpenoid pathways were from mevalonate pathway I (23.16%) which provides the central intermediates for specific terpenoid biosynthesis, and 15.44% were involved in secologanin and strictosidine biosynthesis. Secologanin and strictosidine play important roles in pharmacology [24]. Meanwhile, the monoterpene biosynthesis pathways identified included linalool (3.16%), kaurene (1.75%), and geraniol and geranial (2.11%).
Figure 7. Terpenoid pathways represented in the PlantCyc annotation of the unigenes.

SSR marker discovery
In total, 2,229 sequences containing 2,674 SSRs were identified from 10,270 unigenes, with 1,221 unigene sequences containing more than one SSR. Dinucleotide motifs and trinucleotide motifs were the most abundant, accounting for 50.90 and 46.07%, respectively (Table 3). The most abundant repeat type was AG/CT (1,210), followed by AAG/CTT (524), AGG/CCT (180), ATC/ATG (106), respectively. However, the number of tetra-, penta- and hexa-nucleotide motifs was less than 2% in the unigene sequences. No complex SSR motifs were detected, and not all unigenes had SSRs.
Table 3. Frequency of SSRs in L. cubeba.
| Motif length | Repeat numbers |
Total | % | ||||||
|---|---|---|---|---|---|---|---|---|---|
| 5 | 6 | 7 | 8 | 9 | 10 | >10 | |||
| Di | - | 419 | 270 | 202 | 166 | 198 | 106 | 1361 | 50.90 |
| Tri | 727 | 319 | 165 | 17 | 1 | 1 | 2 | 1232 | 46.07 |
| Tetra | 36 | 10 | 0 | 3 | 0 | 0 | 0 | 49 | 1.83 |
| Penta | 11 | 1 | 0 | 0 | 0 | 1 | 0 | 13 | 0.47 |
| Hexa | 8 | 4 | 5 | 0 | 2 | 0 | 0 | 19 | 0.71 |
| Total | 782 | 753 | 440 | 222 | 169 | 200 | 108 | 2674 | |
| % | 29.24 | 28.16 | 16.45 | 8.30 | 6.32 | 7.48 | 4.04 | ||
Phylogenetic analysis of TPS between L. cubeba and other plants
To examine the phylogenetic relationship between the TPS domain proteins in L. cubeba and other plants, a phylogenetic tree was constructed from alignments of the TPS protein sequences (Figure 8). The phylogenetic tree was constructed using MEGA 4.0. Fourteen unique TPS sequences were identified from the L. cubeba unigenes (Figure 8). These sequences were divided into two clades involved in mono- and sesquiterpenoid biosynthesis. The phylogenetic analysis indicated that 10 unigenes (59387, 57447, 59227, 33144, 61364, 53677, 47566, 24759, 60379, and 2866) were likely to produce monoterpene backbones. The remaining four unigenes (61814, 62098, 40122, and 41975) were likely to be involved in sesquiterpene biosynthesis, and clustered with Warburgia ugandensis and Magnolia grandiflora.
Figure 8. Phylogeny tree of terpene synthases.

The two clades illustrate the likely enzymatic function of 14 L. cubeba unigenes. It also shows a distribution of the L. cubeba sequences throughout the two main clades of the tree. Vitvi Germacrene D (NP_001268213.1, (-)-germacrene D synthase, Vitis vinifera), Vitvi Caryophyllene (AEP17005.1, (E)-beta-caryophyllene synthase, Vitis vinifera), Maldo Germacrene D (AGB14625.1, germacrene-D synthase, Malus domestica), Frave Germacrene D (XP_004308410.1, (-)-germacrene D synthase-like, Fragaria vesca subsp. vesca), Artan Amorphadiene (AAF98444.1, amorpha-4,11-diene synthase, Artemisia annua), Eletr Copaene (ADK94034.1, alpha-copaene synthase, Eleutherococcus trifoliatus), Solly Germacrene C (XP_004242295.1, germacrene C synthase-like, Solanum lycopersicum), Actde Germacrene D (AAX16121.1, germacrene-D synthase, Actinidia deliciosa), Cicar Germacrene D (XP_004505471.1, (-)-germacrene D synthase-like, Cicer arietinum), Ricco Cadinene (XP_002523635.1, (+)-delta-cadinene synthase isozyme A, Ricinus communis), Vitvi Curcumene (ADR74200.1, beta-curcumene synthase, Vitis vinifera), Vitvi Cadinene (ADR74199.1, gamma-cadinene synthase, Vitis vinifera), Vitvi Valencene (NP_001268028.1, valencene synthase-like, Vitis vinifera), Vitvi Germacrene A (ADR66821.1, germacrene A synthase, Vitis vinifera), Citsi Valencene (AF441124_1, valencene synthase, Citrus sinensis), Solca Cascarilladiene (AAT72931.1, cascarilladiene synthase, Solidago canadensis), Goshi Cadinene (AAX44033.1, (+)-delta-cadinene synthase, Gossypium hirsutum), Theca Cadinene (EOY12645.1, delta-cadinene synthase isozyme A, Theobroma cacao), Warug Sesquiterpene (ACJ46047.1, sesquiterpene synthase, Warburgia ugandensis), Maggr Cubebene (B3TPQ6.1, beta-cubebene synthase, Magnolia grandiflora), Nicta Epi-Aristolochene (3M02.A, 5-Epi- Aristolochene Synthase, Nicotiana tabacum), Soltu Vetispiradiene (Q9XJ32.1, vetispiradiene synthase 1, Solanum tuberosum), Solly Vetispiradiene (AAG09949.1, AF171216_1, vetispiradiene synthase, Solanum lycopersicum), Litcu Ocimene (AEJ91554.1, trans-ocimene synthase, Litsea cubeba), Litcu Thujene (AEJ91555.1, alpha-thujene synthase, Litsea cubeba), Litcu Thujene/Sabinene (AEJ91556.1, alpha-thujene synthase/sabinene synthase, Litsea cubeba), Maggr Terpineol (ACC66282.1, alpha-terpineol synthase, Magnolia grandiflora), Maldo Ocimene (AGB14628.1, (E)-beta-ocimene synthase, Malus domestica), Phalu Ocimene (ABY65110.1, beta-ocimene synthase, Phaseolus lunatus), Citli Limonene (AAM53946.1|AF514289_1, (+)-limonene synthase 2, Citrus limon), Pontr Limonene (BAG74774.1, limonene synthase, Poncirus trifoliata), Cucsa Terpineol (XP_004161807.1, (-)-alpha-terpineol synthase-like, Cucumis sativus), Menaq Linalool (AAL99381.1, linalool synthase, Mentha aquatica), Cofar Limonene (CCM43927.1, limonene synthase, Coffea arabica), Vitvi Ocimene/Myrcene (ADR74206.1, (E)-beta-ocimene/myrcene synthase, Vitis vinifera), Vitvi Terpineol (NP_001268216.1, (-)-alpha-terpineol synthase, Vitis vinifera), Ricco Limonene (XP_002533355.1, (R)-limonene synthase, Ricinus communis), Queil Pinene (CAK55186.1, pinene synthase, Quercus ilex), Citun Ocimene (BAD91046.1, (E)-beta-ocimene synthase, Citrus unshiu), Citun Terpinene (BAD27259.1, gamma-terpinene synthase, Citrus unshiu), Artan Linalool (AAF13356.1|AF154124_1, (3R)-linalool synthase, Artemisia annua), Cinos Linalool (AFQ20811.1, S-(+)-linalool synthase, Cinnamomum osmophloeum), Vitvi Linalool/Nerolidol (ADR74212.1, (3S)-linalool/(E)-nerolidol synthase, Vitis vinifera).
Genes related to the terpenoid biosynthesis pathway
The expression profiles were validated through RT-qPCR using 16 genes belonging to terpenoid biosynthetic pathways. All 16 candidates showed significantly differential expression levels in different pathways (Figure 9). The six genes of the MVA pathway (AACT, HMGS, HMGR, MVK, PMVK and MVD) showed the similar trends with their expressions levels being low in leaves but dramatically higher in flowers and young fruits by May 8th. The transcript levels decreased in the fruits on June 4th and June 19th, and then up-regulated on July 4th. The five genes of the MEP pathway (DXS, DXR, CMK, HDS and HDR) showed a great change in the eight L. cubeba samples. In particular, DXS and DXR expression levels, which shared a similar profile, were high in flowers and lower in other samples. In addition, FPPS and IPI exhibited a similar trend with their highest expression levels occurring in young fruits. Geraniol synthase (GES), generating geraniol from geranyl diphosphate, was expressed at its highest level in developing fruits, while the expression analysis of trans-ocimene synthase (OS, generating ocimene from geranyl diphosphate) revealed higher mRNA levels in the leaf and flower tissues, and lower levels in the fruits.
Figure 9. RT-qPCR analysis of 16 terpenoid pathway-related candidate unigenes in L. cubeba.

The gene names, sequences and the primers used for RT-qPCR analysis are shown in File S3. Standard error of the mean for three biological replicates (nested with three technical replicates) is represented by the error bars.
Discussion
Transcriptome sequencing has become an effective tool that is used to discover molecular markers and identify novel genes. In recent years, with the improvement of read length by paired-end sequencing, relatively short reads can be effectively assembled and have been successfully used for studying plants without genomic sequence [25,26]. Prior to this study, very few L. cubeba ESTs had been reported in the public databases and little sequence data were available. Here, we used RNA-Seq technology to profile the L. cubeba transcriptome on the Illumina HiSeqTM 2000 platform. A total of 6.66 Gbp of data with 23,460,490 high-quality reads were obtained from the platform, and 68,648 unigenes were identified by de novo assembly. Among them, about 56% were successfully annotated, indicating their relatively conserved functions. In addition, the functions of the unigenes were classified by COG, GO and the metabolic pathways. The non-annotated unigenes may represent poorly conserved regions [27], such as untranslated regions (UTRs), in L. cubeba. Furthermore, we estimated the gene or protein names and ORFs of the assembled unigenes, putative conserved domains, gene ontology terms, and potential metabolic pathways. Compared with previous transcriptomic studies using the same platform in other plants, such as Chorispora bungeana [28], Carthamus tinctorius [29], Apium graveolens [30] and Boehmeria nivea [31], the average length of L. cubeba unigenes was longer. To the best of our knowledge, this is the first attempt at the de novo sequencing and assembly (without a reference genome) of the L. cubeba transcriptome using Illumina paired-end sequencing technology. The results indicate that our final assembly quality is satisfactory. This work will provide a sequence source and facilitate further studies in gene cloning and functional analyses.
Predicting the number of genes and the potential functions of the unigenes are important parameters in the transcriptome sequencing projects. Due to the lack of a reference genome for L. cubeba, this information has been difficult to estimate the number of genes and predict the potential functions of the unigenes. Here, a large number of unigenes could be matched with unique known proteins in public databases, indicating that the Illumina sequencing project excavated a substantial fraction of gene resources from L. cubeba. However, 44% of the assembled unigenes did not match any known sequences in the public databases. These genes may perform specific roles in L. cubeba and be quite divergent from those of other plant species. L. cubeba has diverse medicinal and economic values, and belongs to the Lauraceae family, which lacks any published genomic resources. Thus, the transcriptome sequence generated in this study will be valuable for further research.
In this study, we presented significant sequence similarities of 48, 13 and 12% between L. cubeba and V. vinifera, P. trichocarpa and R. communis (Figure 3). According to the Angiosperm Phylogeny Group Ⅲ (APG Ⅲ), both P. trichocarpa and R. communis belong to the order Malpighiales in the Rosids family [32], while V. vinifera belongs to Vitalest, which is also classified in the Rosids. Thus, there is a close relationship between Malpighiales and Vitalest. Furthermore, reference genome sequences of V. vinifera, P. trichocarpa and R. communis are available in the public databases [33-35]. Therefore, sequence similarities are high between L. cubeba and the three plant species due to their genome sequences. Here, L. cubeba and V. vinifera showed a close relationship at the molecular level. However, the evidence appears to contradict the taxonomic result. This discrepancy deserves further study.
The number of terpene synthases identified from L. cubeba, ten monoterpene synthases and four sesquiterpene synthases, and the three functionally characterized monoterpene synthase gene sequences have been deposited in the NCBI GenBank database and functionally characterized [11]. At present, a relatively large number of TPSs have been found in plants with genomic sequences. Thirty-two genes encoding putatively active mono-, sesqui-, and di-TPSs were reported in the Arabidopsis genome [36], 69 putatively functional genes in the genome of V. vinifera [37], 44 genes in the cultivated tomato (Solanum lycopersicum) genome [38], and 55 putative genes in cultivated apple (Malus domestica) genome [39]. Whereas, a relatively small number of TPSs were identified in plants without genomic sequences. For example, 17 unique terpene synthase sequences were identified in the Thapsia laciniata transcriptome [40]. The terpene synthases found in L. cubeba were mono- and sesquiterpene synthases, which corresponds well to the dominant components in its essential oils. Additionally, sequence comparisons revealed that all the TPSs had similar properties with respect to their native molecular masses [41]. Many TPSs catalyze the formation of a single product, and some TPSs catalyze the formation of multiple products [42]. The mono-TPS and sesqui-TPS described here are necessary to undergo biochemical characterization.
At present, the terpenoid biosynthetic pathway has been well studied and many genes have been identified that participate in this pathway in other plants [39,43,44]. According to our KEGG pathway enrichment results, 61 unigenes were found to be involved in terpenoid backbone biosynthesis. In this study, many unigene sequences assembled were not the full-length cDNAs and the protein sequences predicted were incomplete. Thus, MCT or gene families involved in terpenoid biosynthesis were not found using the transcriptome results. An analysis of the expression pattern of 16 genes related to terpenoid biosynthesis showed that there are 12 genes with high expression levels in the flowers. DXS, GPPS and OS were hardly detected in the different developing stages of fruits. The expression profile of OS was consistent with the previous results [11], which showed its expression mainly in leaf tissue. Therefore, the identification of these genes will improve our understanding of the mechanisms of terpenoid biosynthesis. This study indicates that Illumina sequencing technology can be applied as a rapid method for de novo transcriptome analysis of non-model plants with unavailable genomic information. We believe that the transcriptome dataset will accelerate research on the gene expression and functional genomics of L. cubeba.
Materials and Methods
Plant materials
All samples of L. cubeba were collected from Fuyang’s Urban Forest Park, Zhejiang Province. No specific permits were required from the park to select samples. The park is not privately-owned and the field studies did not involve protected species. The following plant tissues were sampled: flower buds, full open flowers, young leaves, leaf buds and fruits of seven different ripening stages, 45 days after flowering (DAF) (5/3/2012), 60 DAF (5/18/2012), 77 DAF (6/4/2012), 92 DAF (6/19/2012), 106 DAF (7/4/2012), 118 DAF (7/16/2012), and 132 DAF (7/30/2012). Each tissue was harvested from five different plants. All samples were immediately frozen in liquid nitrogen and stored at −80°C for later use.
RNA extraction, library construction and RNA-seq
Total RNA from each sample was isolated separately using the RN38 EASYspin plus Plant RNA kit (Aidlab Biotech, Beijing, China). The purified RNA concentration was quantified with a spectrophotometer (UV-Vis Spectrophotometer, Quawell Q5000, San Jose, CA, USA), and RNA integrity was evaluated using an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). Samples of 2.0 µg high-quality RNA from each sample were combined into a single large pool in an attempt to maximize the diversity of transcriptional units.
The mRNA-seq library was constructed using Illumina’s TruSeq RNA Sample Preparation Kit (Illumina Inc, San Diego, CA, USA). The isolation of mRNA, fragment interruption, cDNA synthesis, addition of adapters, PCR amplification and RNA-Seq were performed by staff at Beijing BioMarker Technologies (Beijing, China). Poly-A mRNA was isolated using poly-T oligo-attached magnetic beads, and then broken into small pieces using divalent cations under an elevated temperature. The cleaved RNA fragments were copied into first strand cDNA using reverse transcriptase and random primers. This was followed by second strand cDNA synthesis using DNA Polymerase I and RNase H. A single ‘A’ base was ligated to the short fragments after being purified using a MinElute PCR Purification Kit (Qiagen, Dusseldorf, Germany), preparing them for ligation to the sequencing adapters. Fragments (200 bp ± 25 bp) were then separated by agarose gel electrophoresis and selected for PCR amplification as sequencing templates. Finally, the mRNA-seq library was constructed for sequencing on the Illumina HiSeqTM 2000 sequencing platform.
Sequence data analysis and assembly
To obtain high-quality clean read data for de novo assembly, the raw reads from mRNA-seq were filtered by discarding the reads with adaptor contamination, masking low-quality reads with ambiguous ‘N’ bases and removing the reads in which more than 10% bases had a Q-value <20 [20,45]. The clean reads were assembled into contigs using the Trinity method (http://trinityrnaseq.sourceforge.net/), which is efficient in reconstructing full-length transcripts across a broad range of expression levels and sequencing depths [46]. We used the Trinity method with an optimized k-mer length of 25 for de novo assembly. Subsequently, the contigs were linked into transcripts according to the paired-end information of the sequences. Then the transcripts were clustered based on nucleotide sequence identity. The longest transcripts in the cluster units were regarded as unigenes to eliminate redundant sequences, and then the unigenes were combined to produce the final assembly used for annotation.
Functional annotation
To determine the functional annotation of the unigenes, a BLASTX search was performed against the NCBI Nr database with an E-value≤10-5 and other databases, including SwissProt, Protein Information Resource (PIR), Protein Research Foundation (PRF) and Protein Data Bank (PDB). Gene names were assigned based on the annotation of the closest UniProt match, with uninformative descriptions excluded. A BLASTN search was also performed against the NCBI Nt database, GenBank, RefSeq and PDB using a protein query. The ORFs were identified as the nucleotide sequence or as the protein translation provided by the “GetORF” program from the EMBOSS software package [47]. The longest ORF was extracted for each unigene. The expression abundance of the unigenes was represented by the number of reads per kilobase of exon model per million mapped reads (RPKM) [48]. The RPKM method is able to reflect the molar concentration of a transcript by normalizing for RNA length and for the total read number. The Blast2GO program was used to assign GO terms with an E-value≤10-5 including molecular functions, biological processes, and cellular components [49,50]. Annotations of L. cubeba unigenes were then used to predict biochemical pathways using the Pathway Tools [51]. KEGG pathways were retrieved from KEGG web server (http://www.genome.jp/kegg/) [52]. The output of the KEGG analysis includes KO assignments and KEGG pathways that are populated with the KO assignments. The metabolic pathways were annotated by PlantCyc Enzymes database v7.0 (http://www.plantcyc.org), and the terpenoid pathways were highlighted in the PlantCyc annotation [53,54].
Detection of SSR markers
The assembled sequences longer than 1kb were used for the detection of SSR markers. Potential SSR markers were detected among the 10,270 unigenes using MISA software (http://pgrc.ipk-gatersleben.de/misa/). The parameters were designed for the identification of perfect dinucleotide motifs with a minimum of six repeats, and tri-, tetra-, penta-, and hexanucleotide motifs with a minimum of five repeats [26,55].
Alignment and phylogenetic tree building
A Hidden Markov Model (HMM) was employed to identify the terpene synthase genes of the L. cubeba transcriptome. The profile of the terpene synthase N-terminal domain (pfam01397) used for the HMM search (HMMER 3.1, http://hmmer.janelia.org/) was downloaded from the Pfam database (http://pfam.sanger.ac.uk/). A total of 14 terpene synthase sequences were obtained with an E-value threshold of 0.1. The phylogenetic tree was created by MEGA4.0 neighbor-joining software with 1000 bootstrap trials.
Real-time quantitative PCR analysis (RT-qPCR)
The L. cubeba transcriptome database was mined for genes involved in terpenoid biosynthetic pathways. The expression profiles include the genes involved in the MVA pathway (AACT, HMGS, HMGR, MVK, PMVK and MVD), MEP pathway (DXS, DXR, CMK, HDS and HDR) and other genes (IPPI, FPPS, GPPS, GES and OS). cDNA synthesis was performed with 3 µg total RNA from leaves, flowers and six developmental fruit stages (May 18th, June 4th, June 19th, July 4th, July 16th, July 30th) using the Superscript Ш First Strand Synthesis system followed by the RNase H step (Invitrogen, Carlsbad, USA), according to the manufacturer’s protocol in a total volume of 20 µl. Primer pairs were designed using Primer3 (http://frodo.wi.mit.edu/primer3/) with the following parameters: Tm of approximately 60°C, product size range of 120-200 base pairs, primer sequences with a length of 20 nucleotides, and a GC content of 45 to 55%. The gene names, sequences and the primers used for RT-qPCR analysis are listed in File S3. To quantify the expression level the selected genes, the L. cubeba UBC (ubiquitin-conjugating enzyme E2) gene was used as an endogenous control. RT-qPCR reactions were performed in 96-well plates using a 7300 Real Time PCR System (Applied Biosystems, CA, USA) and a SYBR® Premix Ex Taq™ Kit (TaKaRa, Dalian, China). PCR reactions were prepared in 20 µl volumes containing 2 µl of 30-fold diluted synthesized cDNA, 10 µl 2×SYBR® Premix Ex TaqTM, 0.4 µl 10 µM forward primer, 0.4 µl 10 µM reverse primer, 0.4 µl 50× RO× reference dye and 6.8 µl sterile distilled water. The cycling conditions were recommended by the manufacturer (30 s at 95°C, 40 cycles of 95°C for 5 s, and 60°C for 31 s). The specificity of the amplicons was verified by melting curve analysis (60 to 95°C) after 40 PCR cycles. Three biological replicates (nested with three technical replicates) per sample were carried out, and the data analysis was performed as described by Pfaffl [56].
Supporting Information
Summary of Illumina transcriptome sequencing for L. cubeba.
(DOC)
Pathway assignment based on KEGG.
(DOC)
The gene names, sequences and the primers used for RT-qPCR analysis.
(DOC)
Funding Statement
The work presented here was supported by Zhejiang Science and Technology Hall (No. 2011R50030) and Basic Scientific Research Project of Nonprofit Central Research Institutions of Chinese Academy of Forestry (No. 61254). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Wang H, Liu Y (2010) Chemical composition and antibacterial activity of essential oils from different parts of Litsea cubeba . Chem Biodivers 7: 229-235. doi: 10.1002/cbdv.200800349. PubMed: 20087994. [DOI] [PubMed] [Google Scholar]
- 2. Choi EM, Hwang JK (2004) Effects of methanolic extract and fractions from Litsea cubeba bark on the production of inflammatory mediators in RAW264. 7 cells. Fitoterapia 75: 141-148. doi: 10.1016/j.fitote.2003.11.003. PubMed: 15030918. [DOI] [PubMed] [Google Scholar]
- 3. Luo M, Jiang LK, Zou GL (2005) Acute and genetic toxicity of essential oil extracted from Litsea cubeba (Lour.) Pers. J Food Protect 68: 581-588. PubMed: 15771186. [DOI] [PubMed] [Google Scholar]
- 4. Hwang JK, Choi EM, Lee JH (2005) Antioxidant activity of Litsea cubeba . Fitoterapia 76: 684-686. doi: 10.1016/j.fitote.2005.05.007. PubMed: 16239077. [DOI] [PubMed] [Google Scholar]
- 5. Seo SM, Kim J, Lee SG, Shin CH, Shin SC et al. (2009) Fumigant antitermitic activity of plant essential oils and components from Ajowan (Trachyspermum ammi), Allspice (Pimenta dioica), Caraway (Carum carvi), Dill (Anethum graveolens), Geranium (Pelargonium graveolens), and Litsea (Litsea cubeba) oils against Japanese termite (Reticulitermes speratus Kolbe). J Agr Food Chem 57: 6596-6602. [DOI] [PubMed] [Google Scholar]
- 6. Jiang Z, Akhtar Y, Bradbury R, Zhang X, Isman MB (2009) Comparative toxicity of essential oils of Litsea pungens and Litsea cubeba and and blends of their major constituents against the cabbage looper, Trichoplusia ni. J Agr Food Chem 57: 4833-4837. [DOI] [PubMed] [Google Scholar]
- 7. Ho CL, Jie-Pinge O, Liu YC, Hung CP, Tsai MC et al. (2010) Compositions and in vitro anticancer activities of the leaf and fruit oils of Litsea cubeba from Taiwan. Nat Prod Commun 5: 617-620. PubMed: 20433084. [PubMed] [Google Scholar]
- 8. Chen CJ, Tseng YH, Chu FH, Wen TY, Cheng WW et al. (2012) Neuropharmacological activities of fruit essential oil from Litsea cubeba Persoon. J Wood Sci 58: 538-543. doi: 10.1007/s10086-012-1277-3. [DOI] [Google Scholar]
- 9. Liu TT, Yang TS (2012) Antimicrobial impact of the components of essential oil of Litsea cubeba from Taiwan and antimicrobial activity of the oil in food systems. Int J Food Microbiol 156: 68-75. doi: 10.1016/j.ijfoodmicro.2012.03.005. PubMed: 22459760. [DOI] [PubMed] [Google Scholar]
- 10. Kamal KJ, Joshi SD (2006) Medicinal and aromatic plants used in Nepal, Tibet and Trans-Himalayan region: AuthorHouse. USA: Indiana; pp 166-167. [Google Scholar]
- 11. Chang YT, Chu FH (2011) Molecular cloning and characterization of monoterpene synthases from Litsea cubeba (Lour.) Persoon. Tree Genet Genomes 7: 835-844. doi: 10.1007/s11295-011-0377-3. [DOI] [Google Scholar]
- 12. Si L, Chen Y, Han X, Zhan Z, Tian S et al. (2012) Chemical composition of essential oils of Litsea cubeba harvested from its distribution areas in China. Molecules 17: 7057-7066. doi: 10.3390/molecules17067057. PubMed: 22683894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ajikumar PK, Tyo K, Carlsen S, Mucha O, Phon TH et al. (2008) Terpenoids: opportunities for biosynthesis of natural product drugs using engineered microorganisms. Mol Pharm 5: 167-190. doi: 10.1021/mp700151b. PubMed: 18355030. [DOI] [PubMed] [Google Scholar]
- 14. Sapir-Mir M, Mett A, Belausov E, Tal-Meshulam S, Frydman A et al. (2008) Peroxisomal localization of Arabidopsis isopentenyl diphosphate isomerases suggests that part of the plant isoprenoid mevalonic acid pathway is compartmentalized to peroxisomes. Plant Physiol 148: 1219-1228. doi: 10.1104/pp.108.127951. PubMed: 18988695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wouters J, Oudjama Y, Ghosh S, Stalon V, Droogmans L et al. (2003) Structure and mechanism of action of isopentenylpyrophosphate-dimethylallylpyrophosphate isomerase. J Am Chem Soc 125: 3198-3199. doi: 10.1021/ja029171p. PubMed: 12630859. [DOI] [PubMed] [Google Scholar]
- 16. Cheng AX, Lou YG, Mao YB, Lu S, Wang LJ et al. (2007) Plant terpenoids: biosynthesis and ecological functions. J Integr Plant Biol 49: 179-186. doi: 10.1111/j.1744-7909.2007.00395.x. [DOI] [Google Scholar]
- 17. Degenhardt J, Köllner TG, Gershenzon J (2009) Monoterpene and sesquiterpene synthases and the origin of terpene skeletal diversity in plants. Phytochemistry 70: 1621-1637. doi: 10.1016/j.phytochem.2009.07.030. PubMed: 19793600. [DOI] [PubMed] [Google Scholar]
- 18. Tholl D, Lee S (2011) Terpene specialized metabolism in Arabidopsis thaliana . Arabidopsis Book 9: e0143. doi: 10.1199/tab.0143. PubMed: 22303268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Vijay N, Poelstra JW, Künstner A, Wolf JB (2013) Challenges and strategies in transcriptome assembly and differential gene expression quantification. A comprehensive in silico assessment of RNA-seq experiments. Mol Ecol 22: 620-634. doi: 10.1111/mec.12014. PubMed: 22998089. [DOI] [PubMed] [Google Scholar]
- 20. Wang Z, Fang B, Chen J, Zhang X, Luo Z et al. (2010) De novo assembly and characterization of root transcriptome using Illumina paired-end sequencing and development of cSSR markers in sweet potato (Ipomoea batatas). BMC Genomics 11: 726. doi: 10.1186/1471-2164-11-726. PubMed: 21182800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Simpson JT, Wong K, Jackman SD, Schein JE, Jones SJ et al. (2009) ABySS: a parallel assembler for short read sequence data. Genome Res 19: 1117-1123. doi: 10.1101/gr.089532.108. PubMed: 19251739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Miller JR, Koren S, Sutton G (2010) Assembly algorithms for next-generation sequencing data. Genomics 95: 315-327. doi: 10.1016/j.ygeno.2010.03.001. PubMed: 20211242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li R, Zhu H, Ruan J, Qian W, Fang X et al. (2010) De novo assembly of human genomes with massively parallel short read sequencing. Genome Res 20: 265-272. doi: 10.1101/gr.097261.109. PubMed: 20019144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Julsing MK, Koulman A, Woerdenbag HJ, Quax WJ, Kayser O (2006) Combinatorial biosynthesis of medicinal plant secondary metabolites. Biomol Eng 23: 265-279. doi: 10.1016/j.bioeng.2006.08.001. PubMed: 17049920. [DOI] [PubMed] [Google Scholar]
- 25. Zhang J, Liang S, Duan J, Wang J, Chen S et al. (2012) De novo assembly and characterisation of the transcriptome during seed development, and generation of genic-SSR markers in peanut (Arachis hypogaea L.). BMC Genomics 13: 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wei W, Qi X, Wang L, Zhang Y, Hua W et al. (2011) Characterization of the sesame (Sesamum indicum L.) global transcriptome using Illumina paired-end sequencing and development of EST-SSR markers. BMC Genomics 12: 451. doi: 10.1186/1471-2164-12-451. PubMed: 21929789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hou R, Bao Z, Wang S, Su H, Li Y et al. (2011) Transcriptome sequencing and de novo analysis for Yesso scallop (Patinopecten yessoensis) using 454 GS FLX. PLOS ONE 6: e21560. doi: 10.1371/journal.pone.0021560. PubMed: 21720557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhao Z, Tan L, Dang C, Zhang H, Wu Q et al. (2012) Deep-sequencing transcriptome analysis of chilling tolerance mechanisms of a subnival alpine plant, Chorispora bungeana . BMC Plant Biol 12: 222. doi: 10.1186/1471-2229-12-222. PubMed: 23171377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Huang L, Yang X, Sun P, Tong W, Hu S (2012) The first Illumina-based de novo transcriptome sequencing and analysis of safflower flowers. PLOS ONE 7: e38653. doi: 10.1371/journal.pone.0038653. PubMed: 22723874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fu N, Wang Q, Shen HL (2013) De novo assembly, gene annotation and marker development using Illumina paired-end transcriptome sequences in celery (Apium graveolens . p. L.). PloS One 8: e57686 [DOI] [PMC free article] [PubMed]
- 31. Liu T, Zhu S, Tang Q, Chen P, Yu Y et al. (2013) De novo assembly and characterization of transcriptome using Illumina paired-end sequencing and identification of CesA gene in ramie (Boehmeria nivea L Gaud). BMC Genomics 14: 125. doi: 10.1186/1471-2164-14-125. PubMed: 23442184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bremer B, Bremer K, Chase MW, Fay MF, Reveal JL et al. (2009) An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APGIII. Bot J Linn Soc 161: 105-121. doi: 10.1111/j.1095-8339.2009.00996.x. [DOI] [Google Scholar]
- 33. Jaillon O, Aury JM, Noel B, Policriti A, Clepet C et al. (2007) The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 449: 463-467. doi: 10.1038/nature06148. PubMed: 17721507. [DOI] [PubMed] [Google Scholar]
- 34. Wullschleger SD, Weston DJ, DiFazio SP, Tuskan GA (2012) Revisiting the sequencing of the first tree genome: Populus trichocarpa . Tree Physiol 33: 357-364. PubMed: 23100257. [DOI] [PubMed] [Google Scholar]
- 35. Chan AP, Crabtree J, Zhao Q, Lorenzi H, Orvis J et al. (2010) Draft genome sequence of the oilseed species Ricinus communis . Nat Biotechnol 28: 951-956. doi: 10.1038/nbt.1674. PubMed: 20729833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Aubourg S, Lecharny A, Bohlmann J (2002) Genomic analysis of the terpenoid synthase (AtTPS) gene family of Arabidopsis thaliana . Mol Genet Genomics 267: 730-745. doi: 10.1007/s00438-002-0709-y. PubMed: 12207221. [DOI] [PubMed] [Google Scholar]
- 37. Martin DM, Aubourg S, Schouwey MB, Daviet L, Schalk M et al. (2010) Functional annotation, genome organization and phylogeny of the grapevine (Vitis vinifera) terpene synthase gene family based on genome assembly, FLcDNA cloning, and enzyme assays. BMC Plant Biol 10: 226. doi: 10.1186/1471-2229-10-226. PubMed: 20964856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Falara V, Akhtar TA, Nguyen TT, Spyropoulou EA, Bleeker PM et al. (2011) The tomato terpene synthase gene family. Plant Physiol 157: 770-789. doi: 10.1104/pp.111.179648. PubMed: 21813655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nieuwenhuizen NJ, Green SA, Chen X, Bailleul EJ, Matich AJ et al. (2013) Functional genomics reveals that a compact terpene synthase gene family can account for terpene volatile production in apple. Plant Physiol 161: 787-804. doi: 10.1104/pp.112.208249. PubMed: 23256150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Drew DP, Dueholm B, Weitzel C, Zhang Y, Sensen CW et al. (2013) Transcriptome analysis of Thapsia laciniata Rouy provides insights into terpenoid biosynthesis and diversity in Apiaceae. Int J Mol Sci 14: 9080-9098. doi: 10.3390/ijms14059080. PubMed: 23698765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nagegowda DA (2010) Plant volatile terpenoid metabolism: biosynthetic genes, transcriptional regulation and subcellular compartmentation. FEBS Lett 584: 2965-2973. doi: 10.1016/j.febslet.2010.05.045. PubMed: 20553718. [DOI] [PubMed] [Google Scholar]
- 42. Nagegowda AD, Dudareva N (2007) Plant biochemistry and biotechnology of flavor compounds and essential oils. Med Plants Biotechnol From Basic Res Ind Applications: 469-492. [Google Scholar]
- 43. Chen F, Tholl D, Bohlmann J, Pichersky E (2011) The family of terpene synthases in plants: a mid-size family of genes for specialized metabolism that is highly diversified throughout the kingdom. Plant J 66: 212-229. doi: 10.1111/j.1365-313X.2011.04520.x. PubMed: 21443633. [DOI] [PubMed] [Google Scholar]
- 44. Foster AJ, Hall DE, Mortimer L, Abercromby S, Gries R et al. (2013) Identification of genes in Thuja plicata foliar terpenoid defenses. Plant Physiol 161: 1993-2004. doi: 10.1104/pp.112.206383. PubMed: 23388118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Xie F, Burklew CE, Yang Y, Liu M, Xiao P et al. (2012) De novo sequencing and a comprehensive analysis of purple sweet potato (Impomoea batatas L.) transcriptome. Planta 236: 101-113. [DOI] [PubMed] [Google Scholar]
- 46. Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA et al. (2011) Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29: 644-652. doi: 10.1038/nbt.1883. PubMed: 21572440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rice P, Longden I, Bleasby A (2000) EMBOSS: the European molecular biology open software suite. Trends Genet 16: 276-277. doi: 10.1016/S0168-9525(00)02024-2. PubMed: 10827456. [DOI] [PubMed] [Google Scholar]
- 48. Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B (2008) Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 5: 621-628. doi: 10.1038/nmeth.1226. PubMed: 18516045. [DOI] [PubMed] [Google Scholar]
- 49. Conesa A, Götz S, García-Gómez JM, Terol J, Talón M et al. (2005) Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21: 3674-3676. doi: 10.1093/bioinformatics/bti610. PubMed: 16081474. [DOI] [PubMed] [Google Scholar]
- 50. Conesa A, Götz S (2008) Blast2GO: A comprehensive suite for functional analysis in plant genomics. Int J Plants Genom, 2008: 2008. PubMed: 18483572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Karp PD, Paley S, Romero P (2002) The pathway tools software. Bioinformatics 18: S225-S232. doi: 10.1093/bioinformatics/18.suppl_1.S225. PubMed: 12169551. [DOI] [PubMed] [Google Scholar]
- 52. Kanehisa M, Goto S, Kawashima S, Okuno Y, Hattori M (2004) The KEGG resource for deciphering the genome. Nucleic Acids Res 32: D277-D280. doi: 10.1093/nar/gkh063. PubMed: 14681412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Annadurai RS, Jayakumar V, Mugasimangalam RC, Katta MA, Anand S et al. (2012) Next generation sequencing and de novo transcriptome analysis of Costus pictus D Don, a non-model plant with potent anti-diabetic properties. BMC Genomics 13: 663. doi: 10.1186/1471-2164-13-663. PubMed: 23176672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Annadurai RS, Neethiraj R, Jayakumar V, Damodaran AC, Rao SN et al. (2013) De novo transcriptome assembly (NGS) of Curcuma longa L. Rhizome reveals novel transcripts related to anticancer and antimalarial terpenoids. PLOS ONE 8: e56217. doi: 10.1371/journal.pone.0056217. PubMed: 23468859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zeng S, Xiao G, Guo J, Fei Z, Xu Y et al. (2010) Development of a EST dataset and characterization of EST-SSRs in a traditional Chinese medicinal plant, Epimedium sagittatum (Sieb. Et Zucc.) Maxim. BMC Genomics 11: 94. doi: 10.1186/1471-2164-11-94. PubMed: 20141623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Pfaffl M (2001) A new mathematical model for relative quantificaitonin real-time RT-PCR. Nucleic Acids Res 29: 2002-2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Gahlan P, Singh HR, Shankar R, Sharma N, Kumari A et al. (2012) De novo sequencing and characterization of Picrorhiza kurrooa transcriptome at two temperatures showed major transcriptome adjustments. BMC Genomics 13: 126. doi: 10.1186/1471-2164-13-126. PubMed: 22462805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ma Y, Yuan L, Wu B, Li X, Chen S et al. (2012) Genome-wide identification and characterization of novel genes involved in terpenoid biosynthesis in Salvia miltiorrhiza . J Exp Bot 63: 2809-2823. doi: 10.1093/jxb/err466. PubMed: 22291132. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Summary of Illumina transcriptome sequencing for L. cubeba.
(DOC)
Pathway assignment based on KEGG.
(DOC)
The gene names, sequences and the primers used for RT-qPCR analysis.
(DOC)