

Radiologic features of GISTs and non-GIST intramural gastric tumors are described, with emphasis on histopathologic correlation.
Abstract
Intramural gastric masses arise in the wall of the stomach (generally within the submucosa or muscularis propria), often with intact overlying mucosa. These tumors are typically mesenchymal in origin and have overlapping radiologic appearances. A combination of features such as location, attenuation, enhancement, and growth pattern may suggest one diagnosis over another. Gastrointestinal stromal tumors (GISTs) account for the majority of intramural tumors and can vary widely in appearance, from small intraluminal lesions to exophytic masses that protrude into the peritoneal cavity, commonly with areas of hemorrhage or necrosis. A well-circumscribed mass measuring −70 to −120 HU is a lipoma. Leiomyomas usually manifest as low-attenuation masses at the gastric cardia. Homogeneous attenuation is a noteworthy characteristic of schwannomas, particularly for larger lesions that might otherwise be mistaken for GISTs. A hypervascular mass in the antrum is a common manifestation of glomus tumors. Hemangiomas are also hypervascular but often manifest in childhood. Inflammatory fibroid polyps usually arise as a polypoid mass in the antrum. Inflammatory myofibroblastic tumors are infiltrative neoplasms with a propensity for local recurrence. Plexiform fibromyxomas are rare, usually antral tumors. Carcinoid tumors are epithelial in origin, but often submucosal in location, and therefore should be distinguished from other intramural lesions. Multiple carcinoid tumors are associated with hypergastrinemia, either in the setting of chronic atrophic gastritis or Zollinger-Ellison syndrome. Sporadic solitary carcinoid tumors not associated with hypergastrinemia have a higher rate of metastasis. Histopathologic analysis, including immunohistochemistry, is usually required for diagnosis of intramural masses.
© RSNA, 2013
LEARNING OBJECTIVES
After completing this journal-based SA-CME activity, participants will be able to:
List common and uncommon mesenchymal tumors of the stomach.
Discuss pertinent imaging characteristics of each tumor, including common location in the stomach, growth pattern, and degree of enhancement.
Correlate imaging features of gastric mesenchymal tumors with histopathologic findings.
Introduction
The stomach is divided into the cardia, fundus, body, antrum, and pylorus (Fig 1a). According to the World Health Organization, neoplasms of the stomach are classified into two large categories on the basis of the cell of origin: epithelial and nonepithelial (1). Epithelial neoplasms arise from the mucosa and account for the majority of gastric tumors, ranging from benign hyperplastic and adenomatous polyps to malignant adenocarcinomas. In contrast, nonepithelial tumors arise deep to the mucosa, that is, from the submucosa, muscularis propria, or serosa (Fig 1b). Nonepithelial lesions are commonly referred to as “submucosal” or “intramural”; however, the term submucosal can lead to confusion because these lesions do not necessarily arise in the submucosa. For the purposes of this article, nonepithelial tumors arising in the gastric wall will be referred to as “intramural.”
Figure 1a.
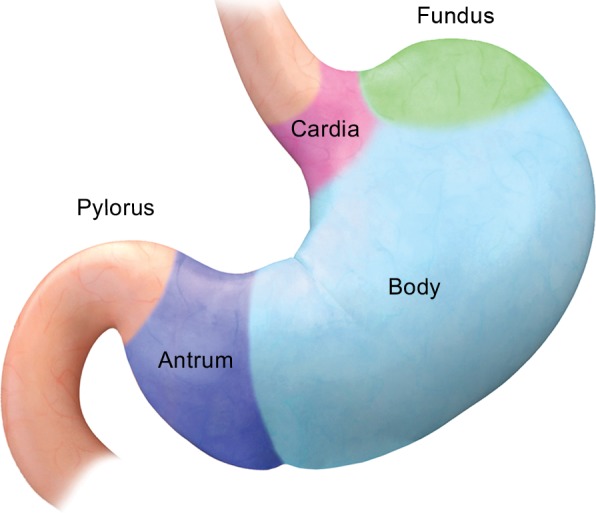
Anatomy of the stomach. (a) Drawing illustrates the divisions of the stomach into the cardia, fundus, body, antrum, and pylorus. (b) Photomicrograph shows a diagrammed cross section of a gastric specimen. Similar to other parts of the gastrointestinal tract, the stomach has four layers: mucosa, submucosa, muscularis propria, and serosa.
Figure 1b.
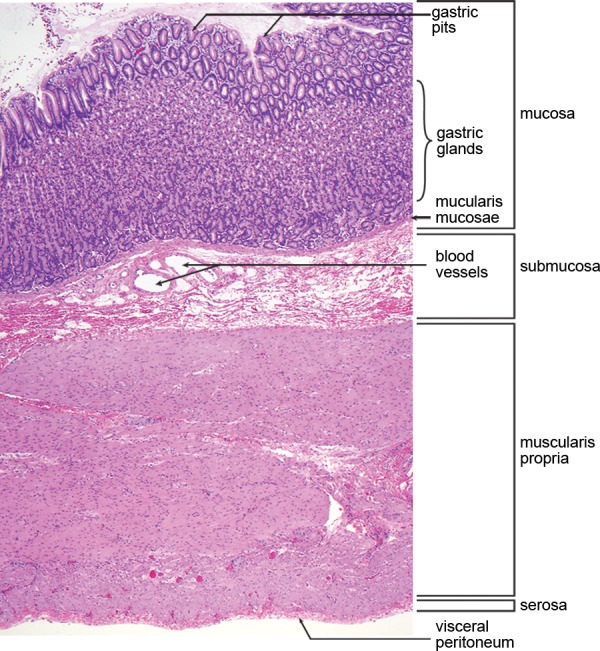
Anatomy of the stomach. (a) Drawing illustrates the divisions of the stomach into the cardia, fundus, body, antrum, and pylorus. (b) Photomicrograph shows a diagrammed cross section of a gastric specimen. Similar to other parts of the gastrointestinal tract, the stomach has four layers: mucosa, submucosa, muscularis propria, and serosa.
Intramural gastric tumors are typically mesenchymal in origin and include gastrointestinal stromal tumors (GISTs), non-GIST sarcomas, lipomas, leiomyomas, schwannomas, glomus tumors, hemangiomas, inflammatory fibroid polyps (IFPs), inflammatory myofibroblastic tumors (IMFTs), and plexiform fibromyxomas. Carcinoid tumors arise from neuroendocrine cells in the mucosa (and are therefore epithelial in origin), but often the bulk of the tumor is submucosal in location; thus, they are included in this discussion of intramural tumors.
In contrast to most epithelial neoplasms, mesenchymal tumors are often well-circumscribed, with intact overlying mucosa. These tumors can grow in an endoluminal, exophytic, or mixed (dumbbell-shaped) pattern (Fig 2). Smaller masses and lesions arising in the submucosa often protrude into the lumen of the stomach. Larger masses and tumors that arise from the deep muscularis propria, such as GISTs, often demonstrate an exophytic pattern of growth, toward the peritoneal cavity.
Figure 2a.
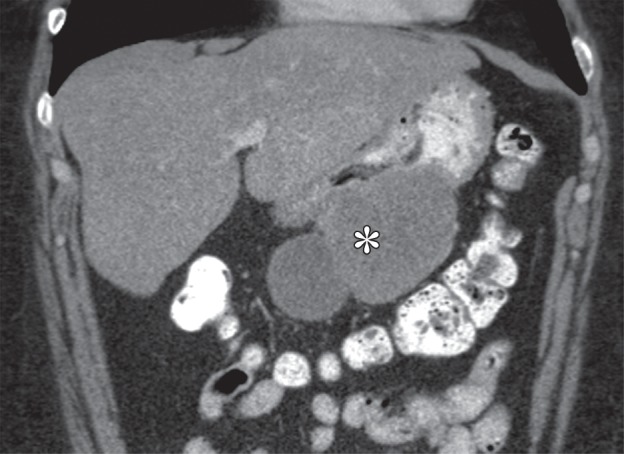
Growth patterns of intramural masses. (a–c) Different growth patterns of GISTs (*) are illustrated on contrast material–enhanced computed tomographic (CT) images from three different cases: exophytic (a), dumbbell-shaped (b), and endoluminal (c). (Image a is from a 51-year-old woman; b, a 56-year-old man; and c, a 64-year-old woman.) (d) Endoscopic image in the same patient as in c shows the intramural lesion with smooth overlying mucosa and central umbilication. (e) Fluoroscopic image in an 81-year-old woman shows a smoothly marginated mass with a central ulcer (arrow), forming obtuse angles with the gastric wall. (f) Axial contrast-enhanced CT image in a 68-year-old man shows an exophytic GIST with focal ulceration (arrow).
Figure 2b.
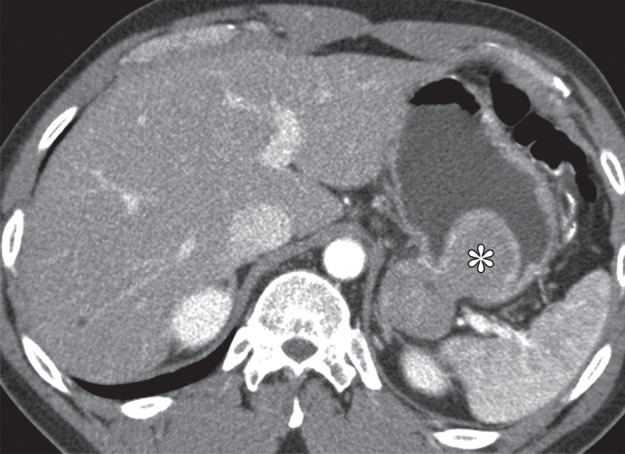
Growth patterns of intramural masses. (a–c) Different growth patterns of GISTs (*) are illustrated on contrast material–enhanced computed tomographic (CT) images from three different cases: exophytic (a), dumbbell-shaped (b), and endoluminal (c). (Image a is from a 51-year-old woman; b, a 56-year-old man; and c, a 64-year-old woman.) (d) Endoscopic image in the same patient as in c shows the intramural lesion with smooth overlying mucosa and central umbilication. (e) Fluoroscopic image in an 81-year-old woman shows a smoothly marginated mass with a central ulcer (arrow), forming obtuse angles with the gastric wall. (f) Axial contrast-enhanced CT image in a 68-year-old man shows an exophytic GIST with focal ulceration (arrow).
Figure 2c.
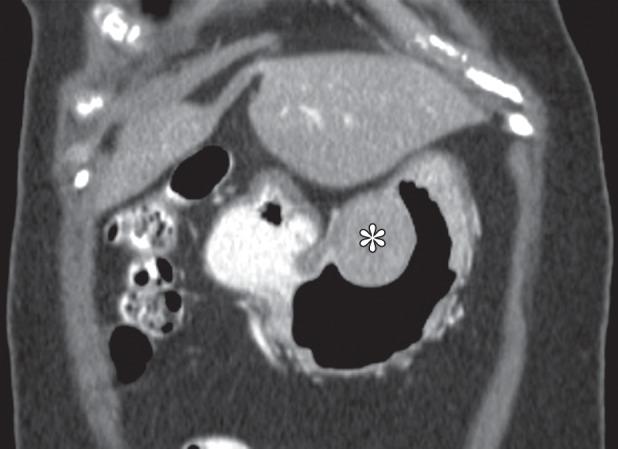
Growth patterns of intramural masses. (a–c) Different growth patterns of GISTs (*) are illustrated on contrast material–enhanced computed tomographic (CT) images from three different cases: exophytic (a), dumbbell-shaped (b), and endoluminal (c). (Image a is from a 51-year-old woman; b, a 56-year-old man; and c, a 64-year-old woman.) (d) Endoscopic image in the same patient as in c shows the intramural lesion with smooth overlying mucosa and central umbilication. (e) Fluoroscopic image in an 81-year-old woman shows a smoothly marginated mass with a central ulcer (arrow), forming obtuse angles with the gastric wall. (f) Axial contrast-enhanced CT image in a 68-year-old man shows an exophytic GIST with focal ulceration (arrow).
Figure 2d.
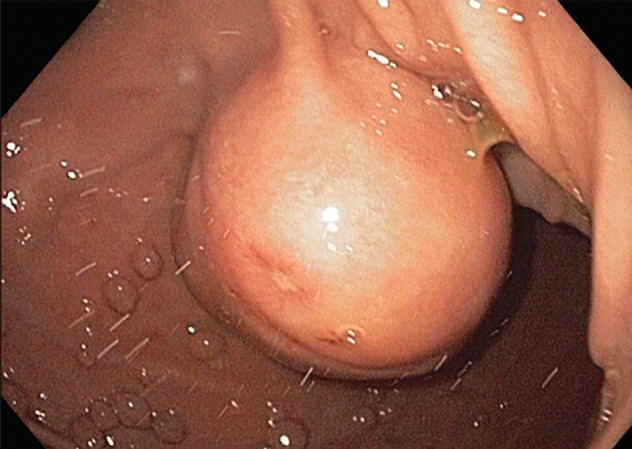
Growth patterns of intramural masses. (a–c) Different growth patterns of GISTs (*) are illustrated on contrast material–enhanced computed tomographic (CT) images from three different cases: exophytic (a), dumbbell-shaped (b), and endoluminal (c). (Image a is from a 51-year-old woman; b, a 56-year-old man; and c, a 64-year-old woman.) (d) Endoscopic image in the same patient as in c shows the intramural lesion with smooth overlying mucosa and central umbilication. (e) Fluoroscopic image in an 81-year-old woman shows a smoothly marginated mass with a central ulcer (arrow), forming obtuse angles with the gastric wall. (f) Axial contrast-enhanced CT image in a 68-year-old man shows an exophytic GIST with focal ulceration (arrow).
Figure 2e.
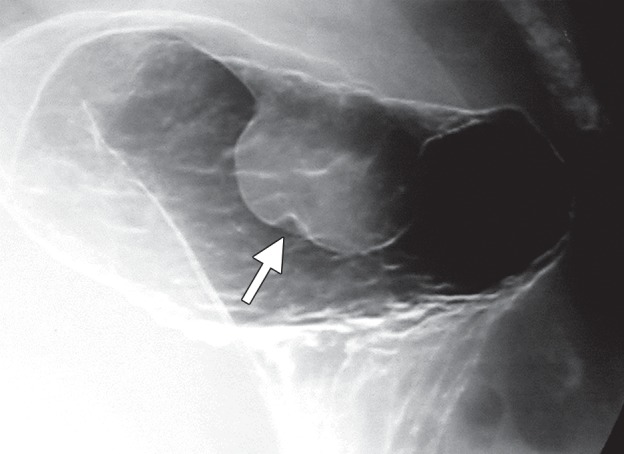
Growth patterns of intramural masses. (a–c) Different growth patterns of GISTs (*) are illustrated on contrast material–enhanced computed tomographic (CT) images from three different cases: exophytic (a), dumbbell-shaped (b), and endoluminal (c). (Image a is from a 51-year-old woman; b, a 56-year-old man; and c, a 64-year-old woman.) (d) Endoscopic image in the same patient as in c shows the intramural lesion with smooth overlying mucosa and central umbilication. (e) Fluoroscopic image in an 81-year-old woman shows a smoothly marginated mass with a central ulcer (arrow), forming obtuse angles with the gastric wall. (f) Axial contrast-enhanced CT image in a 68-year-old man shows an exophytic GIST with focal ulceration (arrow).
Figure 2f.
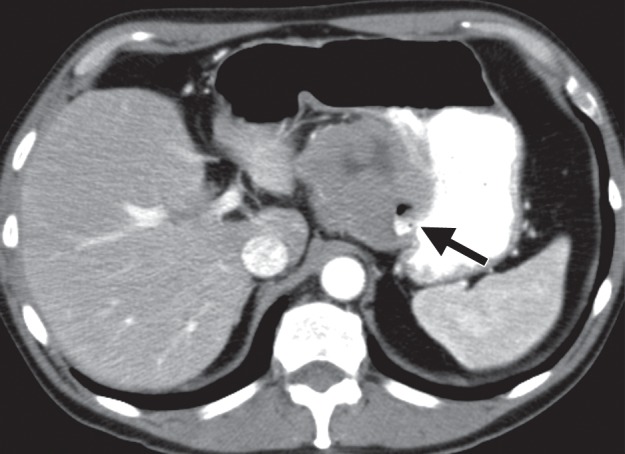
Growth patterns of intramural masses. (a–c) Different growth patterns of GISTs (*) are illustrated on contrast material–enhanced computed tomographic (CT) images from three different cases: exophytic (a), dumbbell-shaped (b), and endoluminal (c). (Image a is from a 51-year-old woman; b, a 56-year-old man; and c, a 64-year-old woman.) (d) Endoscopic image in the same patient as in c shows the intramural lesion with smooth overlying mucosa and central umbilication. (e) Fluoroscopic image in an 81-year-old woman shows a smoothly marginated mass with a central ulcer (arrow), forming obtuse angles with the gastric wall. (f) Axial contrast-enhanced CT image in a 68-year-old man shows an exophytic GIST with focal ulceration (arrow).
Patients with intramural tumors can be asymptomatic or can present with an abdominal mass or pain. In tumors larger than 2 cm, the overlying mucosa can be focally ulcerated because of pressure necrosis from the intramural mass (Fig 2d–2f), resulting in signs of gastrointestinal bleeding, such as hematemesis, melena, and iron-deficiency anemia. Larger masses may result in bowel obstruction or intussusception.
Differentiating between various gastric intramural tumors may be challenging because these lesions often have overlapping radiologic appearances. Attention to a combination of features, such as tumor margin, location, growth pattern, attenuation, and enhancement, may help one suggest a particular diagnosis. In this article, we review the radiologic findings and endoscopic appearances of GIST and non-GIST intramural gastric tumors and correlate them with histopathologic features.
Histologic Analysis
Definitive diagnosis of mesenchymal tumors usually requires histologic analysis, including immunohistochemical studies. Although there is some overlap, many of the mesenchymal tumors that manifest in the stomach have a distinctive histologic appearance; however, immunohistochemical stains are often employed for confirmation of particular tumors. Immunoreactivity to c-KIT (CD117) or DOG1 is used to confirm GIST. Immunoreactivity to desmin helps establish a diagnosis of a leiomyoma. Positivity for S-100 protein, along with distinctive histologic features, is helpful for the diagnosis of schwannomas. The histologic appearance of bland ovoid cells and hypervascularity is suggestive of a glomus tumor, and in this setting, reactivity to smooth muscle actin helps make the diagnosis. A spindle cell tumor with intratumoral plasma cells and immunoreactivity to anaplastic lymphoma kinase is compatible with an IMFT. Chromogranin A and synaptophysin are immunomarkers for carcinoid tumors.
Imaging Technique
Small intramural tumors of the stomach are often discovered incidentally. When dedicated radiologic evaluation of the stomach is desired, adequate gastric distention is important to avoid overlooking small lesions and to differentiate prominent gastric rugae from true masses. Also important is a technique that distinguishes the mucosal surface. On images from a double-contrast barium study, an intramural mass has a smooth mucosal surface and forms obtuse margins with the adjacent gastric wall (Fig 2e). Although superior for visualization of the mucosa, fluoroscopy is limited in the evaluation of extraluminal structures, and CT is routinely performed for further assessment of gastric masses.
At CT, gastric distention can be achieved with oral contrast material. Both positive and negative oral contrast agents have been used (2–4). Although an intramural mass may be seen as a low-attenuation filling defect, mucosal enhancement can be missed if positive oral contrast material is used. Use of negative oral contrast agents (water or low-concentration barium sulfate [VoLumen, E-Z-EM, Lake Success, NY]), on the other hand, improves visualization of enhancing mucosa (Fig 2b). Although the various layers of the gastric wall cannot be differentiated on CT images, the mucosa can usually be distinguished from other layers by virtue of its prominent enhancement during the arterial phase. In a later phase of contrast enhancement, the gastric wall may appear as a single enhancing layer. Dual-phase (arterial, portal venous) or triple-phase (unenhanced, arterial, and portal venous) imaging is helpful not only for determining the enhancement pattern of an intramural mass, but also for detecting hypervascular liver metastases, such as those from GISTs or carcinoid tumors, which could be missed on portal venous phase images (2,5). The unenhanced phase is helpful for assessing the presence of calcifications, especially when positive oral contrast material has been used.
Gastrointestinal Stromal Tumor
GISTs account for 90% of mesenchymal tumors in the gastrointestinal tract and 2%–3% of all gastric malignancies. Although the older medical literature referred to these tumors as leiomyomas, leiomyoblastomas, or leiomyosarcomas, GISTs differ from smooth muscle neoplasms in that they are derived from a precursor of the intestinal pacemaker cells of Cajal, rather than from smooth muscle cells (6). GISTs are characterized by immunoreactivity for c-KIT (CD117), a growth factor receptor tyrosine kinase that is normally expressed on hematopoietic stem cells, mast cells, melanocytes, and the myenteric plexus of the normal adult gastrointestinal tract (7). Development of a drug (imatinib [Gleevec], Novartis, Basel, Switzerland) that targets this receptor tyrosine kinase led to the first breakthrough in the treatment of GIST (8,9) (Fig 3). Five percent of GISTs do not demonstrate c-KIT immunoreactivity; these tumors usually harbor mutations in platelet-derived growth factor receptor α (PDGFR-α ) (10).
Figure 3a.
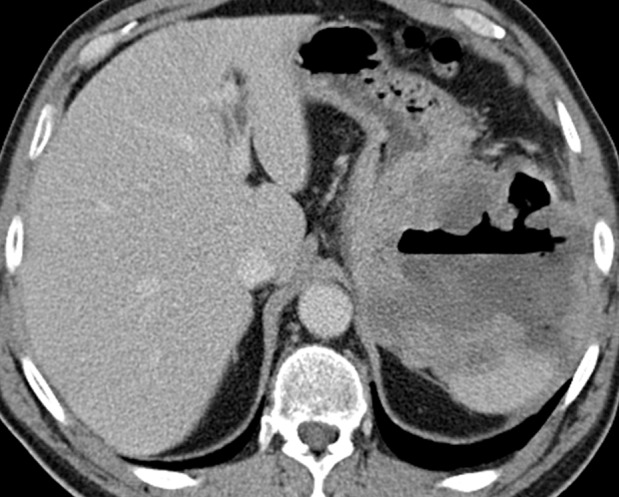
Cavitary GIST in a 64-year-old man with abdominal pain, anemia, and a 20-lb (9-kg) weight loss. (a) Axial contrast-enhanced CT image shows an 11-cm cavitary exophytic tumor. (b) Follow-up contrast-enhanced CT image, obtained after 2 months of imatinib therapy, shows reduction in the size of the tumor, which now measures 6 cm.
Figure 3b.
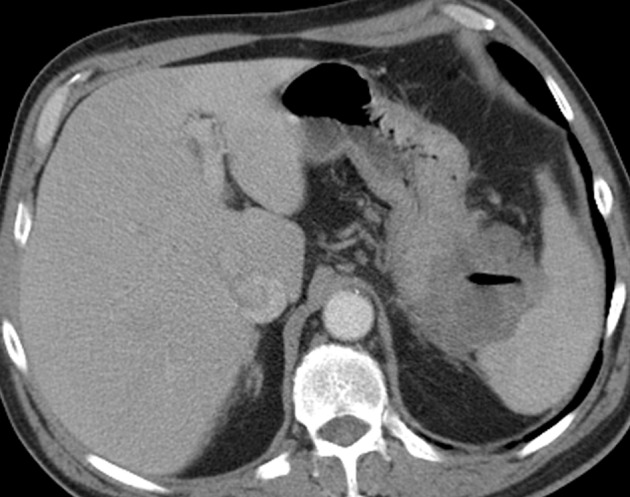
Cavitary GIST in a 64-year-old man with abdominal pain, anemia, and a 20-lb (9-kg) weight loss. (a) Axial contrast-enhanced CT image shows an 11-cm cavitary exophytic tumor. (b) Follow-up contrast-enhanced CT image, obtained after 2 months of imatinib therapy, shows reduction in the size of the tumor, which now measures 6 cm.
GISTs can arise anywhere in the gastrointestinal tract, including the esophagus, stomach, small bowel, or colon, as well as in extravisceral locations such as the mesentery, omentum, or retroperitoneum. The most common location is the stomach (70% of cases), followed by the small intestine, anorectum, colon, and esophagus (11). In the stomach, the body is the most common site, followed by the fundus and then the antrum (12).
GISTs are assessed for risk of progressive disease on the basis of their mitotic rate, size, and location (13,14). Origin in the stomach is a favorable prognostic factor, and gastric GISTs less than 2 cm in size may have no or extremely low malignant potential (11,15).
Clinical manifestations of GISTs depend on their size and location. Small GISTs are usually discovered incidentally. Ulcerated lesions can manifest with symptoms of gastrointestinal bleeding, including hematemesis, melena, and iron-deficiency anemia (12). Patients with larger tumors can present with abdominal pain and early satiety. However, because of the exophytic growth pattern of these tumors, some patients remain asymptomatic until the tumor has become quite large. Bowel obstruction is rare.
Radiologic appearances of GISTs can vary widely depending on tumor size (16). Because they arise from the deep muscularis propria, GISTs frequently have an exophytic or intramural pattern of growth; endoluminal growth is less common. Endoluminal tumors have classic features of intramural masses, being smoothly circumscribed with margins that form obtuse or right angles with the gastric wall (Fig 2e). About 50% of lesions larger than 2 cm develop focal ulceration of the overlying mucosa because of pressure necrosis (Fig 2d–2f) (17), a radiologic feature referred to as the bull’s-eye sign. As they enlarge, exophytic GISTs may invade adjacent structures such as the pancreas, colon, or diaphragm. Areas of hemorrhage, necrosis, or cystic degeneration are common, appearing as focal areas of low attenuation (Fig 2a). Extensive hemorrhage or necrosis may result in formation of a cavity that communicates with the gastric lumen (Fig 3). In rare cases, clumps of calcifications are seen. Nearly half of patients present with metastatic disease, most commonly to the liver and peritoneum. Lymph node metastasis is infrequent (18).
Differential diagnosis of GISTs includes schwannomas, true leiomyomas, and solitary (type 1) carcinoid tumors, particularly for smaller lesions. Occasionally, gastric adenocarcinoma or lymphoma may demonstrate intramural growth and mimic a GIST. However, advanced gastric carcinomas and lymphomas are usually associated with bulky perigastric or celiac lymphadenopathy, which is rare for GISTs. Metastases to the stomach, such as from melanoma or breast cancer, are often submucosal in location and can manifest with bull’s-eye lesions; however, such lesions are usually multiple.
Histologic characteristics of GISTs usually consist of interlacing whorls of spindle-shaped cells with eosinophilic cytoplasm and elongated nuclei, although tumors completely composed of ovoid cells are not uncommon. Immunoreactivity to c-KIT or DOG1 confirms the diagnosis (Fig 4) (7,19).
Figure 4a.
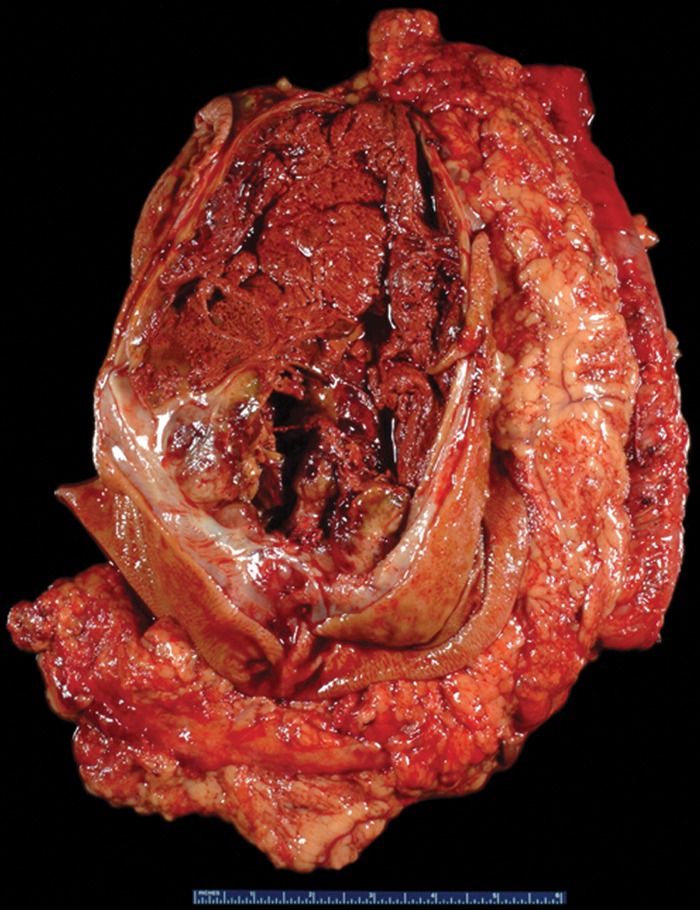
GIST with high risk for progressive disease in a 38-year-old man who had undergone imatinib therapy. (a) Photograph of a gastrectomy specimen, obtained after imatinib therapy, shows a necrotic intramural tumor. Scale is in centimeters. (b) Photomicrograph (original magnification, ×400; hematoxylin-eosin stain) shows that the tumor is composed of spindle cells, which are arranged in intersecting fascicles. Mitotic figures (circles), an indicator of high risk of progression, are easily identified. (c) Photomicrograph (original magnification, ×40; immunohistochemical stain for c-KIT) demonstrates brown staining of the cytoplasm, which confirms that the neoplastic cells are diffusely and strongly immunoreactive for c-KIT.
Figure 4b.
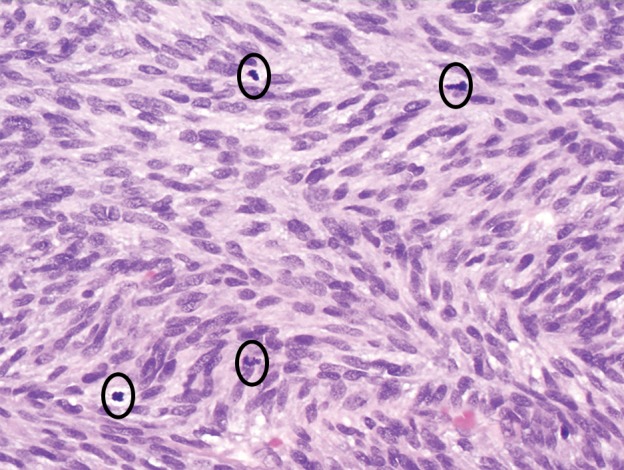
GIST with high risk for progressive disease in a 38-year-old man who had undergone imatinib therapy. (a) Photograph of a gastrectomy specimen, obtained after imatinib therapy, shows a necrotic intramural tumor. Scale is in centimeters. (b) Photomicrograph (original magnification, ×400; hematoxylin-eosin stain) shows that the tumor is composed of spindle cells, which are arranged in intersecting fascicles. Mitotic figures (circles), an indicator of high risk of progression, are easily identified. (c) Photomicrograph (original magnification, ×40; immunohistochemical stain for c-KIT) demonstrates brown staining of the cytoplasm, which confirms that the neoplastic cells are diffusely and strongly immunoreactive for c-KIT.
Figure 4c.
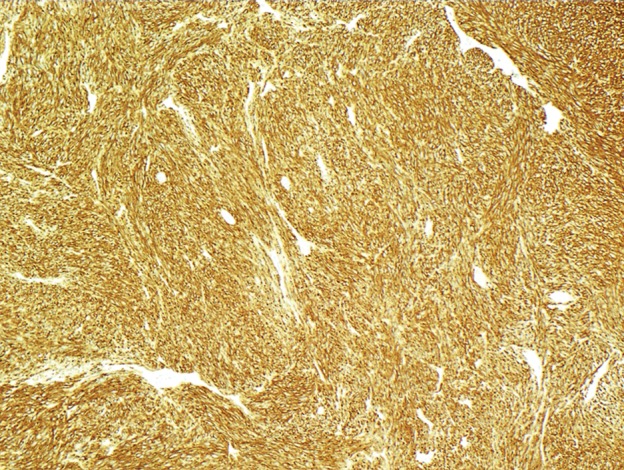
GIST with high risk for progressive disease in a 38-year-old man who had undergone imatinib therapy. (a) Photograph of a gastrectomy specimen, obtained after imatinib therapy, shows a necrotic intramural tumor. Scale is in centimeters. (b) Photomicrograph (original magnification, ×400; hematoxylin-eosin stain) shows that the tumor is composed of spindle cells, which are arranged in intersecting fascicles. Mitotic figures (circles), an indicator of high risk of progression, are easily identified. (c) Photomicrograph (original magnification, ×40; immunohistochemical stain for c-KIT) demonstrates brown staining of the cytoplasm, which confirms that the neoplastic cells are diffusely and strongly immunoreactive for c-KIT.
Non-GIST Sarcoma
Although they are rare, sarcomas other than GIST, such as liposarcomas, leiomyosarcomas, and unclassified sarcomas, may form in the stomach (Fig 5). These lesions are usually large, aggressive tumors with heterogeneous enhancement and areas of necrosis. Their appearance is nonspecific, and histologic confirmation is required for diagnosis. Non-GIST sarcomas are generally negative for CD117, CD34, and DOG1 (20–22).
Figure 5a.
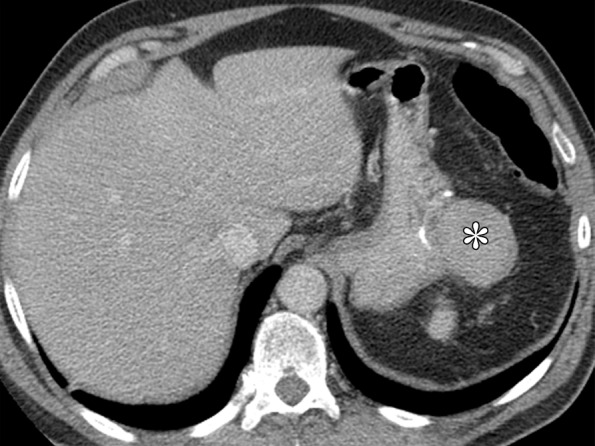
Examples of non-GIST sarcomas. (a) Axial contrast-enhanced CT image in a 62-year-old man demonstrates a dedifferentiated liposarcoma (*) involving the stomach. (b) Coronal contrast-enhanced CT image in a 64-year-old man reveals a tumor (*) for which histologic analysis revealed high-grade spindle cell sarcoma, findings suggestive of leiomyosarcoma.
Figure 5b.
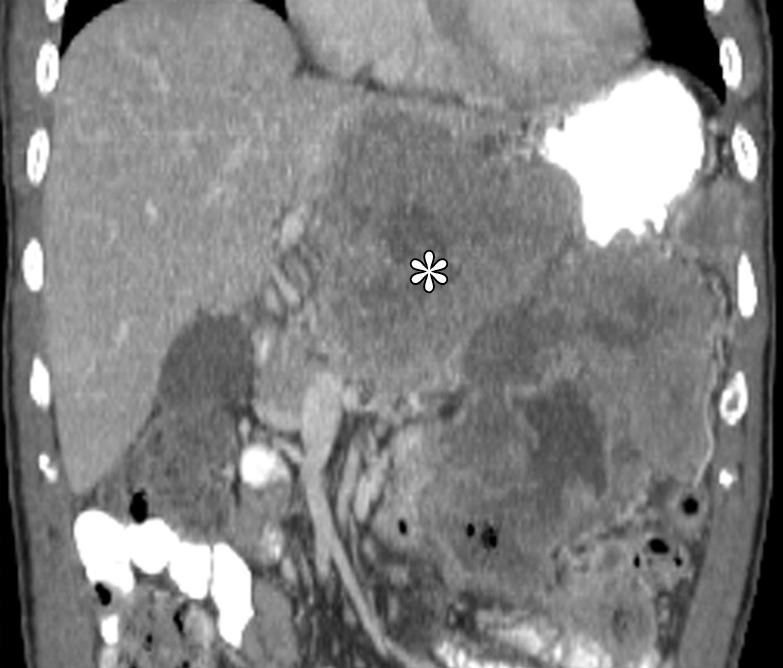
Examples of non-GIST sarcomas. (a) Axial contrast-enhanced CT image in a 62-year-old man demonstrates a dedifferentiated liposarcoma (*) involving the stomach. (b) Coronal contrast-enhanced CT image in a 64-year-old man reveals a tumor (*) for which histologic analysis revealed high-grade spindle cell sarcoma, findings suggestive of leiomyosarcoma.
It is extremely important to accurately differentiate non-GIST sarcomas from GISTs because GISTs respond favorably to imatinib, whereas non-GIST sarcomas have a less predictable response to conventional chemotherapy.
Leiomyosarcomas are differentiated from GIST because they demonstrate positive immunoreactivity for desmin and smooth muscle actin. These tumors consist of intersecting groups of spindle cells and demonstrate high cellularity, abundant mitotic figures, and necrosis (23). Some high-grade sarcomas cannot be further classified despite ancillary studies such as immunohistochemical stains, and they may be diagnosed as “unclassified.”
Lipoma
Gastric lipomas are composed of mature fat cells surrounded by a fibrous capsule (24) (Fig 6). Small lipomas are often asymptomatic. As with other intramural tumors, larger lesions can cause symptoms such as abdominal pain or manifest with gastrointestinal bleeding caused by ulceration of the overlying mucosa. Lipomas infrequently develop into pedunculated lesions, which can prolapse through the pylorus and cause intermittent gastric outlet obstruction. Unless the patient is symptomatic, treatment is not indicated.
Figure 6a.
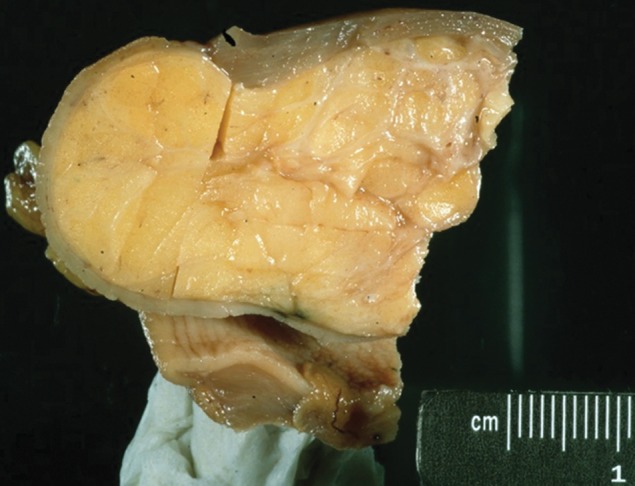
Gastric lipoma. (a) Photograph of a gastrectomy specimen shows a well-circumscribed tumor with a homogeneous, pale-yellow cut surface. (b) Axial contrast-enhanced CT image from a different case shows an endoluminal lesion with uniform fat attenuation (arrow), a finding diagnostic of a lipoma.
Figure 6b.
Gastric lipoma. (a) Photograph of a gastrectomy specimen shows a well-circumscribed tumor with a homogeneous, pale-yellow cut surface. (b) Axial contrast-enhanced CT image from a different case shows an endoluminal lesion with uniform fat attenuation (arrow), a finding diagnostic of a lipoma.
On radiographs, a lipoma can occasionally appear as a radiolucent shadow. During fluoroscopy, a lipoma appears as a smooth well-marginated mass, sometimes with central ulceration. Because lipomas are soft tumors, they can change in size and shape with peristalsis or palpation during fluoroscopy. Three-quarters of lipomas are solitary lesions, with the most common location being the gastric antrum. The majority (95%) of lipomas are endoluminal, arising in the submucosa. On CT images, attenuation values of −70 to −120 HU are diagnostic of a lipoma (24) (Fig 6). One exception to this is the presence of ulceration, which can result in inflammation that manifests with soft-tissue attenuation.
Leiomyoma
True leiomyomas of the stomach are rare. Older literature referred to GISTs as leiomyomas; however, true leiomyomas are negative for c-KIT and strongly and diffusely positive for desmin and smooth muscle actin (11). The distinction between GISTs and leiomyomas is clinically important, because leiomyomas are benign and GISTs are associated with a variable risk of progression and metastasis. In the stomach, small bowel, and colon, GISTs far outnumber leiomyomas. The esophagus is the only site in the gastrointestinal tract where leiomyomas predominate.
Leiomyomas are almost always seen in the gastric cardia as homogeneous, low-attenuation masses with an endoluminal growth pattern (Fig 7) (25,26), often ranging from 1.3 to 4.7 cm in diameter (26). Tumors larger than 2 cm can have central ulceration. Differential diagnosis of leiomyomas includes GISTs and schwannomas.
Figure 7a.
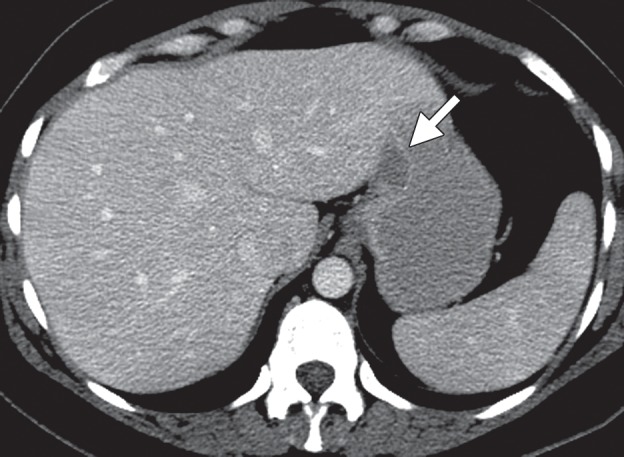
Gastric leiomyoma in a 36-year-old woman. (a) Axial contrast-enhanced CT image shows a leiomyoma (arrow) in the gastric cardia, with intact enhancing mucosa. (b) Low-power photomicrograph (original magnification, ×20; hematoxylin-eosin stain) shows the well-circumscribed tumor, which originated in the muscularis propria, abutting but not invading the overlying submucosa.
Figure 7b.
Gastric leiomyoma in a 36-year-old woman. (a) Axial contrast-enhanced CT image shows a leiomyoma (arrow) in the gastric cardia, with intact enhancing mucosa. (b) Low-power photomicrograph (original magnification, ×20; hematoxylin-eosin stain) shows the well-circumscribed tumor, which originated in the muscularis propria, abutting but not invading the overlying submucosa.
At histologic analysis, leiomyomas usually arise in the muscularis propria. They have low to moderate cellularity, with intersecting fascicles of spindle cells with an eosinophilic cytoplasm (Fig 7). Occasionally, desmin globules are present.
Neurogenic Tumors
Benign nerve sheath tumors, that is, schwannomas, account for 2%–7% of gastrointestinal mesenchymal tumors. Schwannomas are believed to arise from the myenteric plexus within the muscularis propria of the gastrointestinal tract, with the stomach being the most common site (60%–70% of cases), followed by the colon and rectum (27).
At clinical examination, patients may be asymptomatic or present with abdominal pain or gastrointestinal bleeding (if the tumor is ulcerated). Larger lesions can manifest with obstructive symptoms (28,29). There is a female predominance among patients with schwannomas, with a median age of 60 years (30,31).
Schwannomas arise most often in the gastric body, demonstrating an exophytic or intramural pattern of growth (28,29,31). A notable feature of schwannomas is their homogeneous attenuation, particularly for their size (Fig 8). The tumor has low attenuation on unenhanced CT images because of its dense spindle cell composition. Contrast-enhanced CT may show no or minimal enhancement during the arterial phase but delayed enhancement during the equilibrium phase. Although cystic degeneration is a common feature of schwannomas found in other parts of the body, this characteristic is uncommon for gastric schwannomas (28). Calcification is rare. Differential diagnosis of a schwannoma includes GIST, leiomyoma, and lymphoma. Absence of hemorrhage, necrosis, or cavity formation is a useful feature of schwannomas, as it can be used to distinguish schwannomas from the more heterogeneous GISTs.
Figure 8a.
Gastric schwannomas. (a) Axial contrast-enhanced CT image in a 69-year-old woman demonstrates a schwannoma (arrow), incidentally discovered during work-up of a GIST. (b) Endoscopic image in the same patient shows that the mass has smooth, overlying mucosa. (c, d) Axial contrast-enhanced CT images obtained at two levels show a 17-cm schwannoma (*) in a 35-year-old woman with a history of abdominal discomfort for several years. Note the relative homogeneity of the tumor, which would be unusual for a GIST of this size.
Figure 8b.
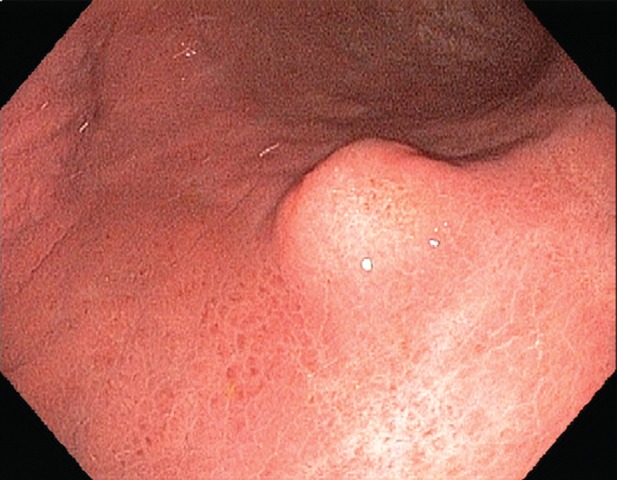
Gastric schwannomas. (a) Axial contrast-enhanced CT image in a 69-year-old woman demonstrates a schwannoma (arrow), incidentally discovered during work-up of a GIST. (b) Endoscopic image in the same patient shows that the mass has smooth, overlying mucosa. (c, d) Axial contrast-enhanced CT images obtained at two levels show a 17-cm schwannoma (*) in a 35-year-old woman with a history of abdominal discomfort for several years. Note the relative homogeneity of the tumor, which would be unusual for a GIST of this size.
Figure 8c.
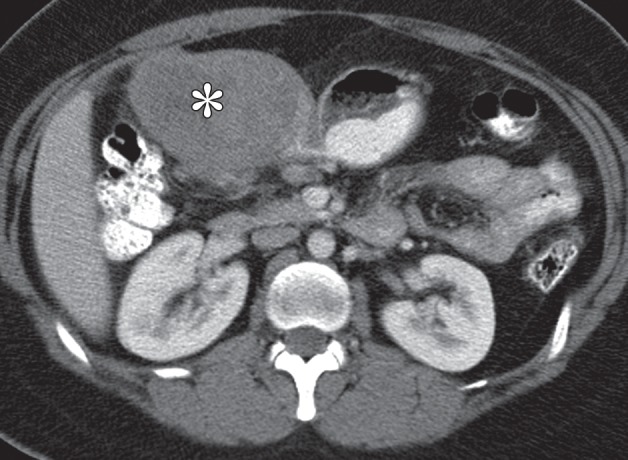
Gastric schwannomas. (a) Axial contrast-enhanced CT image in a 69-year-old woman demonstrates a schwannoma (arrow), incidentally discovered during work-up of a GIST. (b) Endoscopic image in the same patient shows that the mass has smooth, overlying mucosa. (c, d) Axial contrast-enhanced CT images obtained at two levels show a 17-cm schwannoma (*) in a 35-year-old woman with a history of abdominal discomfort for several years. Note the relative homogeneity of the tumor, which would be unusual for a GIST of this size.
Figure 8d.
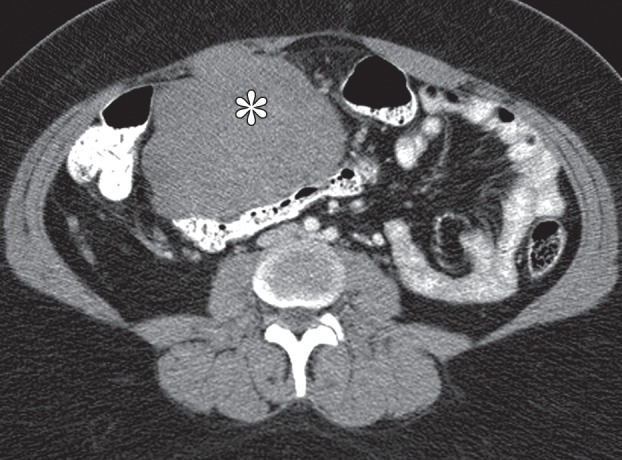
Gastric schwannomas. (a) Axial contrast-enhanced CT image in a 69-year-old woman demonstrates a schwannoma (arrow), incidentally discovered during work-up of a GIST. (b) Endoscopic image in the same patient shows that the mass has smooth, overlying mucosa. (c, d) Axial contrast-enhanced CT images obtained at two levels show a 17-cm schwannoma (*) in a 35-year-old woman with a history of abdominal discomfort for several years. Note the relative homogeneity of the tumor, which would be unusual for a GIST of this size.
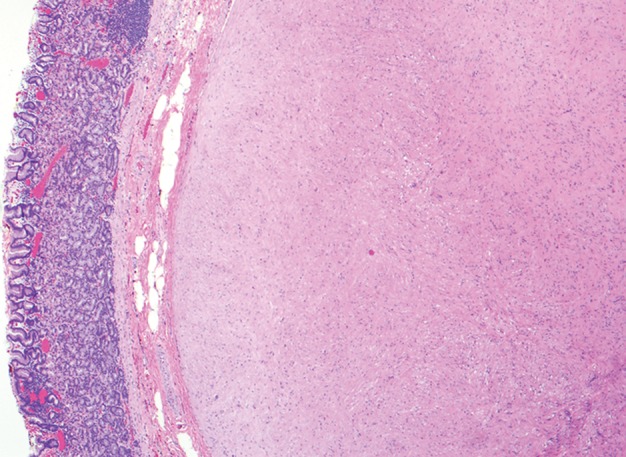
At histologic analysis, gastric schwannomas appear as well-circumscribed but not encapsulated tumors that are characterized by interlacing bundles of spindle cells with wavy cigar-shaped nuclei and collagen. Features characteristic of soft-tissue schwannomas, such as areas of hypo- and hypercelluarity (Antoni A and Antoni B areas) and Verocay bodies (lining up of nuclei), are less frequently seen (30,32). Unique to gastric schwannomas is a peripheral cuff of peritumoral lymphoid aggregates (Fig 9). At immunohistochemical analysis, the tumor cells stain strongly and diffusely for S-100 protein.
Figure 9a.
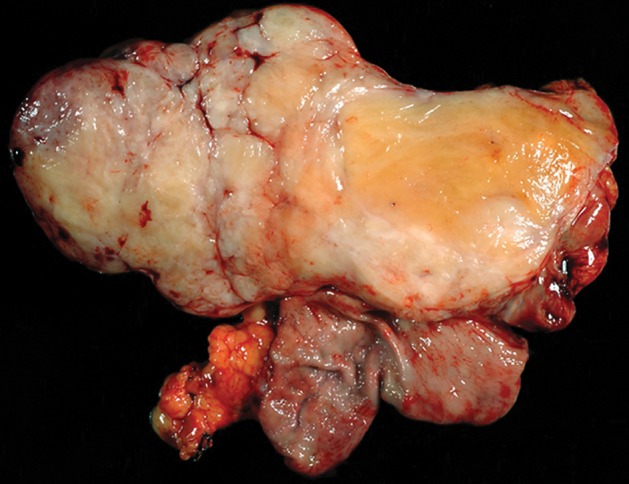
Histopathologic features of schwannoma (specimens from a 36-year-old woman). (a) Photograph of a gastrectomy specimen shows a slightly lobulated, but well-circumscribed, homogeneous intramural tumor. (b) Low-power photomicrograph (original magnification, ×20; hematoxylin-eosin stain) shows that the tumor retains its circumscription and pushes into the submucosa. The tumor is cuffed by lymphoid aggregates (arrowheads), a characteristic feature of schwannomas in this location.
Figure 9b.
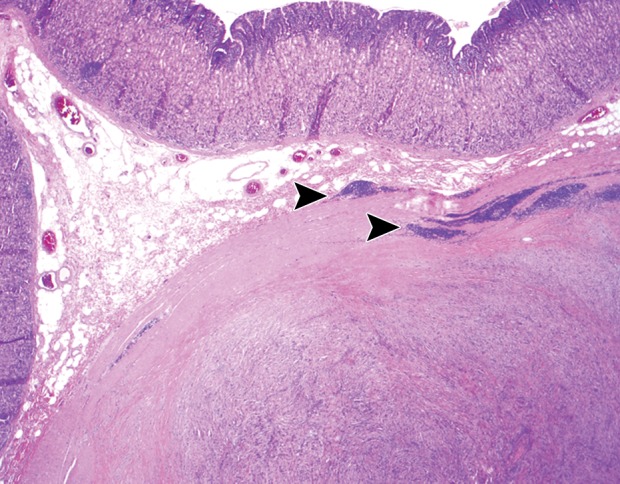
Histopathologic features of schwannoma (specimens from a 36-year-old woman). (a) Photograph of a gastrectomy specimen shows a slightly lobulated, but well-circumscribed, homogeneous intramural tumor. (b) Low-power photomicrograph (original magnification, ×20; hematoxylin-eosin stain) shows that the tumor retains its circumscription and pushes into the submucosa. The tumor is cuffed by lymphoid aggregates (arrowheads), a characteristic feature of schwannomas in this location.
Neurofibromas are rare tumors that arise from the sympathetic nerves of the Auerbach myenteric plexus or, less frequently, the Meissner submucosal plexus. Patients with neurofibromatosis can have multiple neurofibromas and also have increased risk of malignant degeneration (33,34).
Glomus Tumor
Glomus tumors arise from modified smooth muscle cells of the glomus bodies, which are neuromyoarterial receptors commonly seen in the dermis or subcutis of the extremities that regulate body temperature (35). Nearly all gastrointestinal glomus tumors arise in the muscularis propria of the stomach, and they account for 2% of all benign gastric tumors. Rare malignant tumors have been reported for lesions larger than 5 cm (35).
In a study of 32 cases of glomus tumor, 72% of the patients were female and the median age at presentation was 55 years (35). Patients with gastric glomus tumors can be asymptomatic, or they can present with epigastric discomfort, hematemesis, or melena. Surgical resection is usually curative.
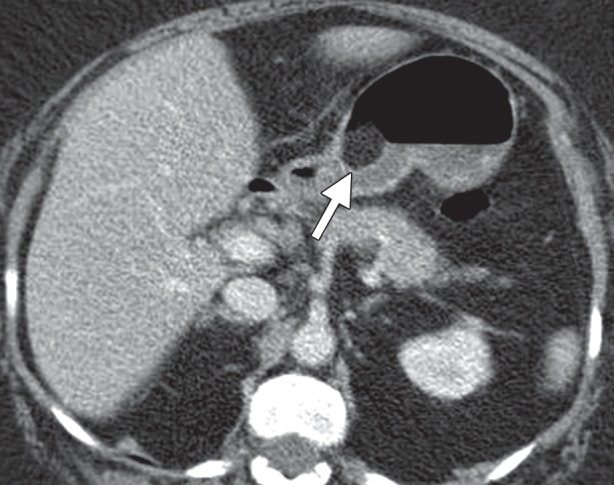
Glomus tumors most commonly manifest as a solitary hypervascular lesion in the gastric antrum and range from 1 to 4 cm in diameter (Fig 10) (25,35). At fluoroscopy, they appear as smooth intramural masses indistinguishable from other mesenchymal tumors. Because they are soft tumors, glomus tumors can change in shape and size during fluoroscopy, similar to lipomas. They are easily differentiated from lipomas on CT images because of their dense arterial phase enhancement, which persists in the delayed phase. A peripheral nodular pattern of enhancement with delayed filling-in, reminiscent of a hemangioma, has been reported as a specific feature of gastric glomus tumors (25,35). Differential diagnosis of glomus tumors includes GIST, carcinoid tumor, metastasis, ectopic pancreas, and hemangioma.
Figure 10a.
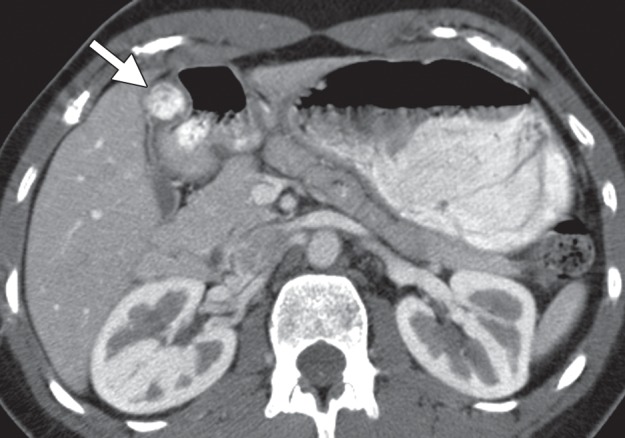
Glomus tumor incidentally found during work-up for fever in a 44-year-old woman. (a, b) Axial contrast-enhanced CT images obtained during the arterial (a) and delayed (b) phases demonstrate an arterially enhancing mass (arrow in a) in the gastric antrum with persistent enhancement (arrow in b). Note the preserved overlying mucosa in both phases. (c) Low-power photomicrograph (original magnification, ×20; hematoxylin-eosin stain) shows the hypervascular tumor originating in the muscularis propria (* denotes vascular spaces).
Figure 10b.
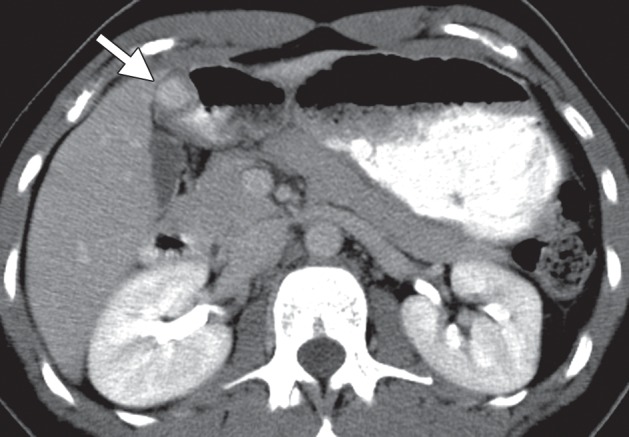
Glomus tumor incidentally found during work-up for fever in a 44-year-old woman. (a, b) Axial contrast-enhanced CT images obtained during the arterial (a) and delayed (b) phases demonstrate an arterially enhancing mass (arrow in a) in the gastric antrum with persistent enhancement (arrow in b). Note the preserved overlying mucosa in both phases. (c) Low-power photomicrograph (original magnification, ×20; hematoxylin-eosin stain) shows the hypervascular tumor originating in the muscularis propria (* denotes vascular spaces).
Figure 10c.
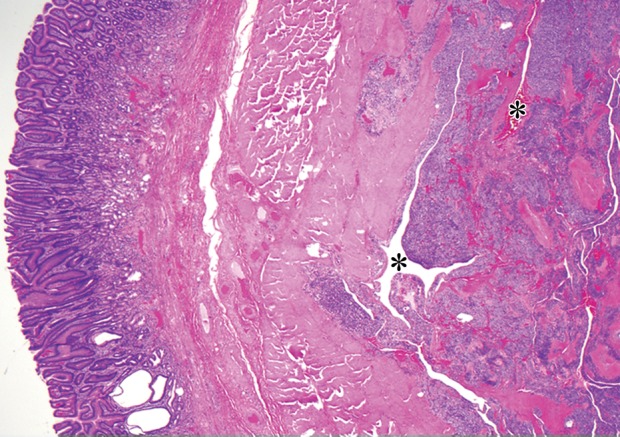
Glomus tumor incidentally found during work-up for fever in a 44-year-old woman. (a, b) Axial contrast-enhanced CT images obtained during the arterial (a) and delayed (b) phases demonstrate an arterially enhancing mass (arrow in a) in the gastric antrum with persistent enhancement (arrow in b). Note the preserved overlying mucosa in both phases. (c) Low-power photomicrograph (original magnification, ×20; hematoxylin-eosin stain) shows the hypervascular tumor originating in the muscularis propria (* denotes vascular spaces).
At histologic analysis, glomus tumors are composed of sheets of monotonous round cells interspersed with numerous delicate vessels (Fig 10). At immunohistochemical analysis, the tumor cells are positive for smooth muscle actin and calponin.
Hemangioma
Hemangiomas are usually solitary, vascular tumors that may occur anywhere in the gastrointestinal tract. Gastric hemangiomas represent 1.6% of all benign gastric tumors (36). Cavernous hemangiomas are composed of large vascular spaces lined by endothelium, whereas capillary hemangiomas consist of multiple small capillaries. It is unknown whether these lesions are true neoplasms or congenital malformations. Occasionally, gastrointestinal hemangiomas can be associated with a cutaneous hemangioma or telangiectasia. The presence of multiple lesions is associated with syndromes such as Klippel-Trénaunay syndrome, Maffucci syndrome, blue rubber-bleb nevus syndrome, or Osler-Rendu-Weber disease (37).
Gastrointestinal hemangiomas can manifest at any age; however, they are commonly seen in early childhood and gradually involute. Most patients present with hemorrhage, abdominal pain, or intussusception. Because of the risk of massive gastrointestinal bleeding, hemangiomas are usually resected (37).
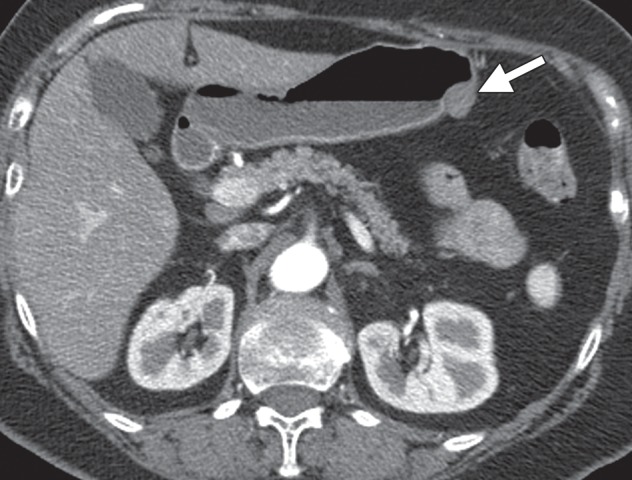
At endoscopy, hemangiomas are seen as bluish black intramural lesions. Fluoroscopy demonstrates an intramural mass similar to a GIST or other mesenchymal tumor. Contrast-enhanced CT shows an avidly enhancing, often intraluminal mass (Fig 11) (36). Phleboliths within the lesion are virtually pathognomonic. The differential diagnosis includes GIST, glomus tumor, and metastasis.
Figure 11.
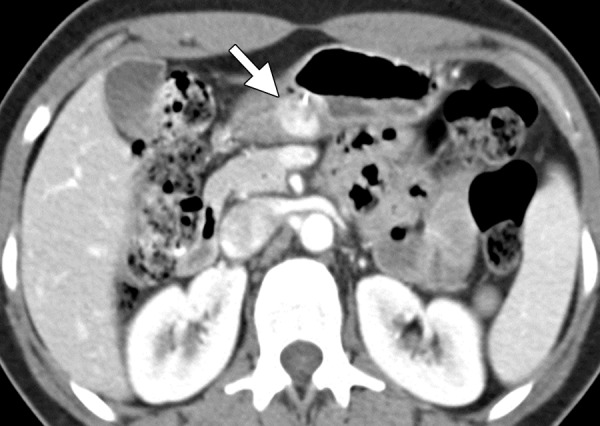
Hemangioma. Axial contrast-enhanced CT image shows an avidly enhancing hemangioma (arrow) in the gastric antrum.
Inflammatory Fibroid Polyp
IFPs have been reported in the literature under a variety of names, including Vanek tumor (38), eosinophilic granuloma, submucosal fibroma, and inflammatory pseudotumor. These lesions are unrelated to eosinophilic granulomas of the lung or bone and are not associated with peripheral eosinophilia.
Usually asymptomatic, IFPs may manifest with abdominal pain or gastrointestinal bleeding because of mucosal ulceration. In rare cases, pedunculated polyps in the gastric antrum may cause intermittent gastric outlet obstruction. Endoscopic resection is curative (39).
IFPs are usually solitary and can be found anywhere in the gastrointestinal tract, but 75% occur in the gastric antrum, followed by the gastric body and fundus. Because IFPs arise in the submucosa (Fig 12), they manifest as intraluminal lesions, which may mimic the appearance of adenomatous polyps, intraluminal GISTs, carcinoid tumors, or schwannomas (40). The tumors are often 2–5 cm in diameter, with a smooth or slightly lobulated contour.
Figure 12a.
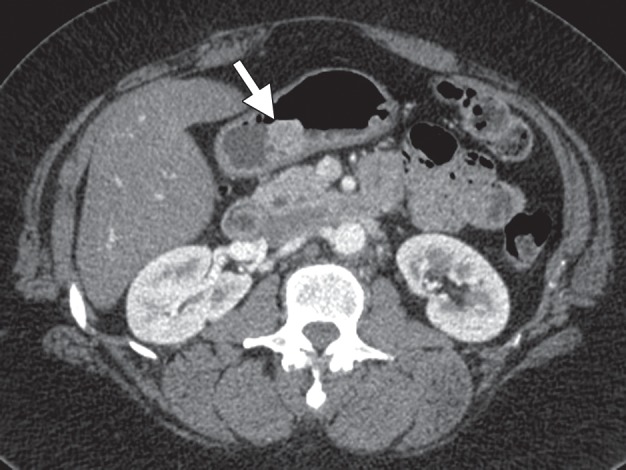
Incidentally discovered IFP in a 53-year-old woman with a history of thyroid carcinoma. (a) Axial contrast-enhanced CT image demonstrates an enhancing mass (arrow) in the gastric body. (b) Low-power photomicrograph (original magnification, ×20; hematoxylin-eosin stain) shows that the tumor arises in the submucosa but infiltrates into the mucosa (arrowheads), forming a polypoid lesion. The tumor is composed of bland spindle cells with an inflammatory component.
Figure 12b.
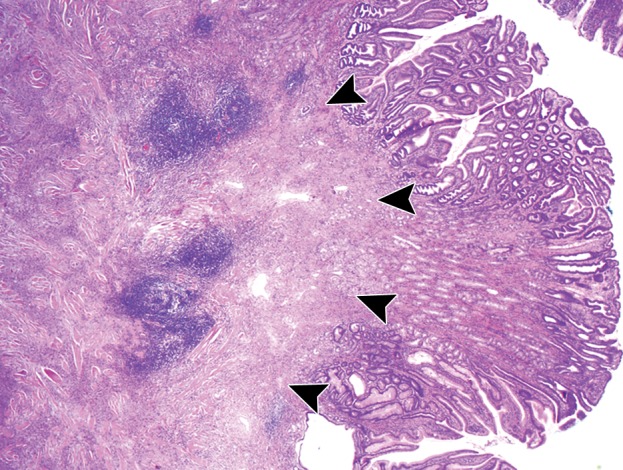
Incidentally discovered IFP in a 53-year-old woman with a history of thyroid carcinoma. (a) Axial contrast-enhanced CT image demonstrates an enhancing mass (arrow) in the gastric body. (b) Low-power photomicrograph (original magnification, ×20; hematoxylin-eosin stain) shows that the tumor arises in the submucosa but infiltrates into the mucosa (arrowheads), forming a polypoid lesion. The tumor is composed of bland spindle cells with an inflammatory component.
At histologic analysis, IFPs are composed of spindle cells arranged in whorls around blood vessels, with prominent inflammatory cells, particularly eosinophils (Fig 12) (41). At immunohistochemical analysis, the tumor stains for CD34 and is diffusely positive for vimentin.
Inflammatory Myofibroblastic Tumor
Originally believed to be a reactive inflammatory process, IMFT is now acknowledged to be a neoplasm of intermediate malignant potential (42), with a propensity for local recurrence and a low rate of metastasis. IMFTs can occur in the abdominal cavity, lung, mediastinum, and retroperitoneum, as well as in solid organs. Young adults and children are most often affected, although the tumor can occur in patients of any age (42,43). Complete resection is the preferred treatment.
On CT images, IMFTs appear as enhancing, aggressive intramural masses (43), which can invade through the gastric wall into the peritoneal cavity or nearby organs (Fig 13) (44). Peritoneal dissemination and lymphadenopathy are rare. Differential diagnosis includes GIST, solitary fibrous tumor, and schwannoma.
Figure 13.
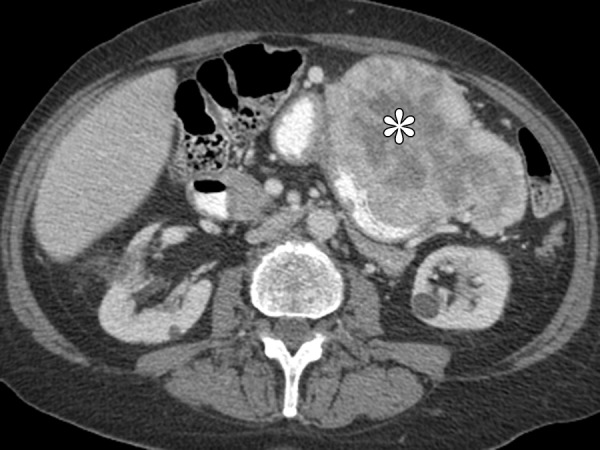
IMFT (pleomorphic variant) in a 69-year-old woman with fever, night sweats, and coagulopathy. Axial contrast-enhanced CT image shows an intramural, heterogeneously enhancing tumor (*) with endoluminal and exophytic growth.
At histopathologic analysis, IMFT is composed of myofibroblastic spindle cells, with a prominent inflammatory cell infiltrate composed of plasma cells, lymphocytes, and eosinophils. The myofibroblastic tumor cells often show reactivity for anaplastic lymphoma kinase (ALK). ALK-negative IMFTs have been associated with older patient age at presentation and can manifest with distant metastases (42). The spindle cells additionally stain with smooth muscle actin, consistent with myofibroblastic differentiation.
Plexiform Fibromyxoma
Also called plexiform angiomyxoid myofibroblastic tumor, plexiform fibromyoma is a benign tumor that appears to be unique to the stomach, almost always occurring in the gastric antrum (Fig 14) (45,46). These intramural tumors range in size from 2 cm to 15 cm and can demonstrate ulceration or mucosal invasion (45,46).
Figure 14a.
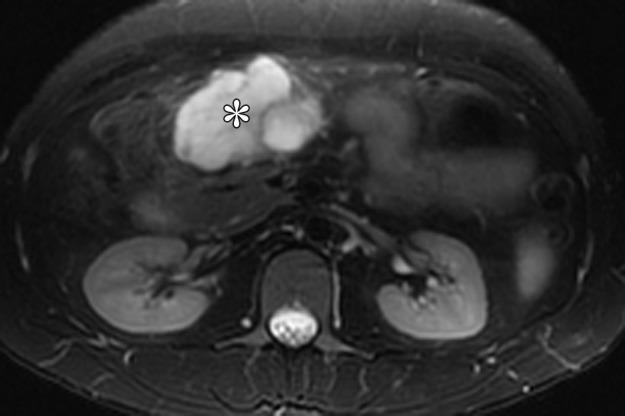
Plexiform fibromyxoma in a 31-year-old man with Peutz-Jeghers syndrome who presented with abdominal pain and melena. (a) Axial T2-weighted magnetic resonance (MR) image shows a hyperintense mass (*) in the gastric antrum. (b) Axial fat-saturated T1-weighted image, obtained after administration of gadolinium contrast material, shows that the mass (*) is avidly enhancing. The mass demonstrated fluid attenuation on CT images (not shown), a finding consistent with myxoid composition.
Figure 14b.
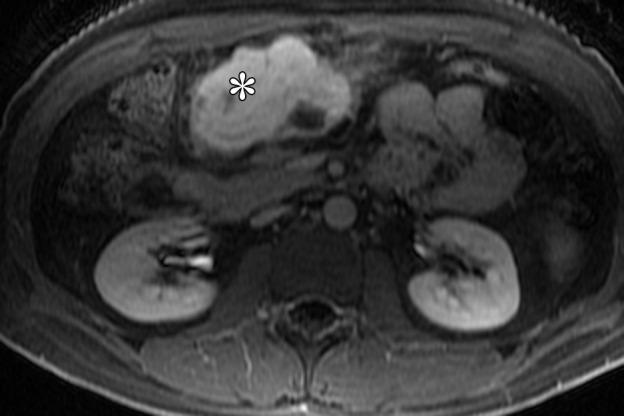
Plexiform fibromyxoma in a 31-year-old man with Peutz-Jeghers syndrome who presented with abdominal pain and melena. (a) Axial T2-weighted magnetic resonance (MR) image shows a hyperintense mass (*) in the gastric antrum. (b) Axial fat-saturated T1-weighted image, obtained after administration of gadolinium contrast material, shows that the mass (*) is avidly enhancing. The mass demonstrated fluid attenuation on CT images (not shown), a finding consistent with myxoid composition.
Patients with plexiform fibromyxoma may present with gastrointestinal bleeding, an abdominal mass, or gastric outlet obstruction (45,46).
On CT images, the tumor demonstrates areas of low attenuation owing to presence of myxoid tissue, interspersed with foci of vascularity. On MR images, the myxoid stroma of the tumor is T2-hyperintense with persistent enhancement after administration of contrast material (Fig 14). The differential diagnosis of plexiform fibromyxoma includes schwannoma, GIST, neurofibroma, and myxoid liposarcoma.
At histologic analysis, the tumors are composed of multiple nodules of myxoid stroma with a prominent capillary network, originating within the muscularis propria. The tumor infiltrates the surrounding muscularis propria and can extend into the submucosa and mucosa. At immunohistochemical analysis, the tumors are positive for smooth muscle actin (45).
Carcinoid Tumor
Carcinoid tumors are well-differentiated neuroendocrine tumors that originate from the diffuse endocrine system. These tumors occur most frequently in the gastrointestinal tract (67% of cases), followed by the tracheobronchial system (24%). In the gastrointestinal tract, the small bowel is the most common location, followed by the rectum, appendix, and stomach (47). Gastric carcinoid tumors are rare, accounting for 1.8% of all gastric malignancies (48).
Carcinoid tumors usually originate from enterochromaffin-like cells (Kulchitsky cells) in the gastric mucosa and are therefore epithelial in origin. However, often the bulk of the tumor is submucosal, and they should be included in the differential diagnosis of a submucosal gastric tumor.
Of the three types of gastric carcinoids, types 1 and 2 are associated with hypergastrinemia, which causes proliferation of enterochromaffin-like cells and thus promotes the development of often multiple carcinoid tumors (49). The majority (75%–80%) of gastric carcinoids are type 1, in which patients have atrophic metaplastic autoimmune gastritis with or without pernicious anemia (Fig 15). Nodal and hepatic metastases are rare in type 1 carcinoid tumors (50). Type 2 tumors account for 5%–10% of gastric carcinoids and are caused by hypergastrinemia from a gastrin-producing tumor in association with multiple endocrine neoplasia (MEN) type 1. Approximately 30% of patients with MEN 1 have gastric carcinoid tumors (51). Patients with type 2 gastric carcinoids are prone to developing local lymph node metastases.
Figure 15a.
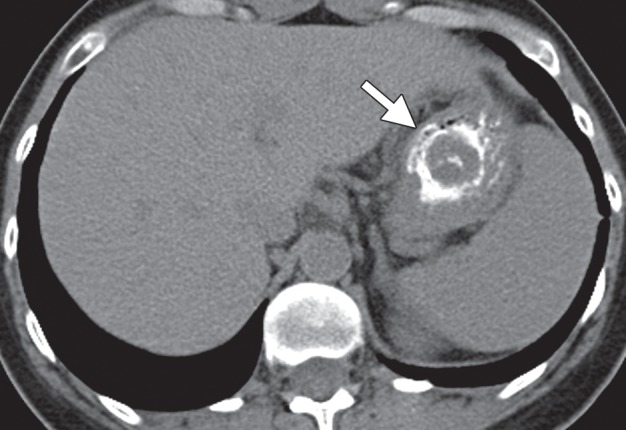
Type 1 gastric carcinoid. (a, b) Precontrast (a) and postcontrast (b) axial CT images, obtained in a 53-year-old woman with a 3-month history of abdominal pain, weight loss, and elevated levels of serum gastrin and chromogranin A, show an avidly enhancing mass (arrow) in the gastric body, with central ulceration. The mass was an unusual, solitary type 1 carcinoid arising in a background of atrophic gastritis. (c) CT image fused with scintigraphy performed after injection of indium 111 pentetreotide (Octreoscan) demonstrates avid radiotracer uptake in the gastric mass (arrow), characteristic of carcinoid tumor. The patient (same as in a and b) remained without evidence of disease 15 months after surgery, without further treatment. (d) Axial contrast-enhanced CT image from a different case shows a more typical example of type 1 gastric carcinoid, with multiple enhancing polypoid lesions (arrowheads).
Figure 15b.
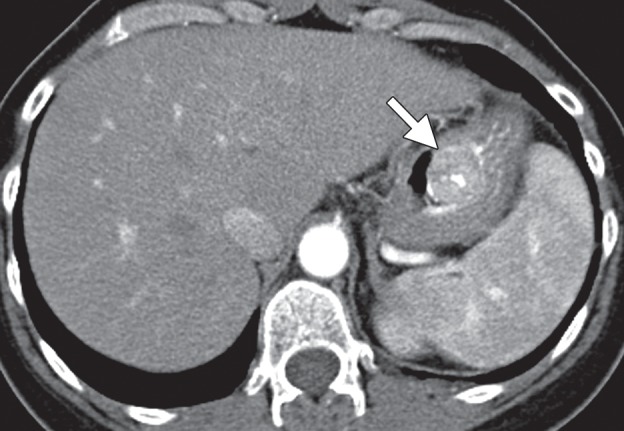
Type 1 gastric carcinoid. (a, b) Precontrast (a) and postcontrast (b) axial CT images, obtained in a 53-year-old woman with a 3-month history of abdominal pain, weight loss, and elevated levels of serum gastrin and chromogranin A, show an avidly enhancing mass (arrow) in the gastric body, with central ulceration. The mass was an unusual, solitary type 1 carcinoid arising in a background of atrophic gastritis. (c) CT image fused with scintigraphy performed after injection of indium 111 pentetreotide (Octreoscan) demonstrates avid radiotracer uptake in the gastric mass (arrow), characteristic of carcinoid tumor. The patient (same as in a and b) remained without evidence of disease 15 months after surgery, without further treatment. (d) Axial contrast-enhanced CT image from a different case shows a more typical example of type 1 gastric carcinoid, with multiple enhancing polypoid lesions (arrowheads).
Figure 15c.
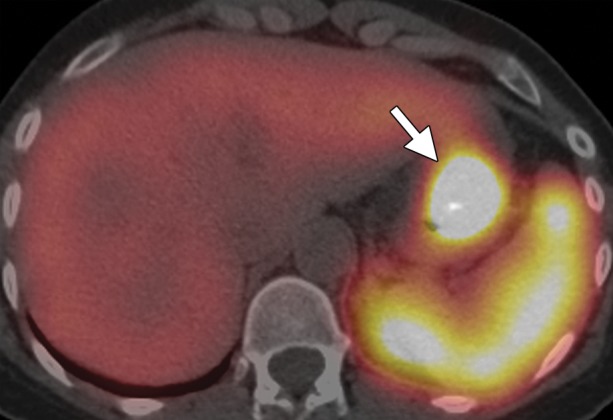
Type 1 gastric carcinoid. (a, b) Precontrast (a) and postcontrast (b) axial CT images, obtained in a 53-year-old woman with a 3-month history of abdominal pain, weight loss, and elevated levels of serum gastrin and chromogranin A, show an avidly enhancing mass (arrow) in the gastric body, with central ulceration. The mass was an unusual, solitary type 1 carcinoid arising in a background of atrophic gastritis. (c) CT image fused with scintigraphy performed after injection of indium 111 pentetreotide (Octreoscan) demonstrates avid radiotracer uptake in the gastric mass (arrow), characteristic of carcinoid tumor. The patient (same as in a and b) remained without evidence of disease 15 months after surgery, without further treatment. (d) Axial contrast-enhanced CT image from a different case shows a more typical example of type 1 gastric carcinoid, with multiple enhancing polypoid lesions (arrowheads).
Figure 15d.
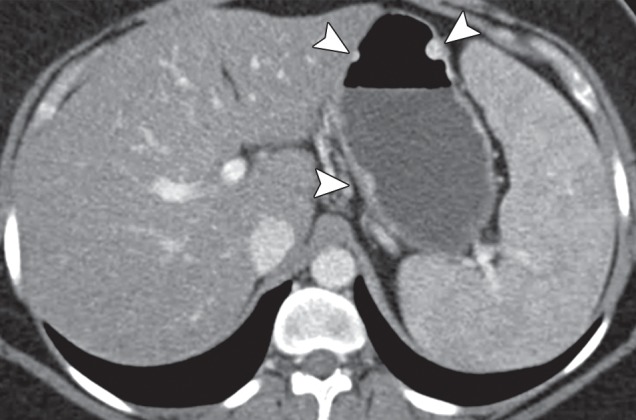
Type 1 gastric carcinoid. (a, b) Precontrast (a) and postcontrast (b) axial CT images, obtained in a 53-year-old woman with a 3-month history of abdominal pain, weight loss, and elevated levels of serum gastrin and chromogranin A, show an avidly enhancing mass (arrow) in the gastric body, with central ulceration. The mass was an unusual, solitary type 1 carcinoid arising in a background of atrophic gastritis. (c) CT image fused with scintigraphy performed after injection of indium 111 pentetreotide (Octreoscan) demonstrates avid radiotracer uptake in the gastric mass (arrow), characteristic of carcinoid tumor. The patient (same as in a and b) remained without evidence of disease 15 months after surgery, without further treatment. (d) Axial contrast-enhanced CT image from a different case shows a more typical example of type 1 gastric carcinoid, with multiple enhancing polypoid lesions (arrowheads).
At radiologic examination, type 1 gastric carcinoid tumors often appear as multiple polyps of less than 2 cm in diameter in the gastric fundus, although they can be solitary. Type 2 carcinoids can appear remarkable because of the presence of multiple masses in the setting of diffuse gastric wall thickening. Differential diagnosis of multiple gastric lesions includes carcinoid tumors, hyperplastic polyps, adenomatous polyps (familial adenomatous polyposis syndrome), hamartomatous polyps (Peutz-Jeghers syndrome), multiple hamartomas (Cowden disease), and metastases (usually intramural).
Type 3 gastric carcinoids (accounting for roughly 13% of all gastric carcinoids) are sporadic tumors that are not associated with hypergastrinemia (50,52). Unlike type 1 and 2 gastric carcinoids, type 3 tumors are usually solitary submucosal tumors that may show ulceration and that are more likely to be recurrent and invasive with distant metastases. The likelihood of metastasis and perigastric lymphadenopathy depends on tumor size. Prognosis is poor, with 20% of patients surviving 5 years. Differential diagnosis of type 3 carcinoid tumors should include adenocarcinoma, lymphoma, and GIST.
Type 1 gastric carcinoid tumors are often incidentally found during imaging studies performed for dyspepsia or chronic atrophic gastritis (53). Patients with type 2 gastric carcinoids have symptoms of Zollinger-Ellison syndrome, with abdominal pain or bleeding from multiple ulcers, diarrhea, and elevated levels of serum gastrin. Type 3 carcinoids have no endocrine manifestations, and patients present with abdominal pain, gastrointestinal bleeding, or weight loss.
At histologic examination, carcinoid tumors usually appear in the mucosa and submucosa. They are composed of monomorphic cells arranged in trabecular or rosette patterns, with inconspicuous nucleoli and a distinctive “salt and pepper” pattern of nuclear chromatin. These tumors show immunoreactivity to chromogranin A and synaptophysin (Fig 16).
Figure 16a.
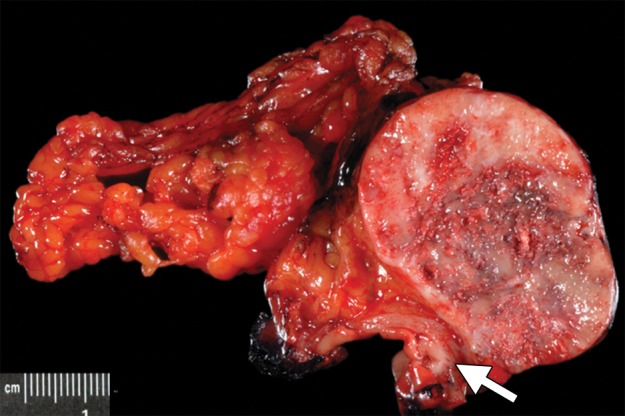
Type 1 gastric carcinoid tumor arising in a background of autoimmune metaplastic atrophic gastritis (same case as in Fig 15a–15c). (a) Photograph of a partial gastrectomy specimen shows an ovoid mass deep to the mucosa (arrow), with a heterogeneous cut surface. (b) Low-power photomicrograph (original magnification, ×20; hematoxylin-eosin stain) demonstrates a hypercellular tumor intermixed with hyalinized stroma. The bulk of the tumor is deep to the mucosa, which is atrophic and inflamed. (c) High-power photomicrograph (original magnification, ×200; hematoxylin-eosin stain) shows that the tumor is composed of round bland cells arranged in an insular growth pattern with intervening hyalinized stroma that contains small vessels. (d) Low-power photomicrograph (original magnification, ×40; immunohistochemical stain for synaptophysin) shows brown staining of cytoplasm, a finding indicative of diffuse positivity for synaptophysin.
Figure 16b.
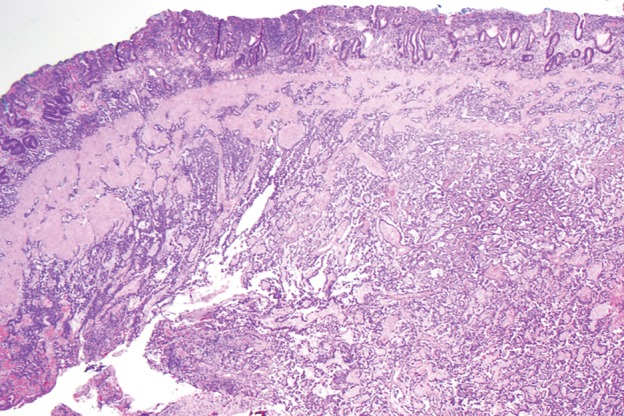
Type 1 gastric carcinoid tumor arising in a background of autoimmune metaplastic atrophic gastritis (same case as in Fig 15a–15c). (a) Photograph of a partial gastrectomy specimen shows an ovoid mass deep to the mucosa (arrow), with a heterogeneous cut surface. (b) Low-power photomicrograph (original magnification, ×20; hematoxylin-eosin stain) demonstrates a hypercellular tumor intermixed with hyalinized stroma. The bulk of the tumor is deep to the mucosa, which is atrophic and inflamed. (c) High-power photomicrograph (original magnification, ×200; hematoxylin-eosin stain) shows that the tumor is composed of round bland cells arranged in an insular growth pattern with intervening hyalinized stroma that contains small vessels. (d) Low-power photomicrograph (original magnification, ×40; immunohistochemical stain for synaptophysin) shows brown staining of cytoplasm, a finding indicative of diffuse positivity for synaptophysin.
Figure 16c.
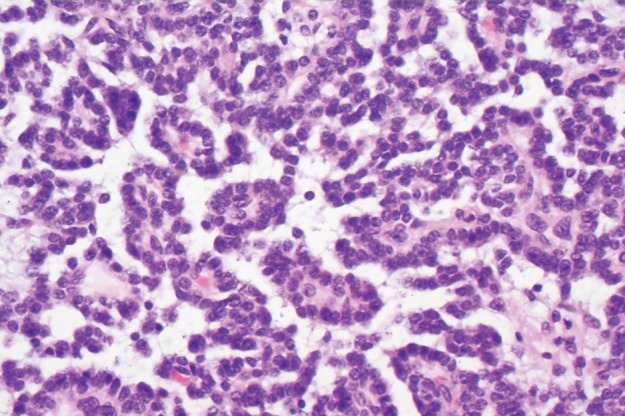
Type 1 gastric carcinoid tumor arising in a background of autoimmune metaplastic atrophic gastritis (same case as in Fig 15a–15c). (a) Photograph of a partial gastrectomy specimen shows an ovoid mass deep to the mucosa (arrow), with a heterogeneous cut surface. (b) Low-power photomicrograph (original magnification, ×20; hematoxylin-eosin stain) demonstrates a hypercellular tumor intermixed with hyalinized stroma. The bulk of the tumor is deep to the mucosa, which is atrophic and inflamed. (c) High-power photomicrograph (original magnification, ×200; hematoxylin-eosin stain) shows that the tumor is composed of round bland cells arranged in an insular growth pattern with intervening hyalinized stroma that contains small vessels. (d) Low-power photomicrograph (original magnification, ×40; immunohistochemical stain for synaptophysin) shows brown staining of cytoplasm, a finding indicative of diffuse positivity for synaptophysin.
Figure 16d.
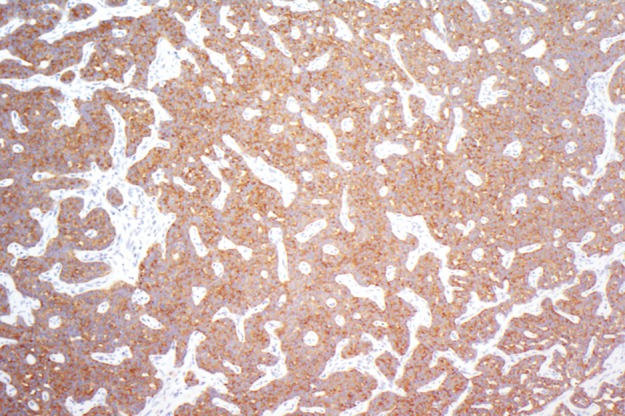
Type 1 gastric carcinoid tumor arising in a background of autoimmune metaplastic atrophic gastritis (same case as in Fig 15a–15c). (a) Photograph of a partial gastrectomy specimen shows an ovoid mass deep to the mucosa (arrow), with a heterogeneous cut surface. (b) Low-power photomicrograph (original magnification, ×20; hematoxylin-eosin stain) demonstrates a hypercellular tumor intermixed with hyalinized stroma. The bulk of the tumor is deep to the mucosa, which is atrophic and inflamed. (c) High-power photomicrograph (original magnification, ×200; hematoxylin-eosin stain) shows that the tumor is composed of round bland cells arranged in an insular growth pattern with intervening hyalinized stroma that contains small vessels. (d) Low-power photomicrograph (original magnification, ×40; immunohistochemical stain for synaptophysin) shows brown staining of cytoplasm, a finding indicative of diffuse positivity for synaptophysin.
Conclusions
Intramural gastric masses commonly have overlapping radiologic appearances. Attention to a combination of imaging features, such as tumor margin, location, attenuation, enhancement, and pattern of growth, may help suggest one diagnosis over another. Evaluation of the tumor margin helps distinguish intramural versus mucosal lesions, as well as potentially malignant from benign intramural tumors. Figure 17 shows one possible approach to characterizing an intramural mass.
Figure 17.

Diagram summarizes an approach to the differential diagnosis of a solitary intramural gastric tumor.
Location of the tumor is important in establishing a differential diagnosis, because some tumors are seen more frequently in certain parts of the stomach; for example, leiomyomas are almost always found in the gastric cardia (the region of the stomach just distal to the lower esophageal sphincter). GISTs and schwannomas often manifest in the body of the stomach. Glomus tumors, IFPs, lipomas, and ectopic pancreas are commonly seen in the gastric antrum.
The finding of a mass with fat attenuation is diagnostic of a lipoma. Leiomyomas and schwannomas are usually low attenuation. Even large schwannomas are characteristically well-circumscribed with homogeneous attenuation, a feature that may be useful in differentiating schwannomas from the more heterogeneous GISTs. Avid enhancement is often seen with glomus tumors, carcinoid tumors, small GISTs, metastases, ectopic pancreas, and hemangiomas. Phleboliths are pathognomonic of a hemangioma, a tumor that rarely occurs in the stomach.
A polypoid growth pattern suggests a differential diagnosis of carcinoid tumor, IFP, GIST, adenomatous polyp, or polypoid cancer. Tumors with exophytic growth patterns include GIST, schwannoma, lymphoma, or poorly differentiated adenocarcinoma. Among this group of tumors, GIST is typically heterogeneous with areas of cystic or necrotic change and lacks lymphadenopathy.
Size is also a consideration, and leiomyomas, glomus tumors, IFPs, and ectopic pancreas are usually less than 5 cm. The presence of multiple lesions should raise the possibility of carcinoid tumors (types 1 and 2), multiple polyps, or metastases. Melanoma, breast cancer, lung cancer, and esophageal carcinoma are the most likely primary malignancies to secondarily involve the stomach.
Lastly, GIST is the most common mesenchymal tumor of the stomach and can have a wide variety of appearances; thus, it will often be included in the differential diagnosis of an intramural tumor. Histologic analysis is ultimately necessary for confirmation.
Acknowledgments
We thank David Bier and Kelly Duggan at M.D. Anderson Cancer Center for producing the medical illustration and for editing the diagnostic images in this article, respectively.
Recipient of a Certificate of Merit award for an education exhibit at the 2012 RSNA Annual Meeting
For this journal-based SA-CME activity, the authors, editor, and reviewers have no financial relationships to disclose
Funding: The work was supported in part by the National Institutes of Health through a grant to M.D. Anderson Cancer Center [grant number CA016672].
Abbreviations:
- GIST
- gastrointestinal stromal tumor
- IFP
- inflammatory fibroid polyp
- IMFT
- inflammatory myofibroblastic tumor
References
- 1.Hamilton SR, Aaltonen LA. World Health Organization classification of tumors: pathology and genetics of tumours of the digestive system. Lyon, France: IARC, 2000 [Google Scholar]
- 2.Lee NK, Kim S, Kim GH, et al. Hypervascular subepithelial gastrointestinal masses: CT-pathologic correlation. RadioGraphics 2010;30(7):1915–1934 [DOI] [PubMed] [Google Scholar]
- 3.Horton KM, Fishman EK. Current role of CT in imaging of the stomach. RadioGraphics 2003;23(1): 75–87 [DOI] [PubMed] [Google Scholar]
- 4.Ba-Ssalamah A, Prokop M, Uffmann M, Pokieser P, Teleky B, Lechner G. Dedicated multidetector CT of the stomach: spectrum of diseases. RadioGraphics 2003;23(3):625–644 [DOI] [PubMed] [Google Scholar]
- 5.Johnson PT, Horton KM, Fishman EK. Hypervascular gastric masses: CT findings and clinical correlates. AJR Am J Roentgenol 2010;195(6): W415–W420 [DOI] [PubMed] [Google Scholar]
- 6.Nishida T, Hirota S. Biological and clinical review of stromal tumors in the gastrointestinal tract. Histol Histopathol 2000;15(4):1293–1301 [DOI] [PubMed] [Google Scholar]
- 7.Sarlomo-Rikala M, Kovatich AJ, Barusevicius A, Miettinen M. CD117: a sensitive marker for gastrointestinal stromal tumors that is more specific than CD34. Mod Pathol 1998;11(8):728–734 [PubMed] [Google Scholar]
- 8.Joensuu H, Roberts PJ, Sarlomo-Rikala M, et al. Effect of the tyrosine kinase inhibitor STI571 in a patient with a metastatic gastrointestinal stromal tumor. N Engl J Med 2001;344(14):1052–1056 [DOI] [PubMed] [Google Scholar]
- 9.Benjamin RS, Choi H, Macapinlac HA, et al. We should desist using RECIST, at least in GIST. J Clin Oncol 2007;25(13):1760–1764 [DOI] [PubMed] [Google Scholar]
- 10.Heinrich MC, Corless CL, Duensing A, et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science 2003;299(5607):708–710 [DOI] [PubMed] [Google Scholar]
- 11.Miettinen M, Lasota J. Gastrointestinal stromal tumors: definition, clinical, histological, immunohistochemical, and molecular genetic features and differential diagnosis. Virchows Arch 2001;438(1):1–12 [DOI] [PubMed] [Google Scholar]
- 12.Levy AD, Remotti HE, Thompson WM, Sobin LH, Miettinen M. Gastrointestinal stromal tumors: radiologic features with pathologic correlation. RadioGraphics 2003;23(2):283–304 [DOI] [PubMed] [Google Scholar]
- 13.Franquemont DW. Differentiation and risk assessment of gastrointestinal stromal tumors. Am J Clin Pathol 1995;103(1):41–47 [DOI] [PubMed] [Google Scholar]
- 14.Fletcher CD, Berman JJ, Corless C, et al. Diagnosis of gastrointestinal stromal tumors: a consensus approach. Hum Pathol 2002;33(5):459–465 [DOI] [PubMed] [Google Scholar]
- 15.Agaimy A. Gastrointestinal stromal tumors (GIST) from risk stratification systems to the new TNM proposal: more questions than answers? A review emphasizing the need for a standardized GIST reporting. Int J Clin Exp Pathol 2010;3(5):461–471 [PMC free article] [PubMed] [Google Scholar]
- 16.Burkill GJ, Badran M, Al-Muderis O, et al. Malignant gastrointestinal stromal tumor: distribution, imaging features, and pattern of metastatic spread. Radiology 2003;226(2):527–532 [DOI] [PubMed] [Google Scholar]
- 17.Suster S. Gastrointestinal stromal tumors. Semin Diagn Pathol 1996;13(4):297–313 [PubMed] [Google Scholar]
- 18.DeMatteo RP, Lewis JJ, Leung D, Mudan SS, Woodruff JM, Brennan MF. Two hundred gastrointestinal stromal tumors: recurrence patterns and prognostic factors for survival. Ann Surg 2000;231(1):51–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Espinosa I, Lee CH, Kim MK, et al. A novel monoclonal antibody against DOG1 is a sensitive and specific marker for gastrointestinal stromal tumors. Am J Surg Pathol 2008;32(2):210–218 [DOI] [PubMed] [Google Scholar]
- 20.Agaimy A, Wünsch PH. True smooth muscle neoplasms of the gastrointestinal tract: morphological spectrum and classification in a series of 85 cases from a single institute. Langenbecks Arch Surg 2007;392(1):75–81 [DOI] [PubMed] [Google Scholar]
- 21.Turner MS, Goldsmith JD. Best practices in diagnostic immunohistochemistry: spindle cell neoplasms of the gastrointestinal tract. Arch Pathol Lab Med 2009;133(9):1370–1374 [DOI] [PubMed] [Google Scholar]
- 22.Miettinen M, Sarlomo-Rikala M, Sobin LH, Lasota J. Gastrointestinal stromal tumors and leiomyosarcomas in the colon: a clinicopathologic, immunohistochemical, and molecular genetic study of 44 cases. Am J Surg Pathol 2000;24(10):1339–1352 [DOI] [PubMed] [Google Scholar]
- 23.Aggarwal G, Sharma S, Zheng M, et al. Primary leiomyosarcomas of the gastrointestinal tract in the post-gastrointestinal stromal tumor era. Ann Diagn Pathol 2012;16(6):532–540 [DOI] [PubMed] [Google Scholar]
- 24.Thompson WM, Kende AI, Levy AD. Imaging characteristics of gastric lipomas in 16 adult and pediatric patients. AJR Am J Roentgenol 2003;181(4): 981–985 [DOI] [PubMed] [Google Scholar]
- 25.Hur BY, Kim SH, Choi JY, et al. Gastroduodenal glomus tumors: differentiation from other subepithelial lesions based on dynamic contrast-enhanced CT findings. AJR Am J Roentgenol 2011;197(6): 1351–1359 [DOI] [PubMed] [Google Scholar]
- 26.Lee MJ, Lim JS, Kwon JE, et al. Gastric true leiomyoma: computed tomographic findings and pathological correlation. J Comput Assist Tomogr 2007; 31(2):204–208 [DOI] [PubMed] [Google Scholar]
- 27.Sarlomo-Rikala M, Miettinen M. Gastric schwannoma: a clinicopathological analysis of six cases. Histopathology 1995;27(4):355–360 [DOI] [PubMed] [Google Scholar]
- 28.Levy AD, Quiles AM, Miettinen M, Sobin LH. Gastrointestinal schwannomas: CT features with clinicopathologic correlation. AJR Am J Roentgenol 2005;184(3):797–802 [DOI] [PubMed] [Google Scholar]
- 29.Hong HS, Ha HK, Won HJ, et al. Gastric schwannomas: radiological features with endoscopic and pathological correlation. Clin Radiol 2008;63(5): 536–542 [DOI] [PubMed] [Google Scholar]
- 30.Voltaggio L, Murray R, Lasota J, Miettinen M. Gastric schwannoma: a clinicopathologic study of 51 cases and critical review of the literature. Hum Pathol 2012;43(5):650–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Agaimy A, Märkl B, Kitz J, et al. Peripheral nerve sheath tumors of the gastrointestinal tract: a multicenter study of 58 patients including NF1-associated gastric schwannoma and unusual morphologic variants. Virchows Arch 2010;456(4):411–422 [DOI] [PubMed] [Google Scholar]
- 32.Williamson JM, Wadley MS, Shepherd NA, Dwerryhouse S. Gastric schwannoma: a benign tumour often mistaken clinically, radiologically and histopathologically for a gastrointestinal stromal tumour—a case series. Ann R Coll Surg Engl 2012;94(4): 245–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.de Rezende NA, Arantes V, de Toledo LL, et al. Plexiform neurofibroma: an unusual cause of GI bleeding and intestinal obstruction. Gastrointest Endosc 2009;70(2):396–398 [DOI] [PubMed] [Google Scholar]
- 34.Bakker JR, Haber MM, Garcia FU. Gastrointestinal neurofibromatosis: an unusual cause of gastric outlet obstruction. Am Surg 2005;71(2):100–105 [PubMed] [Google Scholar]
- 35.Miettinen M, Paal E, Lasota J, Sobin LH. Gastrointestinal glomus tumors: a clinicopathologic, immunohistochemical, and molecular genetic study of 32 cases. Am J Surg Pathol 2002;26(3):301–311 [DOI] [PubMed] [Google Scholar]
- 36.Levy AD, Abbott RM, Rohrmann CA, Jr, Frazier AA, Kende A. Gastrointestinal hemangiomas: imaging findings with pathologic correlation in pediatric and adult patients. AJR Am J Roentgenol 2001;177(5):1073–1081 [DOI] [PubMed] [Google Scholar]
- 37.Fishman SJ, Burrows PE, Leichtner AM, Mulliken JB. Gastrointestinal manifestations of vascular anomalies in childhood: varied etiologies require multiple therapeutic modalities. J Pediatr Surg 1998; 33(7):1163–1167 [DOI] [PubMed] [Google Scholar]
- 38.Vanek J. Gastric submucosal granuloma with eosinophilic infiltration. Am J Pathol 1949;25(3): 397–411 [PMC free article] [PubMed] [Google Scholar]
- 39.Carmack SW, Genta RM, Graham DY, Lauwers GY. Management of gastric polyps: a pathology-based guide for gastroenterologists. Nat Rev Gastroenterol Hepatol 2009;6(6):331–341 [DOI] [PubMed] [Google Scholar]
- 40.Harned RK, Buck JL, Shekitka KM. Inflammatory fibroid polyps of the gastrointestinal tract: radiologic evaluation. Radiology 1992;182(3):863–866 [DOI] [PubMed] [Google Scholar]
- 41.Daum O, Hes O, Vanecek T, et al. Vanek’s tumor (inflammatory fibroid polyp): report of 18 cases and comparison with three cases of original Vanek’s series. Ann Diagn Pathol 2003;7(6):337–347 [DOI] [PubMed] [Google Scholar]
- 42.Coffin CM, Hornick JL, Fletcher CD. Inflammatory myofibroblastic tumor: comparison of clinicopathologic, histologic, and immunohistochemical features including ALK expression in atypical and aggressive cases. Am J Surg Pathol 2007;31(4):509–520 [DOI] [PubMed] [Google Scholar]
- 43.Karnak I, Senocak ME, Ciftci AO, et al. Inflammatory myofibroblastic tumor in children: diagnosis and treatment. J Pediatr Surg 2001;36(6):908–912 [DOI] [PubMed] [Google Scholar]
- 44.Oto A, Akata D, Besim A. Peritoneal inflammatory myofibroblastic pseudotumor metastatic to the liver: CT findings. Eur Radiol 2000;10(9):1501. [DOI] [PubMed] [Google Scholar]
- 45.Miettinen M, Makhlouf HR, Sobin LH, Lasota J. Plexiform fibromyxoma: a distinctive benign gastric antral neoplasm not to be confused with a myxoid GIST. Am J Surg Pathol 2009;33(11):1624–1632 [DOI] [PubMed] [Google Scholar]
- 46.Takahashi Y, Suzuki M, Fukusato T. Plexiform angiomyxoid myofibroblastic tumor of the stomach. World J Gastroenterol 2010;16(23):2835–2840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Levy AD, Sobin LH. Gastrointestinal carcinoids: imaging features with clinicopathologic comparison. RadioGraphics 2007;27(1):237–257 [DOI] [PubMed] [Google Scholar]
- 48.Modlin IM, Lye KD, Kidd MA. 5-decade analysis of 13,715 carcinoid tumors. Cancer 2003;97(4): 934–959 [DOI] [PubMed] [Google Scholar]
- 49.Rindi G, Luinetti O, Cornaggia M, Capella C, Solcia E. Three subtypes of gastric argyrophil carcinoid and the gastric neuroendocrine carcinoma: a clinicopathologic study. Gastroenterology 1993;104(4): 994–1006 [DOI] [PubMed] [Google Scholar]
- 50.Rindi G, Bordi C, Rappel S, La Rosa S, Stolte M, Solcia E. Gastric carcinoids and neuroendocrine carcinomas: pathogenesis, pathology, and behavior. World J Surg 1996;20(2):168–172 [DOI] [PubMed] [Google Scholar]
- 51.Cadiot G, Cattan D, Mignon M. Diagnosis and treatment of ECL cell tumors. Yale J Biol Med 1998; 71(3-4):311–323 [PMC free article] [PubMed] [Google Scholar]
- 52.Binstock AJ, Johnson CD, Stephens DH, Lloyd RV, Fletcher JG. Carcinoid tumors of the stomach: a clinical and radiographic study. AJR Am J Roentgenol 2001;176(4):947–951 [DOI] [PubMed] [Google Scholar]
- 53.Dakin GF, Warner RR, Pomp A, Salky B, Inabnet WB. Presentation, treatment, and outcome of type 1 gastric carcinoid tumors. J Surg Oncol 2006;93(5): 368–372 [DOI] [PubMed] [Google Scholar]