

Abstract
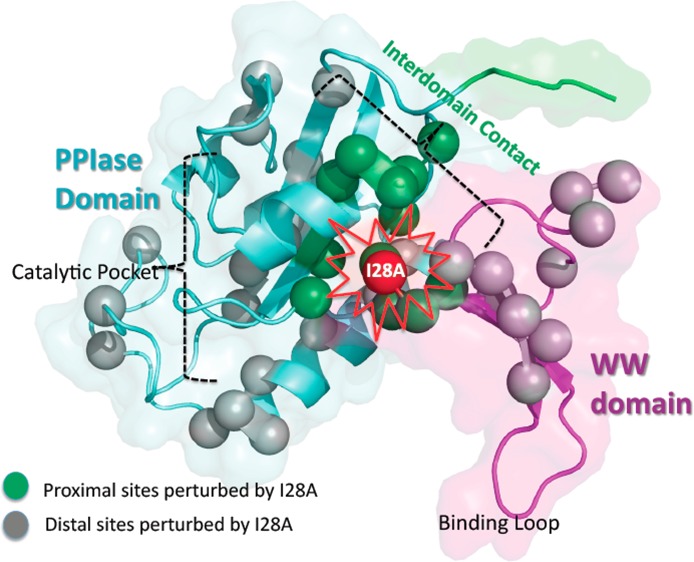
Pin1 is an essential mitotic regulator consisting of a peptidyl–prolyl isomerase (PPIase) domain flexibly tethered to a smaller Trp–Trp (WW) binding domain. Communication between these domains is important for Pin1 in vivo activity; however, the atomic basis for this communication has remained elusive. Our previous nuclear magnetic resonance (NMR) studies of Pin1 functional dynamics suggested that weak interdomain contacts within Pin1 enable allosteric communication between the domain interface and the distal active site of the PPIase domain.1,2 A necessary condition for this hypothesis is that the intrinsic properties of the PPIase domain should be sensitive to interdomain contact. Here, we test this sensitivity by generating a Pin1 mutant, I28A, which weakens the wild-type interdomain contact while maintaining the overall folds of the two domains. Using NMR, we show that I28A leads to altered substrate binding affinity and isomerase activity. Moreover, I28A causes long-range perturbations to conformational flexibility in both domains, for both the apo and substrate-complexed states of the protein. These results show that the distribution of conformations sampled by the PPIase domain is sensitive to interdomain contact and strengthen the hypothesis that such contact supports interdomain allosteric communication in Pin1. Other modular systems may exploit interdomain interactions in a similar manner.
The proteins regulating the cell cycle frequently adopt modular designs that use separate domains to carry out distinct and complementary functions, such as binding and catalysis.3,4 To dissect the mechanisms of these proteins, structural biology has traditionally followed a reductionist approach that focuses on the behavior of isolated domains. But, of course, it is the interdomain interactions that give rise to the rich diversity of protein function. In particular, it is now clear that interdomain interactions provide autoinhibition for many protein functions including kinase activity, transcriptional activation, and nuclear localization (see, e.g., review by Pufall and Graves5). Thus, comprehending protein function requires a scrutiny of interdomain interactions to complement those of the isolated domains. Moreover, at a practical level, these interdomain interactions represent enticing opportunities for novel modes of drug targeting. For example, small molecules that interfere with interdomain interactions are candidates for therapeutic allosteric inhibitors.6,7
The above considerations have motivated our studies of interdomain interactions within human Pin1.8 Pin1 is an essential mitotic regulator that consists of a Trp–Trp (WW) docking domain (residues 1–39) flexibly tethered to a larger peptidyl–prolyl isomerase (PPIase) domain (residues 50–163). Pin1 catalyzes the cis–trans isomerization of phosphorylated Ser/Thr–Pro (pS/T–P) segments in intrinsically disordered regions of other cell cycle proteins9−11 and is a potential therapeutic target for both cancer and Alzheimer’s disease. Both the PPIase and WW domains are specific for pS/T–P segments. The PPIase domain is solely responsible for pS/T–P isomerization, whereas the WW domain functions as a noncatalytic binder of pS/T–P segments.12−14 The “WW” refers to two conserved tryptophans (W), separated by ∼20–22 residues, that are a defining feature of this binding domain family.15
There is compelling evidence for functional interdomain interactions in Pin1. Specifically, while the isolated PPIase and WW domains retain isomerase and binding capability, respectively, in vitro, full-length Pin1 is essential for in vivo activity.16,17 Thus, some form of interdomain communication must exist. Yet, the nature of this communication remains unclear. The most straightforward explanation is that the WW domain, being proximal to the PPIase domain via the interdomain linker, simply increases the local concentration of substrate available to the PPIase active site.18,19 In this scenario, the WW domain acts as an independent binding module. As such, it exerts its influence on the PPIase domain indirectly; it does not alter the distribution of conformations sampled by the PPIase domain, or any properties derived thereof.
Another possible explanation for interdomain communication involves physical contact between the two domains. Previous solution NMR studies have demonstrated weak, transient interdomain interactions for apo Pin120 that intensify upon addition of phosphopeptide substrate.21 Additionally, the original X-ray crystal structure of Pin1 (PDB id 1PIN) depicted a contact interface between the two domains, stabilized in part by an interstitial PEG molecule derived from the crystallization conditions.22 However, how such interdomain contact might serve interdomain communication has been unclear.
Fresh insight linking Pin1 interdomain contact with interdomain communication has come from our previous NMR studies of Pin1 conformational dynamics.1,2 In those studies, we showed that the substrate binding to Pin1 not only enhances its interdomain interactions but also causes a loss of subnanosecond flexibility along a “conduit” of conserved hydrophobic residues in the PPIase domain that link the PPIase–WW domain interface with the distal PPIase active site. We also compared the binding affinity of full-length Pin1 versus the isolated PPIase domain for a phosphoserine peptidomimetic inhibitor locked in this cis conformation.2,23 Critically, that inhibitor bound only to the PPIase catalytic site and not the WW domain. A comparison of the inhibitor binding affinity of full-length Pin1 versus the isolated PPIase domain thus compared the effects of the presence versus absence of WW domain contact. We found 2–4 times higher binding affinity for the isolated PPIase domain compared with full-length Pin1.2 The combined dynamics and binding results led us to hypothesize that interdomain contact allows the WW domain to allosterically regulate the PPIase domain via changes in flexibility among the residues linking the interdomain interface with the PPIase active site.2
These previous findings set the stage for the present work, which is a more direct investigation of Pin1 interdomain contact and its hypothesized role as a mediator for interdomain allostery. In particular, a necessary condition for our allosteric hypothesis is that WW domain contact with the PPIase domain should alter some internal atomic properties of the PPIase domain pertinent to binding, activity, or both. To investigate if this is so, we have conducted NMR studies of a Pin1 mutant containing an alanine substitution designed to weaken Pin1’s capacity for interdomain contact. Specifically, we have generated the isoleucine (I) to alanine (A) substitution mutant, I28A.
Our choice of I28 derives from several previous structural and biophysical studies of Pin1. First, as suggested by the original Pin1 crystal structure (PDB id 1PIN),22 I28 is within the WW domain β2−β3 loop (Loop II, H27–N30), which forms the WW domain side of the domain interface (Figure 1). Also, in our original study of Pin1 side-chain dynamics,1 I28 emerged as part of the aforementioned conduit of conserved hydrophobic residues that lose subnanosecond side-chain flexibility upon substrate binding. Finally, extensive Pin1–WW mutation work by Kelly and co-workers (24) suggested that an I28A substitution would preserve overall folding.
Figure 1.
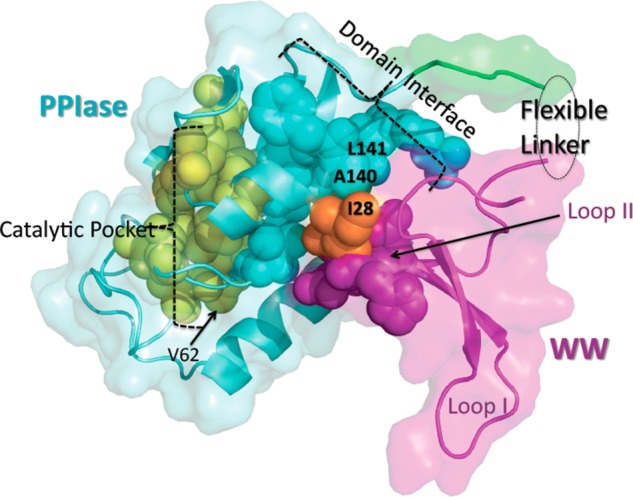
Structure of human Pin1 (PDB id code 1PIN(22)) with key regions color-highlighted. Aquamarine and magenta shading indicate the PPIase and WW domains, respectively. Yellow spheres are selected PPIase domain residues responsible for substrate recognition and catalysis (H59, L61, V62, K63, R68, R69, C113, L122, M130, and F134). Aquamarine spheres are PPIase domain residues at the domain interface (S138, S138, A140, L141, and R142). Magenta spheres are WW domain residues at the domain interface, which include Loop II (H27, I28, T29, and N30). I28, the mutated residue, is in orange. The same Pin1 structure (PDB id code 1PIN(22)) is used for all subsequent figures.
Our main results show that I28A indeed has reduced interdomain contact compared with wild-type Pin1 (WT), which alters substrate binding affinity, isomerase activity, and conformational dynamics (both backbone and side chain). These consequences reveal that (i) WW domain contact with the PPIase domain perturbs the PPIase domain’s intrinsic properties pertinent to substrate binding and activity, (ii) critical mediators of this interdomain contact are I28 and its host WW domain Loop II (H27–N30), as well as the PPIase domain residues S138–R142, and (iii) interdomain contact influences the aforementioned dynamic conduit response, and thus, the means for interdomain allosteric communication between the PPIase domain interface with its catalytic site.
Materials and Methods
Preparation of the I28A Mutant
To make the single-site alanine mutant I28A, we used site-directed mutagenesis by PCR, starting from the wild-type Pin1 construct. The mutated construct was verified by DNA sequencing (Genetics and Bioinformatics Core Facility at the University of Notre Dame). I28A was overexpressed and isotope-labeled using Escherichia coli BL21 (DE3) (Novagen) cells at 25 °C with 50% (v/v) D2O M9 media with 15NH4Cl and d-glucose (13C6) as the sole nitrogen and carbon sources, respectively. Overexpression and purification followed procedures outlined in our previous Pin1 work.1 The final sample was concentrated and dialyzed against the NMR buffer: 30 mM imidazole-d4 (pH 6.6), 5 mM DTT-d10, 30 mM NaCl, 0.03% NaN3, and 90% H2O/10% D2O. Folding was confirmed by comparing the two-dimensional (2-D) 15N–1H heteronuclear single quantum correlation (HSQC) spectra of I28A with wild-type Pin1.
Far-UV Circular Dichroism (CD) Spectroscopy
CD measurements were performed in 20 mM NaH2PO4, pH 7.5 on a Jasco J-815 spectropolarimeter. Far-UV CD spectral acquisitions used a 1 mm path length cuvette with protein concentrations ranging between 2 and 10 μM. Thermal denaturation was monitored at 200 nm over a temperature range of 20–80 °C with 1 min thermal equilibrations for each 1 °C step.
NMR Resonance Assignments and Chemical Shift Perturbations
We recorded NMR spectra at 295 K using Bruker Avance 700 MHz (16.4 T) and 800 MHz (18.8 T) spectrometers equipped with cryogenically cooled probes. I28A backbone assignments (1HN, 15N, 13Cα, and 13Cβ) were confirmed using three-dimensional (3-D) HNCACB,25 HNCOCACB,26 and 2-D 1H–15N HSQC27 pulse schemes. Side-chain aliphatic 13C and 1H resonances were assigned by comparisons with the corresponding wild-type Pin1 spectra. Fourier transformation and NMR resonance assignment used Topspin 1.3 and 2.1 (Bruker Biospin, Inc.) and Sparky (T. D. Goddard and D. G. Kneller, SPARKY 3, University of California, San Francisco). The 15N–1H chemical shift perturbations of the protein in state A relative to state B were defined as
| 1 |
where ΔδNA–B = δN – δNB and ΔδH = δHA – δH. For assessing mutation effects, A was the mutant, whereas B was wild-type Pin1. For assessing ligand-binding effects, A was the protein–ligand complex, whereas B was the apo protein. Backbone 13Cα/β shift changes between apo I28A and apo WT Pin1 were determined from comparing their HNCACB/HNCOCACB spectra.
Kd Values from Chemical Shift Perturbations
For binding studies, the chemical shift perturbations were interpreted in terms of a simple equilibrium
| 2 |
where P, L, and PL are the free protein, free ligand, and protein–ligand complex, respectively. We fitted the chemical shift perturbation versus the ratio of total ligand to total protein (LT/PT) to
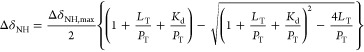 |
3 |
Equation 3 assumes fast binding exchange on the chemical shift time scale. The adjustable parameters were the equilibrium dissociation constant, Kd, and the maximum chemical shift perturbation at saturation, ΔδNH,max. Uncertainties in the fitted parameters were estimated by jack-knife simulations.
Measurements of Cis–Trans Isomerase Activity
We used two methods to measure cis–trans isomerase activity. The first method was the standard chromogenic coupled-assay of Kofron et al.28 The chromogenic substrate, suc-AEPF-pNA was purchased from Sigma. The assay procedures were the same as in our previous Pin1 study.1 The second method was 2-D 1H–1H exchange spectroscopy (EXSY), using a NOESY-based pulse scheme.29 The substrate was a ten-residue phosphothreonine peptide, EQPLpTPVTDL (Anaspec), which is an established proxy for the Pin1 target site in the mitotic phosphatases Cdc25C.14,16 Experiments were carried out at 18.8T (800 MHz 1H Larmor frequency), 295 K. The mixing times for exchange were 1, 20, 30, 40, 50, 60, 80, 100, and 200 (×2) ms. Samples consisted of 50 μM fresh protein (wild-type Pin1, I28A, or the isolated PPIase domain) in the presence of 2 mM Cdc25C phosphopeptide substrate. Exchange rate constants, kEXSY, were estimated by fitting the ratios of the trans-to-cis exchange cross-peaks over the trans diagonal peaks as function of the mixing time tmix, to the two-state expression29,30
| 4 |
The two adjustable parameters were kTC and kCT and kEXSY = kTC + kCT. The exchange cross-peaks were assigned by comparison to 2-D 1H–1H total correlation spectroscopy (TOCSY)31,32 and rotating-frame nuclear Overhauser effect correlation spectroscopy (ROESY)33 spectra. In samples containing just 2 mM Cdc25C phosphopeptide (no protein), the EXSY cross-peaks were absent because the exchange was beyond the limit of detection (too slow).
NMR Spin Relaxation Experiments and Analysis
All backbone R1(15N), R2(15N), and steady-state 1HN–15N NOE (ssNOE) values were measured at 16.4 T (700 MHz 1H Larmor frequency) using previously established 2-D 15N–1H pulse schemes,34,35 at 295 K. The delays for R1(15N) were trelax = 200.5 (2×), 411.4, 633.0, 833.4, 1044.4, 1266.0, 1677.4, and 2099.4 ms. The delays for R2(15N) were multiples of the basic CPMG echo block (8.48 ms) that included pulses and delays to remove unwanted cross-correlated relaxation effects;36,37 this resulted in trelax = 17 (2×), 25.4, 33.9, 42.4, 50.9, 59.4, 76.3, and 93.3 ms. For the ssNOE, we recorded interleaved pairs of spectra in which 1H saturation of 5 s was applied alternately.
Deuterium relaxation rates R1ρ (2D) and R1 (2D) for CH2D methyl groups were measured with established 2-D 13Cmethyl–1Hmethyl pulse schemes38,39 at 16.4 and 18.8 T (700.13 and 800.13 MHz 1H Larmor frequencies), 295 K. 2D hard-pulses were applied at 1.78 kHz, while the spin-locks were applied at reduced strengths of 1.2 kHz. The delays for R1ρ (2D) were trelax = 0.5 (2×), 1.5, 2, 3, 4, 6.5, and 8 ms at 700 and 800 MHz. The R1 (2D) delays had trelax = 0.05 (2×), 10, 15, 21, 31, 42, and 50 ms at both 700 and 800 MHz.
All relaxation analysis used in-house software written in C programming language. Cross-peak intensities were measured by integrating along f2 (1H dimension) through the cross-peak maxima in f1 (13C or 15N), which gave for each resonance a file of cross-peak intensities “I” versus relaxation delay “trelax”. The I(trelax) versus trelax files were fit to two-parameter single-exponential decay functions, I(trelax) = A exp{−Rtrelax}, where R is the desired relaxation rate constant. Statistical errors were estimated using Monte Carlo methods with duplicate spectra furnishing the integral uncertainties.
To describe the backbone NH bond motions, we determined reduced spectral density values40−42, JeffNH(0), JNH(ωN), and ⟨JNH(ωH)⟩, using the relations42
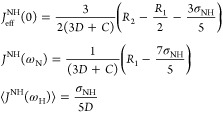 |
5 |
The σNH was extracted from the measured ssNOE and R1(N) via
| 6 |
The C and D constants in eq 5 reflect the 15N chemical-shift-anisotropy and 15N–1H dipolar relaxation mechanisms, respectively, and were C = Δ2ωN2/3 and D = ℏ2γH2γN2/⟨rHN6⟩ (cgs units).
The deuterium (spin-1) relaxation rates are dominated by the quadrupolar relaxation mechanism, resulting in rate constant expressions,43
| 7 |
| 8 |
JCD(ω) is the spectral density function that reports on the reorientational motions of the 13C–2D bond vectors with respect to the external magnetic field, B0. We used a quadrupolar coupling constant QCC = (e2qQ/ℏ) = 2π*167 kHz. To extract dynamics parameters from the relaxation rates, we used eqs 7 and 8 to fit the R1ρ(2D) and R1(2D) rates to an analytical JCD(ω) function given by the Lipari–Szabo formalism39,44,45
| 9 |
Equation 9 assumes that the “twirling” motions of the C–D bond vectors about the methyl symmetry axis are completely averaged out (extreme-narrowing), resulting in the factor of 1/9. Hence, the site-specific motions of the 13C–2D bond vectors are actually those of the corresponding methyl symmetry axes, with amplitudes given by the order parameter SAXIS2. The parameter τ satisfies 1/τ = 1/τm + 1/τe, where τm is the global correlation time for overall tumbling, and τe is a site-specific correlation time related to motions underlying SAXIS.
For the overall tumbling correlation time τm, we used the Levenberg–Marquardt algorithm46 and fit the ratios R2(15N)/R1(15N) of each domain (WW and PPIase) to get domain-specific τm values. The fit included only ratios within one-standard deviation of the raw mean. For each domain, we kept its τm fixed and used the Levenberg–Marquardt algorithm to fit SAXIS2 and τe for the individual methyls.46 Errors in SAXIS and τe were estimated using Monte Carlo simulations based on the estimated uncertainties in the experimental rate constants. Methyls excluded from this fitting because of resonance overlap included V22Cγ2, A31Cβ, L61Cδ2, L88Cδ2, L106Cδ1, L122Cδ2, T152Cγ2, and T162Cγ2. Further details are in our previous studies of Pin1 side chain-dynamics.1,2
Results
Backbone Chemical Shift Perturbations
The goal of the I28A substitution was to weaken interdomain contact within apo Pin1 while maintaining the overall folds of both domains. Comparisons of 2-D 15N–1H HSQC spectra for U–15N/13C, 50% 2D I28A and wild-type Pin1 (WT) indicated we had achieved our goal (Figure 2). The majority of HSQC cross-peaks of I28A were in the same positions as those of WT, indicating unchanged NH chemical shifts. We also compared 13Cα/β chemical shifts for I28A versus WT (Figure S1 of the Supporting Information) because these shifts are sensitive probes to local torsion angles and their fluctuations.47−49 The majority of residues showed small (<0.3 ppm) or no 13Cα/β shift perturbations. Thus, together the 15N and 13Cα/β chemical shifts suggested preservation of the overall WT fold, albeit, with local structural perturbations. Far-UV CD spectra corroborated the preservation of the overall fold (Figure S2 of the Supporting Information). In particular, wave scans of I28A and WT at 20 °C were essentially identical. Moreover, their thermal melts followed by far-UV CD at 200 nm were identical (Tm,I28A = 62.1 ± 0.2 °C versus Tm,WT = 62.5 ± 0.2 °C).
Figure 2.

Comparison of 2-D 15N–1H HSQC NMR spectra for I28A (red cross-peaks) versus wild-type Pin1 (blue cross-peaks) at 16.4 T, 295 K.
We focused mainly on the 2-D 15N–1H spectra to determine the effect of the I28A mutation on interdomain contact. Although the 2-D 15N–1H I28A and WT spectra were overall quite similar, there were important local chemical shift perturbations. We quantified these perturbations, ΔδNHI28A–WT = δI28A – δWT, using eq 1 (Materials and Methods). As expected, I28A showed prominent ΔδNH for its neighboring residues N16 and T29 (Figure 3, upper panel), but there were also perturbations in the PPIase domain. Some were unexpectedly long-ranged, occurring on the far side of the PPIase domain (PPIase catalytic loop, L60, K77, K82, and A107). These 15N–1H shift perturbations were corroborated by similar long-range 13Cα shift perturbations (Figure S1 of the Supporting Information). Specifically, beyond the expected 13Cα shift perturbations at WW domain residues at and flanking the mutation site, there were also a handful of significant 13Cα perturbations in the PPIase domain, including (i) R54 at the C-terminal end of the interdomain linker, (ii) S67, K77 in the catalytic loop, (iii) S105 and F110 in α2, and (iv) I158 at the C-terminus. Most revealing, however, were the 15N–1H shift perturbations at the PPIase residues at the C-terminal end of α4 including S138–R142. These PPIase residues lay across the domain interface from the I28A mutation site and its host Loop II in the WW domain (see Figure 1). Moreover, these perturbations matched those we observed for the isolated WT PPIase domain, relative to full-length Pin1, ΔδNHPPIase–WT (Figure 3, lower panel). In effect, the pattern of chemical shift perturbations in apo I28A matched those caused by the deletion of the WW domain. This was strong evidence that the I28A mutation had weakened the interdomain contact of apo Pin1.
Figure 3.

Backbone 15N–1H chemical shift perturbations for apo protein constructs. Top panel: perturbations ΔδNHI28A–WT for apo I28A relative to apo WT. Bottom panel: Perturbations ΔδNH caused by deletion of the WW domain (i.e., isolated apo PPIase domain relative to apo full-length WT). Green spheres are common chemical shift perturbations >0.05 ppm at the domain interface; red spheres are all other perturbations >0.05 ppm.
Additional evidence for weakened interdomain contact came from comparing the chemical shift perturbations caused by substrate binding (i.e., ΔδNHCOMPLEX–APO) in I28A and WT Pin1. Specifically, previous studies showed that WT binding of the phosphopeptide substrate EQPLpTPVTDL, a proxy for the Pin1 target Cdc25C phosphatase from Xenopus laevis,14,16 caused significant chemical shift perturbations at S138–R1421,21 (Figure 4, lower panel). These WT chemical shift perturbations were the signature response indicating increased interdomain contact stimulated by substrate binding.21 By contrast, adding saturating amounts of Cdc25C substrate to I28A failed to show this response (Figure 4, upper panel). This failure of response in I28A is consistent with its weakened interdomain contact.
Figure 4.

Backbone 15N–1H chemical shift perturbations of I28A (top) and WT (bottom) caused by adding saturating amounts of Cdc25C phosphopeptide substrate, EQPLpTPVTDL. Green spheres highlight domain interface shift perturbations >0.05 ppm for WT, which are mostly absent in I28A. Red spheres indicate all other perturbations >0.05 ppm. Light aquamarine and magenta shading indicate the PPIase and WW domains, respectively.
Cis–Trans Isomerase Activity
We investigated the effect of the I28A mutation on cis–trans isomerase activity using two methods. We first measured activity via the standard chromophoric coupled assay of Kofron et al.,28 which uses the substrate suc-AEPF-pNA. We found that I28A showed an ∼36% reduction of the specificity constant, kcat./KM, relative to WT (I28A, 2724 ± 140 mM–1 s–1; WT, 4250 ± 213 mM–1 s–1). This result echoes that of the isolated WT PPIase domain, which also showed a slight decrease of kcat./KM relative to WT.16
We also measured I28A isomerase activity using 2-D 1H–1H NMR exchange spectroscopy (EXSY),29 which used the Cdc25C phosphopeptide substrate mentioned above. EXSY spectra produced cross-peaks corresponding to pT5 methyl protons exchanging between the cis versus trans chemical shifts. For both I28A and WT, we fitted the time course of these exchange cross-peaks to the two-state exchange expression in eq 4 (Materials and Methods) to get a net exchange rate constant, kEXSY = kTC + kCT.30 It is important to note that kTC and kCT are the apparent rate constants for trans-to-cis and cis-to-trans exchange and are functions of KM and kcat. for the corresponding trans-to-cis and cis-to-trans isomerization processes.50 I28A showed increased kEXSY relative to WT (i.e., kEXSY(I28A) = kCT + kTC = 73 ± 2 s–1 versus kEXSY(WT) = kCT + kTC = 31.3 ± 0.5 s–1) (Figure S3 of the Supporting Information). This increase echoed our previous observation of increased kEXSY for isolated WT PPIase domain relative to full-length WT Pin1.51
Thus, the I28A mutation altered the cis–trans isomerization activity, despite the remote location of I28 from the PPIase active site. The sense of alteration matched that observed when going from full-length Pin1 to the isolated PPIase domain. In particular, both I28A and the isolated PPIase domain show the same trend of slightly increased kEXSY (2-D EXSY) and slightly decreased kcat./KM (chromogenic assay) relative to WT.
Effect of I28A on Substrate Binding Affinity
To assess the mutation’s impact on substrate binding, we titrated I28A with the Cdc25C phosphopeptide and fitted the resulting NH chemical shift perturbations of resolved NH cross-peaks to eq 3 (Materials and Methods). This gave site-specific estimates for binding affinity (i.e., the equilibrium dissociation constant Kd). The values are listed in Table 1, and the corresponding isotherm fits are in Figure S4 of the Supporting Information. Compared with WT, I28A showed weaker binding affinity (i.e., KdI28A > Kd) in both domains. The affinity decrease varied, with the ratios KdI28A > Kd values ranging from ∼5 to ∼10. The spread partly reflected the low signal-to-noise of some cross-peaks; nevertheless, the general trend of an affinity decrease was unambiguous. The decreased binding affinity was striking, given that neither I28 itself nor its host loop directly contact phosphopeptide substrate.13,14
Table 1. Equilibrium Dissociation Constants of the Cdc25C Phosphopeptide Substrate for I28A vs WT from NMR Titrations, 295 K, 16.4 T.
| residue | location | I28A Kd (μM) | WT Kd (μM) |
|---|---|---|---|
| R14 | WW domain | 48.5 ± 4.9 | 2.7 ± 0.7 |
| G20 | WW domain | 43.5 ± 3.0 | 9.1 ± 0.4 |
| R54 | PPIase domain | 110 ± 10 | 7.8 ± 0.4 |
| A140 | PPIase domain | 65 ± 39 | 9.7 ± 2.0 |
Backbone Mobility of apo I28A
We previously characterized the changes in WT Pin1 backbone and side chain flexibility caused by binding the Cdc25C phosphopeptide substrate.1 It was therefore of interest to see whether I28A would have similar responses, in light of its altered substrate binding affinity and cis–trans isomerase activity.
We first characterized the backbone flexibility of apo I28A, by measuring backbone amide 15N relaxation parameters, R1, R2, and steady-state 15N–1H NOE at 16.4 T, 295 K. We analyzed the relaxation data using a reduced NH spectral density mapping procedure42 that produced for each NH a value for JeffNH(0), a local mobility parameter. Jeff(0) is the zero-frequency value for the effective NH spectral density function JeffNH(ω) that describes the reorientational motions of the NH bonds relative to the external magnetic field, B0. For a rigid, isotropically tumbling molecule, Jeff(0) should be uniform across all NH bonds. NH bonds with outlying JeffNH(0) values highlight sites of internal motion. In particular, high Jeff(0) outliers indicate dynamic processes modulating the 15N chemical shift on the microsecond to millisecond time-scale, whereas low JeffNH(0) outliers reflect large amplitude, internal motions that reorient the NH bond on the subnanosecond time scale.42 Comparing Jeff(0) values for apo I28A with those we had previously obtained for apo WT1 allowed us to assess the mutation’s affect on intrinsic backbone dynamics.
We found that the overall profile of JeffNH(0) versus the sequence for apo I28A was similar to apo WT, in that the Jeff(0) values had partitioned into two distinct clusters, corresponding to the WW and PPIase domains. This clustering indicated that the overall molecular tumbling of I28A, like that of WT, could be approximated as two domains tumbling in quasi-independent manner with domain-specific correlation times, τm,WW and τm,PPI.
Nevertheless, the apo I28A showed important local differences in JeffNH(0) compared with apo WT. To highlight these differences, we used the site-specific ratio Jeff(0)/JeffNH,WT(0). Provided the mutation does not affect internal mobility, this ratio should be the same for all NHs within a given domain. Thus, those NH bonds that display outlying ratios represent sites experiencing mutation-induced changes in internal motion. We screened for such outliers by identifying Jeff(0)/JeffNH,WT(0) ratios beyond one standard deviation from the trimmed mean value for the WW or PPIase domain, as appropriate. Figure 5 shows the results; the spheres/bars colored blue and red indicate low and high outliers, respectively. The most prominent high outliers were WW domain residues with Jeff(0)/JeffNH,WT(0) > 1. These are residues whose NH bonds experience microsecond to millisecond exchange dynamics in I28A that are lacking in WT. These residues include N26, T29, and N30, which lie within or adjacent to Loop II, and bracket I28. Surprisingly, they also included WW domain residues outside Loop II, notably K13 and R14 in the β strand, β1′. Possible reasons for these surprising β1′ changes are in the Discussion. Low outliers, Jeff(0)/JeffNH,WT(0) < 1, indicating enhanced subnanosecond mobility of apo I28A versus apo WT, were distal from the mutation site and occurred in the linker, the PPIase domain catalytic pocket, and the flexible PPIase loop (H64–R80) “capping” the catalytic pocket.
Figure 5.

(A) Pin1 colored to highlight changes in backbone NH dynamics in apo I28A compared with apo WT. Ribbons colored aquamarine, magenta, and yellow indicate the PPIase, WW, and PPIase catalytic site regions, respectively. The red/blue spheres are NHs with outlying values of JeffI28A(0)/Jeff(0) (ratio values >1 standard deviation from the domain-specific trimmed mean). Red spheres highlight NH sites showing enhanced exchange dynamics, whereas blue spheres are sites with enhanced subnanosecond flexibility. (B) Bar graph showing the data underlying panel A. The bars are deviations of NH JeffI28A(0)/Jeff(0) from the domain-specific trimmed means. Red/blue bars correspond to the red/blue spheres in (A).
Backbone Mobility of I28A in the Presence of Substrate
We carried out the same backbone 15N relaxation analysis described above for I28A in the presence of saturating amounts of Cdc25C phosphopeptide substrate. To highlight changes in internal motion caused by substrate binding, we used the ratio JeffNH,COMPLEX(0)/Jeff(0), where the JeffNH,APO(0) values were those from the apo measurements described above. If substrate binding simply alters the domain rotational correlation time (e.g., τm,WW or τm,PPI), then the Jeff(0)/JeffNH,APO(0) ratios should be the same across all NHs within a given domain. Thus, those NH bonds that show outlying ratios represent bonds whose local dynamics have changed upon substrate binding. Ratios were identified as outliers if they deviated from the trimmed domain average by more than one standard deviation. We therefore compared the number and location of Jeff(0)/JeffNH,APO(0) outliers for I28A and WT, as a means to compare their dynamic responses to substrate binding.
Comparing these outliers revealed that Cdc25C substrate binding caused backbone dynamic changes in I28A that were absent in WT. Specifically, Figure 6 shows the outliers for both WT and I28A; the spheres/bars colored blue and red indicate low and high deviations, respectively. I28A had many outliers in the WW domain that were absent in WT. These outliers reflect the quenching of the aforementioned microsecond to millisecond exchange dynamics of apo I28A in Loop II in apo I28A, upon substrate binding. Interestingly, I28A also showed outliers at S58, V62, and C113, which are part of the substrate proline binding pockets within the PPIase domain. For these three residues, the ratio JeffNH,COMPLEX(0)/Jeff(0) became smaller, with the denominator JeffNH,APO(0) value close to the domain average; this indicated increased subnanosecond flexibility upon substrate Cdc25 phosphopeptide binding. Such a response was utterly lacking in WT. Thus, the I28A mutation in the WW domain changed the dynamic response of residues in the distal PPIase domain to substrate binding. Other sites outside the substrate binding pocket showing distinctly different dynamic response for I28A included K82 and E83, at the juncture between the catalytic loop and the long helix α1.
Figure 6.

Site-specific changes in NH backbone dynamics caused by Cdc25C substrate for both WT (top panel (A)) and I28A (bottom panel (B)). All structures are from PDB id 1PIN. Ribbon colors of aquamarine, magenta, and yellow ribbon indicate the PPIase, WW, and PPIase catalytic site regions, respectively. The red/blue spheres are NHs with outlying values of JeffNH,COMPLEX(0)/Jeff(0) (ratios >1 standard deviation from the domain-specific trimmed mean) and thus indicate binding-induced changes in local mobility. The red/blue bars in the bar graphs correspond to the red/blue spheres in the structures. In the bottom panel (B), S58, V62, and C113 are PPIase catalytic site residues that show increased subnanosecond mobility upon substrate binding for I28A but not for WT. K82 and E83, at the juncture between the catalytic loop and the long helix α1, also show dynamic changes not found in the WT.
Subnanosecond Side-Chain Mobility in apo I28A
We then investigated the impact of the I28A mutation on side-chain flexibility, specifically, the subnanosecond reorientational motions of methyl symmetry axes with respect to the magnetic field, B0. This involved measuring 2D R1 and R1ρ rate constants for all methyl CH2D isotopomers in U–15N, 13C, 50% 2D enriched I28A at 16.4 and 18.8 T, 295 K. We analyzed the resulting rates using the familiar Lipari–Szabo formalism.44 This produced two methyl-specific dynamics parameters: SAXIS2 and τe, per eq 9 (Materials and Methods). SAXIS is a pure number that measures the amplitude of reorientational dynamics of a methyl symmetry axis, due to subnanosecond internal motions. SAXIS2 ranges from 0 to 1: a value of 0 corresponds to unrestricted internal motion, whereas a value of 1 corresponds to no internal motion (rigid symmetry axis). The τe parameter is an effective correlation time that estimates the rapidity of the internal orientational dynamics but also depends on the amplitude of motion.44
Fitting SAXIS2 and τe relies on prior characterization of the overall molecular tumbling. The backbone NH reduced spectral density analysis above justified approximating the overall I28A tumbling in terms of domain-specific rotational correlation times. Accordingly, we determined τm,WW and τm,PPIase, using the R2(15N)/R1(15N) ratios35 of only those backbone NHs with Jeff(0) within 1 standard deviation of the trimmed mean. For apo I28A, we found τm,WW = 7.8 ± 0.01 ns/r, τm,PPIase = 12.0 ± 0.01 ns/r, and for the Cdc25C complex τm,WW = 7.5 ± 0.01 ns/r and τm,PPIase = 11.4 ± 0.01 ns/r. With the domain-specific overall tumbling times set, we were able to fit the site-specific side-chain internal motion parameters, SAXIS and τe. We then compared SAXIS2 from apo I28A with those we previously determined for apo WT,1 by evaluating the differences ΔSAXIS,APO = SAXIS,APO:I28A2 – SAXIS,APO:WT.
Remarkably, despite the fact that the I28A mutation was in the WW domain, it produced widespread changes in intrinsic side-chain flexibility (ΔSAXIS,APO2) in both domains. In particular, Figure 7, top panel, maps these differences onto the 1PIN crystal structure. Red spheres and positive bars indicate ΔSAXIS,APO > 0, and pinpoint methyl axes for which I28A was more rigid than for WT. Blue spheres and negative bars indicate the opposite trend. In the WW domain, we saw greater I28A flexibility at L7Cδ1; notably L7 has key hydrophobic interactions with W11, one of the two conserved tryptophans of the WW domain. In the PPIase domain, we observed both mobility increases and decreases. Sites where I28A loosened relative to WT (ΔSAXIS,APO2 < 0) included (i) V62Cγ2 in the PPIase β4 strand near the PPIase active site, (ii) T81Cγ2, A85Cβ, and I89Cδ1 in the long PPIase α1 helix, and (iii) A116Cβ in α3 helix. Sites where I28A stiffened compared with WT (ΔSAXIS,APO > 0) included (i) V55Cγ2, L60Cδ1, L60Cδ2, and L61Cδ1 in the PPIase active site, (ii) I93Cδ1 and I96Cδ1 in the long α1 helix, (iii) A140Cβ and L141Cδ2 between α4 and β3, (iv) V150Cγ2 in β3, and (v) I159Cγ2 in β4. Interestingly, the last three locales (A140Cβ, L141Cδ2, V150Cγ2, and I159Cγ2) are all near the domain interface.
Figure 7.

Changes in methyl side-chain order parameters SAXIS2 from deuterium spin relaxation. Top panel: apo states for WT versus I28A, ΔSAXIS,APO = SAXIS,APO:I28A2 – SAXIS,APO:WT. Middle panel: I28A complexed with Cdc25C phosphopeptide versus its apo state, ΔSAXIS,BINDING2 = SAXIS,CMP:I28A – SAXIS,APO:I28A2. Bottom panel: Cdc25C phosphopeptide complexed states for WT versus I28A, ΔSAXIS,COMPLEX = SAXIS,CMP:WT2 – SAXIS,CMP:I28A. The bars denote SAXIS2 with magnitudes greater than or equal to twice (purple) or once (hatched) the estimated statistical errors. The structures to the right of each bar graph shows colored spheres corresponding to the methyls changes highlighted in the bar graphs. The red and blue spheres indicate sites of ΔSAXIS > 0 (more rigid) and ΔSAXIS2 < 0 (more flexible), respectively. Bottom structure: residues colored to contrast the pattern of side-chain flexibility loss in I28A upon Cdc25C substrate binding, with that of the previously defined WT conduit.1 Specifically, the red and deep salmon residues trace the original WT conduit. The red residues are those that lose side-chain flexibility only in WT (i.e., not I28A). The deep salmon residues lose side-chain flexibility in both WT and I28A. The green residues are those that lose side-chain flexibility only in I28A. The red and green residues thus highlight the departure of I28A from the previously defined WT conduit.
Subnanosecond Side Chain Mobility in I28A in the Presence of Substrate
We investigated Cdc25C substrate binding affected I28A side-chain mobility by evaluating ΔSAXIS,BINDING2 = SAXIS,CMP:I28A – SAXIS,APO:I28A2. The ΔSAXIS,CMP:I28A values were from I28A in the presence of saturating amounts of Cdc25C substrate, whereas the ΔSAXIS,APO:I28A2 values were from the apo studies described above. Positive and negative ΔSAXIS,BINDING indicate a loss or gain of side-chain flexibility, respectively, upon Cdc25 binding. The middle panel of Figure 7 maps these differences onto the structure. Losses of flexibility (ΔSAXIS,BINDING2 > 0) occurred at the N-terminus of the WW domain L7Cδ1, the PPIase domain active site V62Cγ2, T81Cγ2, and A85Cβ, I89Cδ1, L106Cδ2, A118Cβ, L122Cδ1, M146Cε, and V150Cγ2. Gains in flexibility (ΔSAXIS,BINDING < 0) occurred at the flexible linker (A53Cβ), the PPIase domain active site L60Cδ1 and L60Cδ2, the domain interface L141Cδ2, and the C-terminal L160Cδ2. Thus, many sites that showed intrinsically different side chain mobility from WT (ΔSAXIS,APO2) also underwent changes in Saxis upon substrate binding.
We wanted to compare how the binding-induced changes in side chain flexibility for I28A compared with what we had already documented for WT. To this end, we compared order parameters from the two Cdc25C complexes, WT/Cdc25C and I28A/Cdc25C, by evaluating the difference ΔSAXIS,CMP2 = SAXIS,CMP:WT – SAXIS,CMP:I28A2. Figure 7, bottom panel, reveals different responses in both domains. Generally, the I28A/Cdc25 complex was stiffer than the WT/Cdc25 complex. The I28A complex showed greater rigidity than the WT complex (ΔSAXIS,CMP < 0) at L7Cδ1 in the WW domain, I89Cδ1 (PPIase α1, domain interface), L106Cδ2 (PPIase α2), L122Cδ1 (PPIase active site), and V150Cγ1 V150Cγ2, I158Cδ1 and I159Cδ1 (PPIase β3 and β4, adjacent to residues comprising the domain interface). On the other hand, the WT complex showed greater rigidity than the I28A complex (ΔSAXIS,CMP2 > 0) at L7Cδ2 (WW domain), A53Cβ (linker), and L60Cδ1 (PPIase β1 active site), and L141Cδ2 (domain interface).
A particularly striking difference in side chain dynamic response occurred at L60–L61–V62, a conserved hydrophobic cluster within the PPIase active site. Specifically, our previous dynamics studies of WT showed a loss of side-chain flexibility for this conserved cluster.1,2 By contrast, the response of I28A upon substrate binding was an increase in flexibility at L60 (Figure 7, middle panel). This flexibility increase also appeared in the subnanosecond backbone flexibility at these sites (Figure 5). The deviant dynamic response in the I28A PPIase active site, for both side chain and backbone, is noteworthy given that its isomerase activity (kcat./KM from the chromogenic assay, and kEXSY from 2-D NMR) differs from WT.
Finally, the structure (PDB id 1PIN) in lower panel (B) of Figure 7 further highlights how the distribution of side-chain flexibility loss in I28A caused by Cdc25C substrate binding deviates from the conduit response we first observed for the WT.1 Specifically, the coloring denotes residues that lose side-chain flexibility (i) only in WT (red), (ii) in both WT and I28A (deep salmon) (iii), and only in I28A (green). The red and deep salmon residues trace the original WT conduit.
Discussion
Pin1 has weak interdomain interactions13,20−22 whose functional significance has not yet been firmly established. At the same time, Pin1 requires some form of interdomain communication for in vivo function.12,17 We recently connected these two observations through our studies of Pin1 functional motions, which led us to propose that interdomain contact allows the WW domain to allosterically regulate the distal PPIase active site.1,2 A necessary condition for this allosteric mechanism is that PPIase domain contact with the WW domain should alter some intrinsic properties of the PPIase domain relevant to binding, activity, or both. Our goal here was to investigate this possibility by weakening the interdomain interaction. Toward this end, we generated I28A, which lies within Loop II (H27–N30) of the Pin1 WW domain. In the 1PIN crystal structure, Loop II makes interdomain contacts with the PPIase domain.22 By observing effects of the I28A mutation on Pin1, we would map the influence of interdomain contact away from the immediate domain interface, and thus gain insight into its relevance for interdomain communication.
I28A Weakens Intrinsic Interdomain Contact
As our 15N–1H NMR chemical shift perturbations and far-UV CD data indicate, the I28A mutation indeed weakens interdomain contact, while preserving the overall structure of WT Pin1 (Figures 2–4, Figure S1 of the Supporting Information). The 15N–1H shift perturbations depict an interdomain contact region consistent with the region depicted in the 1PIN crystal structure22 and include H27–N30 of the WW domain (Loop II) and of the PPIase domain residues S138–R142 (C-terminal residues of α4).
The backbone 15N dynamics study of I28A provides further support for weakened interdomain contact. Loop II (H27–N30) in I28A exhibits greater microsecond to millisecond mobility than Loop II in WT, as evidenced by elevated ratios JeffNH,I28A(0)/Jeff(0) in Figure 5. These elevated ratios indicate enhanced amide proton exchange, conformational exchange, or both. Greater Loop II flexibility in I28A would be consistent with its lowered commitment from Loop II to interdomain contact.
I28A Shows Isomerase Activity Consistent with Weakened Interdomain Contact
The weakened interdomain contact in I28A coincides with changes in isomerase activity. Notably, the changes of I28A relative to WT are similar to those displayed by the isolated PPIase domain. Specifically, both I28A and the isolated PPIase domain show the same trend of slightly increased kEXSY (2-D EXSY assay) and slightly decreased kcat./KM (chromogenic assay) relative to WT. Thus, I28A mutation changes the PPIase activity in a direction that is diagnostic of lost communication with the WW domain. These results suggest that interdomain contact has a functional significance, in that it can fine-tune PPIase activity.
This fine-tuning is intriguing because I28 and the other residues supporting interdomain contact (e.g., H27–N30 in the WW domain Loop II and S138–R142 in the PPIase domain) do not directly contact substrate. Rather, they are spatially removed from those regions that do, namely, the PPIase domain catalytic site and the WW domain substrate binding Loop I (S16–R21) (Figure 1). This physical separation raises the question of how changes in the interdomain contact, either its weakening or elimination, could alter the activity of the distal PPIase catalytic site.
Interdomain Contact Affects the PPIase Domain Properties
The standard explanation emphasizes the WW domain’s role as an independent binding module.9,19 Its proposed influence on the PPIase activity, modulating the local substrate concentration, simply reflects its proximity to the PPIase domain. Interestingly, there appears to be no consensus on what this modulation is: both the enhancement and the depletion of local substrate concentration have been suggested.11,18,52 Mutating the WW domain may compromise its ability to bind substrate, and hence, its ability to modulate local substrate concentration. Two features of this standard explanation are noteworthy. First, it does not explicitly invoke domain contact but merely domain proximity. Second, the view of the WW domain as an independent module implies that a WW domain mutation may perturb local conformation and flexibility within the WW domain but not within the PPIase domain.
Our results from I28A suggest a more complex interdomain relationship. Certainly, the I28A mutation does perturb the WW domain, as evidenced by reduced Cdc25C binding affinity and altered Loop II mobility (Figure 5). But in contrast to the implications of the standard explanation, the WW domain mutation also impacts the intrinsic properties of the PPIase domain. First, comparisons of the apo I28A versus apo WT backbone 15N–1H chemical shifts show perturbations in both domains, including PPIase residues far removed from the PPIase domain interface, such as those of the PPIase domain catalytic loop (Figure 3). Similar remote 13Cα chemical shift perturbations corroborate these remote 15N–1H perturbations (Figure S1 of the Supporting Information). Second, we found weaker Cdc25 phosphopeptide substrate binding affinities (higher Kd = C0 exp(ΔGPPIase,0/kBT)) for both domains (Table 1 and Figure S4 of the Supporting Information). This suggests that the I28A WW domain mutation makes the free energy difference between the complexed and apo states, ΔGPPIase,0 = GCOMPLEXPPIase – GAPO, less negative. Finally, the I28A mutation causes widespread changes in the intrinsic (apo state) backbone and side-chain flexibility of the protein, beyond the domain interface. Figure 5 and the top panel of Figure 7 show distal changes in backbone and side-chain flexibility, respectively, at functional sites of the PPIase domain. In particular, the top panel of Figure 7 shows complementary changes in side-chain flexibility at residues within the PPIase catalytic site (V55Cγ2, L60Cδ1, L60Cδ2, and L61Cδ1). Considering the above points, the view of the WW domain as an independent binding module would appear incomplete. Rather, our data suggest that the internal properties of the PPIase domain are sensitive to WW domain contact and to mutations such as I28A that weaken that contact.
Interdomain Contact Tunes the Dynamic Response of Pin1 to Binding
Our previous work on WT Pin1 functional dynamics in the presence and absence of inhibitors and substrates has led us to propose an additional mechanism for WW domain influence on PPIase activity. Specifically, we proposed that the WW domain also acts as an allosteric effector molecule of the PPIase domain. The allosteric binding site involves the interdomain contact region identified in this study. The mechanism that communicates changes at the interdomain contact region to the distal PPIase catalytic site involves propagated changes in flexibility among the intervening residues. These flexibility changes manifest as a loss of subnanosecond side chain flexibility along a conduit of conserved hydrophobic residues linking the interdomain interface to the active site.1,2
If this dynamic allosteric explanation is tenable, then weakening the interdomain contact via the I28A mutation should produce a different response in side-chain dynamics upon Cdc25C substrate binding. This is what we observe (Figure 7, middle and bottom panels). Critically, I28A lacks key features of the WT dynamic conduit, such as the loss of side-chain flexibility for the PPIase active site residues L60 and L61. In fact, L60 becomes more flexible (Figure 7, middle panel). The colored structure (PDB id 1PIN) at the bottom of Figure 7 further underscores these differences. This differential dynamic response is reinforced by the corresponding 15N backbone dynamics; specifically, I28A showed an enhancement of backbone subnanosecond flexibility for S58, V62, and C113 upon Cdc25C substrate binding (Figure 6) that is absent in the WT. The side chain and backbone results above suggest that the interdomain interface is a critical set of interactions that enable dynamic allosteric regulation of the PPIase domain by the WW domain.
Long-Range Interactions within the WW Domain
I28A weakened both interdomain contact and WW domain binding affinity to the Cdc25C phosphopeptide. This joint effect is intriguing because I28 does not directly contact substrate; rather, it is on the opposite side of the WW domain loop mediating substrate binding, Loop I (S16–R21). The fact that the binding affinity at Loop I is sensitive to a mutation at the far end of the domain points to a yet unremarked mechanism for long-range Loop I–Loop II communication within the Pin1 WW domain.
We speculate that this long-range communication derives from a network of short-range inter-residue interactions within the WW domain. This speculation derives from our unexpected observation that I28A causes changes in mobility beyond its host Loop II and extends to K13 and R14 in the β1′-strand (Figure 5). These long-range perturbations become comprehensible when the Loop II hydrogen bond network in the 1PIN crystal structures is examined (Figure 8).22,53 Of particular interest are hydrogen bonds from the I28A backbone NH to side chains of N26. N26 is highly conserved across Pin1 homologues17 and makes multiple hydrogen bonds that stabilize Loop II and link it to β1′ (cf. Figure 8). In I28A, N26 shows large backbone chemical shift perturbations (Figure 4, Figures S1 of the Supporting Information) and enhanced exchange dynamics (amplified JeffNH(0) in Figure 6) that are absent in WT. Thus, although we did not mutate N26 directly, we have nevertheless changed its local mobility by mutating one of its hydrogen bond partners, I28. Conceivably, the shorter Ala side chain in I28A could decrease side chain steric contacts, both with the PPIase domain and within Loop II itself. Indeed, the perturbations in 13Cα/β chemical shifts for Loop II (apo I28A versus apo WT) suggest local structural perturbations to Loop II, consistent with this notion (Figure S1 of the Supporting Information). This could enable greater backbone mobility at position 28, which would then propagate to N26 and more remote sites, such as K13 and R14, via the network of backbone and side-chain hydrogen bonds. Thus, the long-range mobility perturbations stimulated by I28A make it reasonable to contemplate a network of short-range interactions within the WW domain that could couple perturbations at Loop II to Loop I.
Figure 8.
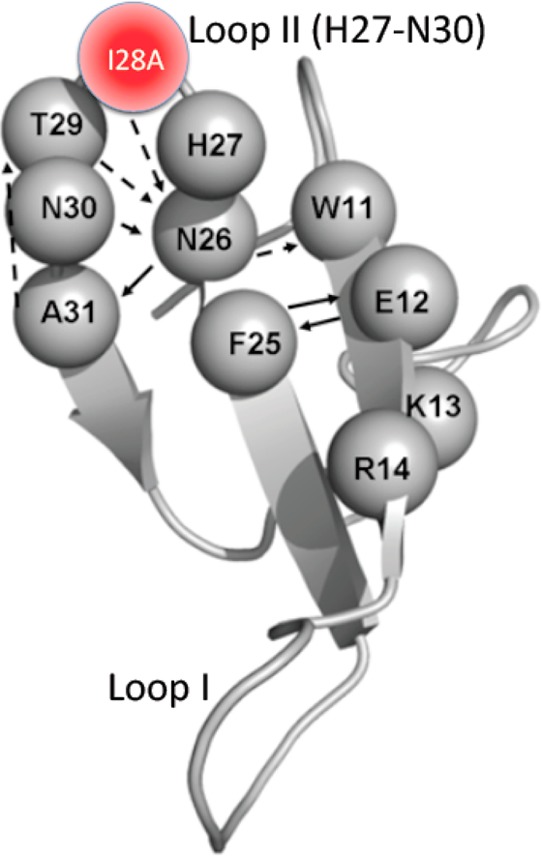
Loop II hydrogen bond pairings of Pin1 WW domain within full-length WT Pin1 (PDB id 1PIN). Solid line black arrows represent backbone–backbone hydrogen bonds, whereas dotted black arrows represent backbone–side-chain hydrogen bonds. The backbone NH of I28 makes hydrogen bonds to the side chain of N26.
If this intra-WW domain network is corroborated by subsequent experiments, it would mean that substrate binding to the WW domain, and its allosteric influence on the PPIase domain, are themselves coupled phenomena. It would also justify the hypothesis of a larger network of interacting residues that couple binding events at the WW domain Loop I (S16–R21) all the way to the PPIase active site, with the interdomain contact surface as a crucial intermediary.
Coevolving Residues
By itself, I28 is not a highly conserved residue across Pin1 homologues.17 Nevertheless, our results show that I28 participates in inter-residue interactions that sustain the weak contacts between the WW and PPIase domain (e.g., the large chemical shift perturbations at PPIase domain residues S138–R142 in Figure 3). Thus, we might expect that I28 would emerge in bioinformatics analyses aimed at finding pairs of coevolving residues. An example is the “protein sector” analysis of Ranganathan and co-workers, which identifies sectors of coevolving residues based on their statistical coupling analysis (SCA) of multiple sequence alignments (MSA).54 Our initial application of this sector/SCA to Pin1 reveals I28/A140 as a coevolving pair. This pair would be consistent with the interdomain contacts identified above (e.g., chemical shift perturbations in Figure 3, side-chain dynamic changes Figure 7), and form the basis for future double mutant studies.
Interdomain Contact Supports Interdomain Allostery in Pin1
In summary, our I28A results reveal that the WW domain Loop II (H27–N30) is critical for establishing transient interdomain contacts with the PPIase domain. Moreover, these contacts influence the distribution of conformations sampled by the PPIase domain. Evidence for this influence consists of the perturbations to backbone chemical shifts, backbone/side-chain mobility, substrate binding affinity, and isomerase activity documented above. These results show that interdomain contact alters the internal properties of the PPIase domain and thus strengthen our hypothesis for allosteric communication between the interdomain interface and the distal active site.
We should reiterate that although this investigation reveals Pin1 interdomain allostery mainly through changes in protein dynamics parameters, it does not exclude the possibility of joint changes in local conformation. Indeed, the backbone chemical shift perturbations indicate that, although the I28A mutation preserves the overall WT fold, it may also instigate local structural changes within the PPIase domain. This possibility is consistent with our main point: a WW domain mutation (I28A) at the domain interface can perturb the conformational sampling of the PPIase domain in such a way that it alters local flexibility, structure, or both. All three scenarios would be consequences of interdomain allostery.
By combining our I28A results with our previous ones, we envision the following underlying scenario. The Pin1 interdomain interactions, although weak, influence the ensemble of conformations sampled by both domains. From the perspective of the PPIase domain, the WW domain acts not only as a binding module but also as an allosteric effector molecule. WW domain contact with the PPIase domain perturbs the conformational sampling by the PPIase domain. These changes in conformational sampling, although stimulated at the domain interface, can propagate away from that interface to the remote catalytic site via correlated internal motions within the PPIase domain. These correlated motions include (but are not limited to) the side chain motions whose perturbations manifest as the dynamic conduit. The results are not gross structural changes but rather subtle changes in local conformation and flexibility at the PPIase catalytic site that fine-tune binding affinity and isomerization activity. Substrate binding stabilizes the subset of conformations involving more intimate interdomain contact and thereby tunes binding affinity and activity. The exact manner of tuning doubtless depends on the details of substrate composition (e.g., residues flanking the pS/T–P segments).
To further investigate the above scenario, we require further mutation studies. In particular, we need mutations that perturb interdomain contact, exclusively, without perturbing the binding affinity of WW domain residues. These mutations could involve those of the PPIase side of the domain interface, and modifications of the flexible linker. Such work is in progress.
Modular proteins are replete in biochemical networks maintaining the cell cycle.4 The weak interdomain interactions investigated here for Pin1 may be present in other modular systems and, perhaps, play similar functional roles. Perturbing these interactions systematically, via small molecule ligands, may be a promising approach for advancing our understanding of the molecules regulating cell growth. Also, as mentioned in the introduction, functional interdomain interactions are attractive target sites for the design of allosteric inhibitors.6,7 Conceivably, fragment-based approaches that target both the catalytic site and interdomain interfaces may enhance target specificity.
Acknowledgments
We are grateful to Dr. Xingsheng Wang, Dr. John S. Zintsmaster, Ms. Petra Rovó, Mr. Thomas E. Frederick, Mr. Michael W. Staude, Prof. Patricia L. Clark, and Dr. Jaroslav Zajicek for valuable suggestions and useful discussions.
Glossary
ABBREVIATIONS USED
- I28A
single site substitution mutant I28A of Pin1
- PPIase
peptidyl–prolyl isomerase
- 2-D
two-dimensional
- MHz
megahertz
- EXSY
exchange spectroscopy
- TOCSY
total correlation spectroscopy
- HSQC
heteronuclear single quantum correlation
- ppm
parts per million
- Cdc25C
phosphothreonine peptide substrate EQPLpTPVTDL
- WW
Trp–Trp
Supporting Information Available
Four figures including the 13Cα/β chemical shift perturbations for apo I28A versus apo WT Pin1, far-UV CD wave scans and thermal melts at 200 nm, fits of 2-D EXSY spectra for cis–trans isomerase activity measurements, and binding isotherms from NH titrations. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
† These authors contributed equally to this work
This work was supported by NIH Grant No. RO1-GM083081 to J.W.P.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Namanja A. T.; Peng T.; Zintsmaster J. S.; Elson A. C.; Shakour M. G.; Peng J. W. (2007) Substrate recognition reduces side-chain flexibility for conserved hydrophobic residues in human Pin1. Structure 15, 313–327. [DOI] [PubMed] [Google Scholar]
- Namanja A. T.; Wang X. J.; Xu B.; Mercedes-Camacho A. Y.; Wilson K. A.; Etzkorn F. A.; Peng J. W. (2011) Stereospecific gating of functional motions in Pin1. Proc. Natl. Acad. Sci. U. S. A. 108, 12289–12294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya R. P.; Remenyi A.; Yeh B. J.; Lim W. A. (2006) Domains, Motifs, and Scaffolds: The Role of Modular Interactions in the Evolution and Wiring of Cell Signaling Circuits. Annu. Rev. Biochem. 75, 655–680. [DOI] [PubMed] [Google Scholar]
- Lim W. A. (2010) Designing customized cell signalling circuits. Nat. Rev. Mol. Cell Biol. 11, 393–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pufall M. A.; Graves B. J. (2002) Autoinhibitory domains: modular effectors of cellular regulation. Annu. Rev. Cell Dev. Biol. 18, 421–462. [DOI] [PubMed] [Google Scholar]
- Peterson J. R.; Golemis E. A. (2004) Autoinhibited proteins as promising drug targets. J. Cell. Biochem. 93, 68–73. [DOI] [PubMed] [Google Scholar]
- Lee G. M.; Craik C. S. (2009) Trapping moving targets with small molecules. Science 324, 213–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu K. P.; Hanes S. D.; Hunter T. (1996) A human peptidyl-prolyl isomerase essential for regulation of mitosis. Nature 380, 544–547. [DOI] [PubMed] [Google Scholar]
- Lu K. P.; Liou Y. C.; Zhou X. Z. (2002) Pinning down proline-directed phosphorylation signaling. Trends Cell Biol. 12, 164–172. [DOI] [PubMed] [Google Scholar]
- Landrieu I.; Smet C.; Wieruszeski J. M.; Sambo A. V.; Wintjens R.; Buee L.; Lippens G. (2006) Exploring the molecular function of PIN1 by nuclear magnetic resonance. Curr. Protein Pept. Sci. 7, 179–194. [DOI] [PubMed] [Google Scholar]
- Lu K. P.; Finn G.; Lee T. H.; Nicholson L. K. (2007) Prolyl cis-trans isomerization as a molecular timer. Nat. Chem. Biol. 3, 619–629. [DOI] [PubMed] [Google Scholar]
- Lu P. J.; Wulf G.; Zhou X. Z.; Davies P.; Lu K. P. (1999) The prolyl isomerase Pin1 restores the function of Alzheimer-associated phosphorylated tau protein. Nature 399, 784–788. [DOI] [PubMed] [Google Scholar]
- Verdecia M. A.; Bowman M. E.; Lu K. P.; Hunter T.; Noel J. P. (2000) Structural Basis for phosphoserine-proline recognition by group IV WW domains. Nat. Struct. Biol. 7, 639–643. [DOI] [PubMed] [Google Scholar]
- Wintjens R.; Wieruszeski J.-M.; Drobecq H.; Rousselot-Pailley P.; Buee L.; Lippens G.; Landrieu I. (2001) 1H NMR Study on the Binding of Pin1 Trp-Trp Domain with Phosphothreonine Peptides. J. Biol. Chem. 276, 25150–25156. [DOI] [PubMed] [Google Scholar]
- Sudol M.; Chen H. I.; Bougeret C.; Einbond A.; Bork P. (1995) Characterization of a novel protein-binding module--the WW domain. FEBS Lett. 369, 67–71. [DOI] [PubMed] [Google Scholar]
- Lu P. J.; Zhou X. Z.; Shen M.; Lu K. P. (1999) Function of WW domains as phosphoserine- or phosphothreonine-binding modules. Science 283, 1325–1328. [DOI] [PubMed] [Google Scholar]
- Behrsin C. D.; Bailey M. L.; Bateman K. S.; Hamilton K. S.; Wahl L. M.; Brandl C. J.; Shilton B. H.; Litchfield D. W. (2007) Functionally Important Residues in the Peptidyl-prolyl Isomerase Pin1 Revealed by Unigenic Evolution. J. Mol. Biol., 365(4), 1143–1162. [DOI] [PubMed] [Google Scholar]
- Lu K. P. (2004) Pinning down cell signaling, cancer and Alzheimer’s disease. Trends Biochem. Sci. 29, 200–209. [DOI] [PubMed] [Google Scholar]
- Lu K. P.; Zhou X. Z. (2007) The prolyl isomerase PIN1: a pivotal new twist in phosphorylation signalling and disease. Nat. Rev. Mol. Cell Biol. 8, 904–916. [DOI] [PubMed] [Google Scholar]
- Bayer E.; Goettsch S.; Mueller J. W.; Griewel B.; Guiberman E.; Mayr L. M.; Bayer P. (2003) Structural analysis of the mitotic regulator hPin1 in solution: insights into domain architecture and substrate binding. J. Biol. Chem. 278, 26183–26193. [DOI] [PubMed] [Google Scholar]
- Jacobs D. M.; Saxena K.; Vogtherr M.; Bernado P.; Pons M.; Fiebig K. M. (2003) Peptide Binding Induces Large Scale Changes in Inter-domain Mobility in Human Pin1. J. Biol. Chem. 278, 26174–26182. [DOI] [PubMed] [Google Scholar]
- Ranganathan R.; Lu K. P.; Hunter T.; Noel J. P. (1997) Structural and Functional Analysis of the Mitotic Rotamase Pin1 Suggests Substrate Recognition Is Phosphorylation Dependent. Cell 89, 875–886. [DOI] [PubMed] [Google Scholar]
- Namanja A. T.; Wang X. J.; Xu B.; Mercedes-Camacho A. Y.; Wilson B. D.; Wilson K. A.; Etzkorn F. A.; Peng J. W. (2010) Toward flexibility-activity relationships by NMR spectroscopy: dynamics of Pin1 ligands. J. Am. Chem. Soc. 132, 5607–5609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaeger M.; Nguyen H.; Crane J. C.; Kelly J. W.; Gruebele M. (2001) The folding mechanism of a beta-sheet: the WW domain. J. Mol. Biol. 311, 373–393. [DOI] [PubMed] [Google Scholar]
- Wittekind M.; Mueller L. (1993) HNCACB, a High-Sensitivity 3D NMR Experiment to Correlate Amide-Proton and Nitrogen Resonances with the Alpha- and Beta-Carbon Resonances in Proteins. J. Magn. Reson., Ser. B 101, 201–205. [Google Scholar]
- Yamazaki T.; Lee W.; Arrowsmith C. H.; Muhandiram D. R.; Kay L. E. (1994) A Suite of Triple Resonance NMR Experiments for the Backbone Assignment of 15N, 13C, 2H Labeled Proteins with High Sensitivity. J. Am. Chem. Soc. 116, 11655–11666. [Google Scholar]
- Bodenhausen G.; Ruben D. J. (1980) Natural abundance nitrogen-15n NMR by enhanced heteronuclear spectroscopy. Chem. Phys. Lett. 69, 185–189. [Google Scholar]
- Kofron J. L.; Kuzmic P.; Kishore V.; Colon-Bonilla E.; Rich D. H. (1991) Determination of kinetic constants for peptidyl prolyl cis-trans isomerases by an improved spectrophotometric assay. Biochemistry 30, 6127–6134. [DOI] [PubMed] [Google Scholar]
- Jeener J.; Meier B. H.; Bachmann P.; Ernst R. R. (1979) Investigation of exchange processes by two-dimensional NMR spectroscopy. J. Chem. Phys. 71, 4546–4553. [Google Scholar]
- Ernst R. R., Bodenhausen G., and Wokaun A. (1987) Principles of Nuclear Magnetic Resonance in One and Two Dimensions, Chapter 9, Section 9.3.1, Oxford Science Publications, Oxford, U.K. [Google Scholar]
- Braunschweiler L.; Ernst R. R. (1983) Coherence transfer by isotropic mixing: application to proton correlation spectroscopy. J. Magn. Reson. 53, 521–528. [Google Scholar]
- Shaka A. J.; Lee C. J.; Pines A. (1988) Isotropic Mixing Sequences. J. Magn. Reson. 77, 274–293. [Google Scholar]
- Bothner-By A. A.; Stephens R. L.; Lee J.-M.; Warren C. D.; Jeanloz R. W. (1984) Structure determination of a tetrasaccharide: transient nuclear Overhauser effects in the rotating frame. J. Am. Chem. Soc. 106, 811–813. [Google Scholar]
- Dayie K. T.; Wagner G. (1994) Relaxation-rate measurements for 15N-1H groups using pulse-field gradients and preservation of coherence pathways. J. Magn. Reson., Ser. A 111, 121–126. [Google Scholar]
- Kay L. E.; Torchia D. A.; Bax A. (1989) Backbone dynamics of proteins as studied by 15N inverse detected heteronuclear NMR spectroscopy: application to staphylococcal nuclease. Biochemistry 28, 8972–8979. [DOI] [PubMed] [Google Scholar]
- Palmer A. G. III.; Skelton N. J.; Chazin W. J.; Wright P. E.; Rance M. (1992) Suppression of the effects of cross-correlation betwee dipolar and anisotropic chemical shift relaxation mechanisms in the measurement of spin-spin relaxation rates. Mol. Phys. 75, 699–711. [Google Scholar]
- Kay L. E.; Nicholson L. K.; Delaglio F.; Bax A.; Torchia D. A. (1992) Pulse Sequences for Removal of the Effects of Cross Correlation between Dipolar and Chemical-Shift Anisotropy Relaxation Mechanisms on the Measurement of Heteronuclear T1 and T2 Values in Proteins. J. Magn. Reson. 97, 359–375. [Google Scholar]
- Millet O.; Muhandiram D. R.; Skrynnikov N. R.; Kay L. E. (2002) Deuterium spin probes of side-chain dynamics in proteins. 1. Measurement of five relaxation rates per deuteron in (13)C-labeled and fractionally (2)H-enriched proteins in solution. J. Am. Chem. Soc. 124, 6439–6448. [DOI] [PubMed] [Google Scholar]
- Muhandiram D. R.; Yamazaki T.; Sykes B. D.; Kay L. E. (1995) Measurement of 2H T1 and T1r Relaxation Times in Uniformly 13C-Labeled and Fractionally 2H-Labeled Proteins in Solution. J. Am. Chem. Soc. 117, 11536–11544. [Google Scholar]
- Farrow N. A.; Zhang O.; Szabo A.; Torchia D. A.; Kay L. E. (1995) Spectral density function mapping using 15N relaxation data exclusively. J. Biomol. NMR 6, 153–162. [DOI] [PubMed] [Google Scholar]
- Ishima R.; Nagayama K. (1995) Protein backbone dynamics revealed by quasi spectral density function analysis of amide N-15 nuclei. Biochemistry 34, 3162–3171. [DOI] [PubMed] [Google Scholar]
- Peng J. W.; Wagner G. (1995) Frequency spectrum of NH bonds in eglin c from spectral density mapping at multiple fields. Biochemistry 34, 16733–16752. [DOI] [PubMed] [Google Scholar]
- Abragam A., (1961) Principles of Nuclear Magnetism, Oxford University Press, Oxford, U.K. [Google Scholar]
- Lipari G.; Szabo A. (1982) Model-Free Approach to the Interpretation of Nuclear Magnetic Resonance Relaxation in Macromolecules. 1. Theory and Range of Validity. J. Am. Chem. Soc. 104, 4546–4559. [Google Scholar]
- Nicholson L. K.; Kay L. E.; Baldisseri D. M.; Arango J.; Young P. E.; Bax A.; Torchia D. A. (1992) Dynamics of methyl groups in proteins as studied by proton-detected 13C NMR spectroscopy. Application to the leucine residues of staphylococcal nuclease. Biochemistry 31, 5253–5263. [DOI] [PubMed] [Google Scholar]
- Press W. H., Teukolsky S. A., Vetterling W. T., and Flannery B. P. (1992) Numerical Recipes in C: The Art of Scientific Computing, Cambridge University Press, Cambridge, U.K. [Google Scholar]
- Wishart D. S.; Sykes B. D. (1994) The 13C chemical-shift index: a simple method for the identification of protein secondary structure using 13C chemical-shift data. J. Biomol. NMR 4, 171–180. [DOI] [PubMed] [Google Scholar]
- Cornilescu G.; Delaglio F.; Bax A. (1999) Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J. Biomol. NMR 13, 289–302. [DOI] [PubMed] [Google Scholar]
- Berjanskii M.; Wishart D. S. (2006) NMR: prediction of protein flexibility. Nat. Protoc. 1, 683–688. [DOI] [PubMed] [Google Scholar]
- Kuchel P. W.; Bulliman B. T.; Chapman B. E. (1988) Mutarotase equilibrium exchange kinetics studied by 13C-NMR. Biophys. Chem. 32, 89–95. [DOI] [PubMed] [Google Scholar]
- Peng J. W.; Wilson B. D.; Namanja A. T. (2009) Mapping the dynamics of ligand reorganization via 13CH3 and 13CH2 relaxation dispersion at natural abundance. J. Biomol. NMR 45, 171–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De S.; Greenwood A. I.; Rogals M. J.; Kovrigin E. L.; Lu K. P.; Nicholson L. K. (2012) Complete thermodynamic and kinetic characterization of the isomer-specific interaction between Pin1-WW domain and the amyloid precursor protein cytoplasmic tail phosphorylated at Thr668. Biochemistry 51, 8583–8596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jager M.; Nguyen H.; Crane J. C.; Kelly J. W.; Gruebele M. (2001) The folding mechanism of a beta-sheet: the WW domain. J. Mol. Biol. 311, 373–393. [DOI] [PubMed] [Google Scholar]
- Halabi N.; Rivoire O.; Leibler S.; Ranganathan R. (2009) Protein sectors: evolutionary units of three-dimensional structure. Cell 138, 774–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.