

Abstract
This study investigated the stability of genetic and environmental effects on the common liability to alcohol, tobacco, and cannabis dependence across adolescence and young adulthood. DSM-IV symptom counts from 2,361 adolescents were obtained using a structured diagnostic interview. Several sex-limited longitudinal common pathway models were used to examine gender differences in the magnitude of additive genetic (A), shared environment, and non-shared environmental effects over time. Model fitting indicated limited gender differences. Among older adolescents (i.e., age >14), the heritability of the latent trait was estimated at 0.43 (0.05, 0.94) during the first wave and 0.63 (0.21, 0.83) during the second wave of assessment. A common genetic factor could account for genetic influences at both assessments, as well as the majority of the stability of SAV over time [rA = 1.00 (0.55, 1.00)]. These results suggest that early genetic factors continue to play a key role at later developmental stages.
Keywords: Longitudinal, Twin study, Tobacco, Cannabis, Alcohol, Drug dependence
Introduction
Substance use disorders are highly comorbid phenomena with high rates of multiple substance use and substance use disorders observed in large national and international epidemiological surveys (Degenhardt et al. 2001; Johnston et al. 2008; Substance Abuse and Mental Health Services Administration 2010). For instance, of the 22.3 million persons aged 12 or older with a past year SUD in the 2007 NSDUH survey, 3.2 million persons were dependent on alcohol and an illicit substance. Both clinical and non-clinical studies highlight the significant overlap in risk for alcohol, tobacco, and other drugs (Hopfer et al. 2001, Malcolm et al. 2006; Young et al. 2006). For instance, Rhee et al. (2006) compared 13 models of comorbidity using a combined clinical and control sample examining comorbidity of dependence on alcohol and any of eight classes of illicit drugs. The best fitting model explaining the comorbidity in the data was the alternate forms (i.e., disorders are the manifestations of an underlying factor).
A number of studies have also demonstrated that phenotypic variation in the liability to use and abuse multiple substances is attributable to common genetic and environmental mechanisms (Cadoret et al. 1986, 1995; Kendler et al. 1997; Merikangas et al. 1998; Young et al. 2000). For example, upwards of 80 % of the observed covariance among alcohol, tobacco, and other drugs is attributable to genetic factors (Button et al. 2007; Hopfer et al. 2001; Kendler et al. 2007, 2008; True et al. 1999; Tsuang et al. 1998; Young et al. 2006). Using community-based twins from the Center for Antisocial Drug Dependence [CADD; a sample whose rate of substance use and dependence is similar to that observed in the general population (Palmer et al. 2009; Young et al. 2002)], we demonstrated that DSM-IV symptoms of alcohol, tobacco, and cannabis dependence are indicative of a latent trait [Substance Addiction Vulnerability (SAV)] in both males and females (Palmer et al. 2012). Furthermore, the study indicated that the magnitude of additive genetic and non-shared environmental influences on SAV was the same across genders. Overall, these studies indicate that, regardless of gender, there are genetic differences that largely predispose individuals to use and abuse multiple substances. Unfortunately, many studies of the development of substance-related problems have focused on individual substances and relied upon either cross-sectional or retrospective information, complicating the interpretations and generalization of the results.
We are aware of two studies that have examined the etiological stability of alcohol and tobacco dependence separately in prospective samples (Malone et al. 2004; Viken et al. 2007) and a single study that has examined the endorsement of Diagnostic and Statistical Manual of Mental Disorders Version III-R (3rd ed., rev.; DSM-III-R; American Psychiatric Association 1987) dependence symptoms across the most commonly used substances during adolescence and young adulthood (i.e., alcohol, tobacco, and cannabis) (Vrieze et al. 2012). Malone et al. (2004) examined the stability of alcohol dependence in a sample of male twins who were assessed DSM-IV dependence symptoms at ages 17, 20, and 24. The researchers were able to demonstrate that early symptoms predicted later symptoms and that an early genetic factor contributes to later substance problems. Viken et al. (2007) investigated the etiological stability of alcohol dependence symptoms but also considered the possibility of varying developmental trends between genders. Their study examined alcohol data collected using the Rutgers Alcohol Problem Index (RAPI) from Finnish twins at ages 18 and 25. Viken and colleagues’ best fitting biometric model at each assessment was an AE model with the additive genetic factor accounting for approximately 50 % of the variance in RAPI scores at each assessment (in males, heritability (h2) at 18 = 0.47, h2 at 25 = 0.48; in females, h2 at 18 = 0.61, h2 at 25 = 0.48). Most notably, genetic correlations across assessments were large (0.95 in males, 0.74 in females) and non-shared environmental correlations were small (0.22 in males, 0.25 in females). More recently, Vrieze et al. (2012) utilized a large longitudinal twin cohort to study changes in the covariance among alcohol, tobacco, and cannabis abuse and dependence symptoms from adolescence into early adulthood. Their analysis showed that the covariance among substance decreased with age. Further, Vrieze et al.’s biometrical analyses showed that the heritability of the latent trait decreased with age while the non-shared environmental effect on the latent trait increased with age.
The goal of this study was to extend our previous report (i.e., Palmer et al. 2012) by examining the contribution of genetic and environmental factors to the common liability to DSM-IV dependence symptoms on alcohol, tobacco, and cannabis across adolescence and young adulthood. In the previous study we identified a heritable latent trait (across males and females) that was indicated by DSM-IV alcohol, tobacco, and cannabis dependence symptoms. The specific goals of this study were to:
Estimate genetic and environmental effects on the latent trait during adolescence and young adulthood, as well as the persistence of their effects over time and,
Determine the contribution of genetic and environmental factors to the stability of the latent trait over time.
Notably, we took the same steps described in our previous report (Palmer et al. 2012) to account for any bias in genetic and environmental estimates that might arise from the treatment of scores from individuals who have never been exposed to alcohol, tobacco, or cannabis. Furthermore, given the developmental nature of substance addiction phenotypes analyses were conducted in informative cohorts of adolescence where the levels of use and problems are more prevalent (Johnston et al. 2008; Palmer et al. 2009).
Methods
Sample
Participants in the current study are part of an ongoing community-based longitudinal study at the CADD at the University of Colorado at Boulder. The CADD was selected for this project because the rates of alcohol, tobacco, and cannabis have already been shown to mirror levels observed in large populations, such as the Monitoring the Future Study and the National Survey on Drug Use and Health (Palmer et al. 2009). The twins utilized in the CADD originate from two community-based twin studies at the University of Colorado that are part of the much larger, Colorado Twin Registry based at the Institute for Behavioral Genetics at the University of Colorado at Boulder. The two studies are the Longitudinal Twin Study (LTS) and the Community Twin Sample (CTS). The CTS study originated in 1984 and was open to all twins born in the state of Colorado from 1982 and onward. The LTS was initiated in 1986 and used a broad set of criteria (see Rhea et al. 2006; 2012) to recruit infant twins to be combined with 59 twin pairs that were part of another project [Infant Twin Sample (approximately 1984)]. Twins from both samples were recruited into the CADD while they were between the ages of 12 and 18 years of age. Twins were required to have no known abnormalities that would prevent participation, such as an inability to provide meaningful consent. A more detailed description of the twin samples in the CADD are available elsewhere (Rhea et al. 2006, 2012).
For the current study, we utilized 2,361 respondents who completed the first two waves of the ongoing study (mean retest lag = 5.22 years, SD = 1.07) and who had zygosity information available. We also identified 383 participants who participated in the study at wave 1 but did not have any data available at wave 2 (further details and the implications of sample attrition are presented in the discussion section). Given the developmental nature of substance use and the very low rates of dependence symptoms generally observed in community samples (Johnston et al. 2008; Young et al. 2002), our sample was assessed as two separate cohorts based on their age of first assessment, which corresponded roughly to the middle school and high school periods. At the first wave of assessment (wave 1), participants in the younger adolescent cohort (N = 1164; 48 % male) were between the ages of 12 and 14 [male mean age = 13.03, standard deviation (SD) = 0.81, female mean age = 12.95, SD = 0.83]. At the wave 2 follow-up twins in the young adolescent cohort were between the ages of 16 and 25 [male mean age = 18.08 (SD = 1.36), female mean age = 18.01 (SD = 1.25)]. At the initial assessment, twins in the older adolescence cohort (N = 1197; 44 % male) were between the ages of 15 and 18 [mean age in males = 16.64 (SD = 1.14), mean age in females = 16.61 (SD = 1.17)]. At the wave 2 follow-up the age range of twins in the older adolescence cohort was from 19 to 29 [mean age in males = 22.11 (SD = 1.81), mean age in females = 22.14 (SD = 1.84)]. Zygosity was determined using a nine-item assessment focused on their physical characteristics and concordance on eleven highly informative short tandem repeat polymorphisms. The number of complete pairs in the younger adolescent cohort was: 134 monozygotic (MZ) male twin pairs (MZM), 147 MZ female twin pairs (MZF), 103 dizygotic male twin pairs (DZM), 103 dizgyotic female twin pairs (DZF), and 78 dizygotic opposite-sex male–female twin pairs (OS). The number of complete pairs in the older adolescent cohort was: 112 MZM, 162 MZF, 61 DZM, 78 DZF, and 140 OS.
Assessments
Our dependent variable of interest in this study was the latent trait indicated by the total amount of DSM-IV drug dependence symptoms on alcohol, tobacco, and cannabis. Symptoms were obtained from a paper version of the Composite International Diagnostic Interview—Substance Abuse Module (CIDI-SAM) at the first wave and a computerized version of the CIDI-SAM during the second wave. The CIDI-SAM is a research-based interview administered by lay interviewers and a reliable diagnostic tool for the collection of dependence data in clinical and general research settings (Cottler et al. 1989; Crowley et al. 2001; Üstun et al. 1997). Respondents reported if they had experienced any of the following symptoms in their lifetime: ‘tolerance’, ‘withdrawal’, ‘substance taken in larger amount and for longer period than intended’, ‘persistent desire or repeated unsuccessful attempt to quit’, ‘much time/activity to obtain, use, recover’, ‘important social, occupational, or recreational activities given up or reduced’, and ‘continued use despite knowledge of adverse consequences’. Lifetime symptom counts were tallied for the first wave and second wave of assessment. Dependence symptoms were assessed amongst individuals who reported use of each substance. Alcohol users were defined as individuals who consumed six of more alcoholic beverages in their lifetime at wave 1, or having used alcohol at least once in their lifetime at wave 2. Adolescent tobacco users were required to have smoked a cigarette, a cigar, a pipe, or used snuff or chewing tobacco at least once in their lifetime; young adult tobacco users were those individuals who had smoked at least 20 cigarettes in their lifetime, or who had used cigars, a pipe, snuff, or chewing tobacco more than five times in their life. Lastly, cannabis users, at both waves of assessment, were defined as those individuals who reported having used marijuana, grass, pot, or hashish more than five times in their lifetime. Since how we score participants who have never been exposed to a drug can bias genetic and environmental estimates, respondents who did not report exposure to a substance received a missing value on or that substance. Consequently, each variable was conditional on drug use. This approach reduces the likelihood that environmental effects that are often observed with substance initiation phenotypes (Hopfer et al. 2003) bias effects observed on dependence in this study.
Analyses
Descriptive statistics were obtained using SAS® and MPlus (Muthén and Muthén, 1998). Due to non-normality in symptom count data, the Mann–Whitney U test was used to test for sex differences in the dependence symptom counts within each age cohort. Twin correlations and confirmatory factor models (that explore the existence of a single latent trait) were conducted using MPlus while accounting for the clustering of individuals within families. Notably, the latent trait (referred to as SAV) was constrained to have a mean of zero and a variance of one.
A Longitudinal Common Pathway Model was used to determine the magnitude of genetic and environmental effects on SAV across waves of assessment. Models were fitted using age and sex corrected log transformed [log(x + 1)] symptom counts in Mx (Neale 1999) (corrections were done within each age cohort). The Longitudinal Common Pathway Model (shown in Fig. 1) is a multivariate extension of the classical twin model. The model decomposes the variance of a latent variable (with direct phenotypic paths to each substance) into latent additive genetic (A), shared environment (C), and non-shared environmental (E) effects, by comparing MZ and DZ twins who, under the equal environments assumption, differ on a phenotype because of differences in genetic relatedness between the pairs of twins. The model also decomposes the residual variance of each observed phenotype into trait-specific genetic and environmental effects. These analyses take advantage of the fact that MZ twins share 100 % of their genetic material while DZ twins share on average 50 % of their genetic information identical by descent. The extent to which MZ twin pairs are more similar than DZ twin pairs is indicative of genetic effects on a phenotype. Using our sample of same and opposite sex twins, we tested whether the magnitude of the genetic effects on the latent trait differed between males and females. In doing so, we compared several sex limitation models. The full model (Model I) allowed all of the pathways for A, C, and E (i.e., on the latent trait and the substance-specific residuals) and the loadings on the latent trait to differ between genders. The full model was compared to a more restricted model (Model II) that equated all the factor loadings across genders, and subsequently to an even more restricted model (Model III) that further constrained the magnitude of A, C, and E on the latent trait to be the same for males and females. Finally, we tested a sex-limited model that constrained the factor loadings and all common and trait specific A, C, and E effects to be the same across genders (this is referred to as Model IV). In each model, the variance of the latent factor was constrained to 1. This allowed us to estimate the factor loadings on the latent trait and to test the significance of the factor structure. To address the main research question of the study we assessed the stability of the genetic and environmental effects on the latent trait across waves of assessment for each cohort by dropping (i.e., fixing at zero) the pathways responsible for the genetic and environmental covariance from the model (i.e., a21, c21, and e21). Since Models II, III, and IV, are nested within Model I, all models were compared by examining the difference in the minus twice log likelihood estimates that are distributed as a Chi square with degrees of freedom equivalent to the differences in degrees of freedom between the models, as well as the change in the comparative fit indices (i.e., the Akaike Information Criterion (Akaike 1987) (AIC) and Sample Size Adjusted Bayesian Information Criterion (SABIC) (Enders and Tofighi 2008) (where lower AIC and SABIC values indicate better fitting/more parsimonious models). Sub models of the best-fitting sex-limitation model were compared using the same statistics. To further illustrate the implications of the model comparisons, a better fitting Model II (compared to Model I) would be indicative of a difference in factor loadings across genders.
Fig. 1.
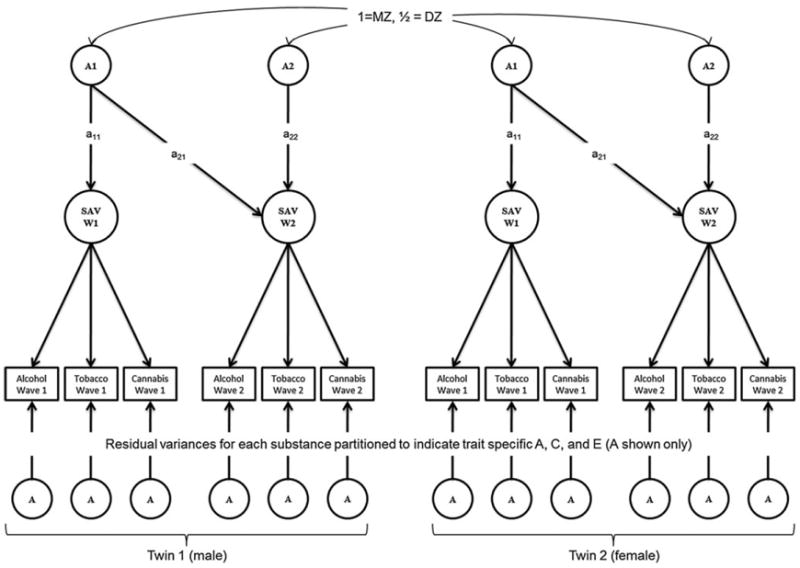
Example of a Longitudinal Common Pathway Model (Twin 1 only). Path diagram illustrating the Longitudinal Common Pathway Model. The variance of the latent trait [Substance Addiction vulnerability (SAV)] at waves 1 and 2 is decomposed into its genetic (A) and shared (C), and non-shared environmental (E) components. The covariance between the genetic (or environmental) factors at each wave is determined by the significance of the path connecting the first factor to the second assessment (i.e., a21, c21, and e21). The model also decomposes the residual variance of each substance into trait specific additive genetic, shared environment, and non-shared environmental effects (not all shown). Abbreviations: A additive genetic factor; a additive genetic path loading (although not shown, the following notations can also be included C shared environment factor; c shared environment path loading; E non-shared environment factor; e non-shared environment path loading)
Results
Sample description
Table 1 presents the frequency of substance use and the mean levels of DSM-IV drug dependence amongst users of each age cohort by gender. As expected, the prevalence of drug use and the mean levels of dependence were higher in the older cohort. Consistent with our earlier report (Palmer et al. 2009), the most frequently used substance at each wave of assessment was alcohol, followed by tobacco, and cannabis. There were several instances in which the mean level of dependence symptoms significantly differed between males and females. For instance, in the young adolescent cohort, the mean level of alcohol dependence symptoms was greater in males at both waves of assessment. Means difference tests within the older cohort indicated no difference in symptom levels between genders during the initial assessment, however, on average, males endorsed more alcohol symptoms while females endorsed more tobacco dependence symptoms at wave 2.
Table 1.
Distribution of use and DSM-IV dependence symptoms by gender and age cohort
| Younger males (564) | Younger females (600) | Older males (529) | Older females (668) | |
|---|---|---|---|---|
| Use | ||||
| Alcohol | 69 (13.27 %) | 61 (11.28 %) | 320 (63.24 %) | 387 (60.47 %) |
| Tobacco | 44 (8.46 %) | 23 (4.25 %) | 229 (45.26 %) | 257 (40.22 %) |
| Cannabis | 13 (2.30 %) | 5 (0.83 %) | 105 (19.84 %) | 141 (21.11 %) |
| Dependence | ||||
| Alcohol | 0.16 (0.41) | 0.03 (0.26) | 0.42 (0.86) | 0.47 (0.94) |
| Tobacco | 0.21 (0.85) | 0.22 (1.04) | 0.81 (1.59) | 1.03 (1.73) |
| Cannabis | 0.92 (1.26) | 2.00 (0.71) | 1.37 (1.64) | 1.32 (1.46) |
| Wave 2 | ||||
| Use | ||||
| Alcohol | 403 (71.45 %) | 337 (62.83 %) | 498 (94.14 %) | 607 (90.87 %) |
| Tobacco | 200 (35.46 %) | 103 (19.03 %) | 329 (62.19 %) | 265 (39.67 %) |
| Cannabis | 185 (32.80 %) | 119 (19.83 %) | 258 (48.77 %) | 245 (36.67 %) |
| Dependence | ||||
| Alcohol | 1.08 (1.55) | 0.76 (1.37) | 1.54 (1.65) | 1.18 (1.61) |
| Tobacco | 2.22 (2.09) | 2.53 (1.97) | 2.24 (2.00) | 2.70 (1.93) |
| Cannabis | 1.23 (1.52) | 1.01 (1.35) | 1.11 (1.51) | 1.01 (1.44) |
Table shows prevalence of use [N (%)] and the mean and SD [mean (SD)] of observed DSM-IV symptom counts for alcohol, tobacco, and cannabis among the identified drug users. Note: Bolded values indicate instances in which the mean level of symptom endorsed in male and female drug users within the same age cohort is significantly different (Mann–Whitney U p-value < 0.05)
Males and females displayed a general tendency to endorse dependence symptoms across alcohol, tobacco, and cannabis at each of assessment. However, this was only the case for participants in the older cohort due to the low frequency of users with dependence problems in the younger cohort. Table 2 shows the correlations between substances at each wave of assessment for males and females separately. In general, the correlations between substances increased over time. For instance, in females, the correlation between substances at wave 1 ranged from 0.22 to 0.29; at wave 2 the correlation between substances ranged from 0.31 to 0.37. Correlations between substances were generally larger in males. Due to the limited information available in the young cohort, we focused on data from the older wave 1 cohort for subsequent analyses.
Table 2.
Phenotypic correlations between substances across waves of assessment in the older cohort by gender
| Alcohol at wave 1 | Tobacco at wave 1 | Cannabis at wave 1 | Alcohol at wave 2 | Tobacco at wave 2 | Cannabis at wave 2 | |
|---|---|---|---|---|---|---|
| Females | ||||||
| Alcohol at wave 1 | 1.00 | 0.34 | 0.35 | 0.21 | 0.17 | 0.20 |
| Tobacco at wave 1 | 0.29 | 1.00 | 0.29 | 0.18 | 0.45 | 0.20 |
| Cannabis at wave 1 | 0.22 | 0.23 | 1.00 | 0.03 | 0.03 | 0.33 |
| Alcohol at wave 2 | 0.26 | 0.11 | 0.12 | 1.00 | 0.31 | 0.33 |
| Tobacco at wave 2 | 0.15 | 0.40 | 0.17 | 0.31 | 1.00 | 0.27 |
| Cannabis at wave 2 | 0.15 | 0.21 | 0.27 | 0.34 | 0.37 | 1.00 |
Table show correlation between log-transformed symptom counts for males and females in the older adolescent cohort. Individuals from the young cohort are not presented due to low rates of dependence, which limited our ability to derive estimates with sufficient confidence. Males are presented above the diagonal and females below the diagonal
Figure 2 describes the results of the confirmatory factor models exploring the structure of the latent trait at each wave of assessment for each gender in the older cohort. Due to the low levels of use and dependence in the young cohort, there was limited evidence of a latent trait in the younger cohort primarily. It was not possible to obtain a model that converged in the younger cohort due to the low covariance coverage within each wave of assessment and across assessments. For example, only 16.8 % of males had non-missing values for alcohol symptoms at wave 1 and only 11.5 % of males have non-missing values for both alcohol symptoms at wave 1 and tobacco symptoms at wave 2. In the older cohort, the latent trait (SAV) was indicated at both waves of assessment and factor loadings could not be fixed at zero without a significant loss in model fit. Furthermore the model fitted the data well [χ2 = 58.913 (Female = 24.844, Male = 34.069), df = 24, p value = 1E–4, RMSEA = 5.1E–3]. The phenotypic correlation between SAV factors at each wave was estimated at 0.69 in females and 0.70 in males, suggesting strong stability of the latent trait. Biometrical analyses were fitted to data from twins in the older cohort.
Fig. 2.
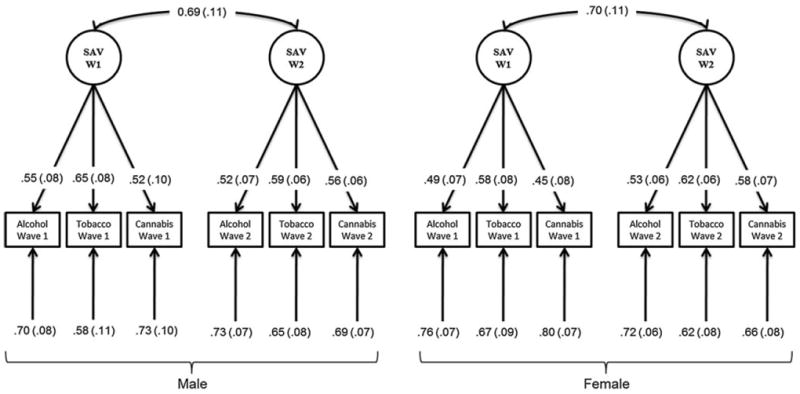
Confirmatory Factor Analysis Results for Older Adolescents. Path diagram illustrating the standardized path loadings (with standard error) from the confirmatory factor analysis conducted using male and female twins in the older adolescent cohort. Abbreviations: SAV Substance Addiction Vulnerability factor
Supplemental Table S1 presents the within and cross trait correlations for individual substances in the older adolescent cohort. In most instances, the same sex MZ twin correlation was greater than or equal to the DZ twin correlation suggesting the role of A, C, and E effects. For tobacco dependence at wave 2, the MZ correlations were more than twice the DZ twin correlation suggesting role of non-additive genetic effects. Given, our interest in the stability of the etiology of the latent trait (derived from the observed traits) and the lack of evidence for non-additive genetic effects in our previous reports (Rhee et al. 2006; Palmer et al. 2012) we decomposed the variance in SAV into additive genetic, shared environment, and non-shared environmental effects.
Longitudinal common pathway model fitting
The Longitudinal Common Pathway model that constrained the factor loadings across genders, as well as the magnitude of genetic and environmental influences on the latent trait (SAV) and the residual variance for each substance, was the best fitting sex-limitation model (i.e., Model IV; as indicated by the improvement in fit in Table 3). As shown in Table 3, many of the additive genetic effects on SAV could not be dropped from the model without a significant loss in fit (i.e., a positive change in the AIC or a significant Chi square difference test). This suggested that a significant proportion of the variation in SAV could be attributed to additive genetic factors. In addition, there was limited evidence for any unique/novel genetic effects emerging during young adulthood/wave 2 as the path a22 could be dropped from the model without any significant loss in model fit. This suggested that a common genetic factor could account for the general liability to alcohol, tobacco, and cannabis dependence over time.
Table 3.
Longitudinal common pathway model fitting and parameter tests on effects on latent trait
| −2LL | df | AIC | SABIC | Δc2 | Δdf | Δp | ΔAIC | ΔBIC | |
|---|---|---|---|---|---|---|---|---|---|
| Comparison of sex-limitation models (I thru IV) | |||||||||
| Model I Common effects sex-limitation model (full model) | 9795.17 | 3567 | 2661.17 | −901.76 | |||||
| Model II Equate factor loadings across genders | 9798.41 | 3573 | 2652.41 | −909.90 | 3.24 | 6 | 0.78 | −8.759 | −8.13 |
| Model III Equate common ACE effects and factor loadings | 9802.52 | 3582 | 2638.52 | −922.47 | 7.35 | 15 | 0.95 | −22.65 | −20.71 |
| Model IV Equate common and specific ACE effects and factor loadings | 9826.96 | 3600 | 2626.96 | −939.52 | 31.79 | 33 | 0.53 | −34.21 | −37.76 |
| Comparisons to best-fitting sex-limitation model (Model IV) | |||||||||
| Drop a11, a21—A effects on SAV at wave 1 | 9833.69 | 3602 | 2629.69 | −939.41 | 6.73 | 2 | 0.04 | 2.73 | 0.12 |
| Drop a22, a21—A effects on SAV at wave 2 | 9835.38 | 3602 | 2631.38 | −938.56 | 8.42 | 2 | 0.02 | 4.42 | 0.96 |
| Drop a21—genetic correlation | 9833.69 | 3601 | 2631.69 | −937.78 | 6.73 | 1 | 0.01 | 4.73 | 1.74 |
| Drop a22—test significance of second A factor on SAV at wave 2 | 9826.96 | 3601 | 2624.96 | −941.15 | 0.00 | 1 | 1.00 | −2.00 | −1.63 |
| Drop a11, a21, a22—A effects on SAV at waves 1 and 2 | 9835.74 | 3603 | 2629.74 | −940.01 | 8.78 | 3 | 0.03 | 2.78 | −0.49 |
| Drop c11, c21—C effects on SAV at wave 1 | 9827.90 | 3602 | 2623.90 | −942.30 | 0.94 | 2 | 0.63 | −3.06 | −2.78 |
| Drop c22, c21—C effects on SAV at wave 2 | 9827.68 | 3602 | 2623.68 | −942.41 | 0.72 | 2 | 0.70 | −3.28 | −2.89 |
| Drop c21—shared environment correlation | 9827.68 | 3601 | 2625.68 | −940.79 | 0.72 | 1 | 0.40 | −1.28 | −1.26 |
| Drop c22—test significance of second C factor on SAV at wave 2 | 9826.96 | 3601 | 2624.96 | −941.15 | 0.00 | 1 | 1.00 | −2.00 | −1.63 |
| Drop c11, c21, c22—C effects on SAV at waves 1 and 2 | 9827.90 | 3603 | 2621.90 | −943.93 | 0.94 | 3 | 0.82 | −5.06 | −4.41 |
| Drop e21—non-shared environmental correlation | 9827.83 | 3601 | 2625.83 | −940.71 | 0.87 | 1 | 0.35 | −1.13 | −1.19 |
| Significance of substance-specific A, C, E effects by wave (compared to Model IV) | |||||||||
| Alcohol at wave 1 | |||||||||
| Drop A specific | 9826.96 | 3601 | 2624.96 | −941.15 | 0.00 | 1 | 1.00 | −2.00 | −1.63 |
| Drop C specific | 9829.12 | 3601 | 2627.12 | −940.06 | 2.16 | 1 | 0.14 | 0.16 | −0.54 |
| Tobacco at wave 1 | |||||||||
| Drop A specific | 9826.97 | 3601 | 2624.97 | −941.14 | 0.01 | 1 | 0.92 | −1.99 | −1.62 |
| Drop C specific | 9828.14 | 3601 | 2626.14 | −940.55 | 1.18 | 1 | 0.28 | −0.82 | −1.03 |
| Cannabis at wave 1 | |||||||||
| Drop A specific | 9826.96 | 3601 | 2624.96 | −941.15 | 0.00 | 1 | 1.00 | −2.00 | −1.63 |
| Drop C specific | 9828.20 | 3601 | 2626.20 | −940.53 | 1.24 | 1 | 0.27 | −0.76 | −1.01 |
| Alcohol at wave 2 | |||||||||
| Drop A specific | 9827.38 | 3601 | 2625.38 | −940.94 | 0.42 | 1 | 0.52 | −1.58 | −1.41 |
| Drop C specific | 9827.15 | 3601 | 2625.15 | −941.05 | 0.19 | 1 | 0.66 | −1.81 | −1.53 |
| Tobacco a wave 2 | |||||||||
| Drop A specific | 9833.91 | 3601 | 2631.91 | −937.67 | 6.95 | 1 | 0.01 | 4.95 | 1.85 |
| Drop C specific | 9826.96 | 3601 | 2624.96 | −941.15 | 0.00 | 1 | 1.00 | −2.00 | −1.63 |
| Cannabis at wave 2 | |||||||||
| Drop A specific | 9827.93 | 3601 | 2625.93 | −940.66 | 0.97 | 1 | 0.33 | −1.03 | −1.14 |
| Drop C specific | 9826.96 | 3601 | 2624.96 | −941.15 | 0.00 | 1 | 1.00 | −2.00 | −1.63 |
Note Table describes the multivariate fit statistics for common pathway models fitted to male and female data at each wave of assessment. Parameter tests are compared to Model IV. Subscript notations for a, c, and e correspond to pathways indicated in Fig. 1
−2LL minus twice log likelihood, A Additive genetic effects, a additive genetic pathway, AIC Akaike Information Criterion, C Shared Environmental effects, c shared environmental pathway, E Non-shared environmental effects, e non-shared environmental pathway, SABIC Sample Size Adjusted Bayesian Information Criterion, df degrees of freedom
The path coefficients corresponding to model IV are shown in Table 4. Using these coefficients we determined the heritability of SAV during adolescence and young adulthood, as well as the relative contribution of shared and non-shared environmental influences. These estimates are displayed in Table 5 which describes the genetic, shared environmental and non-shared environmental effects, correlations across the two waves of assessment, and the proportion of the phenotypic covariance attributed to A, C, and E. The total genetic variance of SAV increased from 0.43 during adolescence to 0.63 during young adulthood. At the same time, the absolute shared environmental variance on SAV decreased (i.e. from 0.30 to 0.05), while the total non-shared environmental variance slightly increased. Almost all of the genetic effect on SAV at young adulthood was due to factors in common with adolescence, indicating a persistent/stable effect of early factors. The phenotypic correlation between the SAV factors at each wave of assessment largely resulted from a common additive genetic factor (74 %), with negligible contribution from shared environment factors (17 %) and non-shared environmental factors (9 %). The estimated correlation between the A1 and A2 genetic factors was 1.00 (0.55, 1.00). Altogether, these results suggest that the covariance between the common liability to drug dependence measured at adolescence and at young adulthood is largely due to a common genetic factor.
Table 4.
Standardized loadings for best-fitting common pathway model (model IV)
| Parameters | Standardized path estimates (95 % CI) |
|---|---|
| Etiology of latent trait | |
| a11 | 0.66 (0.21, 0.96) |
| a21 | 0.79 (0.32, 0.92) |
| a22 | 1E–4 (−0.62, 0.62) |
| c11 | 0.55 (−0.83, 0.83) |
| c21 | 0.22 (−0.60, 0.60) |
| c22 | 1E–4 (−0.43, 0.00) |
| e11 | 0.52 (0.21, 0.70) |
| e21 | 0.13 (−0.16, 0.16) |
| e22 | 0.55 (0.34, 0.72) |
| Factor loadings on latent trait | |
| Alcohol at wave 1 | 0.52 (0.43, 0.62) |
| Tobacco at wave 1 | 0.61 (0.49, 0.71) |
| Cannabis at wave 1 | 0.49 (0.35, 0.62) |
| Alcohol at wave 2 | 0.53 (0.44, 0.61) |
| Tobacco at wave 2 | 0.60 (0.50, 0.68) |
| Cannabis at wave 2 | 0.57 (0.46, 0.66) |
| Etiology of residual variance | |
| a-Alcohol wave 1 | 0.00 (−0.49, 0.49) |
| c-Alcohol wave 1 | 0.40 (−0.51, 0.51) |
| e-Alcohol wave 1 | 0.75 (0.67, 0.82) |
| a-Tobacco wave 1 | 0.14 (−0.59, 0.59) |
| c-Tobacco wave 1 | 0.44 (−0.58, 0.57) |
| e-Tobacco wave 1 | 0.65 (0.54, 0.73) |
| a-Cannabis wave 1 | 0.00 (−0.53, 0.53) |
| c-Cannabis wave 1 | 0.39 (−0.58, 0.55) |
| e-Cannabis wave 1 | 0.78 (0.65, 0.91) |
| a-Alcohol wave 2 | 0.31 (−0.51, 0.51) |
| c-Alcohol wave 2 | 0.23 (−0.46, 0.46) |
| e-Alcohol wave 2 | 0.75 (0.67, 0.82) |
| a-Tobacco wave 2 | 0.57 (0.34, 0.68) |
| c-Tobacco wave 2 | 0.00 (−0.40, 0.40) |
| e-Tobacco wave 2 | 0.56 (0.45, 0.65) |
| a-Cannabis wave 2 | 0.37 (−0.54, 0.54) |
| c-Cannabis wave 2 | 0.00 (−0.44, 0.44) |
| e-Cannabis wave 2 | 0.74 (0.62, 0.85) |
Table shows standardized path loadings and 95 % confidence intervals for the best-fitting sex-limitation model (Model IV)
Table 5.
Magnitude of genetic and environmental influences
| A | C | E | |
|---|---|---|---|
| SAV at adolescence (wave 1) | 0.43 (0.05, 0.94) | 0.30 (0.00, 0.68) | 0.27 (0.04, 0.48) |
| SAV at young adulthood (wave 2) | 0.63 (0.21, 0.83) | 0.05 (0.00, 0.36) | 0.32 (0.14, 0.54) |
| Correlation between factors | 1.00 (0.55, 1.00) | 1.00 (−1.00, 1.00) | 0.23 (−0.29, 0.70) |
| % of covariance explained by: | 74 % | 17 % | 9 % |
Note Table shows the proportion of variation in the common liability to alcohol, tobacco, and cannabis DSM-IV dependence symptoms (as measured by the factor score) attributable to additive genetic (A), shared environment (C) and non-shared environmental factors (E). Covariance and correlation estimates were derived from the full bivariate model
Discussion
Although it is already well established that genetic differences contribute to the variation in susceptibility to substance dependence, the present study makes a unique contribution to the literature in multiple ways. First, it adds to the limited number of studies that examine stability and change of genetic and environmental factors that influence the common liability to drug dependence. Second, to our knowledge, it is the first study to examine gender differences while accounting for age effects on the stability of the liability to drug dependence. Multivariate extensions of the classical twin model are a powerful means to explore the magnitude of common and specific genetic and environmental factors across multiple groups (e.g., age and gender).
For SAV, the magnitude of genetic influences increased from adolescence to young adulthood. Furthermore, almost all of the phenotypic covariance was attributable to genetic influences. These results are consistent with prior longitudinal studies. Although a direct comparison to other studies is not entirely possible, due to our use of a latent trait instead of individual substances, our findings are consistent with Viken et al. (2007), which found large genetic correlations (0.95 in males, 0.74 in females) between alcohol problem scores measured at age 18 and 25. Our findings are also similar to that of Vrieze et al. (2012), which found moderate genetic influences on the latent trait during adolescence into young adulthood. However, unlike Vrieze et al.’s study, our best-fitting sex-limitation model provided point estimates suggesting an increase in the genetic effects on SAV over time which was accompanied by a decrease in shared environmental influences; notably, although Model IV suggests changes in effects over time we were able to constrain the magnitude of ACE effects on SAV to be the same at each wave of assessment without any significant loss in fit (Δχ2 = , Δdf = 4, p = 0.80, ΔAIC = −6.35), however, we prefer to err on the side of caution when over-restricting the parameter space of classical twin models as the resulting estimates may be sample specific. These findings lend support to the biological (i.e., addictive properties of substances) theories that describe the liability to the continuous abuse of substances. For example, neurological models indicated by animal and human addiction research suggest that upon repeated use of substances like tobacco, the reward circuitry that is regulated by dopamine levels becomes dysregulated leading to a pattern of addiction (Gardner 2005; Koob and Le Moal 2001). Because these pathways are genetically determined, individual differences in the efficiency of this system likely account for some of the generalized risk across substances, as well as persistent substance problems over time.
The shared environmental influence on SAV was modest (~0.30) during adolescence and young adulthood. This observation is in agreement with the developmental hypothesis of substance use. Specifically, that substance use during adolescence is driven by genetic and environmental influences. Over time, as use becomes more normative, genetic effects on use phenotypes decrease (i.e., a result of the reduced variance in the population), but on another dimension of addiction (e.g., dependence) informative individual differences exist amongst users, which are more biologically driven. A detailed review of addiction genetics by Agrawal and Lynskey (2008) highlights the fact many twin studies show significant shared environmental influences on drug use, but only additive genetic and non-shared environmental influences on drug dependence.
Gender differences in the liability to SAV were few and inconsistent. Likewise, gender differences in the substance-specific genetic and environmental factors were negligible. These findings are consistent with many other studies of drug dependence (Agrawal and Lynskey 2008). For instance, several studies of alcohol dependence have identified limited differences in the liability to dependence in males and females. For example, using separate samples, Kendler and colleagues (1992) estimated the heritability of alcohol dependence to be approximately 50–60 % in females and 48–58 % in males (Prescott and Kendler 1999).
It is also noteworthy to discuss the significance of genetic effects on the common factor and the persistence of their effects over time. Genetic influences on the SAV factor support the hypothesis that several biological pathways and associated molecules (most likely neuronal) are activated, facilitated, and/or manipulated by drugs of abuse. Thus, if the emergent property of these pathways, as a whole, is for sensitivity to rewarding stimuli, then variation within said pathways would be common to all possible substances of abuse. Neuroimaging studies have provided support for the existence of said pathways. For example, it has been consistently shown that drug-induced changes in dopamine (a natural reinforcer) levels are associated with the reinforcing effects of drugs of abuse (Volkow et al. 2003). Notably, genetic variation within brain circuits (i.e., those related to reward, motivation, learning and memory, and decision-making) related to addiction, are ideal candidates for common sources of variation across substances. The high genetic correlation observed and the strong possibility of a common genetic factor over time confirms the stability of pleiotropic effects of genes across different substances. This is promising for twin and molecular studies as it suggests that the genetic liability is largely stable over time. This suggests that a study attempting to identify genetic variants influencing variation in the liability to alcohol, tobacco, and cannabis dependence may identify the same set of variants in adolescents as a study conducted in young adults. While we did observe an increase in the genetic effect over time, it is important to keep in mind the changes in the environment during this transition period. We observed a decrease in the shared environmental effect over time, while the non-shared environmental effect was relatively stable. Further, the shared environment or non-shared environmental factors did not contribute to the stability of the SAV. No environmental contribution to the stability of SAV reinforces the idea that environmental factors, such as substance using peers, and cultural norms are primarily relevant to early involvement/access to a specific substance or multiple substances. Hence, individual differences in the physiological and associated psychosocial consequences of chronic drug exposure (as measured by the DSM) of any of these three substances appear to be driven by variation in the same biological systems, as there is very little evidence for substance-specific genetic factors in our community sample, with the exception of tobacco dependence.
Several important considerations should be taken into account when referring to these findings. First, this study used only two waves of assessment. Although we were working with small age cohorts of drug users during adolescence (Palmer et al. 2009) and age effects were statistically removed within the younger and older cohorts, there are likely to be considerable differences in the etiology of dependence across early and late adolescence, and late adolescence and young adulthood. This limitation is not unique to the CADD project. It highlights the need for future studies that combine data across multiple samples to obtain the power to test age-to-age differences in the liability to drug addiction. In the future, we hope to extend these approaches to samples with assessments that span adulthood so that we may understand individual differences in patterns of problematic alcohol, tobacco, and cannabis use over time. Second, national longitudinal studies have shown that substance use and problems increases with age, peaks in early 20s (21–24), stabilizes, and then decreases during adulthood. Since some of our participants had not passed through the peak age of drug use and problems (age 23), we cannot discern if all participants have reached their peak level of risk for developing dependence symptoms. Finally, our third limitation is the problem of attrition. Of the 2,745 individuals that were observed at wave 1, 383 did not participate at wave 2. A possible explanation for this dropout is that these individual have more drug related problems (on average) compared to individuals who participated at both assessments. We found this to be the case for alcohol dependence in males, where those who were present at both waves had a mean of 0.37 (SD = 0.80), and those who were present only at wave 1 had a mean of 0.64 (SD = 1.08) (Wilcoxon Two Sample Test Z = 2.59, p < 0.01). Males who did not participate at both waves also reported significantly higher mean levels of tobacco dependence [mean = 1.02 (1.73)] symptoms than males who participated at both waves [mean = 0.71 (1.51)] (Z = 2.07, p = 0.04). Since males participating at both waves of assessment scored lower than males who did not participate at both waves of assessment, it is possible that the observed means in our study are slightly lower than would be expected if there was no attrition in the study.
To summarize, we have demonstrated that the covariance between the adolescent and young adult liability to alcohol, tobacco, and cannabis dependence is largely attributable to a common genetic factor. Our findings add to the growing number of studies that have suggested that persistence of substance problems is largely attributable to additive genetic factors. Our findings are especially useful in the context of measured genetic studies that aim to identify genetic variants common to multiple substances, as it demonstrates that adolescent populations are just as likely as adult populations to identify genetic risk factors for alcohol, tobacco, or cannabis dependence.
Supplementary Material
Acknowledgments
Funding for this study was provided by NIH grants AA021113, MH019927, MH063207, HD010333, AA007464-31, DA011015, and DA021913. We would also like to thank Dr. Nicole R. Nugent of the Division of Behavioral Genetics at Rhode Island Hospital for her input on the MPlus analyses.
Funding This study was funded by NIMH Grants MH019927, and MH063207, NICHD Grant HD010333, NIDA grants DA011015, and DA015522, and NIAAA grant AA021113 (Palmer). Data collection was supported by DA011015. The development and maintenance of the LTS sample was supported by NICHD Grant HD 010333 and MH063207. Individual support for the co-authors was provided by AA021113, DA011015 and DA021913.
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s10519-013-9599-5) contains supplementary material, which is available to authorized users.
Conflict of interest All of the listed authors declare that they have no conflicts of interests.
Contributor Information
R. H. C. Palmer, Email: Rohan_Palmer@Brown.edu, Division of Behavioral Genetics, Rhode Island Hospital, Providence, RI 02903, USA; Department of Psychiatry and Human Behavior, Brown University, Providence, RI 02903, USA; Bradley Hasbro Children’s Research Center, Coro West, 1 Hoppin St., Suite 204, Providence, RI 02903, USA.
S. E. Young, University of Colorado Health Sciences Center, Denver, CO 80262, USA
R. P. Corley, Institute for Behavioral Genetics, University of Colorado, Boulder, CO 80309, USA
C. J. Hopfer, University of Colorado Health Sciences Center, Denver, CO 80262, USA
M. C. Stallings, Institute for Behavioral Genetics, University of Colorado, Boulder, CO 80309, USA
J. K. Hewitt, Institute for Behavioral Genetics, University of Colorado, Boulder, CO 80309, USA
References
- Agrawal A, Lynskey MT. Are there genetic influences on addiction: evidence from family, adoption and twin studies. Addiction. 2008;103(7):1069–1081. doi: 10.1111/j.1360-0443.2008.02213.x. [DOI] [PubMed] [Google Scholar]
- Akaike H. Factor analysis and AIC. Psychometrika. 1987;52:317–332. [Google Scholar]
- American Psychiatric Association. Diagnostic and statistical manual of mental disorders. 3, Revised. American Psychiatric Association; Washington, DC: 1987. [Google Scholar]
- Button TMM, Rhee SH, Hewitt JK, Young SE, Corley RP, Stallings MC. The role of conduct disorder in explaining the comorbidity between alcohol and illicit drug dependence in adolescence. Drug Alcohol Depend. 2007;87(1):46–53. doi: 10.1016/j.drugalcdep.2006.07.012. [DOI] [PubMed] [Google Scholar]
- Cadoret RJ, Troughton E, O’Gorman TW, Heywood E. An adoption study of genetic and environmental factors in drug abuse. Arch Gen Psychiatry. 1986;43(12):1131–1136. doi: 10.1001/archpsyc.1986.01800120017004. [DOI] [PubMed] [Google Scholar]
- Cadoret RJ, Yates WR, Troughton E, Woodworth G, Stewart MA. Adoption study demonstrating two genetic pathways to drug abuse. Arch Gen Psychiatry. 1995;52(1):42–52. doi: 10.1001/archpsyc.1995.03950130042005. [DOI] [PubMed] [Google Scholar]
- Cottler LB, Robins LN, Helzer JE. The reliability of the CIDI-SAM: a comprehensive substance abuse interview. Br J Addict. 1989;84(7):801–814. doi: 10.1111/j.1360-0443.1989.tb03060.x. [DOI] [PubMed] [Google Scholar]
- Crowley TJ, Mikulich SK, Ehlers KM, Whitmore EA, MacDonald MJ. Validity of structured clinical evaluations in adolescents with conduct and substance problems. J Am Acad Child Adolesc Psychiatry. 2001;40(3):265–273. doi: 10.1097/00004583-200103000-00005. [DOI] [PubMed] [Google Scholar]
- Degenhardt L, Hall W, Lynskey M. The relationship between cannabis use and other substance use in the general population. Drug Alcohol Depen. 2001;64:319–327. doi: 10.1016/s0376-8716(01)00130-2. [DOI] [PubMed] [Google Scholar]
- Enders CK, Tofighi D. The impact of misspecifying class-specific residual variances in growth mixture models. Struct Equ Model Multidiscip J. 2008;15:75–95. [Google Scholar]
- Gardner EL. Brain-reward Mechanisms. In: Lowinson JH, Ruiz P, Millman RB, Langrod JG, editors. Substance Abuse: a comprehensive textbook. Lippincott Williams & Wilkins; Philadelphia, Pennsylvania: 2005. pp. 48–97. [Google Scholar]
- Hopfer CJ, Stallings MC, Hewitt JK. Common genetic and environmental vulnerability for alcohol and tobacco use in a volunteer sample of older female twins. J Stud Alcohol. 2001;62(6):717–723. doi: 10.15288/jsa.2001.62.717. [DOI] [PubMed] [Google Scholar]
- Hopfer CJ, Crowley TJ, Hewitt JK. Review of twin and adoption studies of adolescent substance use. J Am Acad Child Adolesc Psychiatry. 2003;42(6):710–719. doi: 10.1097/01.CHI.0000046848.56865.54. [DOI] [PubMed] [Google Scholar]
- Johnston LD, O’Malley PM, Bachman JG, Schulenberg JE. Monitoring the Future national results on adolescent drug use: Overview of key findings, 2007 (NIH Publication No 08-6418) Bethesda, MD: National Institute on Drug Abuse; 2008. [Google Scholar]
- Kendler KS, Heath AC, Neale MC, Kessler R, Eaves LJ. A population-based twin study of alcoholism in women. JAMA. 1992;268(14):1877–1882. [PubMed] [Google Scholar]
- Kendler KS, Davis CG, Kessler RC. The familial aggregation of common psychiatric and substance use disorders in the National Comorbidity Survey: a family history study. Br J Psychiatry. 1997;170:541–548. doi: 10.1192/bjp.170.6.541. [DOI] [PubMed] [Google Scholar]
- Kendler KS, Myers J, Prescott CA. Specificity of genetic and environmental risk factors for symptoms of cannabis, cocaine, alcohol, caffeine, and nicotine dependence. Arch Gen Psychiatry. 2007;64(11):1313–1320. doi: 10.1001/archpsyc.64.11.1313. [DOI] [PubMed] [Google Scholar]
- Kendler KS, Schmitt E, Aggen SH, Prescott CA. Genetic and environmental influences on alcohol, caffeine, cannabis, and nicotine use from early adolescence to middle adulthood. Arch Gen Psychiatry. 2008;65(6):674–682. doi: 10.1001/archpsyc.65.6.674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF, Le Moal M. Drug addiction, dysregulation of reward, and allostasis. Neuropsychopharmacology. 2001;24(2):97–129. doi: 10.1016/S0893-133X(00)00195-0. [DOI] [PubMed] [Google Scholar]
- Malcolm BP, Hesselbrock MN, Segal B. Multiple substance dependence and course of alcoholism among Alaska native men and women. Subst Use Misuse. 2006;41(5):729–741. doi: 10.1080/10826080500391803. [DOI] [PubMed] [Google Scholar]
- Malone SM, Taylor J, Marmorstein NR, McGue M, Iacono WG. Genetic and environmental influences on antisocial behavior and alcohol dependence from adolescence to early adulthood. Dev Psychopathol. 2004;16(4):943–966. doi: 10.1017/s0954579404040088. [DOI] [PubMed] [Google Scholar]
- Merikangas KR, Stolar M, Stevens DE, Goulet J, Preisig MA, Fenton B, Zhang H, O’Malley SS, Rounsaville BJ. Familial transmission of substance use disorders. Arch Gen Psychiatry. 1998;55(11):973–979. doi: 10.1001/archpsyc.55.11.973. [DOI] [PubMed] [Google Scholar]
- Muthén LK, Muthén BO. Mplus User’s Guide. 6. Muthén & Muthén; Los Angeles: 1998–2011. [Google Scholar]
- Neale MC. MX: statistical modeling. 5. Department of Psychiatry; Richmond: 1999. [Google Scholar]
- Palmer RH, Young SE, Hopfer CJ, Corley RP, Stallings MC, Crowley TJ, Hewitt JK. Developmental epidemiology of drug use and abuse in adolescence and young adulthood: evidence of generalized risk. Drug Alcohol Depend. 2009;102(1–3):78–87. doi: 10.1016/j.drugalcdep.2009.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer RH, Button TM, Rhee SH, Corley RP, Young SE, Stallings MC, Hopfer CJ, Hewitt JK. Genetic etiology of the common liability to drug dependence: evidence of common and specific mechanisms for DSM-IV dependence symptoms. Drug Alcohol Depend. 2012;123(Suppl 1):S24–S32. doi: 10.1016/j.drugalcdep.2011.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prescott CA, Kendler KS. Genetic and environmental contributions to alcohol abuse and dependence in a population-based sample of male twins. Am J Psychiatry. 1999;156(1):34–40. doi: 10.1176/ajp.156.1.34. [DOI] [PubMed] [Google Scholar]
- Rhea SA, Gross AA, Haberstick BC, Corley RP. Colorado twin registry. Twin Res Hum Genet. 2006;9(6):941–949. doi: 10.1375/183242706779462895. [DOI] [PubMed] [Google Scholar]
- Rhea SA, Gross AA, Haberstick BC, Corley RP. Colorado twin registry: an update. Twin Res Hum Genet. 2012;23:1–7. doi: 10.1017/thg.2012.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee SH, Hewitt JK, Young SE, Corley RP, Crowley TJ, Neale MC, Stallings MC. Comorbidity between alcohol dependence and illicit drug dependence in adolescents with antisocial behavior and matched controls. Drug Alcohol Depend. 2006;84(1):85–92. doi: 10.1016/j.drugalcdep.2005.12.003. [DOI] [PubMed] [Google Scholar]
- Substance Abuse and Mental Health Services Administration. Results from the 2010 National Survey on Drug Use and Health: Summary of National Findings, NSDUH Series H-41, HHS Publication No (SMA) 11-4658. Rockville, MD: Substance Abuse and Mental Health Services Administration; 2011. [Google Scholar]
- SAS. The output/code/data analysis for this paper was generated using SAS/STAT software, Version 9.1 of the SAS System for Windows. SAS Institute Inc.; Cary, NC, USA: Copyright © 2002–2003. [Google Scholar]
- True WR, Xiang H, Scherrer JF, Madden PAF, Bucholz KK, Heath AC, Eisen SA, Lyons MJ, Goldberg J, Tsuang M. Common genetic vulnerability for nicotine and alcohol dependence in men. Arch Gen Psychiatry. 1999;56(7):655–661. doi: 10.1001/archpsyc.56.7.655. [DOI] [PubMed] [Google Scholar]
- Tsuang MT, Lyons MJ, Meyer JM, Doyle T, Eisen SA, Goldberg J, True W, Lin N, Toomey R, Eaves L. Co-occurrence of abuse of different drugs in men: the role of drug-specific and shared vulnerabilities. Arch Gen Psychiatry. 1998;55(11):967–972. doi: 10.1001/archpsyc.55.11.967. [DOI] [PubMed] [Google Scholar]
- Üstün B, Compton W, Mager D, Babor T, Baiyewu O, Chatterji S, Cottler L, Göġűs A, Mavreas V, Peters L, Pull C, Sauders J, Smeets R, Stipec MR, Vrasti R, Hasin D, Room R, Van den Brink W, Regier D, Blaine J, Grant B, Sartorius N. WHO study on the reliability of the alcohol and drug use disorder instruments: overview of methods and results. Drug Alcohol Depend. 1997;47(3):161–169. doi: 10.1016/s0376-8716(97)00087-2. [DOI] [PubMed] [Google Scholar]
- Viken RJ, Kaprio J, Rose RJ. Personality at ages 16 and 17 and drinking problems at ages 18 and 25: genetic analyses of data from Finn Twin 16–25. Twin Res Hum Genet. 2007;10(1):25–32. doi: 10.1375/twin.10.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkow ND, Fowler JS, Wang GJ. The addicted human brain: insights from imaging studies. J Clin Invest. 2003;111(10):1444–1451. doi: 10.1172/JCI18533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vrieze SI, Hicks BM, Iacono WG, McGue M. Decline in genetic influence on the co-occurrence of alcohol, marijuana, and nicotine dependence symptoms from age 14 to 29. Am J Psychiatry. 2012;169(10):1073–1081. doi: 10.1176/appi.ajp.2012.11081268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young SE, Stallings MC, Corley RP, Krauter KS, Hewitt JK. Genetic and environmental influences on behavioral disinhibition. Am J Med Genet. 2000;96(5):684–695. [PubMed] [Google Scholar]
- Young SE, Corley RP, Stallings MC, Rhee SH, Crowley TJ, Hewitt JK. Substance use, abuse and dependence in adolescence: prevalence, symptom profiles and correlates. Drug Alcohol Depend. 2002;68(3):309–322. doi: 10.1016/s0376-8716(02)00225-9. [DOI] [PubMed] [Google Scholar]
- Young SE, Rhee SH, Stallings MC, Corley RP, Hewitt JK. Genetic and environmental vulnerabilities underlying adolescent substance use and problem use: general or specific? Behav Genet. 2006;36(4):603–615. doi: 10.1007/s10519-006-9066-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.