

Abstract
Amyloid-β peptide (Aβ) is still best known as a molecule to cause Alzheimer’s disease (AD) through accumulation and deposition within the frontal cortex and hippocampus in the brain. Thus, strategies on developing AD drugs have been focused on the reduc-tion of Aβ in the brain. Since accumulation of Aβ depends on the rate of its synthesis and clearance, the metabolic pathway of Aβ in the brain and the whole body should be carefully explored for AD research. Although the synthetic pathway of Aβ is equally important, we summarize primarily the clearance pathway in this paper because the former has been extensively reviewed in previous studies. The clearance of Aβ from the brain is accomplished by several mechanisms which include non-enzymatic and enzymatic pathways. Nonenzymatic pathway includes interstitial fluid drainage, uptake by microglial phagocytosis, and transport across the blood vessel walls into the circulation. Multiple Aβ-degrading enzymes (ADE) implicated in the clearance process have been identified, which include neprilysin, insulin-degrading enzyme, matrix metalloproteinase-9, glutamate carboxypeptidase II and others. A series of studies on Aβ clearance mechanism provide new insight into the pathogenesis of AD at the molecular level and suggest a new target for the development of novel therapeutics.
Keywords: Amyloid-β peptide, Amyloid-β peptide degrading enzyme, Alzheimer’s disease, Clearance, Proteases
INTRODUCTION
Alzheimer’s disease (AD) is a devasting neurodegenerative disease and the most common form of dementia. The prevalence of AD is approximately 5.7% in people over 65 years in South Korea (Kim et al., 2011a). Data from the US shows a rather higher prevalence of 10%. Many countries, including South Korea, are experiencing a fast rate of aging in their population and the prevalence of AD has been continuously increasing along with the rise in life expectancy. Unfortunately, aging is the top risk factor for AD and the total number of patient is expected to double every 20 years in South Korea. Thus, a better understanding of causes of this disease has been urgent and the development of disease-modifying therapy is the biggest issue in the 21st century.
AD is characterized by the accumulation and deposition of amyloid-β peptide peptides (Aβ) within the brain, leading to neuronal cell loss and perturbation of synaptic function (Tanzi and Bertram, 2005). Studies from the last decade revealed that disturbance in Aβ metabolism in the brain is thought to be central to the pathogenesis of the disease. The role of amyloid precursor protein (APP) processing and resulting Aβ production in disease development was established from the genetic analysis of familial forms of AD. However, the blocking of Aβ synthesis does not appear to be effective for reducing the brain Aβ levels as we expected. Recently, the importance of Aβ clearance in AD pathogenesis, especially in late-onset sporadic AD (LOAD) has been raised, and the understanding of Aβ clearance mechanism have provided new therapeutic targets.
REVISIT TO THE AMYLOID HYPOTHESIS
The Aβ cascade hypothesis of AD was originally proposed (Selkoe, 1991; Hardy and Higgins, 1992) by the theory that accumulation of Aβ, in particular Aβ1-42, is the initial trigger for neurodegeneration. However, the chemical nature and the precise biological roles of Aβ in AD pathogenesis have been elusive (Castellani and Smith, 2011). Furthermore, the failure of developing clinically effective disease-modifying drugs has underestimated Aβ-based therapeutic approaches but the genetic studies still strongly place Aβ as a favorable target. Early-onset type of AD (EOAD) occurs as a result of gene mutation involving APP, and presenilin genes (PSEN1, PSEN2). Mutation of these genes showed a common phenomenon of an increase of Aβ1-42 or of the ratio of Aβ1-42 to Aβ1-40 ; Aβ1-42 is more hydrophobic and more prone to aggregate than Aβ1-40 (Jarrett et al., 1993). Furthermore, we also found in Asian population including Korean that beta-site APP cleaving enzyme (BACE)-1 polymorphism in exon 5 infl uences a risk for LOAD in those carrying the ApoE ε4 allele (Jo et al., 2008) although Caucasian may not be the case. These observations strongly suggest a cause-and-effect relationship between Aβ accumulation and AD pathogenesis.
Previously, it has been generally accepted that amyloid plaques, fibrils, or much complicated forms synthesized from Aβ are the major pathological species causing impaired synaptic and neuronal dysfunctions. However, Aβ oligomers are highly toxic and cause synaptic dysfunction (Hardy and Selkoe, 2002), while amyloid plaques or fibrils induce proliferation and activation of glial cells which secret cytotoxic factors and indirectly induce neuronal damage. It was recently proposed that certain receptors were necessary for binding with Aβ oligomers to produce neurotoxicity. For example, Aβ binds to prion proteins (Laurén et al., 2009) or A7 nicotinic acetylcholine receptor (Wang et al., 2000), which causes neuropathy. We found from in silico assay that several neurotransmitters including acetylcholine can bind Aβ more favorably than their corresponding receptors (Hong et al., unpublished data). This result suggests that the binding of neurotransmitters with Aβ might play an important role in AD pathogenesis through the disturbance of the normal signaling of neurotransmitters.
Aβ CLEARANCE
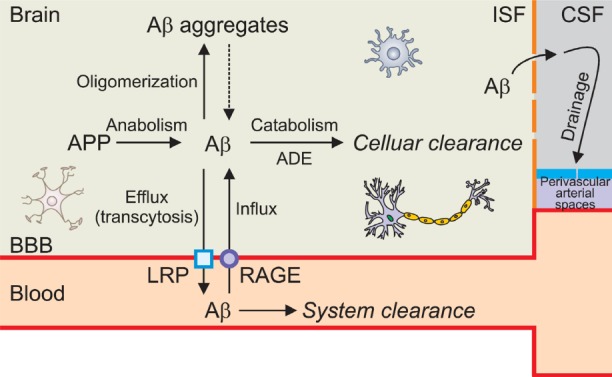
Aβ is generated from APP by sequential cleavages by BACE-1 and the γ-secretase complex (Fig.1). Scientists have focused on this pathway and these enzymes for a long time in order to develop an AD drug with an idea that blocking the activity of these enzymes might reduce the generation of Aβand thus Aβ-mediated cellular toxicity. Recently, however, a new concept of Aβ accumulation has been emerged; Aβ clearance or degradation rather than its synthesis have been found to be more critical in accumulation of Aβ. Furthermore, another mechanism responsible for controlling the brain Aβ levels shows the influx or re-entry into the brain mainly through the receptor for advanced glycation end products. Since the steady state levels of brain Aβ represent a dynamic equilibrium
Fig. 1. Anabolic and catabolic pathways of amyloid-β peptide (Aβ). Aβs are synthesized from amyloid precursor protein (APP) by β-and γ-secretase and then cleared by several mechanisms includingenzyme-mediated degradation.

between synthesis, re-uptake and clearance, any factors that result in the reduced rate of Aβ removal is likely to cause Aβ accumulation. Thus, Aβ clearance pathways including protease-mediated Aβ degradation have been emerged as a new therapeutic target for AD treatment, which are mostly handled in this review.
Aβ synthesis and clearance rates in ordinary adults are measured in the cerebrospinal fluid (CSF) and estimated to be 7.6% and 8.3%, respectively (Bateman et al., 2006) Thus, Aβis unlikely to accumulate in the normal brain. However, small defects in Aβ clearance could be suffi cient to cause Aβ accumulation leading to cell toxicity. Many data clearly suggest that in the central nervous system (CNS), decreased Aβ clearance is more responsible for the development of AD rather than increased Aβ synthesis (Weller et al., 2000). In particular, defects in Aβ clearance process is also likely to be relevant for the accumulation of Aβ in the blood vessel walls in addition to within the brain, resulting in cerebral amyloid angiopathy (CAA) which is present in approximately 90% of AD patients (Love, 2004) and the most common cause of lobar intracerebral hemorrhage in the elderly (Viswanathan and Greenberg, 2011).
Clearance of Aβ from the brain can be accomplished by several mechanisms including non-enzymatic and enzymatic pathways. The non-enzymatic pathway includes 1) the bulk flow of the interstitial fluid (ISF) into the CSF followed by ISF drainage pathway through perivascular basement membranes, 2) the uptake by microglial or astrocytic phagocytosis, and 3) the transport across the blood vessel walls into the blood vessel which is mediated by a series of clearance receptors such as low-density lipoprotein receptor-related protein 1 (LRP1), very low-density lipoprotein receptor (VLDLR)and P-glycoprotein localized predominantly on the abluminal side of the cerebral endothelium (Shibata et al., 2000; Deane et al., 2004). The enzymatic clearance involves several proteases, including neprilysin (NEP), insulin-degrading enzyme (IDE), matrix metalloproteinase (MMP)-9 and glutamate carboxypeptidase II (GCPII).
INTERSTITIAL FLUID DRAINAGE PATHWAY
In addition to accumulation within senile plaques, accumulation of Aβ in AD brain is also found in the walls of capillaries and arteries as characterized in CAA. Several lines of evidence suggested that Aβ deposit in the wall reflects a failure of the elimination of Aβ along the perivascular ISF drainage pathways of the brain (Weller et al., 2000). As shown in Fig. 2, increased production of Aβ or blockage of Aβ drainage through bulk flow of ISF followed by drainage pathways into the blood across the perivascular Virchow-Robin arterial spaces in the brain (Fig.2). As an example, in NEP gene knockout in a human APP (hAPP) mouse model, NEP reduction in cortical blood vessels contributes to the accumulation of Aβ in the vessel walls enough to lead to the development of CAA (Farris et al., 2007). Furthermore this response was observed in gene dosage-dependent manner.
Fig. 2. Schematic presentation showing thebrain and blood vessel compartment and the ways of Aβ clearance pathways. The blood-brain barrier (BBB) is a separation of circulating blood from the brain interstitial fluid (ISF) in the central nervous system (CNS). It is composed of thin and flat endothelial cells inthe capillaries. This barrier also includes athick basement membrane, smooth musclecells and astrocytic endfeet. This barrier restricts the diffusion of most materials into the cerebral ISF. See text for details. APP: Amyloid precursor protein, ADE: Amyloid-β degradation enzyme, LRP: Low-density lipoprotein receptor-related protein, RAGE: Receptor for advanced glycation end product, CSF: Cerebrospinal fluid.
UPTAKE BY MICROGLIAL PHAGOCYTOSIS
Microglia, brain’s resident mononuclear phagocyte is found
within the core of amyloid plaques both in human brain and in rodent transgenic (Tg) AD models. Although the precise role of microglia in AD still remains unclear, microglia play an essential role in Aβ clearance through their ability to take up and degrade soluble and fibrillar forms of Aβ (Rogers et al., 2002).
Microglia cells are activated by Aβ and secrete neurotoxic molecules. In contrast, they have neruroprotective actions by producing neurotrophic factors and by eliminating Aβ from the brain by phagocytosis. In early stage of AD, microglial activation delays disease progression by promoting clearance of Aβby phargocytosis (Frautschy et al., 1998; Wyss-Coray et al., 2003; Wyss-Coray, 2006) before formation of senile plaques.In contrast, with aging microglia tends to be over-activated in response to stress such as amyloids and instigate an inflammatory reaction, which cause neuronal damage. In addition, the ability of microglia to uptake Aβ appears to be dependent on age. Exosome secretion from neurons was reported to enhance Aβ uptake into microglial cells and significantly decreased the extracellular levels of Aβ (Yuyama et al., 2012). However, microglial cells prepared from neonates demonstrated phagocytic ability but this was lost by 6 months (Floden and Combs, 2011). Thus, it is critical to understand the state of microglia activation in different AD stages to determine the effect of potential anti-infl ammatory therapies.
There is still debate regarding the maintenance of the microglial cell population in the CNS. There are two different types of microglia; which are the resident microglial cells and the newly differentiated cells derived from the bone marrow. Although they are both seen near Aβ plaques, bone-marrow derived microglia (BMDM) have been shown to delay or stop the progression of AD (Naert and Rivest, 2011) due to more efficient phagocytic properties compared to their resident counterparts (Simard et al., 2006) and secretion of growth factors (example, glial cell line-derived neurotrophic factor). In this regard, it was demonstrated that transplantation of BMDM or their modified cells reduced Aβ accumulation by enhancing the expression of NEP in microglia to prevent synaptic dysfunctions and improve cognitive functions in AD mouse model (Kim et al., 2012). All together, the recruitment of endogenous stem cells or transplantation of stem cells facilitates Aβ clearance and thus considered as a potential therapeutic strategy for AD.
TRANSPORT ACROSS THE BLOOD VESSEL WALLS INTO THE CIRCULATION
For a long time, many investigators have paid attention to intrinsic neuronal components to understand the causes of neurodegenerative diseases including AD. However, many studies have shown that dysfunction in the blood brain barier (BBB) rather than in neuronal components contributes to the accumulation of neurotoxic materials. Astrocyte, a component of BBB has been paid attention for its role in plaque maintenance and Aβ clearance. Cultured human astrocytes indeed bind to and internalize Aβ (Nielsen et al., 2009). Animal experiments demonstrated that astrocytes internalize Aβby the scavenger receptors, such as low density lipoprotein receptor-related protein 1(LRP1), scavenger receptor class B member 1 (SCARB1) and the macrophage receptor with collagenous structure (MARCO). In addition, albeit there was a controversy whether the prion protein is a receptor for amyloid (Hildebrandt et al., 2009; Laurén et al., 2009), it was found that cellular prion protein participates in Aβ transcytosis across the BBB (Pflanzner et al., 2012).
Among Aβ scavenger receptors, LRP1 has been most extensively studied. LRP1 was originally known to play a role in the transport and metabolism of cholesterol. Later, LRP1 was characterized as a multifunctional scavenger receptor that binds to more than 40 structurally different ligands and has a function to transcytose ligands across BBB. It also serves as a transducing transmembrane cell signaling receptor. A series of evidence suggested that LRP1 expressed in astrocytes regulates brain Aβ levels through endocytic uptake of Aβ (Shibata et al., 2000). LRP1 expressed in brain capillary endothelium (Deane et al., 2004; Bell et al., 2007) and the liver also plays a functional role in systemic Aβ clearance. Earlier studies revealed that LRP1 expressed in neurons were shown to have capability to uptake Aβ bound to alpha-2-macroglobulin or ApoE, ligands for LRP1 (Narita et al., 1997; Bu et al., 2006). In contrast, the recent surface plasma resonance study demonstrated the direct interaction between LRP1 and Aβ in the abluminal side of the brain capillaries (Deane et al., 2004; Bell et al., 2007). Other lipoprotein receptors such as low-density lipoprotein receptor (LDLR), VLDLR and LRP2 are not likely to play a role in the transport of free Aβ across BBB (Deane et al., 2009).
Interestingly, Aβ1-40 is cleared rapidly across the BBB via LRP1 while Aβ1-42 is removed across the BBB at a slower rate(~50%) than Aβ1-40. Furthermore, mutant form of Aβ, Aβ1-40(Dutch), is also cleared less efficiently than Aβ1-40 (Monro et al., 2002). These findings suggest an isoform or mutant form-specific degradation mechanism. In addition to the enhanced toxicity by the mutant Aβ peptides, the mutations in APP resulting in production of aberrant Aβ may increase the accumulation of Aβ due to the slow LRP1-mediated degradation.
Sagare et al. have shown that a soluble form of LRP1(sLRP1) binds to 70-90% of plasma Aβ, preventing its access to the brain (Sagare et al., 2007). Thus, increased sLRP1 expression at the BBB and/or enhanced peripheral Aβ sink activity of sLRP1 has a significant potential to reduce brain Aβaccumulation (Deane et al., 2009). In support of this result, deficient sLRP1-Aβ binding due to the increased level of oxidized sLRP1 which can not bind Aβ showed a failure to reduce AD progression (Sagare et al., 2011). Similarly, dysfunction of LRP1 by antisense of LRP1 reduces BBB clearance, thereby increasing brain Aβ levels and impairing cognition (Jaeger et al., 2009). Other studies demonstrate the role of LRP in AD pa-thogenesis; a semipurified extract of the root of Withania somnifera (withanolides and withanosides) improved AD pathology by enhancing LRP expression in the liver (Sehgal et al., 2012).
DEGRADATION BY Aβ-DEGRADING ENZYME
Multiple Aβ-degrading enzyme family
The proteolytic machinery in the brain certainly contributes to the degradation of Aβ. For the last decade, multiple proteases of Aβ-degrading enzymes (ADE) implicated in this clearance process have been identified. Identification of these enzymes is rather perplexing because of their diversity. These enzymes belong to 1) zinc metalloendopeptidase [NEP-1 and NEP-2, endothelin-converting enzyme (ECE)-1 and -2, angiotensin-converting enzyme (ACE)], 2) thiol-dependent metalloendopeptdiase [insulin-degrading enzyme (IDE)], 3) serine proteases [plasmin, myelin basic protein (MBP), acylpeptide hydrolase], 4) cystein proteases [cathepsin B, D and S], 5) matrix metalloproteinase [MMP-9, MMP-2], and 6) others [GCPII, aminopeptidase A, mitochondrial peptidasome]. Most of them have endopeptidase activity cleaving the amino acid inside Aβ sequences while some (GCPII, MMP-9) contain carboxypeptidase activity cleaving amino acids from the carboxyl teminus. They cleave either at a single site or at multiple sites within Aβ. Specificity for cleaving sites within Aβ and for different Aβ aggregate forms has been well summarized in a recent paper (Nalivaeva et al., 2012) and thus this review will not list the aspects in their cleavage functions.
The enzymes produce smaller-sized enzymatic products. In vitro studies showed that they cleave the full-length Aβ, producing fragments that are less neurotoxic and more easily cleared. However, it can’t be certain whether the variety of Aβ products cleaved by ADE are benefi cial to the cells or not;some products such as Aβ25-35 and Aβ22-35 have similar toxicity and aggregation property as the full-length Aβ1-40 or Aβ1-42 monomer (Pike et al., 1995). In fact, Aβs exist in systemic equilibrium of many heterogeneous Aβ forms, including soluble monomeric, oligomeric, protofibrillar, and fibrillar forms. Studies have suggested that toxicity of the Aβ fragments is probably attributable to the topology of the cleaved Aβ products. In this regard (Numata and Kaplan, 2010), demonstrated that a linking region between the two sheets in Aβ is the key determinant. Therefore, the toxicity of various Aβ forms and their cleaved products is dependent on what they are composed of and/or how they are assembled. Recently, Aβ oligomer structures have been reported by nuclear magnetic resonance spectroscopy (Ahmed et al., 2010).
Validation of ADE function in cleaving Aβ
The biological function of the enzymes in clearance of Aβ was validated by many in vivo studies. Although gene deletion of a few ADE such as plasmin, urokinase plasminogen activator, or tissue plasminogen activator caused no alteration in the endogenous Aβ levels (Tucker et al., 2004; Eckman et al., 2006), knockout experiment in mice or rats of the specific ADE clearly demonstrated the increased steady-state levels of Aβ in the brain (Iwata et al., 2001; Eckman et al., 2003; Farris et al., 2003; Miller et al., 2003; Hafez et al., 2011). For example, deletion of NEP gene in hAPP mice increased Aβ oligomers and impaired hippocampal synaptic plasticity and cognitive dysfunction before the appearance of amyloid plaque load (Huang et al., 2006; Madani et al., 2006). Delivery of NEP inhibitors into hippocampus also caused an accumulation of Aβ and impairment of learning and memory (Mouri et al., 2006; Zou et al., 2006). Mice lacking other ADE such as ECE-1, ECE-2 or IDE gene also showed a significant but modest increase in endogenous Aβ amounts, suggesting that they are physiologically involved in Aβ metabolism.
The role for ADE in Aβ degradation is also ascertained by overexpression studies. Overexpression of the ADE gene in the Tg AD model mice showed reduction of Aβ level in the brain and improved cognitive function. In this regard, hNEP gene-overexpressed AD model mice (APP Swed/Ind) showed a reduced cerebral Aβ level and plaque formation, and significantly improved life expectancy (Leissring et al., 2003; Poirier et al., 2006), although this result was not reproducible in another separate experiment using the same double Tg mice (Meilandt et al., 2009). Nevertheless, other studies demonstrated the evidence of clinical benefit of intracerebral NEP increase. Viral delivery of hNEP into the hippocampus of hAPP Tg mice reduced both intracellular and extracellular Aβ levels and plaque pathology, oxidative stress, inflammation, and synaptic and dendritic damage as well as improved behavior and memory (Marr et al., 2003; Iwata et al., 2004; El-Amouri et al., 2008; Spencer et al., 2008). Therefore, agents which are able to selectively increase ADE levels and activities have the potential as a candidate for AD treatment.
Paradox of increased ADE expression or activity in AD
The rise of brain Aβ levels has been widely accepted as an important pathogenic factor for development of AD. Thus, it was assumed particularly in late-onset AD brain, that age-related decline of ADE activity would contribute to Aβ accumulation and this decline should be sharper in AD. Studies of ADE expression levels or activity with aging in human and mouse brains have been undertaken from various laboratories but no conclusive results have been obtained. Earlier studies supported the reduction of mRNAs and proteins of NEP and IDE in AD brain (Akiyama et al., 2001; Russo et al., 2005; Miners et al., 2006). Other laboratories also reported the reduction of NEP protein in AD and with age (Hellström-Lindahl et al., 2008). However, in those studies, the immunohistochemical analysis of NEP with human brain sections and its quantification in brain tissue homogenates was poor and less specific to the particular enzyme, raising a question that this reduction may result from the secondary phenomena of neuronal death rather than a primary cause of the disease development. To this end, a highly sensitive fluorescence immunocapture method was developed using a specific enzyme inhibitors to measure the specific enzyme activity which can discriminate between closely related enzymes, for example NEP and IDE-1 (Miners et al., 2008a, 2008b). The results showed that NEP
and IDE activities rather increase, but not decrease, with normal aging (Miners et al., 2010a), this rise progressively with increasing disease severity (Miners et al., 2009). In addition, those of NEP were also found to be elevated in brains of Down syndrome patients (Miners et al., 2011); the levels increased with disease progression.
These controversies extend to other ADE as well. The ACE activity was increased in AD (Savaskan et al., 2001; Miners et al., 2008c, 2010b). The levels of ECE-1 (Wang et al., 2009a), ECE-2 (Palmer et al., 2009), MMP-2 (Yan et al., 2006), MMP-3, MMP-9 (Bruno et al., 2009) and ACE-2 were reported to increase in AD, but other studies showed no alterations in those of MMP-2, MMP-3, and MMP-9 (Baig et al., 2008). Similarly, ADE activity was also found to be increased in the cortex of aged Tg2576 mice (Deb et al., 1999; Tucker et al., 2000; Leal et al., 2006; Palmer et al., 2009). We also found that GCPII, a newly-identified ADE by our group (Kim et al., 2010), was increased in the aged brain and further increased in AD model mice brain (APPswedish/presenilin exon 9 deletion mutant; unpublished data).
Although these unexpected findings are not yet to be fully understood, it is likely that ADE is increased in response to Aβ through a compensatory mechanism of the body; ADE induction by Aβ may refl ect a protection of cells against the Aβtoxicity. This hypothesis is supported by other in vitro and in vivo results; the induction of NEP in AD Tg mouse brain after injection of Aβ1-42 (Mohajeri et al., 2002) in a dose-dependent manner. Similarly, the activity of ADE including GCPII is increased in cells treated with aggregated Aβ (Deb et al., 1999; Lee et al., 2003; Jung et al., 2003; Leal et al., 2006; Mueller-Steiner et al., 2006; Wang et al., 2009a, 2009b; Palmer et al., 2009; Miners et al., 2010b). Taken together, these findings argue strongly against a notion that deficit of ADE is associated with AD.
THERAPEUTIC APPROACHES OF ADE IN TREATMENT FOR AD
Although it is too early to conclude that a decline in ADE activity plays a major role in the accumulation of Aβ in AD brain (Miners et al., 2009, 2010b), increase or over-expression of these enzymes at least could significantly reduce amyloid deposit and enhance cognitive function. The strategy to translate ADE into therapeutic applications is as follows: 1) The administration of compounds that enhance the ADE activity, 2) the gene therapy using the ADE genes, and 3) the cell therapies based on stem cell transplantation.
Compounds that enhance the ADE activity
Several agents have been identified to increase NEP expression. The neuropeptide somatostatin has been found to upregulate NEP activity through the concerted action with its receptors, somatostatin receptors (SSTR)-2 and SSTR-4 (Saito et al., 2005). In AD brain, the reduction of somatostatin and SSTR levels were found and infusion of Aβ caused impairment of somatostatin signaling (Aguado-Llera et al., 2005) and reduction of NEP expression (Burgos-Ramos et al., 2009a). Although the exact mechanism involved in this signaling pathway is yet to be clarified, this result tells that Aβ levels may be associated with or even controlled by somatostatin agonists. Concurrently, studies with minocycline (Burgos-Ramos et al., 2009b) and erythropoietin (Danielyan et al., 2009) showed increased NEP expression, preventing AD abnormalties.
Another agent is APP intracellular domain which is reported to increase the NEP promoter activity in human neuroblastoma cells. A tyrosine kinase inhibitor, imatinib (Gleevec) increased both APP intracellular domain and NEP (Eisele et al., 2007). Epigenetic regulators such as valproic acid also increased NEP activity (Belyaev et al., 2009) as well as plasmin (Pulukuri et al., 2007). Estrogen (Xiao et al., 2009; Liang et al., 2010), gensenoside Rg3, a major component of ginseng (Yang et al., 2009), green tea extracts (Melzig and Janka, 2003; Ayoub and Melzig, 2006) and red wine (Melzig and Escher, 2002) have all been reported to increase NEP activity. Table 1 summarizes the regulatory molecules that regulate the ADE levels or activity.
Table 1.
The regulatory molecules that are known to modulate the ADE level and or activity
| ADEs | Regulator | Effects | References |
|---|---|---|---|
|
| |||
| Neprilysin (NEP) | Somatostatin | Upregulation of NEP activity in primary cortical neurons | Saito et al., 2005 |
| Minocycline | Prevention of toxic effects of Aβ (25-35) by enhancing NEP expression in rat temporal cortex | Burgos-Ramos et al., 2009b | |
| Intracellular domain of APP and APLP (AICD) | Upregulation of NEP expression by AICD | Pardossi-Piquard et al., 2005 | |
| Gleevec (Tyrosine kinase inhibitor) | Elevation of NEP mRNA and protein levels | Eisele et al., 2007 | |
| Valproate and trichostatin A (Histone deacetylase inhibitors) | Increase in NEP expression and activity in SHSY-5Y cell | Belyaev et al., 2009 | |
| Estrogen | Regulation of NEP expression through physical interactions between estrogen receptor and estrogen response elements in the NEP gene | Xiao et al., 2009; Liang et al., 2010 | |
| Ginsenoside Rg3 | Promotion of Aβ degradation by enhancing gene expression of NEP | Yang et al., 2009 | |
| Green tea extract (EFLA®85942) | Strong enhancement of cellular NEP activity without change of cellular ACE activity | Melzig and Janka, 2003 | |
| GW742 (Selective PPARδ agonist) | Upregulation of NEP in 5xFAD mice | Kalinin et al., 2009 | |
| Polyphenols (Epilobium angustifolium) | Induction of NEP activity in SK-N-SH and PC-3 cells | Kiss et al., 2006 | |
| sICAM-1 | Induction of NEP expression in BV2 cells and in wild-type mice brains Decrease of Aβ plaques by hUCB-MSC-derived sICAM-1 which induces NEP expression in microglia | Kim et al., 2012 | |
| Erythropoietin | Enhanced metabolism of Aβ in MSCs by increasing their NEP content | Danielyan et al., 2009 | |
| Endothelin-converting enzyme (ECE-1) | PKCε | Promotion of Aβ clearance and reduction of AD neuropathology through increased ECE enzyme activity | Choi et al., 2006; Kim et al. 2011b |
| 4-hydroxy-nonenal (HNE) | Upregulation of ECE-1 mRNA and protein | Wang et al., 2009a, b | |
| Aβ | Elevation of endothelin-1 in AD and upregulated by Aβ | Palmer et al., 2012 | |
| Insulin degrading enzyme (IDE) | Retinoic acid | Upregulation by retinoic acid, a well-known inducer of neuronal differentiation and/or programmed cell death | Melino et al., 1996 |
| PPARγ | Activation of an IDE like Aβ degrading activity | Espuny-Camacho et al., 2010 | |
| HES-1 and Hey-1 (Notch signaling proteins) | Binding to IDE proximal promoter and repression of transcription and its activity | Leal et al., 2012 | |
| U0126 (ERK1/2 inhibitor) | Blocking of increased IDE protein level induced by fAβ | Leal et al., 2006 | |
| Angiotensin converting enzyme (ACE) | Perindopril (ACE inhibitor) | Cognitive impairment and brain injury in a mouse model of AD induced by intracerebroventricular injection of Aβ | Dong et al., 2011 |
| MMP-2 | Aβ | Enhancement of MMP-2 and membrane-type-MMP expression in U87 human glioma cell | Deb et al., 1999 |
| MMP-3 (Stromelysin-1) | Aβ | Increase of MMP-3 in enriched astrocytes and mixed hippocampal cultures | Deb and Gottschall, 1996 |
| MMP-9 | Yin Yang 1 | Binding to MMP-9 promoter to repress MMP-9 transcription | Rylski et al., 2008 |
| JunB | Repression of MMP-9 transcription in depolarized rat brain neuron | Rylski et al., 2009 | |
| Small molecule inhibitor of PAI-1 | Enhanced clearance of Aβ in brain by sustaining the plasmin proteolysis cascade | Jacobsen et al., 2008 | |
| Plasmin | PAI-1 (neuroserpin) | Inhibition of tissue plasminogen activator activity leading to reduced plasmin activity | Fabbro and Seeds, 2009 |
Aβ: Amyloid-β peptide, APLP: Amyloid precursor-like protein, APP: Amyloid precursor protein, HUCB-MSC: Human umbilical cord blood-derived mesenchymal stem cell, MMP: Matrix metalloproteinase, PAI-1: Plasminogen activator inhibitor-1, sICAM-1: Soluble in-tercellular adhesion molecule-1.
Therapy for ADE gene delivery
Gene therapy approaches has employed viral-vector mediated transfection of ADE genes to increase their expression in AD model animals. Both in vitro and in vivo studies have shown that gene delivery of NEP is effective in reducing Aβ level; NEP expression using Sindbis viral vector in murine primary cortical neurons effectively reduced the Aβ levels (Hama et al., 2001). Similarly, lentiviral NEP injected into the hippocampus of hAPP Tg mice reduced the plaque burden (Marr et al., 2003) and improved memory performance (Spencer et al., 2008). In addition, injection of adeno-associated viral NEP to NEP knockout mice abolished the increase in Aβ levels in hippocampus, and led to efficient degradation of soluble and insoluble Aβ in hAPP AD mice (Iwata et al., 2004). Recently the peripheral delivery of ADE, instead of the brain delivery was attempted and the results showed the promising results. Adeno-associated virus transfection of NEP into the hind limb of triple Tg AD model mice produced 60 % reduction in soluble Aβ and 50 % reduction in plaque deposit within the brain at 6 months (Liu et al., 2010).
A technology called convection-enhanced delivery has been developed to improve the brain delivery of the proteins. This technique is a novel neurosurgical method of direct drug delivery to the brain through ultrafine microcatheters. The application of this technology might be effective for brain diseases showing local pathology, such as Parkinson’s disease. Further studies demonstrated that drugs delivered with this method are accumulated near blood vessels and perivascular spaces (Krauze et al., 2005), suggesting the possibility of using CAA treatment (Weller et al., 2000; Carare et al., 2008; Weller et al., 2008).
Stem cell therapy
Stem cells have the potential to directly substitute damaged cells and as a vehicle for delivering ADE into the CNS. Studies demonstrated that transplantation of adult mesenchymal stem cells reduced the brain Aβ, which was mediated by increased NEP mRNA and protein levels (Miners et al., 2011). In other study using with human umbilical cord blood mesenchymal stem cells (hUCB-MSCs) NEP expression in transplanted cells into the hippocampus of APP AD mice was increased and amyloid plaques in that region and other regions were decreased by the active migration of hUCB-MSCs toward Aβdeposits (Kim et al., 2012). Although the exact mechanism is not clear yet, cytokines such as intracellular adhesion molecule-1 which is released from the transplanted mesenchymal stem cells were involved (Kim et al., 2012).
CONCLUSIONS AND PERSPECTIVES
Understanding of Aβ metabolic pathway is uppermost to enlighten the pathogenesis and cure for AD. Although there are huge publications dealing with signaling pathways of Aβ synthesis and related enzymes, the identification of molecules responsible for Aβ clearance pathways and their mechanistic links to AD is still underway. In addition to nonenzymatic pathway, enzymatic pathway by ADE serves Aβ clearance. The results have suggested a pivotal role for ADE in reducing AD symptoms in both cell and animal models. Thus, the modulation of ADE expression and activity provides a simple strategy of whether clearance of Aβ offers the therapeutic potential for AD. It is of great interest to find that peripheral delivery of ADE gene gives a significant efficacy to reduce AD symptoms as compared to the direct delivery to the brain, which has been a longtime obstacle for curing brain diseases. In addition, it is valuable to develop modifiers of ADE as therapeutics for AD. However, several studies also found that, unexpectedly, ADE expression and activity increase with normal aging and rise progressively with increasing severity of AD, which is likely to occur through a compensatory mechanism against increased levels of Aβ. Thus further researches will answer a question of what the benefits of ADE overdose are.
Acknowledgments
This work is supported by National Research Foundation of Korea (NRF) funded by the Korea government (No: 2011-0016127).
References
- 1.Aguado-Llera D. Arilla-Ferreiro E. Campos-Barros A. Puebla-Jiménez L. Barrios V. Protective effects of insulin-like growth factor-I on the somatostatinergic system in the temporal cortex of beta-amyloid-treated rats. J. Neurochem. (2005);92:607–615. doi: 10.1111/j.1471-4159.2004.02889.x. [DOI] [PubMed] [Google Scholar]
- 2.Ahmed M. Davis J. Aucoin D. Sato T. Ahuja S. Aimoto S. Elliott J. I. Van Nostrand W. E. Smith S. O. Structural conversion of neurotoxic amyloid-beta(1-42) oligomers to fibrils. Nat.Struct. Mol. Biol. (2010);17:561–567. doi: 10.1038/nsmb.1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Akiyama H. Kondo H. Ikeda K. Kato M. McGeer P. L. Immunohistochemical localization of neprilysin in the human cerebral cortex: inverse association with vulnerability to amyloid beta-protein(Abeta) deposition. Brain Res. (2001);902:277–281. doi: 10.1016/s0006-8993(01)02390-3. [DOI] [PubMed] [Google Scholar]
- 4.Ayoub S. Melzig M. F. Induction of neutral endopeptidase(NEP) activity of SK-N-SH cells by natural compounds from green tea. J. Pharm. Pharmacol. (2006);58:495–501. doi: 10.1211/jpp.58.4.0009. [DOI] [PubMed] [Google Scholar]
- 5.Baig S. Kehoe P. G. Love S. MMP-2, -3 and -9 levels and activity are not related to Abeta load in the frontal cortex in Alzheimer's disease. Neuropathol. Appl. Neurobiol. (2008);34:205–215. doi: 10.1111/j.1365-2990.2007.00897.x. [DOI] [PubMed] [Google Scholar]
- 6.Bateman R. J. Munsell L. Y. Morris J. C. Swarm R. Yarasheski K. E. Holtzman D. M. Human amyloid-beta synthesis and clearance rates as measured in cerebrospinal fluid in vivo. Nat. Med. (2006);12:856–861. doi: 10.1038/nm1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bell R. D. Sagare A.P. Friedman A. E. Bedi G. S. Holtzman D.M. Deane R. Zlokovic B. V. Transport pathways for clearance of human Alzheimer's amyloid beta-peptide and apolipoproteins E and J in the mouse central nervous system. J. Cereb. Blood Flow Metab. (2007);27:909–918. doi: 10.1038/sj.jcbfm.9600419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Belyaev N. D. Nalivaeva N. N. Makova N. Z. Turner A. J. Neprilysin gene expression requires binding of the amyloid precursor protein intracellular domain to its promoter: implications for Alzheimer disease. EMBO Rep. (2009);10:94–100. doi: 10.1038/embor.2008.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bruno M. A. Mufson E. J. Wuu J. Cuello A. C. Increased matrix metalloproteinase 9 activity in mild cognitive impairment. J. Neuropathol. Exp. Neurol. (2009);68:1309–1318. doi: 10.1097/NEN.0b013e3181c22569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bu G. Cam J. Zerbinatti C. LRP in amyloid-beta production and metabolism. Ann. N. Y. Acad. Sci. (2006);1086:35–53. doi: 10.1196/annals.1377.005. [DOI] [PubMed] [Google Scholar]
- 11.Burgos-Ramos E. Martos-Moreno G. A. López M. G. Herranz R. Aguado-Llera D. Egea J. Frechilla D. Cenarruzabeitia E. León R. Arilla-Ferreiro E. Argente J. Barrios V. The N-terminal tripeptide of insulin-like growth factor-I protects against beta-amyloid-induced somatostatin depletion by calcium and glycogen synthase kinase 3 beta modulation. J. Neurochem. (2009a);109:360–370. doi: 10.1111/j.1471-4159.2009.05980.x. [DOI] [PubMed] [Google Scholar]
- 12.Burgos-Ramos E. Puebla-Jiménez L. Arilla-Ferreiro E. Minocycline prevents Abeta(25-35)-induced reduction of soma-tostatin and neprilysin content in rat temporal cortex. Life Sci. (2009b);84:205–210. doi: 10.1016/j.lfs.2008.11.019. [DOI] [PubMed] [Google Scholar]
- 13.Carare R. O. Bernardes-Silva M. Newman T. A. Page A. M. Nicoll J. A. Perry V. H. Weller R. O. Solutes, but not cells, drain from the brain parenchyma along basement membranes of capillaries and arteries: signifi cance for cerebral amyloid angiopathy and neuroimmunology. Neuropathol. Appl. Neurobiol. (2008);34:131–144. doi: 10.1111/j.1365-2990.2007.00926.x. [DOI] [PubMed] [Google Scholar]
- 14.Castellani R. J. Smith M. A. Compounding artefacts with uncertainty and an amyloid cascade hypothesis that is 'too big to fail'. J. Pathol. (2011);224:147–152. doi: 10.1002/path.2885. [DOI] [PubMed] [Google Scholar]
- 15.Choi D. S. Wang D. Yu G. Q. Zhu G. Kharazia V. N. Paredes J.P. Chang W. S. Deitchman J. K. Mucke L. Messing R. O. PKCepsilon increases endothelin converting enzyme activity and reduces amyloid plaque pathology in transgenic mice. Proc. Natl. Acad. Sci. USA. (2006);103:8215–8220. doi: 10.1073/pnas.0509725103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Danielyan L. Schäfer R. Schulz A. Ladewig T. Lourhmati A. Buadze M. Schmitt A. L. Verleysdonk S. Kabisch D. Koeppen K. Siegel G. Proksch B. Kluba T. Eckert A. Köhle C. Schöneberg T. Northoff H. Schwab M. Gleiter C. H. Survival, neuron-like differentiation and functionality of mesenchymal stem cells in neurotoxic environment: the critical role of erythropoietin. Cell Death Differ. (2009);16:1599–1614. doi: 10.1038/cdd.2009.95. [DOI] [PubMed] [Google Scholar]
- 17.Deane R. Bell R. D. Sagare A. Zlokovic B. V. Clearance of amyloid-beta peptide across the blood-brain barrier: implication for therapies in Alzheimer's disease. CNS Neurol. Disord. Drug Targets. (2009);8:16–30. doi: 10.2174/187152709787601867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deane R. Wu Z. Sagare A. Davis J. Du Yan S. Hamm K. Xu F. Parisi M. LaRue B. Hu H. W. Spijkers P. Guo H. Song X. Lenting P. J. Van Nostrand W. E. Zlokovic B. V. LRP/amyloid beta-peptide interaction mediates differential brain effl ux of Abeta isoforms. Neuron. (2004);43:333–344. doi: 10.1016/j.neuron.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 19.Deb S. Gottschall P. E. Increased production of matrix metalloproteinases in enriched astrocyte and mixed hippocampal cultures treated with beta-amyloid peptides. J. Neurochem. (1996);66:1641–1647. doi: 10.1046/j.1471-4159.1996.66041641.x. [DOI] [PubMed] [Google Scholar]
- 20.Deb S. Zhang J. W. Gottschall P. E. Activated isoforms of MMP-2 are induced in U87 human glioma cells in response to beta-amyloid peptide. J. Neurosci. Res. (1999);55:44–53. doi: 10.1002/(SICI)1097-4547(19990101)55:1<44::AID-JNR6>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 21.Dong Y. F. Kataoka K. Tokutomi Y. Nako H. Nakamura T. Toyama K. Sueta D. Koibuchi N. Yamamoto E. Ogawa H. Kim-Mitsuyama S. Perindopril, a centrally active angiotensin-converting enzyme inhibitor, prevents cognitive impairment in mouse models of Alzheimer's disease. FASEB J. (2011);25:2911–2920. doi: 10.1096/fj.11-182873. [DOI] [PubMed] [Google Scholar]
- 22.Eckman E. A. Adams S. K. Troendle F. J. Stodola B. A. Kahn M.A. Fauq A. H. Xiao H. D. Bernstein K. E. Eckman C. B. Regulation of steady-state beta-amyloid levels in the brain by neprilysin and endothelin-converting enzyme but not angiotensin-converting enzyme. J. Biol. Chem. (2006);281:30471–30478. doi: 10.1074/jbc.M605827200. [DOI] [PubMed] [Google Scholar]
- 23.Eckman E. A. Watson M. Marlow L. Sambamurti K. Eckman C. B. Alzheimer's disease beta-amyloid peptide is increased in mice deficient in endothelin-converting enzyme. J. Biol. Chem. (2003);278:2081–2084. doi: 10.1074/jbc.C200642200. [DOI] [PubMed] [Google Scholar]
- 24.Eisele Y. S. Baumann M. Klebl B. Nordhammer C. Jucker M. Kilger E. Gleevec increases levels of the amyloid precursor protein intracellular domain and of the amyloid-beta degrading enzyme neprilysin. Mol. Biol. Cell. (2007);18:3591–3600. doi: 10.1091/mbc.E07-01-0035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.El-Amouri S. S. Zhu H. Yu J. Marr R. Verma I. M. Kindy M.S. Neprilysin: an enzyme candidate to slow the progression of Alzheimer's disease. Am. J. Pathol. (2008);172:1342–1354. doi: 10.2353/ajpath.2008.070620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Espuny-Camacho I. Dominguez D. Merchiers P. Van Rompaey L. Selkoe D. De Strooper B. Peroxisome proliferator-activated receptor gamma enhances the activity of an insulin degrading enzyme-like metalloprotease for amyloid-beta clearance. J. Alzheimers Dis. (2010);20:1119–1132. doi: 10.3233/JAD-2010-091633. [DOI] [PubMed] [Google Scholar]
- 27.Fabbro S. Seeds N. W. Plasminogen activator activity is inhibited while neuroserpin is up-regulated in the Alzheimer disease brain. J. Neurochem. (2009);109:303–315. doi: 10.1111/j.1471-4159.2009.05894.x. [DOI] [PubMed] [Google Scholar]
- 28.Farris W. Mansourian S. Chang Y. Lindsley L. Eckman E. A. Frosch M. P. Eckman C. B. Tanzi R. E. Selkoe D. J. Guenette S. Insulin-degrading enzyme regulates the levels of insulin amyloid beta-protein and the beta-amyloid precursor protein, intracellular domain in vivo. Proc. Natl. Acad. Sci. USA. (2003);100:4162–4167. doi: 10.1073/pnas.0230450100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Farris W. Schütz S. G. Cirrito J. R. Shankar G. M. Sun X. George A. Leissring M. A. Walsh D. M. Qiu W. Q. Holtzman D. M. Selkoe D. J. Loss of neprilysin function promotes amyloid plaque formation and causes cerebral amyloid angiopathy. Am. J. Pathol. (2007);171:241–251. doi: 10.2353/ajpath.2007.070105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Floden A. M. Combs C. K. Microglia demonstrate age-dependent interaction with amyloid-β fibrils. J. Alzheimers Dis. (2011);25:279–293. doi: 10.3233/JAD-2011-101014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Frautschy S. A. Yang F. Irrizarry M. Hyman B. Saido T. C. Hsiao K. Cole G. M. Microglial response to amyloid plaques in APPsw transgenic mice. Am. J. Pathol. (1998);152:307–317. [PMC free article] [PubMed] [Google Scholar]
- 32.Hafez D. Huang J. Y. Huynh A. M. Valtierra S. Rockenstein E. Bruno A. M. Lu B. DesGroseillers L. Masliah E. Marr R. A. Neprilysin-2 is an important β-amyloid degrading enzyme. Am. J. Pathol. (2011);178:306–312. doi: 10.1016/j.ajpath.2010.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hama E. Shirotani K. Masumoto H. Sekine-Aizawa Y. Aizawa H. Saido T. C. Clearance of extracellular and cell-associated amyloid beta peptide through viral expression of neprilysin in primary neurons. J. Biochem. (2001);130:721–726. doi: 10.1093/oxfordjournals.jbchem.a003040. [DOI] [PubMed] [Google Scholar]
- 34.Hardy J. A. Higgins G. A. Alzheimer's disease: the amyloid cascade hypothesis. Science. (1992);256:184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- 35.Hardy J. Selkoe D. J. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. (2002);297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 36.Hellström-Lindahl E. Ravid R. Nordberg A. Age-dependent decline of neprilysin in Alzheimer's disease and normal brain: inverse correlation with A beta levels. Neurobiol. Aging. (2008);29:210–221. doi: 10.1016/j.neurobiolaging.2006.10.010. [DOI] [PubMed] [Google Scholar]
- 37.Hildebrandt H. Haldenwanger A. Eling P. False recognition correlates with amyloid-beta (1-42) but not with total tau in cerebrospinal fluid of patients with dementia and mild cognitive impairment. J. Alzheimers Dis. (2009);16:157–165. doi: 10.3233/JAD-2009-0931. [DOI] [PubMed] [Google Scholar]
- 38.Hong H. Baik T. Song K. Nam I. Chung M. Jo S. In silico study of interaction between neurotransmitters and β-amyloid peptide (Aβ): a novel working hypothesis of Aβ-mediated pathogenesis of Alzheimer’s disease. (Submitted) [Google Scholar]
- 39.Huang S. M. Mouri A. Kokubo H. Nakajima R. Suemoto T. Higuchi M. Staufenbiel M. Noda Y. Yamaguchi H. Nabeshima T. Saido T. C. Iwata N. Neprilysin-sensitive synapse-associated amyloid-beta peptide oligomers impair neuronal plasticity and cognitive function. J. Biol. Chem. (2006);281:17941–17951. doi: 10.1074/jbc.M601372200. [DOI] [PubMed] [Google Scholar]
- 40.Iwata N. Mizukami H. Shirotani K. Takaki Y. Muramatsu S. Lu B. Gerard N. P. Gerard C. Ozawa K. Saido T. C. Presynaptic localization of neprilysin contributes to efficient clearance of amyloid-beta peptide in mouse brain. J. Neurosci. (2004);24:991–998. doi: 10.1523/JNEUROSCI.4792-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Iwata N. Tsubuki S. Takaki Y. Shirotani K. Lu B. Gerard N. P. Gerard C. Hama E. Lee H. J. Saido T. C. Metabolic regulation of brain Abeta by neprilysin. Science. (2001);292:1550–1552. doi: 10.1126/science.1059946. [DOI] [PubMed] [Google Scholar]
- 42.Jacobsen J. S. Comery T. A. Martone R. L. Elokdah H. Crandall D. L. Oganesian A. Aschmies S. Kirksey Y. Gonzales C. Xu J. Zhou H. Atchison K. Wagner E. Zaleska M. M. Das I. Arias R. L. Bard J. Riddell D. Gardell S. J. Abou-Gharbia M. Robichaud A. Magolda R. Vlasuk G. P. Bjornsson T. Reinhart P. H. Pangalos M. N. Enhanced clearance of Abeta in brain by sustaining the plasmin proteolysis cascade. Proc. Natl. Acad. Sci. USA. (2008);105:8754–8759. doi: 10.1073/pnas.0710823105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jaeger L. B. Dohgu S. Hwang M. C. Farr S. A. Murphy M. P. Fleegal-DeMotta M. A. Lynch J. L. Robinson S. M. Niehoff M.L. Johnson S. N. Kumar V. B. Banks W. A. Testing the neurovascular hypothesis of Alzheimer's disease: LRP-1 antisense reduces blood-brain barrier clearance, increases brain levels of amyloid-beta protein, and impairs cognition. J. Alzheimers Dis. (2009);17:553–570. doi: 10.3233/JAD-2009-1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jarrett J. T. Berger E. P. Lansbury P. T. Jr. The carboxy terminus of the beta amyloid protein is critical for the seeding of amyloid formation: implications for the pathogenesis of Alzheimer's disease. Biochemistry. (1993);32:4693–4697. doi: 10.1021/bi00069a001. [DOI] [PubMed] [Google Scholar]
- 45.Jo S. A. Ahn K. Kim E. Kim H. S. Jo I. Kim D. K. Han C. Park M. H. Association of BACE1 gene polymorphism with Alzheimer's disease in Asian populations: meta-analysis including Korean samples. Dement. Geriatr. Cogn. Disord. (2008);25:165–169. doi: 10.1159/000112918. [DOI] [PubMed] [Google Scholar]
- 46.Jung S. S. Zhang W. Van Nostrand W. E. Pathogenic A beta induces the expression and activation of matrix metalloproteinase-2 in human cerebrovascular smooth muscle cells. J. Neurochem. (2003);85:1208–1215. doi: 10.1046/j.1471-4159.2003.01745.x. [DOI] [PubMed] [Google Scholar]
- 47.Kalinin S. Richardson J. C. Feinstein D. L. A PPARdelta agonist reduces amyloid burden and brain inflammation in a transgenic mouse model of Alzheimer's disease. Curr. Alzheimer. Res. (2009);6:431–437. doi: 10.2174/156720509789207949. [DOI] [PubMed] [Google Scholar]
- 48.Kim J. Y. Kim D. H. Kim J. H. Lee D. Jeon H. B. Kwon S. J. Kim S. M. Yoo Y. J. Lee E. H. Choi S. J. Seo S. W. Lee J. I. Na D. L. Yang Y. S. Oh W. Chang J. W. Soluble intracellular adhesion molecule-1 secreted by human umbilical cord blood-derived mesenchymal stem cell reduces amyloid-β plaques. Cell Death Differ. (2012);19:680–691. doi: 10.1038/cdd.2011.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim K. W. Kim M. H. Kim M. D. Kim B. J. Kim S. K. Kim J. L. Moon S. W. Bae J. N. Woo J. I. Ryu S. H. Yoon J.C. Lee N. J. Lee D. Y. Lee D. W. Lee S. B. Lee J. J. Lee JY. Lee CU. Chang S. M. Jhoo J. H. Cho M. J. A nationwide survey on the prevalence of dementia and mild cognitive impairment in South Korea. J. Alzheimers Dis. (2011a);23:281–291. doi: 10.3233/JAD-2010-101221. [DOI] [PubMed] [Google Scholar]
- 50.Kim M. J. Chae S. S. Koh Y. H. Lee S. K. Jo S. A. Glutamate carboxypeptidase II: an amyloid peptide-degrading enzyme with physiological function in the brain. FASEB J. (2010);24:4491–4502. doi: 10.1096/fj.09-148825. [DOI] [PubMed] [Google Scholar]
- 51.Kim T. Hinton D. J. Choi D. S. Protein kinase C-regulated aβ production and clearance. Int. J. Alzheimers Dis. (2011b):857368. doi: 10.4061/2011/857368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kiss A. Kowalski J. Melzig M. F. Effect of Epilobium angustifolium L. extracts and polyphenols on cell proliferation and neutral endopeptidase activity in selected cell lines. Pharmazie. (2006);61:66–69. [PubMed] [Google Scholar]
- 53.Krauze M. T. Saito R. Noble C. Bringas J. Forsayeth J. McKnight T. R. Park J. Bankiewicz K. S. Effects of the perivascular space on convection-enhanced delivery of liposomes in primate putamen. Exp. Neurol. (2005);196:104–111. doi: 10.1016/j.expneurol.2005.07.009. [DOI] [PubMed] [Google Scholar]
- 54.Laurén J. Gimbel D. A. Nygaard H. B. Gilbert J. W. Strittmatter S. M. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature. (2009);457:1128–1132. doi: 10.1038/nature07761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Leal M. C. Dorfman V. B. Gamba A. F. Frangione B. Wisniewski T. Castaño E. M. Sigurdsson E. M. Morelli L. Plaque-associated overexpression of insulin-degrading enzyme in the cerebral cortex of aged transgenic tg2576 mice with Alzheimer pathology. J. Neuropathol. Exp. Neurol. (2006);65:976–987. doi: 10.1097/01.jnen.0000235853.70092.ba. [DOI] [PubMed] [Google Scholar]
- 56.Leal M. C. Surace E. I. Holgado M. P. Ferrari C. C. Tarelli R. Pitossi F. Wisniewski T. Castaño E. M. Morelli L. Notch signaling proteins HES-1 and Hey-1 bind to insulin degrading enzyme (IDE) proximal promoter and repress its transcription and activity: implications for cellular Aβ metabolism. Biochim. Biophys. Acta. (2012);1823:227–235. doi: 10.1016/j.bbamcr.2011.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lee J. M. Yin K. J. Hsin I. Chen S. Fryer J. D. Holtzman D. M. Hsu C. Y. Xu J. Matrix metalloproteinase-9 and spontaneous hemorrhage in an animal model of cerebral amyloid angiopathy. Ann. Neurol. (2003);54:379–382. doi: 10.1002/ana.10671. [DOI] [PubMed] [Google Scholar]
- 58.Leissring M. A. Farris W. Chang A. Y. Walsh D. M. Wu X. Sun X. Frosch M. P. Selkoe D. J. Enhanced proteolysis of beta-amyloid in APP transgenic mice prevents plaque formationsecondary pathology, and premature death. Neuron. (2003);40:1087–1093. doi: 10.1016/s0896-6273(03)00787-6. [DOI] [PubMed] [Google Scholar]
- 59.Liang K. Yang L. Yin C. Xiao Z. Zhang J. Liu Y. Huang J. Estrogen stimulates degradation of beta-amyloid peptide by up-regulating neprilysin. J. Biol. Chem. (2010);285:935–942. doi: 10.1074/jbc.M109.051664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu Y. Studzinski C. Beckett T. Murphy M. P. Klein R. L. Hersh L. B. Circulating neprilysin clears brain amyloid. Mol. Cell Neurosci. (2010);45:101–107. doi: 10.1016/j.mcn.2010.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Love S. Contribution of cerebral amyloid angiopathy to Alzheimer's disease. J. Neurol. Neurosurg. Psychiatry. (2004);75:1–4. [PMC free article] [PubMed] [Google Scholar]
- 62.Madani R. Poirier R. Wolfer D. P. Welzl H. Groscurth P. Lipp H. P. Lu B. El Mouedden M. Mercken M. Nitsch R. M. Mohajeri M. H. Lack of neprilysin suffices to generate murine amyloid-like deposits in the brain and behavioral defi cit in vivo. J. Neurosci. Res. (2006);84:1871–1878. doi: 10.1002/jnr.21074. [DOI] [PubMed] [Google Scholar]
- 63.Marr R. A. Rockenstein E. Mukherjee A. Kindy M. S. Hersh L. B. Gage F. H. Verma I. M. Masliah E. Neprilysin gene transfer reduces human amyloid pathology in transgenic mice. J. Neurosci. (2003);23:1992–1996. doi: 10.1523/JNEUROSCI.23-06-01992.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Meilandt W. J. Cisse M. Ho K. Wu T. Esposito L. A. Scearce-Levie K. Cheng I. H. Yu G. Q. Mucke L. Neprilysin overexpression inhibits plaque formation but fails to reduce pathogenic Abeta oligomers and associated cognitive deficits in human amyloid precursor protein transgenic mice. J. Neurosci. (2009);29:1977–1986. doi: 10.1523/JNEUROSCI.2984-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Melino G. Draoui M. Bernardini S. Bellincampi L. Reichert U. Cohen P. Regulation by retinoic acid of insulin-degrading enzyme and of a related endoprotease in human neuroblastoma cell lines. Cell Growth Differ. (1996);7:787–796. [PubMed] [Google Scholar]
- 66.Melzig M. F. Escher F. Induction of neutral endopeptidase and angiotensin-converting enzyme activity of SK-N-SH cells in vitro by quercetin and resveratrol. Pharmazie. (2002);57:556–558. [PubMed] [Google Scholar]
- 67.Melzig M. F. Janka M. Enhancement of neutral endopeptidase activity in SK-N-SH cells by green tea extract. Phytomedicine. (2003);10:494–498. doi: 10.1078/094471103322331449. [DOI] [PubMed] [Google Scholar]
- 68.Miller B. C. Eckman E. A. Sambamurti K. Dobbs N. Chow K. M. Eckman C. B. Hersh L. B. Thiele D. L. Amyloid-beta peptide levels in brain are inversely correlated with insulysin activity levels in vivo. Proc. Natl. Acad. Sci. USA. (2003);100:6221–6226. doi: 10.1073/pnas.1031520100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Miners J. S. Ashby E. Van Helmond Z. Chalmers K. A. Palmer L. E. Love S. Kehoe P. G. Angiotensin-converting enzyme (ACE) levels and activity in Alzheimer's disease, and relationship of perivascular ACE-1 to cerebral amyloid angiopathy. Neuropathol. Appl. Neurobiol. (2008a);34:181–193. doi: 10.1111/j.1365-2990.2007.00885.x. [DOI] [PubMed] [Google Scholar]
- 70.Miners J. S. Baig S. Tayler H. Kehoe P. G. Love S. Neprilysin and insulin-degrading enzyme levels are increased in Alzheimer disease in relation to disease severity. J. Neuropathol. Exp. Neurol. (2009);68:902–914. doi: 10.1097/NEN.0b013e3181afe475. [DOI] [PubMed] [Google Scholar]
- 71.Miners J. S. Kehoe P. G. Love S. Immunocapture-based fluorometric assay for the measurement of insulin-degrading enzyme activity in brain tissue homogenates. J. Neurosci. Methods. (2008b);169:177–181. doi: 10.1016/j.jneumeth.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 72.Miners J. S. Morris S. Love S. Kehoe P. G. Accumulation of insoluble amyloid-β in down's syndrome is associated with increased BACE-1 and neprilysin activities. J. Alzheimers Dis. (2011);23:101–108. doi: 10.3233/JAD-2010-101395. [DOI] [PubMed] [Google Scholar]
- 73.Miners J. S. van Helmond Z. Chalmers K. Wilcock G. Love S. Kehoe P. G. Decreased expression and activity of neprilysin in Alzheimer disease are associated with cerebral amyloid angiopathy. J. Neuropathol. Exp. Neurol. (2006);65:1012–1021. doi: 10.1097/01.jnen.0000240463.87886.9a. [DOI] [PubMed] [Google Scholar]
- 74.Miners J. S. van Helmond Z. Kehoe P. G. Love S. Changes with age in the activities of beta-secretase and the Abeta-degrading enzymes neprilysin, insulin-degrading enzyme and angiotensin-converting enzyme. Brain Pathol. (2010a);20:794–802. doi: 10.1111/j.1750-3639.2010.00375.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Miners J. S. van Helmond Z. Raiker M. Love S. Kehoe P. G. ACE variants and association with brain Aβ levels in Alzheimer's disease. Am. J. Transl. Res. (2010b);3:73–80. [PMC free article] [PubMed] [Google Scholar]
- 76.Miners J. S. Verbeek M. M. Rikkert M. O. Kehoe P. G. Love S. Immunocapture-based fluorometric assay for the measurement of neprilysin-specific enzyme activity in brain tissue homogenates and cerebrospinal fluid. J. Neurosci. Methods. (2008c);167:229–236. doi: 10.1016/j.jneumeth.2007.08.012. [DOI] [PubMed] [Google Scholar]
- 77.Mohajeri M. H. Wollmer M. A. Nitsch R. M. Abeta 42-induced increase in neprilysin is associated with prevention of amyloid plaque formation in vivo. J. Biol. Chem. (2002);277:35460–35465. doi: 10.1074/jbc.M202899200. [DOI] [PubMed] [Google Scholar]
- 78.Monro O. R. Mackic J. B. Yamada S. Segal M. B. Ghiso J. Maurer C. Calero M. Frangione B. Zlokovic B. V. Substitution at codon 22 reduces clearance of Alzheimer's amyloid-beta peptide from the cerebrospinal fluid and prevents its transport from the central nervous system into blood. Neurobiol. Aging. (2002);23:405–412. doi: 10.1016/s0197-4580(01)00317-7. [DOI] [PubMed] [Google Scholar]
- 79.Mouri A. Zou L. B. Iwata N. Saido T. C. Wang D. Wang M. W. Noda Y. Nabeshima T. Inhibition of neprilysin by thiorphan (i.c.v.) causes an accumulation of amyloid beta and impairment of learning and memory. Behav. Brain Res. (2006);168:83–91. doi: 10.1016/j.bbr.2005.10.014. [DOI] [PubMed] [Google Scholar]
- 80.Mueller-Steiner S. Zhou Y. Arai H. Roberson E. D. Sun B. Chen J. Wang X. Yu G. Esposito L. Mucke L. Gan L. Antiamyloidogenic and neuroprotective functions of cathepsin B: implications for Alzheimer's disease. Neuron. (2006);51:703–714. doi: 10.1016/j.neuron.2006.07.027. [DOI] [PubMed] [Google Scholar]
- 81.Naert G. Rivest S. The role of microglial cell subsets in Alzheimer's disease. Curr. Alzheimer Res. (2011);8:151–155. doi: 10.2174/156720511795256035. [DOI] [PubMed] [Google Scholar]
- 82.Nalivaeva N. N. Beckett C. Belyaev N. D. Turner A. J. Are amyloid-degrading enzymes viable therapeutic targets in Alzheimer's disease? J. Neurochem. (2012);120(Suppl 1):167–185. doi: 10.1111/j.1471-4159.2011.07510.x. [DOI] [PubMed] [Google Scholar]
- 83.Narita M. Holtzman D. M. Schwartz A. L. Bu G. Alpha2-macroglobulin complexes with and mediates the endocytosis of beta-amyloid peptide via cell surface low-density lipoprotein receptor-related protein. J. Neurochem. (1997);69:1904–1911. doi: 10.1046/j.1471-4159.1997.69051904.x. [DOI] [PubMed] [Google Scholar]
- 84.Nielsen H. M. Veerhuis R. Holmqvist B. Janciauskiene S. Binding and uptake of A beta1-42 by primary human astrocytes in vitro. Glia. (2009);57:978–988. doi: 10.1002/glia.20822. [DOI] [PubMed] [Google Scholar]
- 85.Numata K. Kaplan D. L. Mechanisms of enzymatic degradation of amyloid Beta microfibrils generating nanofilaments and nanospheres related to cytotoxicity. Biochemistry. (2010);49:3254–3260. doi: 10.1021/bi902134p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Palmer J. C. Baig S. Kehoe P. G. Love S. Endothelin-converting enzyme-2 is increased in Alzheimer's disease and up-regulated by Abeta. Am. J. Pathol. (2009);175:262–270. doi: 10.2353/ajpath.2009.081054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Palmer J. C. Barker R. Kehoe P. G. Love S. Endothelin-1 is Elevated in Alzheimer's Disease and Upregulated byAmyloid-β. J. Alzheimers Dis. (2012);29:853–861. doi: 10.3233/JAD-2012-111760. [DOI] [PubMed] [Google Scholar]
- 88.Pardossi-Piquard R. Petit A. Kawarai T. Sunyach C. Alves da Costa C. Vincent B. Ring S. D'Adamio L. Shen J. Müller U. St George Hyslop P. Checler F. Presenilin-dependent transcriptional control of the Abeta-degrading enzyme neprilysin by intracellular domains of betaAPP and APLP. Neuron. (2005);46:541–554. doi: 10.1016/j.neuron.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 89.Pflanzner T. Petsch B. André-Dohmen B. Müller-Schiffmann A. Tschickardt S. Weggen S. Stitz L. Korth C. Pietrzik C. U. Cellular prion protein participates in amyloid-β transcytosis across the blood-brain barrier. J. Cereb. Blood Flow Metab. (2012);32:628–632. doi: 10.1038/jcbfm.2012.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Pike C. J. Walencewicz-Wasserman A. J. Kosmoski J. Cribbs D. H. Glabe C. G. Cotman C. W. Structure-activity analyses of beta-amyloid peptides: contributions of the beta 25-35 region to aggregation and neurotoxicity. J. Neurochem. (1995);64:253–265. doi: 10.1046/j.1471-4159.1995.64010253.x. [DOI] [PubMed] [Google Scholar]
- 91.Poirier R. Wolfer D. P. Welzl H. Tracy J. Galsworthy M. J. Nitsch R. M. Mohajeri M. H. Neuronal neprilysin overexpression is associated with attenuation of Abeta-related spatial memory deficit. Neurobiol. Dis. (2006);24:475–483. doi: 10.1016/j.nbd.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 92.Pulukuri S. M. Estes N. Patel J. Rao J. S. Demethylation-linked activation of urokinase plasminogen activator is involved in progression of prostate cancer. Cancer Res. (2007);67:930–939. doi: 10.1158/0008-5472.CAN-06-2892. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 93.Rogers J. Strohmeyer R. Kovelowski C. J. Li R. Microglia and inflammatory mechanisms in the clearance of amyloid beta peptide. Glia. (2002);40:260–269. doi: 10.1002/glia.10153. [DOI] [PubMed] [Google Scholar]
- 94.Russo R. Borghi R. Markesbery W. Tabaton M. Piccini A. Neprylisin decreases uniformly in Alzheimer's disease and in normal aging. FEBS Lett. (2005);579:6027–6030. doi: 10.1016/j.febslet.2005.09.054. [DOI] [PubMed] [Google Scholar]
- 95.Rylski M. Amborska R. Zybura K. Michaluk P. Bielinska B. Konopacki F. A. Wilczynski G. M. Kaczmarek L. JunB is a repressor of MMP-9 transcription in depolarized rat brain neurons. Mol. Cell Neurosci. (2009);40:98–110. doi: 10.1016/j.mcn.2008.09.005. [DOI] [PubMed] [Google Scholar]
- 96.Rylski M. Amborska R. Zybura K. Mioduszewska B. Michaluk P. Jaworski J. Kaczmarek L. Yin Yang 1 is a critical repressor of matrix metalloproteinase-9 expression in brain neurons. J. Biol. Chem. (2008);283:35140–35153. doi: 10.1074/jbc.M804540200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sagare A. P. Deane R. Zetterberg H. Wallin A. Blennow K. Zlokovic B. V. Impaired lipoprotein receptor-mediated peripheral binding of plasma amyloid-β is an early biomarker for mild cognitive impairment preceding Alzheimer's disease. J. Alzheimers Dis. (2011);24:25–34. doi: 10.3233/JAD-2010-101248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sagare A. Deane R. Bell R. D. Johnson B. Hamm K. Pendu R. Marky A. Lenting P. J. Wu Z. Zarcone T. Goate A. Mayo K. Perlmutter D. Coma M. Zhong Z. Zlokovic B. V. Clearance of amyloid-beta by circulating lipoprotein receptors. Nat. Med. (2007);13:1029–1031. doi: 10.1038/nm1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Saito T. Iwata N. Tsubuki S. Takaki Y. Takano J. Huang S. M. Suemoto T. Higuchi M. Saido T. C. Somatostatin regulates brain amyloid beta peptide Abeta42 through modulation of proteolytic degradation. Nat. Med. (2005);11:434–439. doi: 10.1038/nm1206. [DOI] [PubMed] [Google Scholar]
- 100.Savaskan E. Hock C. Olivieri G. Bruttel S. Rosenberg C. Hulette C. Müller-Spahn F. Cortical alterations of angiotensin converting enzyme angiotensin II and AT1 receptor in Alzheimer's dementia. Neurobiol. Aging. (2001);22:541–546. doi: 10.1016/s0197-4580(00)00259-1. [DOI] [PubMed] [Google Scholar]
- 101.Sehgal N. Gupta A. Valli R. K. Joshi S. D. Mills J. T. Hamel E. Khanna P. Jain S. C. Thakur S. S. Ravindranath V. Withania somnifera reverses Alzheimer's disease pathology by enhancing low-density lipoprotein receptor-related protein in liver. Proc. Natl. Acad. Sci. USA. (2012);109:3510–3515. doi: 10.1073/pnas.1112209109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Selkoe D. J. Alzheimer's disease. In the beginning... Nature. (1991);35:432–433. doi: 10.1038/354432a0. [DOI] [PubMed] [Google Scholar]
- 103.Shibata M. Yamada S. Kumar S. R. Calero M. Bading J. Frangione B. Holtzman D. M. Miller C. A. Strickland D. K. Ghiso J. Zlokovic B. V. Clearance of Alzheimer's amyloid-ss(1-40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J. Clin. Invest. (2000);106:1489–1499. doi: 10.1172/JCI10498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Simard A. R. Soulet D. Gowing G. Julien J. P. Rivest S. Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer's disease. Neuron. (2006);49:489–502. doi: 10.1016/j.neuron.2006.01.022. [DOI] [PubMed] [Google Scholar]
- 105.Spencer B. Marr R. A. Rockenstein E. Crews L. Adame A. Potkar R. Patrick C. Gage F. H. Verma I. M. Masliah E. Long-term neprilysin gene transfer is associated with reduced levels of intracellular Abeta and behavioral improvement in APP transgenic mice. BMC Neurosci. (2008);9:109. doi: 10.1186/1471-2202-9-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Tanzi R. E. Bertram L. Twenty years of the Alzheimer's disease amyloid hypothesis: a genetic perspective. Cell. (2005);120:545–555. doi: 10.1016/j.cell.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 107.Tucker H. M. Kihiko M. Caldwell J. N. Wright S. Kawarabayashi T. Price D. Walker D. Scheff S. McGillis J. P. Rydel R. E. Estus S. The plasmin system is induced by and degrades amyloid-beta aggregates. J. Neurosci. (2000);20:3937–3946. doi: 10.1523/JNEUROSCI.20-11-03937.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Tucker H. M. Simpson J. Kihiko-Ehmann M. Younkin L. H. McGillis J. P. Younkin S. G. Degen J. L. Estus S. Plasmin defi ciency does not alter endogenous murine amyloid beta levels in mice. Neurosci. Lett. (2004);368:285–289. doi: 10.1016/j.neulet.2004.07.011. [DOI] [PubMed] [Google Scholar]
- 109.Viswanathan A. Greenberg S. M. Cerebral amyloid angiopathy in the elderly. Ann. Neurol. (2011);70:871–880. doi: 10.1002/ana.22516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wang H. Y. Lee D. H. Davis C. B. Shank R. P. Amyloid peptide Abeta(1-42) binds selectively and with picomolar affinity to alpha7 nicotinic acetylcholine receptors. J. Neurochem. (2000);75:1155–1161. doi: 10.1046/j.1471-4159.2000.0751155.x. [DOI] [PubMed] [Google Scholar]
- 111.Wang R. Wang S. Malter J. S. Wang D. S. Effects of 4-hydroxy-nonenal and Amyloid-beta on expression and activity of endothelin converting enzyme and insulin degrading enzyme in SH-SY5Y cells. J. Alzheimers Dis. (2009a);17:489–501. doi: 10.3233/JAD-2009-1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wang R. Wang S. Malter J. S. Wang D. S. Effects of HNE-modifi cation induced by Abeta on neprilysin expression and activity in SH-SY5Y cells. J. Neurochem. (2009b);108:1072–1082. doi: 10.1111/j.1471-4159.2008.05855.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Weller R. O. Massey A. Kuo Y. M. Roher A. E. Cerebral amyloid angiopathy: accumulation of A beta in interstitial fluid drainage pathways in Alzheimer's disease. Ann. N. Y. Acad. Sci. (2000);903:110–117. doi: 10.1111/j.1749-6632.2000.tb06356.x. [DOI] [PubMed] [Google Scholar]
- 114.Weller R. O. Subash M. Preston S. D. Mazanti I. Carare R. O. Perivascular drainage of amyloid-beta peptides from the brain and its failure in cerebral amyloid angiopathy and Alzheimer's disease. Brain Pathol. (2008);18:253–266. doi: 10.1111/j.1750-3639.2008.00133.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wyss-Coray T. Infl ammation in Alzheimer disease: driving force bystander or benefi cial response? Nat. Med. (2006);12:1005–1015. doi: 10.1038/nm1484. [DOI] [PubMed] [Google Scholar]
- 116.Wyss-Coray T. Loike J. D. Brionne T. C. Lu E. Anankov R. Yan F. Silverstein S. C. Husemann J. Adult mouse astrocytes degrade amyloid-beta in vitro and in situ. Nat. Med. (2003);9:453–457. doi: 10.1038/nm838. [DOI] [PubMed] [Google Scholar]
- 117.Xiao Z. M. Sun L. Liu Y. M. Zhang J. J. Huang J. Estrogen regulation of the neprilysin gene through a hormone-responsive element. J. Mol. Neurosci. (2009);39:22–26. doi: 10.1007/s12031-008-9168-1. [DOI] [PubMed] [Google Scholar]
- 118.Yan P. Hu X. Song H. Yin K. Bateman R. J. Cirrito J. R. Xiao Q. Hsu F. F. Turk J. W. Xu J. Hsu C. Y. Holtzman D. M. Lee J. M. Matrix metalloproteinase-9 degrades amyloid-beta fibrils in vitro and compact plaques in situ. J. Biol. Chem. (2006);281:24566–24574. doi: 10.1074/jbc.M602440200. [DOI] [PubMed] [Google Scholar]
- 119.Yang L. Xu S. Liu C. Su Z. In vivo metabolism study of ginsenoside Re in rat using high-performance liquid chromatography coupled with tandem mass spectrometry. Anal. Bioanal. Chem. (2009);395:1441–1451. doi: 10.1007/s00216-009-3121-1. [DOI] [PubMed] [Google Scholar]
- 120.Yuyama K. Sun H. Mitsutake S. Igarashi Y. Sphingolipid-modulated exosome secretion promotes the clearance of amyloid-β by microglia. J. Biol. Chem. (2012) doi: 10.1074/jbc.M111.324616. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Zou L. B. Mouri A. Iwata N. Saido T. C. Wang D. Wang M. W. Mizoguchi H. Noda Y. Nabeshima T. Inhibition of neprilysin by infusion of thiorphan into the hippocampus causes an accumulation of amyloid Beta and impairment of learning and memory. J. Pharmacol. Exp. Ther. (2006);317:334–340. doi: 10.1124/jpet.105.095687. [DOI] [PubMed] [Google Scholar]