


Abstract
DNA repair is the first barrier in the defense against genotoxic stress. In recent years, mechanisms that recognize DNA damage and activate DNA repair functions through transcriptional upregulation and post-translational modification were the focus of intensive research. Most DNA repair pathways are complex, involving many proteins working in discrete consecutive steps. Therefore, their balanced expression is important for avoiding erroneous repair that might result from excessive base removal and DNA cleavage. Amelioration of DNA repair requires both a fine-tuned system of lesion recognition and transcription factors that regulate repair genes in a balanced way. Transcriptional upregulation of DNA repair genes by genotoxic stress is counteracted by DNA damage that blocks transcription. Therefore, induction of DNA repair resulting in an adaptive response is only visible through a narrow window of dose. Here, we review transcriptional regulation of DNA repair genes in normal and cancer cells and describe mechanisms of promoter activation following genotoxic exposures through environmental carcinogens and anticancer drugs. The data available to date indicate that 25 DNA repair genes are subject to regulation following genotoxic stress in rodent and human cells, but for only a few of them, the data are solid as to the mechanism, homeostatic regulation and involvement in an adaptive response to genotoxic stress.
INTRODUCTION
Genotoxic agents cause DNA damage that, if not repaired, results in chromosomal changes, gene mutations, cancer formation or cell death. To counteract the disastrous effects of genotoxic stress, cells have evolved sophisticated DNA repair mechanisms that remove or tolerate DNA lesions and thus maintain genomic stability. More than 130 different DNA repair proteins have been identified, and their role in DNA repair has been elucidated in great detail (1). Most of the DNA repair mechanisms comprise nucleases, which by themselves represent a danger to the genome. Therefore, DNA repair has to be tightly regulated in unexposed cells and, in case of genotoxic insults, has to be appropriately activated. The first discovered example of an inducible repair system is the SOS response of Escherichia coli (2), in which on DNA damage various nucleotide excision repair (NER) genes (uvrA, uvrB, uvrD) and translesion polymerases (umuD and umuC) become simultaneously upregulated. Subsequently, again in E. coli, the adaptive response was discovered (3,4) and mechanistically resolved by showing that it results from activation of the ada gene that encodes the inducible Ada alkyltransferase, which acts both as a repair protein and transcriptional activator (5–7).
In mammalian cells, regulation of DNA repair mechanisms is controlled at multiple levels. These include post-translational modification by acetylation, phoshorylation, ubiquitination and sumoylation, as well as the control by histone modification. The synthesis of repair proteins and the corresponding mRNAs is strictly regulated. Activation by genotoxin-induced DNA damage has been reported for 25 repair genes (July 2013) (Table 1). Their induction involves multiple players of the DNA damage response such as ataxia telangiectasia mutated (ATM), ATM and Rad3 related (ATR) and PARP1 as well as key transcription factors (Figure 1A). In this review, we focus on the transcriptional regulation of DNA repair genes and highlight the available data concerning DNA damage triggered promoter activation and its role in cellular protection and adaptation against genotoxic stress.
Table 1.
Genotoxin inducible DNA repair genes
| Gene | Repair mechanism | Function | Inducing genotoxic agens | Transcription-factor | Reference |
|---|---|---|---|---|---|
| apex1 | BER | Endonuclease | H2O2, ionizing radiation | AP-1 | (8–10) |
| ddb1 | NER | Damage recognition | ionizing radiation | ? | (11) |
| ddb2 | NER | Damage recognition | UV-C, adriamycin, cisplatin, BPDE, ACNU, BCNU, ionizing radiation, fotemustine | p53, BRCA1, p63 | (12–20) |
| ercc1 | NER | Co-Factor of XPF | ionizing radiation, arsenic com., B[a]P | AP-1 | (21–26) |
| fen1 | BER | Flap-Endonuclease | UV-C, MMS | p53 | (27,28) |
| karp1 | NHEJ | Ku-Binding | ionizing radiation, UV-C, etoposide, MMS | p53 | (29) |
| lig1 | BER | Ligase | UV-C | ? | (30) |
| mgmt | Damage reversal | Alkyltransferase | ionizing radiation,UV-C, MMS | p53, AP-1,NF-κB | (31–37) |
| mpg | BER | Glycosylase | UV-C, TPA, EMS | Sp1, AP-2 | (38,39) |
| mlh1 | MMR | ATPase | cisplatin | p53 | (40) |
| msh2 | MMR | Damage recognition | UV-B, TPA | p53, AP-1 | (41,42) |
| neil1 | BER | Glycosylase | ROS | AP-1 | (43) |
| ogg1 | BER | Glycosylase | MMS, ionizing radiation | NF-YA | (44,45) |
| pcna | several | Replication clamp | ionizing radiation, UV-C | p53, AP-1 | (46–49) |
| pms2 | MMR | Endonuclease | cisplatin | p53 | (40) |
| polI | TLS | Polymerase | UV-C, MNNG | Sp1 | (50,51) |
| polβ | BER | Polymerase | MNNG, MMS | TFEIF, CREB-1, ATF-1 | (52–56) |
| polH | TLS | Polymerase | ionizing radiation, CPT | p53 | (57) |
| polK | TLS | Polymerase | BPDE | HSF1 | (58) |
| rev3 | TLS | Polymerase | MNNG | ? | (59,60) |
| trex1 | several? | Exonuclease | UV-C, BPDE, CPT, ACNU, TPT, fotemustine | AP-1 | (61–63) |
| xrcc1 | BER, SSBR | Scaffold protein | ionizing radiation, MMS | E2F1 | (21,22,64) |
| xpc | NER | Damage recognition | UV-C, γ-ray , MMS, B[a]P, ACNU, BCNU, fotemustine | p53, BRCA1 | (14,15,19,20,65–69) |
| xpf | NER | Endonuclease | UV-C, B[a]P, ionizing radiation | AP-1 | (11,70–72) |
| xpg | NER | Endonuclease | UV-C, B[a]P | AP-1, E2F1 | (70–72) |
Figure 1.

Transcription factors and signalling involved in repair gene regulation. (A) Transcription factors involved in genotoxin-triggered transcriptional activation of DNA repair genes. (B) Growth factor and DDR triggered activation of transcription factors involved in repair gene regulation.
DNA damage-triggered activation of kinases and transcription factors involved in the regulation of DNA repair genes
Genotoxic stress-triggered PI3 kinases
Transcriptional regulation of genes is dependent on the activation of transcription factors and their binding to the promoter region of a given gene. The activation of transcription factors following genotoxic stress results from activation of the DNA damage response (DDR). In this pathway, the most important sensors of DNA damage, notably DNA double-strand breaks (DSB) and replication blocking lesions, are the phosphatidylinositol-3-kinases ATM and ATR. They are at the heart of the DDR and are activated within minutes after exposure of mammalian cells to genotoxins [for review see (73,74)]. In brief, activation of ATM requires the MRN complex consisting of MRE11, NBS1 and RAD50 that triggers ATM autophosphorylation. Phosphorylation of ATM occurs at Ser1981 (75), leading to dissociation of inactive ATM dimers to active monomers. ATM monomerization requires the interaction with the MRN complex and single-stranded DNA (ssDNA) (76). MRN by itself recognizes and migrates to DSBs induced by ionizing radiation (77) and to ssDNA on replication blockage (78) and remains at the site of DNA damage. In the absence of the MRN complex, the autophosphorylation ability of ATM is reduced (79), and ATM is not recruited to the DSB (76). ATR is activated following blockage of DNA polymerases and the formation of large stretches of ssDNA, which are generated through uncoupling of the MCM helicase from the replication fork and subsequent binding of replication protein A (RPA) to ssDNA (80). RPA labelled ssDNA induces the recruitment of ATR complexed with ATR-interacting protein (ATRIP) and the 9–1–1 complex consisting of Rad9, Hus1 and Rad1 to the site of damage (81). Rad9 interacts with TopBP1 (82) and recruits it to the stalled replication fork (83,84) where it directly activates the ATR-ATRIP complex (85). In addition, ATR becomes autophosphorylated on DNA damage at Thr1989, which was shown to be crucial for its activation (86).
Following activation of ATM and ATR, both proteins phosphorylate and thereby activate multiple proteins involved in DNA repair, cell cycle control, apoptosis and autophagy. Among them are also proteins that serve as transcription factors. The most important transcription factors activated by the DDR pathway are p53, breast cancer-associated protein 1 (BRCA1), NF-κB and AP-1 (Figure 1A), which will be briefly described as to their mode of activation following genotoxic stress (summarized in Figure 1B) before their role in repair gene regulation is being discussed.
Genotoxic stress-triggered NF-κB activation
NF-κB represents a dimeric transcription factor composed of various homo- and hetero-dimers formed by the proteins RelA (p65), RelB, c-Rel, p50/p105 (NF-κB1) and p52/p100 (NF-κB2) (87,88). NF-κB is activated on multiple insults including anticancer drugs and ionizing radiation (89). In unexposed cells, NF-κB is present in the cytoplasm owing to interaction with members of the inhibitory IκB family (IκBα, IκBβ and IκBε) (90,91). Activation of NF-κB is mediated via proteosomal degradation of the inhibitory proteins and subsequent nuclear translocation (Figure 1B). Upon genotoxic stress, activation of NF-κB is provoked by the IκB-kinase (IKK) complex. The IKK complex consists of IKKα and IKKβ, forming the catalytic subunit, and the regulatory subunit IKKγ (NEMO). NEMO can shuttle between the nucleus and the cytoplasm. Upon genotoxic stress, it becomes modified by the DDR. Thus, it has been shown that PARP1 assembles ATM and the SUMO-1 ligase PIASy, which results in sumoylation followed by ATM-mediated phosphorylation of NEMO (92,93). NEMO sumoylation is then replaced by Lys63-linked mono-ubiquitination, leading to the nuclear export of NEMO as a complex with ATM. In addition, ubiquitination of NEMO allows the recruitment of additional kinases, which phosphorylate IKKβ in its activation loop at Ser177 and Ser181 (94). The activated IKK complex then phosphorylates IκBα on Ser32 and Ser36, which marks it for ubiquitination and proteasomal degradation, thereby releasing NF-κB. Activation of NF-κB can also occur independent of IKK. In this case, genotoxic stress activates CK2, which phosphorylates IκBα and targets it for degradation (95).
Genotoxic stress-triggered p53 activation
p53 is a sequence-specific transcription factor (96) that plays a major role in the regulation of DNA repair, apoptosis and cell cycle progression. p53 becomes activated on DNA replication arrest and DSB induced by chemical genotoxins and irradiation via the ATM/ATR pathway (Figure 1B). Thus, following DSB formation, ATM phosphorylates the checkpoint kinase-2 (CHK2) at Thr 68 (97,98) while, following replication blockage, ATR phosphorylates CHK1 at Ser 345 (99,100). In turn, CHK2 and CHK1 phosphorylate p53 at Ser20, thereby activating it (101). ATM and ATR can also directly phosphorylate p53 at Ser15, thereby increasing its transactivation activity (102,103). ATM/ATR also phosphorylate MDM2, which results in MDM2 degradation by ubiquitination and stabilization of the p53 protein (104). As a consequence, the cellular amount of p53, its nuclear translocation and its DNA-binding activity all become enhanced and p53 target genes become transcriptionally activated.
Genotoxic stress-triggered BRCA1 activation
Another transcription factor that interacts with p53 and shares common target genes is BRCA1. BRCA1 is implicated in the regulation of several cellular functions such as chromatin remodelling and DNA repair (105). BRCA1 becomes phosphorylated and, in turn, activated by ATM and ATR (106,107) and is involved in transcriptional activation of some DNA repair genes (see later in the text). BRCA1 was found to be associated with RNA polymerase II holoenzyme complex (108), thus having an impact on transcription in vitro (109). Additionally, BRCA1 interacts with p53 and stimulates its transcriptional activity (110,111). A gene transcriptionally activated by BRCA1 is GADD45a, which is one of the first discovered genotoxic stress-inducible genes (112). It was believed to be involved in DNA repair, but until now no evidence is available that GADD45a directly increases the activity of repair functions. GADD45a, however, activates MTK1/MEKK4 by binding to it (113,114). MTK1/MEKK4 is the major upstream regulatory kinase of MKK4 and MKK7 that control the activity of the stress-activated protein kinases/c-Jun-N-terminal kinases (SAPK/JNK) (Figure 1B). By activating this pathway, GADD45a may indirectly stimulate DNA repair via AP-1 triggered upregulation of repair genes.
Genotoxic stress-triggered AP-1 activation
AP-1 consists of different dimeric complexes containing proteins of the Jun (c-Jun, JunB and JunD), Fos (c-Fos, FosB, Fra1, Fra2) and CREB/ATF (ATF1, ATF2) family, which can exert different specificities and functions (115). Depending on the composition of the dimeric protein and the target sequence, AP-1 binds with different affinity to the AP-1 consensus sequence in the promoter, hereby regulating target genes with different strengths. Binding of Jun/Fos occurs mainly onto heptameric (TGAGTCA) sites, and Jun/ATF-2 binds to octameric CRE (cAMP response element) binding sites (TGACGTCA) (115). Activation of AP-1 depends mainly on the MAPK (mitogen-activated protein kinase) pathway and is mediated via increased expression and post-translational activation of the AP-1 components. Stimulation of the MAPK cascade is either provoked by activated growth factor receptors, such as the epidermal growth factor (EGF) receptor (116,117), or by DNA damage which is dependent on the activated DDR. Thus, ATM-dependent activation of the Jun kinase (JNK) (118–121) and ATR-dependent activation of p38 kinase (p38K) have been reported (122). On activation, JNK, ERK1/ERK2 (extracellular signal regulated kinase 1/2) and p38K phosphorylate and thereby stimulate the DNA binding and transactivating activity of AP-1 (Figure 1B). Several DNA repair genes harbour one or more functional AP-1 binding sites in their promoter and have been identified as targets of this DNA damage-triggered response (Table 1).
Regulation of DNA repair genes by promoter activation
Upon genotoxic stress, transcription factors listed earlier in the text become activated, bind to a corresponding promoter, stimulate RNA polymerase II binding, assembly of transcription factors and finally mRNA synthesis (called gene activation following genotoxic stress or, briefly, ‘gene induction’). In the following, we will describe the DNA repair genes (to our knowledge, 25 of ∼130 DNA repair genes) that were shown to be inducible on promoter level in mammalian cells following exposure to genotoxic agents. We should note that for some of the repair genes listed in Table 1, evidence for induction was only provided in a single rodent or human cell line, which could not be confirmed in other lines. For some repair genes, conflicting data were reported, and for only a few of them data are solid and reproducible in rodent and human experimental systems. It is noteworthy that clear evidence for induction by genotoxic stress in the normal tissue of the human body is lacking for all repair genes. We should also note that in some studies, the regulation of repair genes was addressed in co-transfection experiments without the exposure to genotoxic stress. Thus, BRCA2 (123), Ku70 and Ku80 (124) were shown to be positively regulated by NF-κB, and Rad51 negatively regulated by p53 (125). As it is unclear whether this artificial situation can be translated to genotoxin-exposed cells, these experiments will not be discussed further.
Single step repair by O6-methylguanine-DNA methyltransferase
O6-methylguanine-DNA methyltransferase (MGMT), alias alkyltransferase, does not need cofactors or another ‘helper’ protein; it represents a one-step repair mechanism, which is responsible for the removal of alkyl groups from the O6-position of guanine and the O4-position of thymine [for recent review see (126)] (Figure 2A). The question of induction of MGMT is highly important. First, if MGMT upregulation occurred, it would protect against O6-alkylating environmental and tobacco smoke carcinogens (127). Second, it would have a high impact on cancer therapy, as tumours like glioblastoma and metastatic melanoma are treated with O6-alkylating agents (temozolomide, dacarbazine, chloroethylating nitrosoureas) against which MGMT offers protection. Notably, in glioma therapy, ionizing radiation is applied concomitantly with temozolomide (nearly daily for a period of 30 days, total 60 Gy). In addition, corticosteroids are administered to reduce oedema and inflammation. An upregulation of MGMT in tumour cells provoked either by temozolomide, ionizing radiation or corticosteroid treatment would render the therapy with O6-alkylating agents inefficient. Therefore, it is of upmost importance to understand how MGMT is regulated by genotoxic stress and also by non-genotoxic gene activators.
Figure 2.

Regulation of MGMT transcription. (A) Regulation of MGMT promoter and MGMT-mediated repair of DNA alkylation damage. (B) Structure of the human MGMT promoter showing the positions of transcription factor-binding sites.
Transcriptional activation of MGMT was repeatedly shown in vitro in primary rat hepatocytes and in rat hepatoma cell lines on treatment with ultraviolet (UV)-C light, ionizing radiation or alkylating agents (31,32). It is a delayed and transient response giving rise to increased transcript levels 12–24 h after treatment, which result in a higher level of MGMT protein and repair activity. The level of induction correlated with the degree of differentiation of the lines and was especially high in rat H4IIE cells grown as spheroids (128). Induction of MGMT was also observed in vivo in the rat liver (up to 20-fold) (33) and several organs of mice after ionising radiation (34). While MGMT induction on genotoxic stress was shown conclusively in rodents, there is as yet no convincing evidence for human cells, despite intensive research. Thus, the MGMT gene was found to be non-inducible in human fibroblasts (32,129) and glioma cells (130) following treatment with alkylating agents and ionizing radiation. Furthermore, there was no increase in MGMT repair activity following these treatments. This is surprising, as the human MGMT promoter contains potential transcription factor binding sites (131) that render it susceptible to induction (Figure 2B). The cloned MGMT promoter was shown to be strongly active following transfection in rat, mouse and human cell lines (132). Interestingly, following transfection in rat H4IIE cells, the human MGMT promoter was expressed at a higher level if the cells were pre-treated with ionizing radiation (128), indicating the human MGMT promoter has the potential to be inducible by genotoxic stress. The endogenous gene, however, was not responding under the same treatment conditions in human cells (unpublished data).
In rodent cells, p53 appears to be required for MGMT gene induction, as p53 knockout mice did not show upregulation of MGMT following whole-body ionizing radiation (34). Also on transfection of the human MGMT promoter in mouse fibroblasts treated with ionising radiation, p53 is required for enhanced promoter activation (35). The role of p53 in MGMT regulation appears to be a complex matter as p53 also seems to influence the basal expression level. Thus, knockout of p53 in murine astrocytes strongly reduced the expression of MGMT (133), and, paradoxically, enforced expression of wild-type p53 in human tumour cells also reduced the transcription of the MGMT gene (35,134). Attention should be paid to the fact that the human MGMT promoter does not harbour a p53 consensus binding site. Thus, the negative regulation of MGMT by p53 seems to be independent of p53 binding to the promoter. A reasonable explanation was provided by studies that draw attention to the transcription factor Sp1. The promoter of MGMT is rich in Sp1-binding sites (Figure 2B), which have a strong impact on the basal MGMT expression level. In this study, high expression of p53 sequestered Sp1 and prevented their binding to the MGMT promoter, finally leading to a reduced expression of MGMT (135).
Although studies on genotoxin-induced MGMT induction largely failed, evidence was provided that the MGMT expression level was increased in human HeLa S3 cells following the treatment with activators of protein kinase C such as phorbol-12-myristate-13-acetate (TPA) and 1,2-diacylglycerol. This induction was regulated via c-Fos and c-Jun mediated activation of AP-1, which binds to two AP-1 binding sites within the MGMT promoter (36). The MGMT promoter also contains glucocorticoide responsive elements (Figure 2B) and was shown to be inducible by corticosteroids in rat hepatoma H4IIE cells (128) and in a human glioma cell line by treatment with dexamethasone (136). We should note that in the glioma study with U87MG and U138MG cells, a high concentration of dexamethasone (10 µM) was applied, which is far above the physiological level. Treatment with dexamethasone at a <100 nM level did not induce MGMT in these and other glioma cells (own unpublished data). We should also note that induction of MGMT by dexamethasone was reported to occur in U87MG cells (136), which are promoter methylated and therefore silenced for MGMT. Overall, although the MGMT promoter contains glucocorticoide responsive elements and is potentially subject to glucocorticoid regulation, convincing evidence is still lacking that glucocorticoids upregulate MGMT in gliomas and other cancers. Another transcription factor that was shown to bind to and activate the MGMT promoter is NF-κB (37). It has yet to be proven, however, whether NF-κB regulation of MGMT is of biological importance. There are also data showing that MGMT transcription is downregulated by interferon β (IFN-β), which causes p53 activation in human glioblastoma cells (137,138) and by cisplatin in human gallbladder cancer cells (139). The biological relevance of these findings is unclear.
Base excision repair (BER)
BER removes single bases damaged by oxidation, methylation and other small chemical modifications from DNA. The first step in BER is executed by glycosylases, such as 8-oxoguanine-DNA glycosylase (OGG1), 3-methyladenine-DNA glycosylase (MPG) or endonuclease VIII-like 1 (NEIL1), which remove modified purines and pyrimidines from DNA leaving apurinic/apyrimidinic sites. These sites are converted into DNA single-strand breaks, which is catalysed by the apurinic/apyrimidinic endonuclease (APE1, APEX, REF-1). During short patch BER, the remaining sugar backbone is removed by DNA polymerase β, which also inserts a new nucleotide. During long-patch BER, the flap endonuclease 1 (FEN1) stimulates strand displacement and repair synthesis via DNA polymerase β. The X-ray repair cross-complementing protein-1 (XRCC1), which interacts with DNA ligase III and polymerase β, is involved in the ligation step during short-patch BER, and the DNA ligase I (LIG1) performs the ligation step during long-patch BER (Figure 3A). The following BER genes have been described to be subject to transcriptional upregulation.
Figure 3.
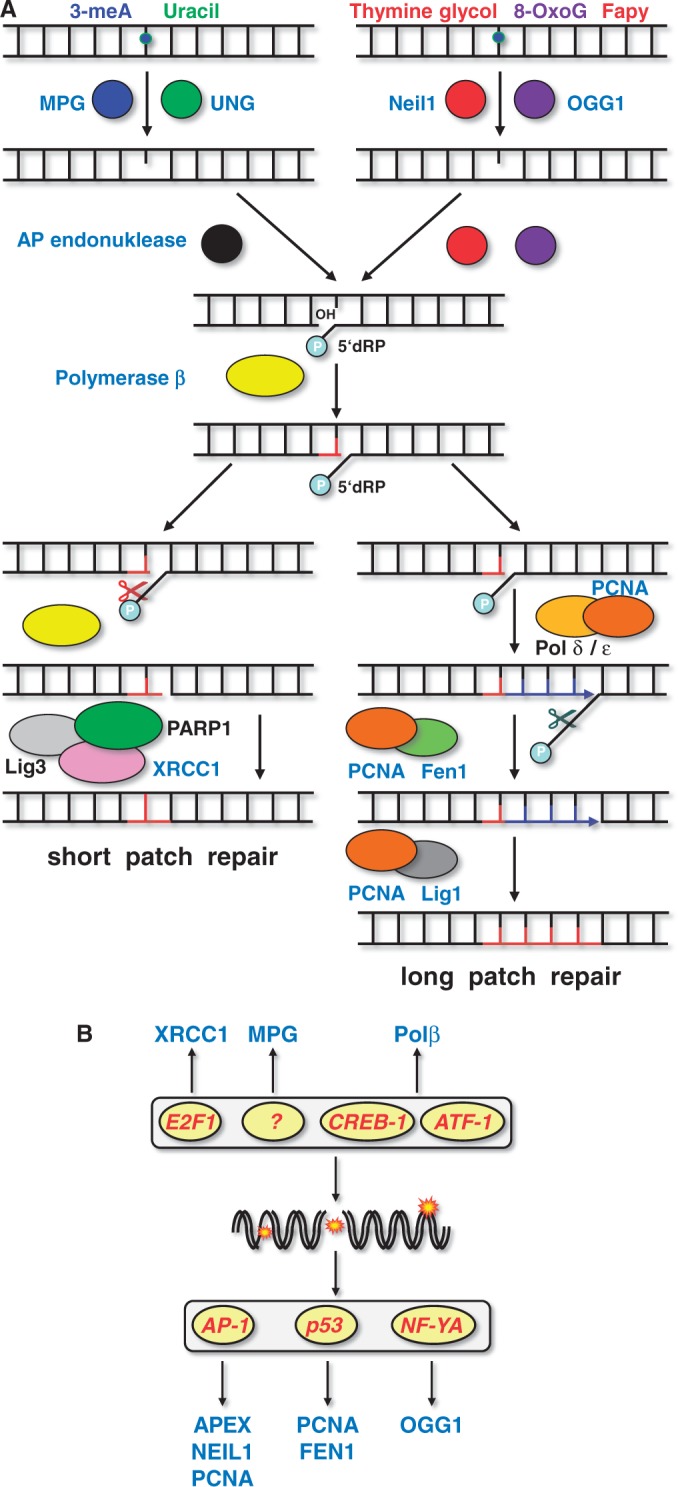
Regulation of BER genes. (A) Mechanism of BER. (B) BER genes and transcription factors that were reported to be regulated by genotoxic stress.
OGG1: Transcriptional activation of OGG1 was described for HCT116 colorectal carcinoma cells that were exposed to methyl methanesulfonate (MMS) (44). Two inverted CCAAT motifs in the ogg1 promoter were required that were activated by the transcription factor NF-YA, finally leading to enhanced expression of the mRNA and protein. The ogg1 gene was also shown to be induced after treatment with ionizing radiation in lung tissue of mice (45). However, this induction was only observed on mRNA, but not on protein level. NEIL1: Transcriptional induction of NEIL1 was observed on exposure to reactive oxygen species (ROS) in human HCT116 colon cancer cells, which resulted in enhanced expression of the mRNA and protein. The induction rests on the binding of the transcription factors c-Jun and CREB/ATF2 to two identical CRE/AP-1-binding sites in the NEIL1 promoter (43). MPG: Induction of the rat MPG promoter was observed on transfection in H4IIE rat hepatoma cells that were exposed to the tumour promoter TPA or UV light (38). However, induction of the endogenous mRNA or protein was not observed (128). The human MPG promoter was cloned and shown to be regulated by Sp1 and AP-2 (140). Induction of the human mpg gene was shown following ethyl methanesulfonate (EMS) exposure in the human lymphoblastoid cell line AHH-1, but again no induction of the corresponding protein was reported (39). APE1: Transcriptional activation of APE1 occurs in rat liver hepatoma cells (H4) upon exposure to γ-rays and in CHO cells and V79 fibroblasts upon treatment with agents generating ROS such as hydrogen peroxide and sodium hypochloride. It was accompanied by enhanced expression of the APE1 protein (8,9). Oxidative stress-induced transcriptional activation of the human APE1 promoter transfected in CHO cells was mediated via a CREB binding site, which is recognized by c-Jun and ATF-2 (10). In human cells exposed to genotoxic stress, APE1 expression appears to be negatively regulated similar to the mechanism described earlier in the text for MGMT. Also in this case, p53 sequesters Sp1 and thus represses APE1 expression (141). Interestingly, APE1 is expressed at a higher level in the inflammatory tissue of colon (in patients suffering from colitis ulcerosa), indicating that ROS generated during chronic inflammation upregulated the APE1 gene in vivo (142). APE1 was also shown to be induced in neurons of rats treated with kainic acid (143). Polβ: Transcriptional upregulation of polymerase β (Polβ) was shown to be stimulated in CHO cells by simple methylating agents such as N-methyl-N′-nitro-N-nitrosoguanidine (MNNG) and MMS (52). Induction is mediated by the decanucleotide palindromic element GTGACGTCAC at positions −49 to −40 of the core promoter (53), which is recognized by the transcription factors CREB-1 and ATF-1 (54). However, an enhanced expression of the protein was not reported on genotoxin exposure. Other transcription factors involved in the regulation of Polβ in unexposed cells are telomerase transcriptional element-interacting factor (TFEIF) that, following ectopic expression in HeLa cells, upregulated Polβ mRNA and protein (55), and NF-κB that regulates Polβ in EBV-immortalized B cells by binding to a proximal NF-κB binding site (56). Fen1: Transcriptional upregulation of FEN1 mRNA and protein was observed in mouse embryonic fibroblasts after exposure to UV-C light, which required p53 (27). In addition to UV-C, FEN1 expression was also induced by MMS as shown in microarray analyses of L5178Y mouse lymphoma cells (28). In human cells, Fen1 was not induced under the same treatment conditions (our unpublished data), indicating cell type specificity of the response. XRCC1: Transcriptional activation of XRCC1 was shown following ionizing radiation and the mitogen EGF via the EGFR and MAPK pathway in DU145 prostate carcinoma cells (21,22). XRCC1 was also induced on MMS treatment in mouse embryonic fibroblasts, which was triggered by the transcription factor E2F1 (64). In this case, induction of XRCC1 was observed on mRNA and protein level. LIG1: Transcriptional upregulation of the lig1 mRNA was reported in human primary fibroblasts after exposure to UV-C light, where it was associated with enhanced ligase activity (30).
In summary, the transcription factors involved in BER gene regulation are E2F1, CREB-1, ATF-1, AP-1, p53 and NF-YA (Figure 3B). We should note that there is no convincing evidence that upregulation of either one of the BER genes described earlier in the text results in a higher BER capacity. BER is significantly post-translationally regulated, e.g. by ubiquitination and phosphorylation (144,145). It is of note that transcriptional regulation of BER genes (XRCC1 and LIG1) occurs during maturation of macrophages and dendritic cells, which impacts their response to genotoxic stress (146).
NER
The repair of bulky DNA adducts is accomplished by the NER pathway (Figure 4A). In the global genomic repair sub-pathway of NER, the DNA damage binding protein 2 (DDB2, p48) forms a complex with DDB1 yielding the DNA damage recognition complex XPE, which recognizes UV-induced cyclobutan pyrimidine dimers (CPDs) and, only marginally, (6–4) photoproducts (6-4PP). A second heterodimeric recognition complex is formed by the xeroderma pigmentosum C (XPC) protein and hRad23B that recognizes mainly UV light-induced 6-4PP, but not CPDs. A third recognition complex (RPA-XPA) recognizes 6-4PP and cisplatin lesions and verifies the damage detected by XPE or XPC/HR23B. After recognizing a bulky lesion, the transcription factor TFIIH is recruited to the site of DNA damage and unwinds the DNA via its XPB and XPD subunits. The excision of the adduct-containing polynucleotide requires dual incision 5′ and 3′ from the adduct. The excision repair cross-complementing protein 1 (ERCC1) forms a complex with the Xeroderma pigmentosum F (XPF) protein and performs the 5′-incision step during NER. The 3′ incision is mediated by Xeroderma pigmentosum G (XPG) protein. The resulting DNA gap is filled by DNA polymerase (Pol δ and Pol ε) and sealed by LIG1. Several of the NER components were reported to be subject to transcriptional regulation following genotoxic stress (Figure 4B), as outlined below.
Figure 4.
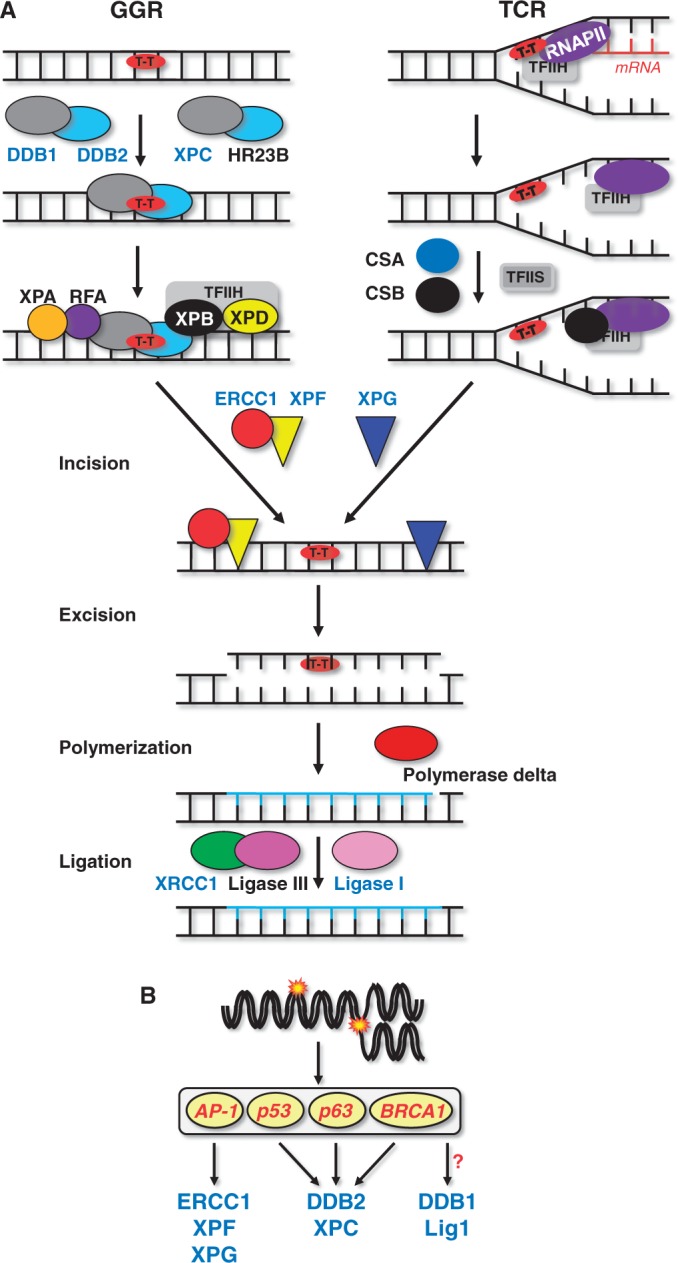
Regulation of NER. (A) Mechanism of NER and key proteins involved. (B) NER genes and transcription factors regulated by genotoxic stress.
DDB1: Transcriptional activation of DDB1 was shown to be induced by ionizing radiation in MCF10F human breast epithelial cells, leading to enhanced expression of mRNA (11). No evidence was provided, however, for an upregulation of the protein. The underlying mechanism and responsible transcription factors remained unknown. DDB2: There are several consistent reports demonstrating that DDB2 is induced after exposure of human cells to UV-C light and ionizing radiation. The induction is mediated by p53 (12,13) and TAp63γ (14). Beside p53, BRCA1 can also transcriptionally activate DDB2 (15,16). DDB2 expression was also reported to be enhanced after exposure to adriamycin (doxorubicin) and cisplatin in HCT116 and H460 cells (16), on benzo(a)pyrene in HepG2 and MCF-7 cells (17,18) and on the chloroethylating anticancer drugs carmustine and nimustine in glioblastoma cells (19). Recently, we showed that DDB2 is upregulated in metastatic melanoma cells following treatment with the chloroethylating anticancer drug fotemustine. Upregulation was p53 dependent. It occurred on mRNA and protein level, enhanced crosslink repair and conferred killing protection, demonstrating acquired drug resistance of cancer cells by upregulation of NER (20). In mouse fibroblasts, DDB2 is non-inducible owing to alteration of the p53 binding site in the DDB2 promoter (13). The lack of upregulation of DDB2 in mouse cells is supposed to be a reason for their low NER capacity. DDB2 is presumably the best example of a genotoxic stress-inducible DNA repair gene (12–19) that gives rise to an adaptive response (20). XPC: Transcriptional activation of xpc was observed following UV-C treatment, which was reported to require p53 in human WI38 fibroblasts and HCT116 colorectal cancer cells (65), TAp63γ in Saos-2 cells (14) and BRCA1 in U2OS osteosarcoma cells (15). XPC expression was also induced after ionizing radiation in human lymphoblastoid cells (66), after UV-C, ionizing radiation and MMS in peripheral blood lymphocytes and different tumour cell lines (67), and after benzo[a]pyrene diol epoxide (BPDE) treatment in human mammary epithelial (68) and MCF-7 cells (69). Similar to DDB2, XPC was upregulated in glioblastoma cells following carmustine and nimustine treatment (19) and in metastatic melanoma cells after fotemustine treatment (20). The response was p53 dependent and a long lasting (up to 4 days following a single treatment) and was related to acquired drug resistance of the cancer cells (20). ERCC1: Transcriptional activation of ercc1 was observed using real-time PCR on treatment with arsenic compounds in liver (23) and after treatment with ionizing radiation in murine lung (45). In human prostate cancer cells, ERCC1 expression was induced by ionizing radiation via the EGFR and MAPK pathway (21,22). ERCC1 expression was also found to be upregulated in NIH3T3 cells, which was triggered by activated c-Ha-RAS and AP-1 (24). In human ovarian cancer cells, ERCC1 was induced by cisplatin and phorbol ester treatment via AP-1 (25,26), and this induction was blocked by interleukin-1α (147). ERCC1 binds to XPF and stabilizes the protein. Therefore, increase of ERCC1 protein might also have an indirect effect on the XPF level. XPF/XPG: Transcriptional activation of XPF was reported for MCF10F human breast epithelial cells by ionizing radiation (11). In murine and human fibroblasts, XPF and XPG were induced on UV-C treatment. The transcription factors responsible were c-Fos/AP-1 (70,71). Enhanced expression of XPG mRNA was detected on exposure of Cockayne syndrome cells to interferon β (148). In human bronchial epithelial cells, the XPG expression was reported to be regulated by the transcription factors CEBPG and E2F1/YY1 (149). A concerted upregulation of NER proteins has been shown in human bronchial epithelial cells following exposure to B[a]P. In this case, the level of XPA, XPC, XPF, XPG and ERCC1 protein was concomitantly upregulated (72).
Mismatch repair
Mismatch repair (MMR) is initiated following recognition of the mismatch by the dimeric MutSα complex, composed of MSH2 and MSH6. MutSα is responsible for the detection and repair of single base mismatches and small insertion/deletion mismatches generated during replication. MSH2 can also form a complex with MSH3, designated as MutSβ, which is only capable of binding to insertion/deletion mismatches, but not base-base mismatches. On binding to the mismatch, MutSα interacts with the MutLα complex, formed by the MutL homologous protein 1 (MLH1) and PMS2, which exerts ATPase and endonuclease activity and mediates together with the exonuclease I (Exo1) the removal of the DNA strand carrying the mispaired base.
MSH2: The msh2 promoter was reported to be activated upon UV-B exposure in a p53 and c-Jun-dependent manner in Saos-2 and HaCat cells (41). Different potential p53 binding sites were identified in the human msh2 promoter in Saos-2 (150) and A2780 ovarian cancer cells (151). All these reports rest on experiments performed using MSH2 promoter constructs, whereas induction of the endogenous mRNA and protein was not shown. Beside p53, AP-1 is also involved in the regulation of msh2 gene expression. However, this regulation appears to be independent of genotoxic stress. Thus, in myeloid leukemic U937 cells, the expression of MSH2 protein was increased on treatment with TPA, which occurred via protein kinase C activation (42). Mutation of the AP-1 binding sites in the msh2 promoter or expression of dominant negative c-Jun abrogated the TPA-triggered induction of MSH2 (152). In our studies, we did not observe upregulation of the endogenous msh2 gene following mutagenic treatment (unpublished data), and, to our knowledge, there is no report showing convincingly that MSH2 is transcriptionally upregulated by genotoxic stress. This provides a good example that experiments with the cloned promoter should be complemented with analysis of the endogenous gene. Upon MNNG exposure, phosphorylation and nuclear translocation of MSH2 and MSH6 was observed (153,154) indicating post-translational mechanisms involved in regulation of MMR. MLH1/PMS2: Using a screening approach of serial analysis of binding elements, PMS2 and MLH1 have been shown to be under p53 control. In contrast to MSH2, induction of the endogenous mRNA and protein of MLH1 and PMS2 was observed. Both genes were rapidly induced upon cisplatin treatment in human fibroblasts (40). Whether upregulation of these MMR genes following genotoxic stress ameliorates the cells MMR capacity remains to be seen.
Translesion synthesis
Translesion synthesis represents a DNA repair mechanism associated with bypassing and tolerating replication blocking lesions. The mechanism rests on specialized DNA polymerases, which bypass replication blocking lesions in an error-free or error-prone way. PolH: For DNA polymerase eta (PolH), p53 mediated induction has been reported following ionizing radiation and camptothecin, but not UV-C treatment in human cell lines on mRNA and protein level (57). PolI: Polymerase iota (PolI) was found to be upregulated on protein level in breast cancer cells after treatment with UV-C light (50). PolI was also shown to be upregulated following MNNG treatment in human amnion follicular lymphoma (FL) cells (51). PolK: Polymerase kappa (PolK) was found to be upregulated on mRNA level in human amnion FL cells on exposure to BPDE and analysis of the PolK promoter suggests the transcription factor HSF1 to be involved (58). PolZ: In promoter studies, polymerase zeta (PolZ) subunit REV3 was activated upon transfection into human cells treated with MNNG (59,60). The promoter region responsible for this induction (−404 to −102) contains binding sites for CREB, NF-κB and AP-2. Whether induction of translesion polymerases confers genotoxin resistance or impacts the mutation rate of cells is unknown.
Miscellaneous DNA repair genes
Several DNA repair associated genes were reported to be subject to genotoxin-induced transcriptional regulation. KARP1: Ku86 autoantigen-related protein-1 (KARP-1) has been shown to work as a heterodimer with Ku70. Although supporting DNA binding of Ku70, it cannot completely replace Ku80 in DSB repair (155). On treatment with ionizing radiation, UV-C, etoposide and MMS, KARP1 was shown to be p53-dependently upregulated in HCT116 cells (29). PCNA: PCNA plays an important role in regulating various DNA metabolic functions. At the transcriptional level, PCNA gets activated by ionizing radiation in human lung epithelial cells (46) and human fibroblasts (47). In both cases, induction is triggered by p53, which activates the PCNA promoter (48). Contrary to ionizing radiation, UV light upregulates PCNA in human fibroblasts in a p53-independent manner (49). The rat promoter of PCNA was shown to be inducible following UV exposure via AP-1 (156). The biological consequences of PCNA upregulation have not yet been addressed. TREX1: The three prime exonuclease 1 (TREX1) is a 3′-5′ exonuclease that catalyzes the excision of nucleoside monophosphates from the 3′ termini of DNA (157). TREX1 prefers a partial duplex DNA with multiple mispaired 3′ termini (158). The exact role of TREX1 in DNA repair is still unclear. Induction of TREX1 was reported in mouse and human fibroblasts on exposure to UV-C and BPDE, which is a result of promoter activation involving c-Fos/AP-1 (61,62). In addition, TREX1 was found to be induced in murine macrophages on IFN-γ treatment via c-Jun/AP-1 (159). Induction was also reported in nasopharyngeal carcinoma KB cells by the anticancer drug camptothecin (63), in glioma cells by nimustine and topotecan and in malignant melanoma cells by fotemustine (62). Knockdown of TREX1 gave rise to sensitization of cells to UV light and alkylating agents (62). Therefore, it is reasonable to suppose that induction of TREX1 is part of the cellular defense against genotoxins.
Coordinated induction of repair genes?
As can be seen from Table 1, the genotoxic stress-inducible transcription factors AP-1 and p53 appear to be the most important players involved in the transcriptional activation of DNA repair genes. According to available data, AP-1 appears to regulate 9 of 25 DNA repair genes (listed in Table 1) (apex1, ercc1, mgmt, msh2, neil1, pcna, trex1, xpf and xpc) and p53 regulates 10 of 25 DNA repair genes (ddb2, fen1, karp1, mgmt, mlh1, msh2, pcna, pms2, polh and xpc). As genotoxic stress induces both AP-1 and p53, multiple DNA repair genes may be upregulated by the same type of DNA damage that finally ameliorates the DNA repair capacity of a cell. This is most obvious in the case of UV light, which predominantly induces genes involved in NER. Thus, following UV exposure six members of NER are induced: ddb2 and xpc via p53, ercc1, xpf and xpg via AP-1, and ligI for which the transcription factor is unknown. The combined activation of these genes leads to enhanced DNA repair, reduced cytotoxicity and, following repeated treatments, provokes an adaptive response at the cellular level (71).
Most studies focused on a single gene and, therefore, it is not clear whether all repair genes or a subgroup of them can be upregulated at the same time following genotoxic stress. As a matter of fact, simultaneous induction of all p53/AP-1 regulated repair genes has not been described in the literature. In array studies, we observed upregulation of individual genes, but not all of them which are listed in Table 1 at the same time (unpublished data). There could be several reasons: (i) Cancer cell lines are widely used in these studies. Most cancer lines harbour alterations in the p53 and/or MAPK/AP-1 pathway, which may abrogate the induction of subsets of DNA repair genes. (ii) Depending on the genotoxic insult, activation of p53 and AP-1 occurs at different time points. For example, ionizing radiation directly induces DSB and immediately activates DDR, but most chemical agents induce lesions that must be converted into secondary lesions (e.g. AP sites, DSB) to activate the DDR. (iii) Although activation of p53 is mainly dependent on phosphorylation, activation of AP-1 requires synthesis of transcription factors like c-Fos, FosB, c-Jun, JunB, Fra1-2 or ATF1, which involves complex pathways. It is conceivable that these pathways are activated only by some genotoxins. (iv) Most genotoxins inhibit transcription, notably at high dose levels. At low doses, the DNA damage level might not be high enough to activate DDR, and at high doses, transcriptional inhibition provoked by DNA adducts nullifies the effect of gene activation. Therefore, DNA repair gene induction can only be seen at moderate genotoxin dose levels. Given the time course of activation of repair genes (immediate early versus late response genes), it is obvious that extensive background experiments as to dose and exposure time are required to uncover repair gene induction following genotoxic exposures.
Epigenetic changes, Sp1 and HIF-1
Besides promoter activation by transcription factors, an alternative process to achieve upregulation of DNA repair genes rests on de-repression of a silenced promoter. Silencing may occur by promoter hypermethylation or by proteins acting as a repressor. An example for epigenetic silencing of repair genes is given by MGMT (126). Silencing of MGMT appears to be a rather stable trait (160), and reactivation of the hypermethylated MGMT promoter has not been achieved after single treatment with a DNA damaging agent. Therefore, it is reasonable to conclude that epigenetic silencing prevents genotoxic stress-induced repair gene induction. MGMT, once activated, is also an example of strong basal regulation. Key factor is Sp1, which can be sequestered by p53, thus reducing MGMT basal expression (135). This pertains to other Sp1-regulated repair genes as well. Thus, the RECQ4 helicase is negatively regulated by p53, which relates to a lower amount of Sp1 that binds to the promoter (161). Other DNA repair genes regulated by Sp1 are MSH2 (162), MSH6 (163), EXO1 (164), APE1 (165), XPB (166), DDB1 and DDB2 (167).
Repression of DNA repair genes can also result from stress that impacts the cellular ROS level. As an example, MLH1 is transcriptionally repressed by hypoxia, which regulates HIF-1 (168,169), whereas ABH5, CSB and XPA are positively regulated by HIF-1 (170–172). Striking examples of an interaction between Sp1 and HIF-1 are given by MMR and NER. In case of MMR, the MSH2 promoter is targeted by Sp1, which functions as a molecular switch. Under normoxic conditions, Sp1 recruits c-Myc, which acts as transcriptional stimulator, whereas under hypoxia, c-Myc is replaced by HIF-1, which acts as a repressor of MSH2 (162). For NER, it was shown that under normal conditions, non-phosphorylated HIF-1 binds to the hypoxia response element in the promoter of XPC. As this site overlaps with the SP1-binding site, binding of Sp1 and thereby expression of XPC is reduced. On UV-B exposure, HIF-1 becomes degraded and SP1 can again bind to the promoter, which leads to an increase of XPC expression (173). HIF-1 can also become phosphorylated by MAP kinase (174), which further enhances XPC expression.
Repair gene induction contributes to homeostatic repair regulation in genotoxin stressed cells
Compared with bacteria, in which the induction of DNA repair genes such as ada is ∼1000-fold following MNNG exposure (3), the induction of DNA repair genes in mammalian cells is rather low (e.g. for MGMT up to 15-fold in rat liver, which is a comparatively high level; in rat hepatoma cells in vitro 2–4-fold) (33). This, however, does not necessarily mean that the biological significance is negligible. One should keep in mind that mammalian cells express DNA repair genes at detectable basal level and that even slight upregulation may significantly ameliorate repair capacity. Thus, a 2-fold increase in the basal expression of MGMT in a human hepatocyte, which already expresses 100 000 molecules, will significantly increase the capacity of the liver for removing mutagenic and toxic O6-alkylguanine adducts from DNA.
It should also be recalled that genotoxins induce a broad spectrum of lesions, many of which block transcription. Therefore, an adverse side effect of genotoxin exposure is downregulation of DNA repair itself. To counteract genotoxin-triggered transcriptional inhibition, cells are equipped with maintenance functions. An example is provided by UV light and bulky DNA damage-generating agents that induce c-Fos as immediate-early and late response (71) following treatment. This induction of c-Fos/AP-1 is important for cell cycle and repair regulation following DNA damage, as shown in experiments with c-Fos-lacking cells. c-fos knockout and knockdown cells are hypersensitive to UV light and chemical genotoxins. Cells show a higher than normal DNA adduct level and enhanced replication and transcription blockage (70,175,176). Repair of DNA adducts is slow in c-Fos-lacking cells because of a decline in short-lived mRNAs of genes encoding NER proteins, notably XPF and XPG (Figure 5A). These mRNAs are declining due to the UV-induced block to transcription. In normal cells, c-Fos is induced immediately after UV irradiation, which triggers xpf and xpg resynthesis and stimulates NER (70,71). Therefore, the genotoxin-triggered c-Fos response counteracts genotoxin-induced downregulation of these NER genes (Figure 5A). Of note, XPF and XPG triggered by c-Fos/AP-1 are only slightly upregulated above the basal level after UV treatment (∼2-fold). Nevertheless, the stimulation of the xpf and xpg promoter following genotoxic stress is important, as it maintains the NER capacity of the cell following genotoxic stress, resulting in better removal of CPD lesions and survival. This type of induction might be designated as homeostatic or maintenance regulation (Figure 5B). It might be generally important for repair proteins whose expression above a critical level is deleterious, which seems to be the case for endonucleases like XPF and XPG, and DNA glycosylases like MPG (177).
Figure 5.
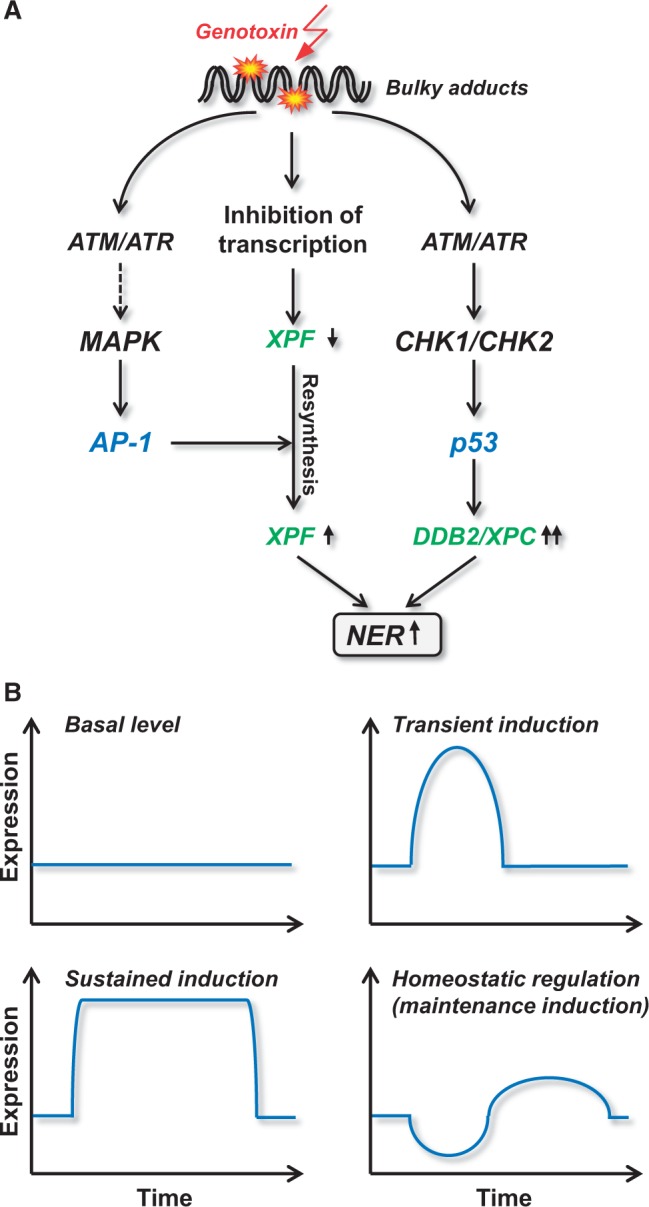
Genotoxin-triggered transcriptional repair gene regulation. (A) Pathways of upregulation of DDB2, XPC and XPF. (B) Different modes of repair gene regulation; for explanation, see text.
Studies with c-Fos deficient cells also provided insight into the interplay between AP-1, repair gene induction and cell death pathways. In c-Fos-lacking cells, impaired NER gives rise to sustained transcription blockage, which results in a decline in the expression of genes like MAPK-phosphatase 1 (MKP-1) (178,179). MPK-1 is responsible for dephosphorylation of c-Jun, which, similar to c-Fos, becomes activated following genotoxic stress. It contributes to the regulation of a subset of genes regulated by AP-1 containing c-Jun (c-Jun homo- or heterodimers). Reduced expression of MKP-1 leads to a sustained activation of JNK and thereby to a higher and sustained c-Jun/AP-1 activity level. c-Jun/AP-1 in turn upregulates the Fas ligand, which initiates apoptosis by activation of the Fas receptor pathway (179). This was taken to explain why cells impaired in homeostatic repair gene regulation are vulnerable and destined to die following genotoxic stress.
Transient versus sustained upregulation of repair genes
As outlined in Figure 5B, DNA repair genes may be expressed at basal (non-inducible) level or display short and transient induction, maintenance induction (hardly exceeding the control level) or sustained upregulation. An example for sustained upregulation is provided by DDB2 and XPC, which are upregulated in melanoma cells following a single dose of the crosslink inducing anticancer drug fotemustine for up to 1 week. This long-lasting upregulation is triggered by p53, which is stabilized during this long post-exposure period (20). The mechanism behind is not yet clear. It is conceivable that some DNA adducts are highly persistent and signal long-term DDR. This model is supported by the finding that cisplatin triggers sustained upregulation of MAP kinase and c-Jun (180), which upregulates both DNA repair and cell death functions. In addition, sustained upregulation of MAP kinases and JNK activation was also observed upon UV-exposure in c-Fos-deficient cells that showed abrogation of NER and consequently persisting DNA damage (179). The decision between transient versus sustained regulation of DNA repair genes appears to be cell-type specific. Thus, c-Fos activation in normal mouse fibroblasts following UV-exposure is part of an immediate early response, whereas its induction in human fibroblasts occurs in a biphasic manner (181), and only the late and sustained expression of c-Fos mRNA is translated into XPF and XPG protein accumulation (71).
Induction of DNA repair genes and adaptive response
The definition of adaptive response, in the narrow sense, rests on the finding that exposure of cells to a low priming/conditioning dose of a genotoxin leads to their enhanced protection against a subsequent higher (challenge) dose of the same genotoxin. In a broader sense, the genotoxic adaptive response is characterized by protection of pretreated cells against a wide range of genotoxicants. The adaptive response was first observed in E. coli that were continuously grown in medium containing the methylating agent MNNG. Following exposure, the mutation and killing frequency declined, which was shown to be a result of induction of repair genes that are controlled by Ada (3,4).
Genotoxin-induced adaptive responses resulting from upregulation of DNA repair functions have also been observed in mammalian cells. MGMT upregulation following methylation in rat hepatoma H4IIE cells caused reduction in HPRT mutation frequency induced by a challenge dose of MNNG (32). As aforementioned, an intriguing question that still awaits resolution is whether MGMT is subject to upregulation in humans exposed to genotoxic stress, causing adaptation to O6-alkylating agents. For tobacco smoke and MGMT, the issue has been discussed recently; the data are controversial (127). During chemotherapy of gliomas, MGMT levels were found to increase stepwise during therapy (182,183), which likely renders the tumour resistant to O6-alkylating drugs (184,185). It is unclear whether this increase is a consequence of upregulation of MGMT on gene or post-transcriptional level [e.g. by miRNA regulation (186)], or merely a therapy-related selection of preexisting tumour cells expressing high MGMT.
Besides MGMT, the most intensively studied example of adaptation is the radiation-induced adaptive response (187) where exposure to low doses of ionizing radiation result in a decreased amount of chromosomal aberrations induced by a subsequent challenge dose. This was shown for human lymphocytes (188,189), lymphoblastoid cells (190) and hepatoma cell lines (191) in a mechanism requiring protein de novo synthesis (192). It was also shown that the radiation-induced adaptive response involves the induction of repair mechanisms (193). Thus, low-ionizing radiation doses stimulate the repair of chromosomal breaks in human skin fibroblasts (194). More recently, it was reported that ionizing radiation doses >100 mGy increase the NHEJ efficiency in mouse embryonic fibroblasts and human cancer cells (195). As outlined earlier in the text, several DNA repair genes have been reported to be upregulated following ionizing radiation (Table 1); however, convincing evidence that this results in killing or mutagenic adaptation is still lacking.
An adaptive response was also observed following treatment with bleomycin, mitomycin C (196) and ENU (197). Whether this rests on upregulation of DNA repair has not been shown convincingly. For UV-C light, an adaptive response has been demonstrated in human fibroblasts that resulted from amelioration of NER (198). Recent data showed that increase in NER activity following UV-C is the result of transcriptional upregulation of the NER genes XPF and XPG, which was triggered by the MAPK/AP-1 pathway and resulted in protection from cell death of a subsequently applied UV-C dose (71) (Figure 5A). Likewise, sustained upregulation of DDB2/XPC (triggered by p53) in malignant melanoma cells resulted in a lower killing response of a subsequent dose of fotemustine (20). Whether upregulation of XPF/XPG and DDB2/XPC is a general response provoked by genotoxins, which causes adaptation and occurs in human tissues in vivo is a challenging question that needs to be answered in the future.
CONCLUSIONS
DNA is so precious and fragile and under constant threat by genotoxins that it requires continuous proof-reading and repair. Most of the DNA repair mechanisms are constitutively expressed. Some of them, however, are subject to upregulation following genotoxic stress, which was shown for experimental mutagens, environmental carcinogens and anticancer drugs. A couple of DNA repair genes were reported to be inducible on mutagen treatment, but only for few of them the data are solid, reproducible and pertain to both rodent and human cell systems. Some genes are induced above the normal level (2–15-fold in mammalian systems), others are transcriptionally stimulated to compensate transcription inhibition and to maintain the basal expression level, which is in balance with other repair factors in complex repair pathways (here defined as maintenance or homeostatic regulation). For many repair genes, data are conflicting. The only DNA repair mechanism for which robust data for human cells exist is NER, where induction of ddb2, xpc, xpf and xpg gives rise to an increase in NER activity and adaptive response. Transcriptional activation of the NER system is of high biological relevance, as most environmental and man-made carcinogens induce DNA damage that is repaired by NER. For MGMT, data in rodent cells are robust while induction and adaptive response in human cells still awaits conclusive proof. The highly important question of an adaptive response in cancer prevention clearly warrants more intensive exploration. The same is true for upregulation of DNA repair genes during cancer therapy. It is conceivable that chronic or even single dose treatment of patients upregulates NER in cancer cells, which thereby acquire drug resistance as recently shown with glioma and malignant melanoma cells in vitro. Induced DNA repair is a key response following environmental and man-made carcinogens. It is reasonable to posit that upregulation of DNA repair by natural compounds and lifestyle drugs may enhance the cellular repair capacity and thus ameliorate protection against carcinogenic exposures through ionizing radiation, sun light and chemical genotoxicants. This highly important issue needs more intense exploration.
ACKNOWLEDGEMENTS
The authors are grateful to M.T. Tomicic for suggestions and A.D. Thomas and C. Strauch for critical reading the manuscript.
FUNDING
Deutsche Forschungsgemeinschaft [CH665-2 and KA725]; and the German Cancer Foundation. Funding for open access charge: University Medical Center, Mainz.
Conflict of interest statement. None declared.
REFERENCES
- 1.Christmann M, Tomicic MT, Roos WP, Kaina B. Mechanisms of human DNA repair: an update. Toxicology. 2003;193:3–34. doi: 10.1016/s0300-483x(03)00287-7. [DOI] [PubMed] [Google Scholar]
- 2.Michel B. After 30 years of study, the bacterial SOS response still surprises us. PLoS Biol. 2005;3:e255. doi: 10.1371/journal.pbio.0030255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Samson L, Cairns J. A new pathway for DNA repair in Escherichia coli. Nature. 1977;267:281–283. doi: 10.1038/267281a0. [DOI] [PubMed] [Google Scholar]
- 4.Jeggo P, Defais TM, Samson L, Schendel P. An adaptive response of E. coli to low levels of alkylating agent: comparison with previously characterised DNA repair pathways. Mol. Gen. Genet. 1977;157:1–9. doi: 10.1007/BF00268680. [DOI] [PubMed] [Google Scholar]
- 5.Mitra S, Pal BC, Foote RS. O6-methylguanine-DNA methyltransferase in wild-type and ada mutants of Escherichia coli. J. Bacteriol. 1982;152:534–537. doi: 10.1128/jb.152.1.534-537.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sedgwick B. Molecular cloning of a gene which regulates the adaptive response to alkylating agents in Escherichia coli. Mol. Gen. Genet. 1983;191:466–472. doi: 10.1007/BF00425764. [DOI] [PubMed] [Google Scholar]
- 7.Teo I, Sedgwick B, Demple B, Li B, Lindahl T. Induction of resistance to alkylating agents in E. coli: the ada+ gene product serves both as a regulatory protein and as an enzyme for repair of mutagenic damage. EMBO J. 1984;3:2151–2157. doi: 10.1002/j.1460-2075.1984.tb02105.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grosch S, Fritz G, Kaina B. Apurinic endonuclease (Ref-1) is induced in mammalian cells by oxidative stress and involved in clastogenic adaptation. Cancer Res. 1998;58:4410–4416. [PubMed] [Google Scholar]
- 9.Ramana CV, Boldogh I, Izumi T, Mitra S. Activation of apurinic/apyrimidinic endonuclease in human cells by reactive oxygen species and its correlation with their adaptive response to genotoxicity of free radicals. Proc. Natl Acad. Sci. USA. 1998;95:5061–5066. doi: 10.1073/pnas.95.9.5061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grosch S, Kaina B. Transcriptional activation of apurinic/apyrimidinic endonuclease (Ape, Ref-1) by oxidative stress requires CREB. Biochem. Biophys. Res. Commun. 1999;261:859–863. doi: 10.1006/bbrc.1999.1125. [DOI] [PubMed] [Google Scholar]
- 11.Roy D, Guida P, Zhou G, Echiburu-Chau C, Calaf GM. Gene expression profiling of breast cells induced by X-rays and heavy ions. Int. J. Mol. Med. 2008;21:627–636. doi: 10.3892/ijmm.21.5.627. [DOI] [PubMed] [Google Scholar]
- 12.Hwang BJ, Ford JM, Hanawalt PC, Chu G. Expression of the p48 xeroderma pigmentosum gene is p53-dependent and is involved in global genomic repair. Proc. Natl Acad. Sci. USA. 1999;96:424–428. doi: 10.1073/pnas.96.2.424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tan T, Chu G. p53 Binds and activates the xeroderma pigmentosum DDB2 gene in humans but not mice. Mol. Cell. Biol. 2002;22:3247–3254. doi: 10.1128/MCB.22.10.3247-3254.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu J, Lin M, Zhang C, Wang D, Feng Z, Hu W. TAp63gamma enhances nucleotide excision repair through transcriptional regulation of DNA repair genes. DNA Repair. 2012;11:167–176. doi: 10.1016/j.dnarep.2011.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hartman AR, Ford JM. BRCA1 induces DNA damage recognition factors and enhances nucleotide excision repair. Nat. Genet. 2002;32:180–184. doi: 10.1038/ng953. [DOI] [PubMed] [Google Scholar]
- 16.Takimoto R, MacLachlan TK, Dicker DT, Niitsu Y, Mori T, el-Deiry WS. BRCA1 transcriptionally regulates damaged DNA binding protein (DDB2) in the DNA repair response following UV-irradiation. Cancer Biol. Ther. 2002;1:177–186. doi: 10.4161/cbt.65. [DOI] [PubMed] [Google Scholar]
- 17.Hockley SL, Arlt VM, Brewer D, Te Poele R, Workman P, Giddings I, Phillips DH. AHR- and DNA-damage-mediated gene expression responses induced by Benzo(a)pyrene in human cell lines. Chem. Res. Toxicol. 2007;20:1797–1810. doi: 10.1021/tx700252n. [DOI] [PubMed] [Google Scholar]
- 18.Hockley SL, Arlt VM, Jahnke G, Hartwig A, Giddings I, Phillips DH. Identification through microarray gene expression analysis of cellular responses to benzo(a)pyrene and its diol-epoxide that are dependent or independent of p53. Carcinogenesis. 2007;29:202–210. doi: 10.1093/carcin/bgm227. [DOI] [PubMed] [Google Scholar]
- 19.Batista LF, Roos WP, Christmann M, Menck CF, Kaina B. Differential sensitivity of malignant glioma cells to methylating and chloroethylating anticancer drugs: p53 determines the switch by regulating xpc, ddb2, and DNA double-strand breaks. Cancer Res. 2007;67:11886–11895. doi: 10.1158/0008-5472.CAN-07-2964. [DOI] [PubMed] [Google Scholar]
- 20.Barckhausen C, Roos WP, Naumann SC, Kaina B. Malignant melanoma cells acquire resistance to DNA interstrand cross-linking chemotherapeutics by p53-triggered upregulation of DDB2/XPC-mediated DNA repair. Oncogene. 2013 doi: 10.1038/onc.2013.141. April 22 (doi:10.1038/onc.2013.141; epub ahead of print) [DOI] [PubMed] [Google Scholar]
- 21.Yacoub A, Park JS, Qiao L, Dent P, Hagan MP. MAPK dependence of DNA damage repair: ionizing radiation and the induction of expression of the DNA repair genes XRCC1 and ERCC1 in DU145 human prostate carcinoma cells in a MEK1/2 dependent fashion. Int. J. Radiat. Biol. 2001;77:1067–1078. doi: 10.1080/09553000110069317. [DOI] [PubMed] [Google Scholar]
- 22.Yacoub A, McKinstry R, Hinman D, Chung T, Dent P, Hagan MP. Epidermal growth factor and ionizing radiation up-regulate the DNA repair genes XRCC1 and ERCC1 in DU145 and LNCaP prostate carcinoma through MAPK signaling. Radiat Res. 2003;159:439–452. doi: 10.1667/0033-7587(2003)159[0439:egfair]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 23.Liu J, Kadiiska MB, Liu Y, Lu T, Qu W, Waalkes MP. Stress-related gene expression in mice treated with inorganic arsenicals. Toxicol Sci. 2001;61:314–320. doi: 10.1093/toxsci/61.2.314. [DOI] [PubMed] [Google Scholar]
- 24.Youn CK, Kim MH, Cho HJ, Kim HB, Chang IY, Chung MH, You HJ. Oncogenic H-Ras up-regulates expression of ERCC1 to protect cells from platinum-based anticancer agents. Cancer Res. 2004;64:4849–4857. doi: 10.1158/0008-5472.CAN-04-0348. [DOI] [PubMed] [Google Scholar]
- 25.Li Q, Gardner K, Zhang L, Tsang B, Bostick-Bruton F, Reed E. Cisplatin induction of ERCC-1 mRNA expression in A2780/CP70 human ovarian cancer cells. J. Biol. Chem. 1998;273:23419–23425. doi: 10.1074/jbc.273.36.23419. [DOI] [PubMed] [Google Scholar]
- 26.Li Q, Zhang L, Tsang B, Gardner K, Bostick-Bruton F, Reed E. Phorbol ester exposure activates an AP-1-mediated increase in ERCC-1 messenger RNA expression in human ovarian tumor cells. Cell Mol. Life Sci. 1999;55:456–466. doi: 10.1007/s000180050302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Christmann M, Tomicic MT, Origer J, Kaina B. Fen1 is induced p53 dependently and involved in the recovery from UV-light-induced replication inhibition. Oncogene. 2005;24:8304–8313. doi: 10.1038/sj.onc.1208994. [DOI] [PubMed] [Google Scholar]
- 28.Islaih M, Li B, Kadura IA, Reid-Hubbard JL, Deahl JT, Altizer JL, Watson DE, Newton RK. Comparison of gene expression changes induced in mouse and human cells treated with direct-acting mutagens. Environ. Mol. Mutagen. 2004;44:401–419. doi: 10.1002/em.20065. [DOI] [PubMed] [Google Scholar]
- 29.Myung K, Braastad C, He DM, Hendrickson EA. KARP-1 is induced by DNA damage in a p53- and ataxia telangiectasia mutated-dependent fashion. Proc. Natl Acad. Sci. USA. 1998;95:7664–7669. doi: 10.1073/pnas.95.13.7664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Montecucco A, Savini E, Biamonti G, Stefanini M, Focher F, Ciarrocchi G. Late induction of human DNA ligase I after UV-C irradiation. Nucleic Acids Res. 1995;23:962–966. doi: 10.1093/nar/23.6.962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grombacher T, Kaina B. Constitutive expression and inducibility of O6-methylguanine-DNA methyltransferase and N-methylpurine-DNA glycosylase in rat liver cells exhibiting different status of differentiation. Biochim. Biophys. Acta. 1995;1270:63–72. doi: 10.1016/0925-4439(94)00073-y. [DOI] [PubMed] [Google Scholar]
- 32.Fritz G, Tano K, Mitra S, Kaina B. Inducibility of the DNA repair gene encoding O6-methylguanine-DNA methyltransferase in mammalian cells by DNA-damaging treatments. Mol. Cell. Biol. 1991;11:4660–4668. doi: 10.1128/mcb.11.9.4660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chan CL, Wu Z, Eastman A, Bresnick E. Irradiation-induced expression of O6-methylguanine-DNA methyltransferase in mammalian cells. Cancer Res. 1992;52:1804–1809. [PubMed] [Google Scholar]
- 34.Rafferty JA, Clarke AR, Sellappan D, Koref MS, Frayling IM, Margison GP. Induction of murine O6-alkylguanine-DNA-alkyltransferase in response to ionising radiation is p53 gene dose dependent. Oncogene. 1996;12:693–697. [PubMed] [Google Scholar]
- 35.Grombacher T, Eichhorn U, Kaina B. p53 is involved in regulation of the DNA repair gene O6-methylguanine-DNA methyltransferase (MGMT) by DNA damaging agents. Oncogene. 1998;17:845–851. doi: 10.1038/sj.onc.1202000. [DOI] [PubMed] [Google Scholar]
- 36.Boldogh I, Ramana CV, Chen Z, Biswas T, Hazra TK, Grosch S, Grombacher T, Mitra S, Kaina B. Regulation of expression of the DNA repair gene O6-methylguanine-DNA methyltransferase via protein kinase C-mediated signaling. Cancer Res. 1998;58:3950–3956. [PubMed] [Google Scholar]
- 37.Lavon I, Fuchs D, Zrihan D, Efroni G, Zelikovitch B, Fellig Y, Siegal T. Novel mechanism whereby nuclear factor kappaB mediates DNA damage repair through regulation of O(6)-methylguanine-DNA-methyltransferase. Cancer Res. 2007;67:8952–8959. doi: 10.1158/0008-5472.CAN-06-3820. [DOI] [PubMed] [Google Scholar]
- 38.Grombacher T, Kaina B. Isolation and analysis of inducibility of the rat N-methylpurine-DNA glycosylase promoter. DNA Cell Biol. 1996;15:581–588. doi: 10.1089/dna.1996.15.581. [DOI] [PubMed] [Google Scholar]
- 39.Zair ZM, Jenkins GJ, Doak SH, Singh R, Brown K, Johnson GE. N-methylpurine DNA glycosylase plays a pivotal role in the threshold response of ethyl methanesulfonate-induced chromosome damage. Toxicol Sci. 2011;119:346–358. doi: 10.1093/toxsci/kfq341. [DOI] [PubMed] [Google Scholar]
- 40.Chen J, Sadowski I. Identification of the mismatch repair genes PMS2 and MLH1 as p53 target genes by using serial analysis of binding elements. Proc. Natl Acad. Sci. USA. 2005;102:4813–4818. doi: 10.1073/pnas.0407069102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Scherer SJ, Maier SM, Seifert M, Hanselmann RG, Zang KD, Muller-Hermelink HK, Angel P, Welter C, Schartl M. p53 and c-Jun functionally synergize in the regulation of the DNA repair gene hMSH2 in response to UV. J. Biol. Chem. 2000;275:37469–37473. doi: 10.1074/jbc.M006990200. [DOI] [PubMed] [Google Scholar]
- 42.Humbert O, Hermine T, Hernandez H, Bouget T, Selves J, Laurent G, Salles B, Lautier D. Implication of protein kinase C in the regulation of DNA mismatch repair protein expression and function. J. Biol. Chem. 2002;277:18061–18068. doi: 10.1074/jbc.M103451200. [DOI] [PubMed] [Google Scholar]
- 43.Das A, Hazra TK, Boldogh I, Mitra S, Bhakat KK. Induction of the human oxidized base-specific DNA glycosylase NEIL1 by reactive oxygen species. J. Biol. Chem. 2005;280:35272–35280. doi: 10.1074/jbc.M505526200. [DOI] [PubMed] [Google Scholar]
- 44.Lee MR, Kim SH, Cho HJ, Lee KY, Moon AR, Jeong HG, Lee JS, Hyun JW, Chung MH, You HJ. Transcription factors NF-YA regulate the induction of human OGG1 following DNA-alkylating agent methylmethane sulfonate (MMS) treatment. J. Biol. Chem. 2004;279:9857–9866. doi: 10.1074/jbc.M311132200. [DOI] [PubMed] [Google Scholar]
- 45.Risom L, Moller P, Vogel U, Kristjansen PE, Loft S. X-ray-induced oxidative stress: DNA damage and gene expression of HO-1, ERCC1 and OGG1 in mouse lung. Free Radic. Res. 2003;37:957–966. doi: 10.1080/1071576031000150788. [DOI] [PubMed] [Google Scholar]
- 46.Shan B, Morris GF. Binding sequence-dependent regulation of the human proliferating cell nuclear antigen promoter by p53. Exp. Cell. Res. 2005;305:10–22. doi: 10.1016/j.yexcr.2004.09.033. [DOI] [PubMed] [Google Scholar]
- 47.Shan B, Xu J, Zhuo Y, Morris CA, Morris GF. Induction of p53-dependent activation of the human proliferating cell nuclear antigen gene in chromatin by ionizing radiation. J. Biol. Chem. 2003;278:44009–44017. doi: 10.1074/jbc.M302671200. [DOI] [PubMed] [Google Scholar]
- 48.Shivakumar CV, Brown DR, Deb S, Deb SP. Wild-type human p53 transactivates the human proliferating cell nuclear antigen promoter. Mol. Cell. Biol. 1995;15:6785–6793. doi: 10.1128/mcb.15.12.6785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chang YC, Chang HW, Liao CB, Liu YC. The roles of p53, DNA repair, and oxidative stress in ultraviolet C induction of proliferating cell nuclear antigen expression. Ann. NY Acad. Sci. 2002;973:384–391. doi: 10.1111/j.1749-6632.2002.tb04670.x. [DOI] [PubMed] [Google Scholar]
- 50.Yang J, Chen Z, Liu Y, Hickey RJ, Malkas LH. Altered DNA polymerase iota expression in breast cancer cells leads to a reduction in DNA replication fidelity and a higher rate of mutagenesis. Cancer Res. 2004;64:5597–5607. doi: 10.1158/0008-5472.CAN-04-0603. [DOI] [PubMed] [Google Scholar]
- 51.Zhu H, Fan Y, Jiang H, Shen J, Qi H, Mei R, Shao J. Response of human DNA polymerase iota promoter to N-methyl-N'-nitro-N-nitrosoguanidine. Environ. Toxicol. Pharmacol. 2010;29:79–86. doi: 10.1016/j.etap.2009.11.001. [DOI] [PubMed] [Google Scholar]
- 52.Fornace AJ, Jr, Zmudzka B, Hollander MC, Wilson SH. Induction of beta-polymerase mRNA by DNA-damaging agents in Chinese hamster ovary cells. Mol. Cell. Biol. 1989;9:851–853. doi: 10.1128/mcb.9.2.851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kedar PS, Widen SG, Englander EW, Fornace AJ, Jr, Wilson SH. The ATF/CREB transcription factor-binding site in the polymerase beta promoter mediates the positive effect of N-methyl-N'-nitro-N-nitrosoguanidine on transcription. Proc. Natl Acad. Sci. USA. 1991;88:3729–3733. doi: 10.1073/pnas.88.9.3729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.He F, Yang XP, Srivastava DK, Wilson SH. DNA polymerase beta gene expression: the promoter activator CREB-1 is upregulated in Chinese hamster ovary cells by DNA alkylating agent-induced stress. Biol. Chem. 2003;384:19–23. doi: 10.1515/BC.2003.003. [DOI] [PubMed] [Google Scholar]
- 55.Zhao Y, Zheng J, Ling Y, Hou L, Zhang B. Transcriptional upregulation of DNA polymerase beta by TEIF. Biochem. Biophys. Res. Commun. 2005;333:908–916. doi: 10.1016/j.bbrc.2005.05.172. [DOI] [PubMed] [Google Scholar]
- 56.Faumont N, Le Clorennec C, Teira P, Goormachtigh G, Coll J, Canitrot Y, Cazaux C, Hoffmann JS, Brousset P, Delsol G, et al. Regulation of DNA polymerase beta by the LMP1 oncoprotein of EBV through the nuclear factor-kappaB pathway. Cancer Res. 2009;69:5177–5185. doi: 10.1158/0008-5472.CAN-08-2866. [DOI] [PubMed] [Google Scholar]
- 57.Liu G, Chen X. DNA polymerase eta, the product of the xeroderma pigmentosum variant gene and a target of p53, modulates the DNA damage checkpoint and p53 activation. Mol. Cell. Biol. 2006;26:1398–1413. doi: 10.1128/MCB.26.4.1398-1413.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhu H, Fan Y, Shen J, Qi H, Shao J. Characterization of human DNA polymerase kappa promoter in response to benzo[a]pyrene diol epoxide. Environ. Toxicol. Pharmacol. 2012;33:205–211. doi: 10.1016/j.etap.2011.12.002. [DOI] [PubMed] [Google Scholar]
- 59.Yu Y, Yang J, Zhu F, Xu F. Response of REV3 promoter to N-methyl-N'-nitro-N-nitrosoguanidine. Mutat. Res. 2004;550:49–58. doi: 10.1016/j.mrfmmm.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 60.Zhu F, Yang J, Xu F, Yu YN. Cloning and bioinformatics of human REV3 gene promoter region and its response to carcinogen N-methyl-N'-nitro-N-nitrosoguanidine. Zhejiang Da Xue Xue Bao. Yi Xue Ban. 2003;32:393–397. doi: 10.3785/j.issn.1008-9292.2003.05.006. [DOI] [PubMed] [Google Scholar]
- 61.Christmann M, Tomicic MT, Aasland D, Berdelle N, Kaina B. Three prime exonuclease I (TREX1) is Fos/AP-1 regulated by genotoxic stress and protects against ultraviolet light and benzo(a)pyrene-induced DNA damage. Nucleic Acids Res. 2010;38:6418–6432. doi: 10.1093/nar/gkq455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tomicic MT, Aasland D, Nikolova T, Kaina B, Christmann M. Human three prime exonuclease TREX1 is induced by genotoxic stress and involved in protection of glioma and melanoma cells to anticancer drugs. Biochim. Biophys. Acta. 2013;1833:1832–1843. doi: 10.1016/j.bbamcr.2013.03.029. [DOI] [PubMed] [Google Scholar]
- 63.Wang CJ, Lam W, Bussom S, Chang HM, Cheng YC. TREX1 acts in degrading damaged DNA from drug-treated tumor cells. DNA Repair. 2009;8:1179–1189. doi: 10.1016/j.dnarep.2009.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chen D, Yu Z, Zhu Z, Lopez CD. E2F1 regulates the base excision repair gene XRCC1 and promotes DNA repair. J. Biol. Chem. 2008;283:15381–15389. doi: 10.1074/jbc.M710296200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Adimoolam S, Ford JM. p53 and DNA damage-inducible expression of the xeroderma pigmentosum group C gene. Proc. Natl Acad. Sci. USA. 2002;99:12985–12990. doi: 10.1073/pnas.202485699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Long XH, Zhao ZQ, He XP, Wang HP, Xu QZ, An J, Bai B, Sui JL, Zhou PK. Dose-dependent expression changes of early response genes to ionizing radiation in human lymphoblastoid cells. Int. J. Mol. Med. 2007;19:607–615. [PubMed] [Google Scholar]
- 67.Amundson SA, Patterson A, Do KT, Fornace AJ., Jr A nucleotide excision repair master-switch: p53 regulated coordinate induction of global genomic repair genes. Cancer Biol. Ther. 2002;1:145–149. doi: 10.4161/cbt.59. [DOI] [PubMed] [Google Scholar]
- 68.Wang A, Gu J, Judson-Kremer K, Powell KL, Mistry H, Simhambhatla P, Aldaz CM, Gaddis S, MacLeod MC. Response of human mammary epithelial cells to DNA damage induced by BPDE: involvement of novel regulatory pathways. Carcinogenesis. 2003;24:225–234. doi: 10.1093/carcin/24.2.225. [DOI] [PubMed] [Google Scholar]
- 69.Mahadevan B, Keshava C, Musafia-Jeknic T, Pecaj A, Weston A, Baird WM. Altered gene expression patterns in MCF-7 cells induced by the urban dust particulate complex mixture standard reference material 1649a. Cancer Res. 2005;65:1251–1258. doi: 10.1158/0008-5472.CAN-04-2357. [DOI] [PubMed] [Google Scholar]
- 70.Christmann M, Tomicic MT, Origer J, Aasland D, Kaina B. c-Fos is required for excision repair of UV-light induced DNA lesions by triggering the re-synthesis of XPF. Nucleic Acids Res. 2006;34:6530–6539. doi: 10.1093/nar/gkl895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tomicic MT, Reischmann P, Rasenberger B, Meise R, Kaina B, Christmann M. Delayed c-Fos activation in human cells triggers XPF induction and an adaptive response to UVC-induced DNA damage and cytotoxicity. Cell. Mol. Life Sci. 2011;68:1785–1798. doi: 10.1007/s00018-010-0546-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yang J, Liu X, Niu P, Zou Y, Gong Z, Yuan J, Wu T. Dynamic changes of XPA, XPC, XPF, XPG and ERCC1 protein expression and their correlations with levels of DNA damage in human bronchial epithelia cells exposed to benzo[a]pyrene. Toxicol. Lett. 2007;174:10–17. doi: 10.1016/j.toxlet.2007.08.004. [DOI] [PubMed] [Google Scholar]
- 73.Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol. Cell. 2010;40:179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Harper JW, Elledge SJ. The DNA damage response: ten years after. Mol. Cell. 2007;28:739–745. doi: 10.1016/j.molcel.2007.11.015. [DOI] [PubMed] [Google Scholar]
- 75.Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421:499–506. doi: 10.1038/nature01368. [DOI] [PubMed] [Google Scholar]
- 76.Lee JH, Paull TT. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science. 2005;308:551–554. doi: 10.1126/science.1108297. [DOI] [PubMed] [Google Scholar]
- 77.Mirzoeva OK, Petrini JH. DNA damage-dependent nuclear dynamics of the Mre11 complex. Mol. Cell. Biol. 2001;21:281–288. doi: 10.1128/MCB.21.1.281-288.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mirzoeva OK, Petrini JH. DNA replication-dependent nuclear dynamics of the Mre11 complex. Mol. Cancer Res. 2003;1:207–218. [PubMed] [Google Scholar]
- 79.Uziel T, Lerenthal Y, Moyal L, Andegeko Y, Mittelman L, Shiloh Y. Requirement of the MRN complex for ATM activation by DNA damage. EMBO J. 2003;22:5612–5621. doi: 10.1093/emboj/cdg541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Byun TS, Pacek M, Yee MC, Walter JC, Cimprich KA. Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes Dev. 2005;19:1040–1052. doi: 10.1101/gad.1301205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Parrilla-Castellar ER, Arlander SJ, Karnitz L. Dial 9-1-1 for DNA damage: the Rad9-Hus1-Rad1 (9-1-1) clamp complex. DNA Repair. 2004;3:1009–1014. doi: 10.1016/j.dnarep.2004.03.032. [DOI] [PubMed] [Google Scholar]
- 82.Makiniemi M, Hillukkala T, Tuusa J, Reini K, Vaara M, Huang D, Pospiech H, Majuri I, Westerling T, Makela TP, et al. BRCT domain-containing protein TopBP1 functions in DNA replication and damage response. J. Biol. Chem. 2001;276:30399–30406. doi: 10.1074/jbc.M102245200. [DOI] [PubMed] [Google Scholar]
- 83.Delacroix S, Wagner JM, Kobayashi M, Yamamoto K, Karnitz LM. The Rad9-Hus1-Rad1 (9-1-1) clamp activates checkpoint signaling via TopBP1. Genes Dev. 2007;21:1472–1477. doi: 10.1101/gad.1547007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lee J, Kumagai A, Dunphy WG. The Rad9-Hus1-Rad1 checkpoint clamp regulates interaction of TopBP1 with ATR. J. Biol. Chem. 2007;282:28036–28044. doi: 10.1074/jbc.M704635200. [DOI] [PubMed] [Google Scholar]
- 85.Kumagai A, Lee J, Yoo HY, Dunphy WG. TopBP1 activates the ATR-ATRIP complex. Cell. 2006;124:943–955. doi: 10.1016/j.cell.2005.12.041. [DOI] [PubMed] [Google Scholar]
- 86.Liu S, Shiotani B, Lahiri M, Marechal A, Tse A, Leung CC, Glover JN, Yang XH, Zou L. ATR autophosphorylation as a molecular switch for checkpoint activation. Mol. Cell. 2011;43:192–202. doi: 10.1016/j.molcel.2011.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Perkins ND. Post-translational modifications regulating the activity and function of the nuclear factor kappa B pathway. Oncogene. 2006;25:6717–6730. doi: 10.1038/sj.onc.1209937. [DOI] [PubMed] [Google Scholar]
- 88.Perkins ND. Integrating cell-signalling pathways with NF-kappaB and IKK function. Nat. Rev. Mol. Cell Biol. 2007;8:49–62. doi: 10.1038/nrm2083. [DOI] [PubMed] [Google Scholar]
- 89.Habraken Y, Piette J. NF-kappaB activation by double-strand breaks. Biochem. Pharmacol. 2006;72:1132–1141. doi: 10.1016/j.bcp.2006.07.015. [DOI] [PubMed] [Google Scholar]
- 90.Huang TT, Kudo N, Yoshida M, Miyamoto S. A nuclear export signal in the N-terminal regulatory domain of IkappaBalpha controls cytoplasmic localization of inactive NF-kappaB/IkappaBalpha complexes. Proc. Natl Acad. Sci. USA. 2000;97:1014–1019. doi: 10.1073/pnas.97.3.1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Johnson C, Van Antwerp D, Hope TJ. An N-terminal nuclear export signal is required for the nucleocytoplasmic shuttling of IkappaBalpha. EMBO J. 1999;18:6682–6693. doi: 10.1093/emboj/18.23.6682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Stilmann M, Hinz M, Arslan SC, Zimmer A, Schreiber V, Scheidereit C. A nuclear poly(ADP-ribose)-dependent signalosome confers DNA damage-induced IkappaB kinase activation. Mol. Cell. 2009;36:365–378. doi: 10.1016/j.molcel.2009.09.032. [DOI] [PubMed] [Google Scholar]
- 93.Wu ZH, Shi Y, Tibbetts RS, Miyamoto S. Molecular linkage between the kinase ATM and NF-kappaB signaling in response to genotoxic stimuli. Science. 2006;311:1141–1146. doi: 10.1126/science.1121513. [DOI] [PubMed] [Google Scholar]
- 94.Krappmann D, Scheidereit C. A pervasive role of ubiquitin conjugation in activation and termination of IkappaB kinase pathways. EMBO Rep. 2005;6:321–326. doi: 10.1038/sj.embor.7400380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kato T, Jr, Delhase M, Hoffmann A, Karin M. CK2 is a C-terminal IkappaB kinase responsible for NF-kappaB activation during the UV response. Mol. Cell. 2003;12:829–839. doi: 10.1016/s1097-2765(03)00358-7. [DOI] [PubMed] [Google Scholar]
- 96.el-Deiry WS, Kern SE, Pietenpol JA, Kinzler KW, Vogelstein B. Definition of a consensus binding site for p53. Nat. Genet. 1992;1:45–49. doi: 10.1038/ng0492-45. [DOI] [PubMed] [Google Scholar]
- 97.Zhou BB, Chaturvedi P, Spring K, Scott SP, Johanson RA, Mishra R, Mattern MR, Winkler JD, Khanna KK. Caffeine abolishes the mammalian G(2)/M DNA damage checkpoint by inhibiting ataxia-telangiectasia-mutated kinase activity. J. Biol. Chem. 2000;275:10342–10348. doi: 10.1074/jbc.275.14.10342. [DOI] [PubMed] [Google Scholar]
- 98.Matsuoka S, Rotman G, Ogawa A, Shiloh Y, Tamai K, Elledge SJ. Ataxia telangiectasia-mutated phosphorylates Chk2 in vivo and in vitro. Proc. Natl Acad. Sci. USA. 2000;97:10389–10394. doi: 10.1073/pnas.190030497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Liu Q, Guntuku S, Cui XS, Matsuoka S, Cortez D, Tamai K, Luo G, Carattini-Rivera S, DeMayo F, Bradley A, et al. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev. 2000;14:1448–1459. [PMC free article] [PubMed] [Google Scholar]
- 100.Guo Z, Kumagai A, Wang SX, Dunphy WG. Requirement for Atr in phosphorylation of Chk1 and cell cycle regulation in response to DNA replication blocks and UV-damaged DNA in Xenopus egg extracts. Genes Dev. 2000;14:2745–2756. doi: 10.1101/gad.842500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Shieh SY, Ahn J, Tamai K, Taya Y, Prives C. The human homologs of checkpoint kinases Chk1 and Cds1 (Chk2) phosphorylate p53 at multiple DNA damage-inducible sites. Genes Dev. 2000;14:289–300. [PMC free article] [PubMed] [Google Scholar]
- 102.Banin S, Moyal L, Shieh S, Taya Y, Anderson CW, Chessa L, Smorodinsky NI, Prives C, Reiss Y, Shiloh Y, et al. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science. 1998;281:1674–1677. doi: 10.1126/science.281.5383.1674. [DOI] [PubMed] [Google Scholar]
- 103.Canman CE, Lim DS, Cimprich KA, Taya Y, Tamai K, Sakaguchi K, Appella E, Kastan MB, Siliciano JD. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science. 1998;281:1677–1679. doi: 10.1126/science.281.5383.1677. [DOI] [PubMed] [Google Scholar]
- 104.Khosravi R, Maya R, Gottlieb T, Oren M, Shiloh Y, Shkedy D. Rapid ATM-dependent phosphorylation of MDM2 precedes p53 accumulation in response to DNA damage. Proc. Natl Acad. Sci. USA. 1999;96:14973–14977. doi: 10.1073/pnas.96.26.14973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Starita LM, Parvin JD. The multiple nuclear functions of BRCA1: transcription, ubiquitination and DNA repair. Curr. Opin. Cell Biol. 2003;15:345–350. doi: 10.1016/s0955-0674(03)00042-5. [DOI] [PubMed] [Google Scholar]
- 106.Xu B, O'Donnell AH, Kim ST, Kastan MB. Phosphorylation of serine 1387 in Brca1 is specifically required for the Atm-mediated S-phase checkpoint after ionizing irradiation. Cancer Res. 2002;62:4588–4591. [PubMed] [Google Scholar]
- 107.Tibbetts RS, Cortez D, Brumbaugh KM, Scully R, Livingston D, Elledge SJ, Abraham RT. Functional interactions between BRCA1 and the checkpoint kinase ATR during genotoxic stress. Genes Dev. 2000;14:2989–3002. doi: 10.1101/gad.851000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Anderson SF, Schlegel BP, Nakajima T, Wolpin ES, Parvin JD. BRCA1 protein is linked to the RNA polymerase II holoenzyme complex via RNA helicase A. Nat. Genet. 1998;19:254–256. doi: 10.1038/930. [DOI] [PubMed] [Google Scholar]
- 109.Haile DT, Parvin JD. Activation of transcription in vitro by the BRCA1 carboxyl-terminal domain. J. Biol. Chem. 1999;274:2113–2117. doi: 10.1074/jbc.274.4.2113. [DOI] [PubMed] [Google Scholar]
- 110.Ouchi T, Monteiro AN, August A, Aaronson SA, Hanafusa H. BRCA1 regulates p53-dependent gene expression. Proc. Natl Acad. Sci. USA. 1998;95:2302–2306. doi: 10.1073/pnas.95.5.2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Zhang H, Somasundaram K, Peng Y, Tian H, Bi D, Weber BL, El-Deiry WS. BRCA1 physically associates with p53 and stimulates its transcriptional activity. Oncogene. 1998;16:1713–1721. doi: 10.1038/sj.onc.1201932. [DOI] [PubMed] [Google Scholar]
- 112.Fornace AJ, Jr, Nebert DW, Hollander MC, Luethy JD, Papathanasiou M, Fargnoli J, Holbrook NJ. Mammalian genes coordinately regulated by growth arrest signals and DNA-damaging agents. Mol. Cell. Biol. 1989;9:4196–4203. doi: 10.1128/mcb.9.10.4196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Takekawa M, Saito H. A family of stress-inducible GADD45-like proteins mediate activation of the stress-responsive MTK1/MEKK4 MAPKKK. Cell. 1998;95:521–530. doi: 10.1016/s0092-8674(00)81619-0. [DOI] [PubMed] [Google Scholar]
- 114.Mita H, Tsutsui J, Takekawa M, Witten EA, Saito H. Regulation of MTK1/MEKK4 kinase activity by its N-terminal autoinhibitory domain and GADD45 binding. Mol. Cell. Biol. 2002;22:4544–4555. doi: 10.1128/MCB.22.13.4544-4555.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.van Dam H, Castellazzi M. Distinct roles of Jun : Fos and Jun : ATF dimers in oncogenesis. Oncogene. 2001;20:2453–2464. doi: 10.1038/sj.onc.1204239. [DOI] [PubMed] [Google Scholar]
- 116.Knebel A, Rahmsdorf HJ, Ullrich A, Herrlich P. Dephosphorylation of receptor tyrosine kinases as target of regulation by radiation, oxidants or alkylating agents. EMBO J. 1996;15:5314–5325. [PMC free article] [PubMed] [Google Scholar]
- 117.Blattner C, Knebel A, Radler-Pohl A, Sachsenmaier C, Herrlich P, Rahmsdorf HJ. DNA damaging agents and growth factors induce changes in the program of expressed gene products through common routes. Environ. Mol. Mutagen. 1994;24:3–10. doi: 10.1002/em.2850240103. [DOI] [PubMed] [Google Scholar]
- 118.Fritz G, Kaina B. Activation of c-Jun N-terminal kinase 1 by UV irradiation is inhibited by wortmannin without affecting c-iun expression. Mol. Cell. Biol. 1999;19:1768–1774. doi: 10.1128/mcb.19.3.1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Damrot J, Helbig L, Roos WP, Barrantes SQ, Kaina B, Fritz G. DNA replication arrest in response to genotoxic stress provokes early activation of stress-activated protein kinases (SAPK/JNK) J. Mol. Biol. 2008;385:1409–1421. doi: 10.1016/j.jmb.2008.12.015. [DOI] [PubMed] [Google Scholar]
- 120.Kim WJ, Rajasekaran B, Brown KD. MLH1- and ATM-dependent MAPK signaling is activated through c-Abl in response to the alkylator N-methyl-N'-nitro-N'-nitrosoguanidine. J. Biol. Chem. 2007;282:32021–32031. doi: 10.1074/jbc.M701451200. [DOI] [PubMed] [Google Scholar]
- 121.Shafman TD, Saleem A, Kyriakis J, Weichselbaum R, Kharbanda S, Kufe DW. Defective induction of stress-activated protein kinase activity in ataxia-telangiectasia cells exposed to ionizing radiation. Cancer Res. 1995;55:3242–3245. [PubMed] [Google Scholar]
- 122.Im JS, Lee JK. ATR-dependent activation of p38 MAP kinase is responsible for apoptotic cell death in cells depleted of Cdc7. J. Biol. Chem. 2008;283:25171–25177. doi: 10.1074/jbc.M802851200. [DOI] [PubMed] [Google Scholar]
- 123.Wu K, Jiang SW, Thangaraju M, Wu G, Couch FJ. Induction of the BRCA2 promoter by nuclear factor-kappa B. J. Biol. Chem. 2000;275:35548–35556. doi: 10.1074/jbc.M004390200. [DOI] [PubMed] [Google Scholar]
- 124.Lim JW, Kim H, Kim KH. Expression of Ku70 and Ku80 mediated by NF-kappa B and cyclooxygenase-2 is related to proliferation of human gastric cancer cells. J. Biol. Chem. 2002;277:46093–46100. doi: 10.1074/jbc.M206603200. [DOI] [PubMed] [Google Scholar]
- 125.Arias-Lopez C, Lazaro-Trueba I, Kerr P, Lord CJ, Dexter T, Iravani M, Ashworth A, Silva A. p53 modulates homologous recombination by transcriptional regulation of the RAD51 gene. EMBO Rep. 2006;7:219–224. doi: 10.1038/sj.embor.7400587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Christmann M, Verbeek B, Roos WP, Kaina B. O(6)-Methylguanine-DNA methyltransferase (MGMT) in normal tissues and tumors: enzyme activity, promoter methylation and immunohistochemistry. Biochim. Biophys. Acta. 2011;1816:179–190. doi: 10.1016/j.bbcan.2011.06.002. [DOI] [PubMed] [Google Scholar]
- 127.Christmann M, Kaina B. O(6)-methylguanine-DNA methyltransferase (MGMT): impact on cancer risk in response to tobacco smoke. Mutat. Res. 2012;736:64–74. doi: 10.1016/j.mrfmmm.2011.06.004. [DOI] [PubMed] [Google Scholar]
- 128.Grombacher T, Mitra S, Kaina B. Induction of the alkyltransferase (MGMT) gene by DNA damaging agents and the glucocorticoid dexamethasone and comparison with the response of base excision repair genes. Carcinogenesis. 1996;17:2329–2336. doi: 10.1093/carcin/17.11.2329. [DOI] [PubMed] [Google Scholar]
- 129.Fritz G, Kaina B. Stress factors affecting expression of O6-methylguanine-DNA methyltransferase mRNA in rat hepatoma cells. Biochim. Biophys. Acta. 1992;1171:35–40. doi: 10.1016/0167-4781(92)90137-o. [DOI] [PubMed] [Google Scholar]
- 130.Hermisson M, Klumpp A, Wick W, Wischhusen J, Nagel G, Roos W, Kaina B, Weller M. O6-methylguanine DNA methyltransferase and p53 status predict temozolomide sensitivity in human malignant glioma cells. J. Neurochem. 2006;96:766–776. doi: 10.1111/j.1471-4159.2005.03583.x. [DOI] [PubMed] [Google Scholar]
- 131.Harris LC, Potter PM, Tano K, Shiota S, Mitra S, Brent TP. Characterization of the promoter region of the human O6-methylguanine-DNA methyltransferase gene. Nucleic Acids Res. 1991;19:6163–6167. doi: 10.1093/nar/19.22.6163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Harris LC, Potter PM, Remack JS, Brent TP. A comparison of human O6-methylguanine-DNA methyltransferase promoter activity in Mer+ and Mer- cells. Cancer Res. 1992;52:6404–6406. [PubMed] [Google Scholar]
- 133.Blough MD, Zlatescu MC, Cairncross JG. O6-methylguanine-DNA methyltransferase regulation by p53 in astrocytic cells. Cancer Res. 2007;67:580–584. doi: 10.1158/0008-5472.CAN-06-2782. [DOI] [PubMed] [Google Scholar]
- 134.Srivenugopal KS, Shou J, Mullapudi SR, Lang FF, Jr, Rao JS, Ali-Osman F. Enforced expression of wild-type p53 curtails the transcription of the O(6)-methylguanine-DNA methyltransferase gene in human tumor cells and enhances their sensitivity to alkylating agents. Clin. Cancer Res. 2001;7:1398–1409. [PubMed] [Google Scholar]
- 135.Bocangel D, Sengupta S, Mitra S, Bhakat KK. p53-Mediated down-regulation of the human DNA repair gene O6-methylguanine-DNA methyltransferase (MGMT) via interaction with Sp1 transcription factor. Anticancer Res. 2009;29:3741–3750. [PMC free article] [PubMed] [Google Scholar]
- 136.Ueda S, Mineta T, Nakahara Y, Okamoto H, Shiraishi T, Tabuchi K. Induction of the DNA repair gene O6-methylguanine-DNA methyltransferase by dexamethasone in glioblastomas. J. Neurosurg. 2004;101:659–663. doi: 10.3171/jns.2004.101.4.0659. [DOI] [PubMed] [Google Scholar]
- 137.Natsume A, Ishii D, Wakabayashi T, Tsuno T, Hatano H, Mizuno M, Yoshida J. IFN-beta down-regulates the expression of DNA repair gene MGMT and sensitizes resistant glioma cells to temozolomide. Cancer Res. 2005;65:7573–7579. doi: 10.1158/0008-5472.CAN-05-0036. [DOI] [PubMed] [Google Scholar]
- 138.Natsume A, Wakabayashi T, Ishii D, Maruta H, Fujii M, Shimato S, Ito M, Yoshida J. A combination of IFN-beta and temozolomide in human glioma xenograft models: implication of p53-mediated MGMT downregulation. Cancer Chemother. Pharmacol. 2007;61:653–659. doi: 10.1007/s00280-007-0520-x. [DOI] [PubMed] [Google Scholar]
- 139.Sato K, Kitajima Y, Nakagawachi T, Soejima H, Miyoshi A, Koga Y, Miyazaki K. Cisplatin represses transcriptional activity from the minimal promoter of the O6-methylguanine methyltransferase gene and increases sensitivity of human gallbladder cancer cells to 1-(4-amino-2-methyl-5-pyrimidinyl) methyl-3-2-chloroethyl)-3-nitrosourea. Oncol. Rep. 2005;13:899–906. [PubMed] [Google Scholar]
- 140.Cerda SR, Chu SS, Garcia P, Chung J, Grumet JD, Thimmapaya B, Weitzman SA. Regulation of expression of N-methylpurine DNA glycosylase in human mammary epithelial cells: role of transcription factor AP-2. Chem. Res. Toxicol. 1999;12:1098–1109. doi: 10.1021/tx9901027. [DOI] [PubMed] [Google Scholar]
- 141.Zaky A, Busso C, Izumi T, Chattopadhyay R, Bassiouny A, Mitra S, Bhakat KK. Regulation of the human AP-endonuclease (APE1/Ref-1) expression by the tumor suppressor p53 in response to DNA damage. Nucleic Acids Res. 2008;36:1555–1566. doi: 10.1093/nar/gkm1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Hofseth LJ, Khan MA, Ambrose M, Nikolayeva O, Xu-Welliver M, Kartalou M, Hussain SP, Roth RB, Zhou X, Mechanic LE, et al. The adaptive imbalance in base excision-repair enzymes generates microsatellite instability in chronic inflammation. J. Clin. Invest. 2003;112:1887–1894. doi: 10.1172/JCI19757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Quach N, Chan T, Lu TA, Schreiber SS, Tan Z. Induction of DNA repair proteins, Ref-1 and XRCC1, in adult rat brain following kainic acid-induced seizures. Brain Res. 2005;1042:236–240. doi: 10.1016/j.brainres.2005.02.053. [DOI] [PubMed] [Google Scholar]
- 144.Dianov GL, Hubscher U. Mammalian base excision repair: the forgotten archangel. Nucleic Acids Res. 2013;41:3483–3490. doi: 10.1093/nar/gkt076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Parsons JL, Dianov GL. Co-ordination of base excision repair and genome stability. DNA Repair. 2013;12:326–333. doi: 10.1016/j.dnarep.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 146.Bauer M, Goldstein M, Christmann M, Becker H, Heylmann D, Kaina B. Human monocytes are severely impaired in base and DNA double-strand break repair that renders them vulnerable to oxidative stress. Proc. Natl Acad. Sci. USA. 2011;108:21105–21110. doi: 10.1073/pnas.1111919109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Li Q, Bostick-Bruton F, Reed E. Effect of interleukin-1 alpha and tumour necrosis factor-alpha on cisplatin-induced ERCC-1 mRNA expression in a human ovarian carcinoma cell line. Anticancer Res. 1998;18:2283–2287. [PubMed] [Google Scholar]
- 148.Sugita K, Suzuki N, Higuchi Y, Kita K, Suzuki Y, Lehmann A. Enhancement of XPG mRNA expression by human interferon-beta in Cockayne syndrome cells. Mutat Res. 1998;408:67–72. doi: 10.1016/s0921-8777(98)00020-2. [DOI] [PubMed] [Google Scholar]
- 149.Crawford EL, Blomquist T, Mullins DN, Yoon Y, Hernandez DR, Al-Bagdhadi M, Ruiz J, Hammersley J, Willey JC. CEBPG regulates ERCC5/XPG expression in human bronchial epithelial cells and this regulation is modified by E2F1/YY1 interactions. Carcinogenesis. 2007;28:2552–2559. doi: 10.1093/carcin/bgm214. [DOI] [PubMed] [Google Scholar]
- 150.Scherer SJ, Welter C, Zang KD, Dooley S. Specific in vitro binding of p53 to the promoter region of the human mismatch repair gene hMSH2. Biochem. Biophys. Res. Commun. 1996;221:722–728. doi: 10.1006/bbrc.1996.0663. [DOI] [PubMed] [Google Scholar]
- 151.Warnick CT, Dabbas B, Ford CD, Strait KA. Identification of a p53 response element in the promoter region of the hMSH2 gene required for expression in A2780 ovarian cancer cells. J. Biol. Chem. 2001;276:27363–27370. doi: 10.1074/jbc.M103088200. [DOI] [PubMed] [Google Scholar]
- 152.Humbert O, Achour I, Lautier D, Laurent G, Salles B. hMSH2 expression is driven by AP1-dependent regulation through phorbol-ester exposure. Nucleic Acids Res. 2003;31:5627–5634. doi: 10.1093/nar/gkg781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Christmann M, Tomicic MT, Kaina B. Phosphorylation of mismatch repair proteins MSH2 and MSH6 affecting MutS{alpha} mismatch-sbinding activity. Nucleic Acids Res. 2002;30:1959–1966. doi: 10.1093/nar/30.9.1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Christmann M, Kaina B. Nuclear translocation of mismatch repair proteins MSH2 and MSH6 as a response of cells to alkylating agents. J. Biol. Chem. 2000;275:36256–36262. doi: 10.1074/jbc.M005377200. [DOI] [PubMed] [Google Scholar]
- 155.Koike M, Yutoku Y, Koike A. KARP-1 works as a heterodimer with Ku70, but the function of KARP-1 cannot perfectly replace that of Ku80 in DSB repair. Exp. Cell Res. 2011;317:2267–2275. doi: 10.1016/j.yexcr.2011.06.015. [DOI] [PubMed] [Google Scholar]
- 156.Chang HW, Lai YC, Cheng CY, Ho JL, Ding ST, Liu YC. UV inducibility of rat proliferating cell nuclear antigen gene promoter. J. Cell Biochem. 1999;73:423–432. [PubMed] [Google Scholar]
- 157.Mazur DJ, Perrino FW. Identification and expression of the TREX1 and TREX2 cDNA sequences encoding mammalian 3′—>5′ exonucleases. J. Biol. Chem. 1999;274:19655–19660. doi: 10.1074/jbc.274.28.19655. [DOI] [PubMed] [Google Scholar]
- 158.Mazur DJ, Perrino FW. Excision of 3′ termini by the Trex1 and TREX2 3′—>5′ exonucleases. Characterization of the recombinant proteins. J. Biol. Chem. 2001;276:17022–17029. doi: 10.1074/jbc.M100623200. [DOI] [PubMed] [Google Scholar]
- 159.Serra M, Forcales SV, Pereira-Lopes S, Lloberas J, Celada A. Characterization of Trex1 induction by IFN-gamma in murine macrophages. J. Immunol. 2011;186:2299–2308. doi: 10.4049/jimmunol.1002364. [DOI] [PubMed] [Google Scholar]
- 160.Felsberg J, Thon N, Eigenbrod S, Hentschel B, Sabel MC, Westphal M, Schackert G, Kreth FW, Pietsch T, Loffler M, et al. Promoter methylation and expression of MGMT and the DNA mismatch repair genes MLH1, MSH2, MSH6 and PMS2 in paired primary and recurrent glioblastomas. Int. J. Cancer. 2011;129:659–670. doi: 10.1002/ijc.26083. [DOI] [PubMed] [Google Scholar]
- 161.Sengupta S, Shimamoto A, Koshiji M, Pedeux R, Rusin M, Spillare EA, Shen JC, Huang LE, Lindor NM, Furuichi Y, et al. Tumor suppressor p53 represses transcription of RECQ4 helicase. Oncogene. 2005;24:1738–1748. doi: 10.1038/sj.onc.1208380. [DOI] [PubMed] [Google Scholar]
- 162.Koshiji M, To KK, Hammer S, Kumamoto K, Harris AL, Modrich P, Huang LE. HIF-1alpha induces genetic instability by transcriptionally downregulating MutSalpha expression. Mol. Cell. 2005;17:793–803. doi: 10.1016/j.molcel.2005.02.015. [DOI] [PubMed] [Google Scholar]
- 163.Gazzoli I, Kolodner RD. Regulation of the human MSH6 gene by the Sp1 transcription factor and alteration of promoter activity and expression by polymorphisms. Mol. Cell. Biol. 2003;23:7992–8007. doi: 10.1128/MCB.23.22.7992-8007.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Ladd PD, Wilson DM, III, Kelley MR, Skalnik DG. Identification of the human HEX1/hExo1 gene promoter and characterization of elements responsible for promoter activity. DNA Repair. 2003;2:187–198. doi: 10.1016/s1568-7864(02)00195-7. [DOI] [PubMed] [Google Scholar]
- 165.Fung H, Bennett RA, Demple B. Key role of a downstream specificity protein 1 site in cell cycle-regulated transcription of the AP endonuclease gene APE1/APEX in NIH3T3 cells. J. Biol. Chem. 2001;276:42011–42017. doi: 10.1074/jbc.M106423200. [DOI] [PubMed] [Google Scholar]
- 166.Ma L, Weeda G, Jochemsen AG, Bootsma D, Hoeijmakers JH, van der Eb AJ. Molecular and functional analysis of the XPBC/ERCC-3 promoter: transcription activity is dependent on the integrity of an Sp1-binding site. Nucleic Acids Res. 1992;20:217–224. doi: 10.1093/nar/20.2.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Liu W, Nichols AF, Graham JA, Dualan R, Abbas A, Linn S. Nuclear transport of human DDB protein induced by ultraviolet light. J. Biol. Chem. 2000;275:21429–21434. doi: 10.1074/jbc.M000961200. [DOI] [PubMed] [Google Scholar]
- 168.Nakamura H, Tanimoto K, Hiyama K, Yunokawa M, Kawamoto T, Kato Y, Yoshiga K, Poellinger L, Hiyama E, Nishiyama M. Human mismatch repair gene, MLH1, is transcriptionally repressed by the hypoxia-inducible transcription factors, DEC1 and DEC2. Oncogene. 2008;27:4200–4209. doi: 10.1038/onc.2008.58. [DOI] [PubMed] [Google Scholar]
- 169.Mihaylova VT, Bindra RS, Yuan J, Campisi D, Narayanan L, Jensen R, Giordano F, Johnson RS, Rockwell S, Glazer PM. Decreased expression of the DNA mismatch repair gene Mlh1 under hypoxic stress in mammalian cells. Mol. Cell. Biol. 2003;23:3265–3273. doi: 10.1128/MCB.23.9.3265-3273.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Thalhammer A, Bencokova Z, Poole R, Loenarz C, Adam J, O'Flaherty L, Schodel J, Mole D, Giaslakiotis K, Schofield CJ, et al. Human AlkB homologue 5 is a nuclear 2-oxoglutarate dependent oxygenase and a direct target of hypoxia-inducible factor 1alpha (HIF-1alpha) PLoS One. 2011;6:e16210. doi: 10.1371/journal.pone.0016210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Liu Y, Bernauer AM, Yingling CM, Belinsky SA. HIF1alpha regulated expression of XPA contributes to cisplatin resistance in lung cancer. Carcinogenesis. 2012;33:1187–1192. doi: 10.1093/carcin/bgs142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Filippi S, Latini P, Frontini M, Palitti F, Egly JM, Proietti-De-Santis L. CSB protein is (a direct target of HIF-1 and) a critical mediator of the hypoxic response. Embo J. 2008;27:2545–2556. doi: 10.1038/emboj.2008.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Rezvani HR, Mahfouf W, Ali N, Chemin C, Ged C, Kim AL, de Verneuil H, Taieb A, Bickers DR, Mazurier F. Hypoxia-inducible factor-1alpha regulates the expression of nucleotide excision repair proteins in keratinocytes. Nucleic Acids Res. 2009;38:797–809. doi: 10.1093/nar/gkp1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Rezvani HR, Dedieu S, North S, Belloc F, Rossignol R, Letellier T, de Verneuil H, Taieb A, Mazurier F. Hypoxia-inducible factor-1alpha, a key factor in the keratinocyte response to UVB exposure. J. Biol. Chem. 2007;282:16413–16422. doi: 10.1074/jbc.M611397200. [DOI] [PubMed] [Google Scholar]
- 175.Haas S, Kaina B. c-Fos is involved in the cellular defence against the genotoxic effect of UV radiation. Carcinogenesis. 1995;16:985–991. doi: 10.1093/carcin/16.5.985. [DOI] [PubMed] [Google Scholar]
- 176.Kaina B, Haas S, Kappes H. A general role for c-Fos in cellular protection against DNA-damaging carcinogens and cytostatic drugs. Cancer Res. 1997;57:2721–2731. [PubMed] [Google Scholar]
- 177.Coquerelle T, Dosch J, Kaina B. Overexpression of N-methylpurine-DNA glycosylase in Chinese hamster ovary cells renders them more sensitive to the production of chromosomal aberrations by methylating agents—a case of imbalanced DNA repair. Mutat. Res. 1995;336:9–17. doi: 10.1016/0921-8777(94)00035-5. [DOI] [PubMed] [Google Scholar]
- 178.Hamdi M, Kool J, Cornelissen-Steijger P, Carlotti F, Popeijus HE, van der Burgt C, Janssen JM, Yasui A, Hoeben RC, Terleth C, et al. DNA damage in transcribed genes induces apoptosis via the JNK pathway and the JNK-phosphatase MKP-1. Oncogene. 2005;24:7135–7144. doi: 10.1038/sj.onc.1208875. [DOI] [PubMed] [Google Scholar]
- 179.Christmann M, Tomicic MT, Aasland D, Kaina B. A role for UV-light-induced c-Fos: Stimulation of nucleotide excision repair and protection against sustained JNK activation and apoptosis. Carcinogenesis. 2007;28:183–190. doi: 10.1093/carcin/bgl119. [DOI] [PubMed] [Google Scholar]
- 180.Brozovic A, Fritz G, Christmann M, Zisowsky J, Jaehde U, Osmak M, Kaina B. Long-term activation of SAPK/JNK, p38 kinase and fas-L expression by cisplatin is attenuated in human carcinoma cells that acquired drug resistance. Int. J. Cancer. 2004;112:974–985. doi: 10.1002/ijc.20522. [DOI] [PubMed] [Google Scholar]
- 181.Blattner C, Kannouche P, Litfin M, Bender K, Rahmsdorf HJ, Angulo JF, Herrlich P. UV-Induced stabilization of c-fos and other short-lived mRNAs. Mol. Cell. Biol. 2000;20:3616–3625. doi: 10.1128/mcb.20.10.3616-3625.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Christmann M, Nagel G, Horn S, Krahn U, Wiewrodt D, Sommer C, Kaina B. MGMT activity, promoter methylation and immunohistochemistry of pretreatment and recurrent malignant gliomas: a comparative study on astrocytoma and glioblastoma. Int. J. Cancer. 2010;127:2106–2118. doi: 10.1002/ijc.25229. [DOI] [PubMed] [Google Scholar]
- 183.Wiewrodt D, Nagel G, Dreimuller N, Hundsberger T, Perneczky A, Kaina B. MGMT in primary and recurrent human glioblastomas after radiation and chemotherapy and comparison with p53 status and clinical outcome. Int. J. Cancer. 2008;122:1391–1399. doi: 10.1002/ijc.23219. [DOI] [PubMed] [Google Scholar]
- 184.Middleton MR, Lunn JM, Morris C, Rustin G, Wedge SR, Brampton MH, Lind MJ, Lee SM, Newell DR, Bleehen NM, et al. O6-methylguanine-DNA methyltransferase in pretreatment tumour biopsies as a predictor of response to temozolomide in melanoma. Br. J. Cancer. 1998;78:1199–1202. doi: 10.1038/bjc.1998.654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Belanich M, Pastor M, Randall T, Guerra D, Kibitel J, Alas L, Li B, Citron M, Wasserman P, White A, et al. Retrospective study of the correlation between the DNA repair protein alkyltransferase and survival of brain tumor patients treated with carmustine. Cancer Res. 1996;56:783–788. [PubMed] [Google Scholar]
- 186.Zhang W, Zhang J, Hoadley K, Kushwaha D, Ramakrishnan V, Li S, Kang C, You Y, Jiang C, Song SW, et al. miR-181d: a predictive glioblastoma biomarker that downregulates MGMT expression. Neuro. Oncol. 2012;14:712–719. doi: 10.1093/neuonc/nos089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Mitchel RE. Low doses of radiation are protective in vitro and in vivo: evolutionary origins. Dose Response. 2006;4:75–90. doi: 10.2203/dose-response.04-002.Mitchel. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Olivieri G, Bodycote J, Wolff S. Adaptive response of human lymphocytes to low concentrations of radioactive thymidine. Science. 1984;223:594–597. doi: 10.1126/science.6695170. [DOI] [PubMed] [Google Scholar]
- 189.Wolff S, Afzal V, Wiencke JK, Olivieri G, Michaeli A. Human lymphocytes exposed to low doses of ionizing radiations become refractory to high doses of radiation as well as to chemical mutagens that induce double-strand breaks in DNA. Int. J. Radiat. Biol. Relat. Stud. Phys. Chem. Med. 1988;53:39–47. doi: 10.1080/09553008814550401. [DOI] [PubMed] [Google Scholar]
- 190.Rigaud O, Moustacchi E. Radioadaptation to the mutagenic effect of ionizing radiation in human lymphoblasts: molecular analysis of HPRT mutants. Cancer Res. 1994;54:1924s–1928s. [PubMed] [Google Scholar]
- 191.Seong J, Suh CO, Kim GE. Adaptive response to ionizing radiation induced by low doses of gamma rays in human cell lines. Int. J. Radiat. Oncol. Biol. Phys. 1995;33:869–874. doi: 10.1016/0360-3016(95)00085-X. [DOI] [PubMed] [Google Scholar]
- 192.Youngblom JH, Wiencke JK, Wolff S. Inhibition of the adaptive response of human lymphocytes to very low doses of ionizing radiation by the protein synthesis inhibitor cycloheximide. Mutat. Res. 1989;227:257–261. doi: 10.1016/0165-7992(89)90107-3. [DOI] [PubMed] [Google Scholar]
- 193.Wiencke JK, Afzal V, Olivieri G, Wolff S. Evidence that the [3H]thymidine-induced adaptive response of human lymphocytes to subsequent doses of X-rays involves the induction of a chromosomal repair mechanism. Mutagenesis. 1986;1:375–380. doi: 10.1093/mutage/1.5.375. [DOI] [PubMed] [Google Scholar]
- 194.Azzam EI, Raaphorst GP, Mitchel RE. Radiation-induced adaptive response for protection against micronucleus formation and neoplastic transformation in C3H 10T1/2 mouse embryo cells. Radiat Res. 1994;138:S28–S31. [PubMed] [Google Scholar]
- 195.Klammer H, Kadhim M, Iliakis G. Evidence of an adaptive response targeting DNA nonhomologous end joining and its transmission to bystander cells. Cancer Res. 2010;70:8498–8506. doi: 10.1158/0008-5472.CAN-10-1181. [DOI] [PubMed] [Google Scholar]
- 196.Schlade-Bartusiak K, Stembalska-Kozlowska A, Bernady M, Kudyba M, Sasiadek M. Analysis of adaptive response to bleomycin and mitomycin C. Mutat. Res. 2002;513:75–81. doi: 10.1016/s1383-5718(01)00288-1. [DOI] [PubMed] [Google Scholar]
- 197.Nikolova T, Huttner E. Adaptive and synergistic effects of a low-dose ENU pretreatment on the frequency of chromosomal aberrations induced by a challenge dose of ENU in human peripheral blood lymphocytes in vitro. Mutat. Res. 1996;357:131–141. doi: 10.1016/0027-5107(96)00094-2. [DOI] [PubMed] [Google Scholar]
- 198.Ye N, Bianchi MS, Bianchi NO, Holmquist GP. Adaptive enhancement and kinetics of nucleotide excision repair in humans. Mutat. Res. 1999;435:43–61. doi: 10.1016/s0921-8777(99)00022-1. [DOI] [PubMed] [Google Scholar]