

Abstract
The Forkhead Box O (Foxo) proteins represent an evolutionarily conserved family of transcription factors that play an important role in regulating processes including metabolism, longevity, and cell death/survival. How is it that a single transcription factor can initiate such divergent cellular responses? We will review the evidence that specific patterns of post-translational modifications play a key role in directing Foxo into various transcriptional readouts. This regulation appears to take on a two tiered regulatory model; with a group of well defined post-translational modifications regulating nuclear localization and transcriptional activity while a second set of modifications regulate the transcriptional specificity of Foxo.
Keywords: Foxo, post-translational modifications, phosphorylation, acetylation, ubiquitination
INTRODUCTION
The Forkead Box O (Foxo) transcription factors are members of the Forkhead superfamily of winged helix transcription factors. The Foxo family regulates cellular processes including metabolism, stress response, DNA damage repair and cell death. The Foxo family is highly conserved evolutionarily, with Foxo orthologs exhibiting similar physiological functions in model organisms such as Caenorhabditis elegans, Drosophila melanogaster, and in vertebrates. There are four mammalian Foxos, FOXO1, FOXO3, FOXO4 and FOXO6. FOXO2 is identical to FOXO3 and FoxO5 is the fish ortholog of FOXO3. While all Foxo members are capable of binding the same core DNA sequence, 5’-TTGTTTAC-3’, the expression of individual Foxos is tissue specific. In addition, some Foxos undergo specific regulation or lack the regulatory interactions of other isoforms. In most cases groups study the regulation of a single Foxo isoform. Frequently regulatory pathways that are discovered in a single Foxo isoform are conserved between isoforms, therefore, for the sake of simplicity, the general term Foxo will be used in this article instead of listing each individual isoform.
While much is known about signaling pathways that influence its nuclear localization, the necessary antecedent for transcriptional activity of Foxo, less is known about signaling pathways that control Foxo transcription of specific genes in specific contexts. The ability of Foxo to regulate so many cellular processes would suggest that such signaling pathways exist in order to integrate cellular context and direct Foxo to specific targets. For example nuclear localization of Foxo through Paclitaxel treatment causes cell death in breast cancer cells [1], while nuclear localization of Foxo through Psammaplysene A treatment is neuroprotective in multiple models of motorneuron disease [2]. Even within the same cell type Foxo can have opposing actions (mediating either apoptosis or survival of neurons [2, 3] depending on the cellular context), raising the question of how its opposing activities are regulated. One mechanism of regulating Foxo transcriptional output is through its pattern of posttranslational modifications (i.e. phosphorylation, acetylation and ubiquitination). We will review the evidence that these patterns of posttranslational modification are responsible for regulating both transcriptional specificity and transcriptional activation. In order to fully understand Foxo signaling it is important to consider both pathways – those that generally activate or inhibit Foxo (by regulating its subcellular localization), and those that direct Foxo to transcribe specific genes.
FOXO INHIBITORY KINASES
The following kinases are known, general inhibitors of Foxo signaling. Their signaling activities are responsible for promoting translocation of Foxo out of the nucleus and regulating its degradation. Because of their ability to turn off Foxo transcription, and therefore repress, for example, Foxo’s pro-apototic activity, these kinases are often considered in terms of Foxo’s role in cancer biology. While it is clear that these signaling pathways play a role in controlling Foxo’s subcellular localization, the extent to which these posttranslational modifications (at any of these sites) play a role in the specificity of Foxo transactivation is unclear (see Fig. 1).
Fig. (1).

Numerous residues on Foxo are regulated via phosphorylation. These sites influence Foxo’s subcellular localization, transcriptional potency and specify gene targets for activation. *This SGK site is not present in Foxo6. **This JNK site is only present in Foxo4.
PI3’K Regulates Foxo Activation through Its Downstream Kinases AKT and SGK
The Phosphoinositide 3 Kinase (PI3’K) pathway is a major regulator of Foxo activation. Activation of PI3’K leads to phosphorylation and activation of downstream targets including Protein Kinase B (also known as AKT) and Serum and Glucocorticoid inducible Kinase (SGK). AKT and SGK are both capable of recognizing the same motifs for substrate phosphorylation, RXRXXS/T, and are responsible for phosphorylation of Foxo at 3 key residues: Thr32, Ser253 and Ser315 (of Foxo3). When Foxo is phosphorylated at these residues it associates with 14-3-3 proteins and is exported from the nucleus, where it is transcriptionally inactive [4-6].
While AKT and SGK are capable of phosphorylating the same residues, there does seem to be some preference for specific sites between these two kinases. In particular, the C-terminal most phosphorylation site, Ser315, seems to be preferentially phosphorylated by SGK, while Ser253 is preferentially phosphorylated by AKT [7]. The N-terminal Thr32 is efficiently phosphorylated by both kinases. Experiments using dominant negative forms of these two kinases have demonstrated that disruption of either AKT or SGK kinase activity leads to nuclear Foxo, suggesting a non-redundant role for these kinases in efficiently phosphorylating Foxo at these three sites and targeting it to the cytoplasm.
Each of these three phosphorylation sites appears to mediate its own important function in the nuclear export of Foxo. The most N-terminal phosphorylation site, Thr32, is involved in Foxo association with 14-3-3, which is essential for Foxo nuclear export. The central phosphorylation site at Ser253 is located in the Foxo Nuclear Localization Sequence (NLS) and acts to disrupt NLS activity by introducing a negative charge to the basic NLS region. This prevents re-entry of Foxo into the nucleus. The C-terminal most phosphorylation site, Ser315, plays a key role in the rate of nuclear export. Phosphorylation of Ser315 unmasks the Foxo Nuclear Export Sequence (NES) thus increasing its rate of nuclear export. The importance of this residue is demonstrated by Foxo6, which lacks this phosphorylation site, and unlike the other three Foxos, is predominantly nuclear [8]. Nuclear export of Foxo is an active process that depends on the association between CRM1 and Foxo. Treatment of cells with the CRM1 specific inhibitor leptomycin B (which covalently modifies the CRM1 NES binding domain) results in a robust nuclear localization of Foxo even when Foxo is phosphorylated at Thr32, Ser253, and Ser315 [4].
Acetylation of Foxo influences AKT phosphorylation at Ser-253. Foxo mutants that are acetylation-mimetic at Foxo lysine residues 242, 245, and 262 become highly phosphorylated under basal growth conditions. Similarly, phosphorylation of wild type Foxo (Ser253) increases with deacetylase inhibitor treatment, while phosphorylation at Ser253 of non-acetylatable mutants is unaffected by deacetylase inhibitors [9]. Acetylation of Foxo by the histone acetyl transeferases CBP and p300 inhibit Foxo DNA binding capacity in vitro. Phosphorylation of Foxo at Thr32 disrupts the interaction between Foxo and CBP/p300. These data suggest a model in which acetylation inhibits Foxo transcription in a sequential manner. Once Foxo is acetylated on lysine 242, 245, and 262, its DNA binding capacity decreases. Acetylated Foxo, which does not interact with DNA, is a superior substrate for phosphorylation by AKT at nuclear export sites. Phospho Thr32/Ser253 Foxo loses the interaction between Foxo and CBP/p300 and thus negates the favorable chromatin remodeling that this interaction promotes.
The ability of PI3’K to activate AKT and SGK is negatively regulated by the lipid phosphatase, phosphatase and tensin homolog (PTEN) which degrades PIP3 thus inhibiting PI3’K. Inactivation of PTEN is common in glioblastomas and prostate cancer [10, 11]. Similarly, decreased PTEN activity has been characterized in breast cancer [12, 13]. The ability of this pathway to robustly control Foxo nuclear localization helps explain the oncogenic properties of PTEN. Normally, PTEN hydrolyzes PIP3, the product of PI3’K, and therefore decreases the activity of AKT and SGK. PTEN deficiency results in a constitutively cytoplasmic Foxo. The lack of nuclear Foxo in these cells could contribute to oncogenic transformation through a loss of the cell cycle arrest and proapoptotic capabilities of Foxo.
Phosphorylation of Ser315 by SGK Primes Foxo for CK1 Phosphorylation and Increases the Rate of Nuclear Export
Casein Kinase 1 (CK1) plays a role in cell differentiation and circadian rhythm control and its dysregulation can contribute to neurodegeneration and cancer development [14]. CK1 can phosphorylate Ser318 and Ser321 but only after phosphorylation of Ser315 by SGK. Phosphorylation of Ser315 by SGK creates a consensus sequence for CK1 phosphorylation, S/T(P)XXS/T, at Ser318 which in turn creates a CK1 consensus sequence at Ser321. Ser to Ala mutations and phosphospecific antibodies have been used to demonstrate that phosphorylation at these residues is, in fact, hierarchical, with phosphorylation of Ser315 by SGK being necessary for the subsequent phosphorylation of Ser318 and Ser321 by CK1. As mutations at any of these sites have no effect on nuclear import rates; they probably function by increasing nuclear export. It is thought that in conjunction with Ser325 this grouping of highly phosphorylated residues creates an acidic patch that increases Foxo’s association with its nuclear export complex [15].
Dual Specificity Tyrosine Phosphorylated and Regulated Kinase 1A (DYRK1A) Increases Nuclear Export of Foxo
DYRK1A is a serine/threonine kinase found in both the nuclear and cytoplasmic compartments. While some DYRK1A targets are known, a detailed understanding of its cellular functions is lacking. Decreased levels of the Drosphila ortholog, MiniBrain Kinase, results in decreased brain size suggesting a role in the regulation of neurogenesis. Based on its chromosomal location, and the role of MiniBrain, DYRK1A has been implicated in the pathogennesis of Down syndrome [16]. DYRK1A has been shown to phosphorylate Foxo at Ser325. Phosphorylation at this residue decreases the amount of Foxo in the nuclear fraction and accordingly decreases Foxo transcriptional activity. DYRK1A activity is SGK-and-CK1-independent and a Ser to Ala mutation at Ser325 increases the amount of nuclear Foxo [17]. Although DYRK1A activity is PI3’K independent, phosphorylation of Foxo at Ser325 by DYRK1A seems to have a synergistic effect with SGK and CK1 perhaps by enhancing the acidic patch effect that promotes Foxo to interact with its nuclear export complex.
IKK Regulates Foxo Inactivation and Degradation in an AKT Independent Fashion
The IκB Kinase (IKK) is best known for its regulation of the Nuclear Factor-κB family of transcription factors [18]. Activation of the IKK has also been shown to inactivate Foxo. Constitutive activation of IKK has been linked to breast carcinoma, suggesting a model where constitutive activation of IKK increases pro-survival factor expression and inhibits the pro-apoptotic functions of Foxo. Hu et al. demonstrated that the ability of IKK to induce cytoplasmic localization of Foxo is AKT independent [19]. This AKT independence was characterized using mutant forms of Foxo that are non-phosphorylatable at the three AKT/SGK sites described above, as well as cell lines lacking AKT activity. Activation of IKK has been shown to induce phosphorylation of Foxo at ser644. Phosphorylation at this residue results in both nuclear exclusion and degradation of Foxo. Phosphorylation at ser644 has been shown to induce ubiquitination of Foxo and a subsequent decrease in Foxo levels that can be inhibited using the proteasomal inhibitor MG132.
ERK Inhibits and Degrades Foxo
The RAS-ERK pathway is a signaling cascade that regulates cellular proliferation and survival [20]. Many tumors are RAS overexpressors or express a mutant, constitutively active, form of RAS. When this is the case, ERK activity is upregulated and there is an accompanying decrease in Foxo transcriptional activity in two ways. First, ERK has been shown to phosphorylate Foxo (at Ser294, Ser344, and Ser425 on Foxo3) and this phosphorylation results in increasing amounts of cytoplasmic Foxo. Second, Foxo phosphorylated at these three residues is less stable, and is degraded in a proteasome dependent fashion. This decreased stability may be based on its increased interaction with the E3-ligase, MDM2 [21].
FOXO ACTIVATING KINASES
Stressors Induce Nuclear Accumulation of Foxo
A number of different kinases (MST1, JNK, and CDK1) are capable of promoting translocation of Foxo into the nucleus under conditions of cellular stress. Once localized to the nucleus, Foxo seems to execute either a pro-survival or pro-death transcriptional pattern dependent on the cellular context and severity of the insult. While phosphorylation at any of the sites described below seems to be sufficient to induce nuclear localization, little is known about the consequences of activating these pathways together. Do these multiple stress dependent kinases represent redundant pathways of Foxo activation, thus ensuring that Foxo responds to stressors, or might they act to integrate data to help determine the severity of the stressor and thus signal the appropriate transcriptional pattern to execute?
Oxidative Stress Activates JNK which Activates Foxo
c-Jun N-terminal Kinase (JNK) is a kinase which is activated by cell stress, including inflammatory signals, ultraviolet light and Reactive Oxygen Species (ROS), and upon activation can increase Foxo transcription. ROS activate the small GTPase Ral, and Ral, in turn activates JNK. JNK then phosphorylates Foxo at Thr447 and Thr451 leading to an increase in its transcriptional activity [22]. The JNK dependent effect on Foxo transcription appears to be antagonized by the AKT/SGK signaling pathway. ROS are known to induce nuclear localization of Foxo even in the presence of serum (when AKT is active and would otherwise lead to cytoplasmic localization of Foxo). Nevertheless, Foxo proteins which are phosphomimetic at the JNK sites show similar association with 14-3-3 upon insulin treatment as wild type Foxo, and undergo cytoplasmic localization. This suggests that the nuclear localization of Foxo after ROS treatment is not regulated by JNK. While Foxo is regulated normally by PI3’K after JNK phosphorylation, JNK phosphorylation of Foxo makes Foxo transcriptionally more active.
CDK1 Activates Foxo as a Key Check in the Cell Cycle
Cyclin-Dependent Kinase 1 (CDK1) phosphorylates Foxo in the forkhead domain at Ser249. CDK1 is a kinase associated with cell cycle progression, predominantly the G2/M transition. As opposed to the PI3’K/AKT pathway, phosphorylation at Ser249 by CDK1 disrupts the interaction between Foxo and 14-3-3, thus leading to its nuclear localization. Phosphorylation at this site varies with the cell cycle, with Foxo being highly phosphorylated at Ser249 during the G2/M transition (when CDK1 is most active) and relatively unphosphorylated during the G0/G1 transition. The ability of CDK1 to increase Foxo transcription likely correlates with Foxo’s ability to induce expression of DNA damage repair genes such as GADD45 and genes that eliminate ROS. These genes are turned on at the G2/M transition and ensure the cell is in an optimal state before the cell cycle continues [23].
Another interesting aspect of this pathway is that aberrant activation of the cell cycle has been associated with cell death in post-mitotic neurons. For example depriving cerebellar granule neurons of action potential firing (through blockage of calcium flux) causes cell death. This activity deprivation is also associated with an increase in CDK1 activity, leading to Foxo activation [23]. Foxo-dependent cell death is mediated by the expression of pro-apoptotic factors such as Bim. There is some disagreement about this as some have suggested that phosphorylation at Ser249 results in inhibition of Foxo potentially leading to tumorogenesis [24].
MST1 Phosphorylates Foxo Leading to its Activation
Mammalian Sterile 20-like kinase 1 (MST1) is known to control cell proliferation, survival and morphology [25]. The fly ortholog of MST1, Hippo, is a known tumor suppressor and it is suspected that MST1 has a tumor suppressor capacity as well. MST1 is capable of phosphorylating Foxo at Ser207 [26]. Once phosphorylated at this site the interaction between Foxo and 14-3-3 is disrupted and Foxo localizes to the nucleus. Phosphorylation at this site can be initiated by ROS or activity/trophic factor deprivation in neurons. Neuronal death induced by trophic factor/activity withdrawal or ROS exhibit a dependence on MST1 and Foxo [27]. While not characterized, it is likely that this signaling pathway may also be adaptive allowing for survival depending on the severity of the insult. Insults resulting in cell death are generally studied due to the fact that they result in an easily quantifiable phenotype (i.e. death/survival). For this reason it is difficult to say definitively that this pathway is adaptive when less severe insults are administered, but the Foxo transcriptome would suggest that this is the case [28].
KINASES THAT DICTATE A TRANSCRIPTIONAL PROFILE
Foxo is the transcriptional regulator of a group of proteins whose cellular functions, when considered as a whole, are not directed towards a single purpose. For example Foxo is the regulator of GADD45a and manganese superoxide dismutase, which are involved in DNA damage repair and ROS scavenging respectively. Yet Foxo also regulates expression of FAS-ligand and BIM which are involved in apoptosis [29]. These four proteins would likely never be activated in unison as there is no clear purpose in clearing cellular ROS and repairing DNA in a cell which has initiated an apoptotic pathway. This suggests that Foxo is capable of integrating cellular signals in order to select the transcriptional profile suited to affect a specific outcome. This is analogous to the cellular function of the transcription factor p53, which acts to initiate cell survival and DNA damage repair in the case of mild insults but initiates cell death when insults are severe [30].
It is clear that post-translational modifications play an important role in specifying the transcriptional outputs of Foxo. It has been technically challenging thus far to profile the Foxo transcriptome in the case of every post translational modification due to the sheer number of potential modifications and pathways that act upon Foxo. While it is likely that some of the aforementioned inhibitory and activating kinases are specifying more than an on/off type signal, AMPK is the only kinase whose effect on Foxo’s transcriptional profile has been well characterized.
AMPK Acts to Integrate Data on Energy Levels into Foxo Transcriptional Activity
The AMP-activated Protein Kinase (AMPK) is a key regulator of energy homeostasis in cells. It is regulated by the ratio of AMP to ATP in a cell, with higher AMP/ATP ratios resulting in AMPK activation. AMPK can phosphorylate Foxo at six sites. While phosphorylation at these sites appears not to have any effect on the localization or overall activity of Foxo, these sites do influence the transcriptional profile of Foxo. Microarray studies of cells expressing mutant Foxo that is non-phosphorylatable at the six AMPK sites has revealed a transcript profile in which genes involved in ROS resistance are up regulated [31]. Simultaneously, others are turned down and many remain unchanged. This is particularly interesting because it represents a second layer of control dictated by post translational modification. AMPK does not regulate Foxo in a binary manner (i.e. transcripttionaly active versus inactive) instead it is dictating a specific transcriptional output. The mechanism by which phosphorylation at these sites alters Foxo’s transcriptional pattern have not been well explored. Potential mechanisms for AMPK’s control of Foxo dependent transcription include: recruitment of additional proteins to a transcriptional complex or direction of Foxo to specific promoter regions.
ACETYLATION OF FOXO
The effect of acetylation on Foxo activity represents a complex balance between histone deacetylases (HDACs) such as Silent Information Regulator 2 (SIRT1) and (SIRT2) and the Histone Acetyl Transferases (HATs) CREB Binding Protein (CBP) and p300. The histone acetyltransferase activity of the coactivators CBP/p300 could enhance Foxo transcription indirectly through their ability to acetylate histones and favorably remodel chromatin. In contrast, acetylation of Foxo has been shown to inhibit its DNA binding capacity [9]. This negative feedback mechanism is most likely offset by the activity of SIRT1 and 2 which are capable of deacetylating Foxo. Monoubiquitination of Foxo lysines which can prevent acetylation, should both keep Foxo non-acetylated and extend its association with DNA promoter regions (Fig. 2).
Fig. (2).
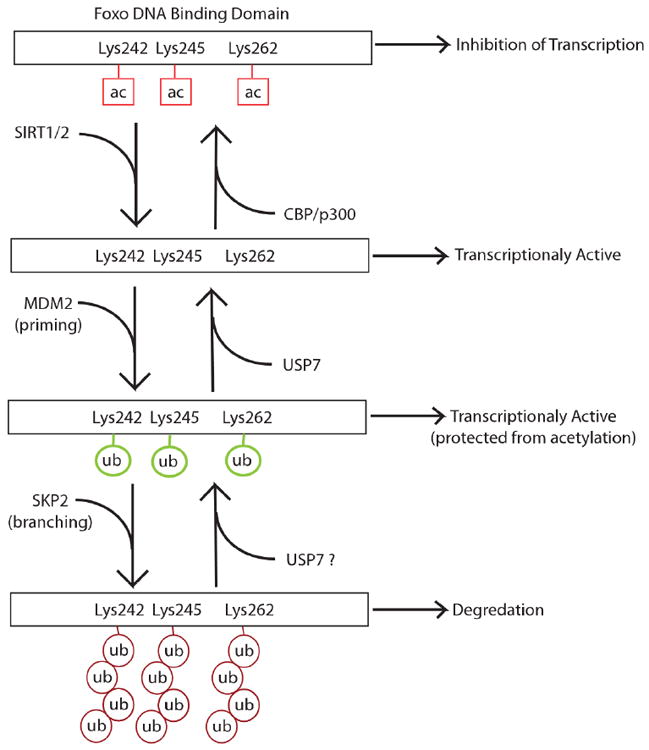
The acetylases CBP and p300 play an important role in chromatin remodeling, allowing Foxo to effectively transcribe its targets. Interestingly CBP/p300 also have an inhibitory effect on Foxo DNA binding through acetylation of the Foxo DNA binding domain. This negative regulatory effect is counteracted by the histone deaeetylases SIRTl and SIRT2. Foxo acetylation can also potentially be prevented through monoubiquitination of the lysines in Foxo’s DNA bidning domain. Monoubiquitinaiton also primes Foxo for potential polyubiquitination and eventual degradation.
SIRT1 and 2
SIRT1 and SIRT2 are members of the sirtuin family of decacetylases and are the mammalian homologs of the yeast deacetylase Sir2. In yeast, Sir2 plays an important role in organismal longevity through histone deacetylation and gene silencing. Deacetylation of other contributory proteins by Sir2, such as acetyl-coenzyme A, may also play a role in longevity through regulation of intermediary metabolism [32]. In mammals, SIRT1 and SIRT2 interact with Foxo in a ROS-dependent manner. ROS promotes nuclear localization of Foxo and nuclear localization of Foxo is necessary for the Foxo SIRT1 interaction since SIRT1 is constitutively nuclear. Yet, nuclear localization alone is not sufficient to initiate the Foxo-SIRT1 interaction; cells must be exposed to increased levels of ROS to induce this protein-protein interaction. SIRT2 on the other hand shuttles between the nucleus and the cytoplasm, so it is conceivable that SIRT2 could deacetylate Foxo in the nucleus or the cytoplasm.
Based on the work of several groups, it appears that deacetylation of Foxo by SIRT1 imparts transcriptional specificity, shifting Foxo’s transcriptional profile away from apoptosis and towards expression of survival promoting genes [33-36]. This is consistent with findings that inhibition of SIRT1 or disruption of its interaction with Foxo promotes the expression of apoptotic genes controlled by Foxo in prostate cancer derived cell lines [37, 38]. Experiments in mouse embryonic fibroblasts that are null for SIRT1 suggest that deacetylation of Foxo by SIRT1 selectively increases the expression of ROS resistance genes while having little effect on pro-apoptotic gene expression [33]. SIRT1 deacetylation of Foxo also seems to increase the ability of Foxo to induce cell cycle arrest. These two findings taken together suggest the role for deacetylation of Foxo, by SIRT1, is to pause the cell cycle in order to allow cells to repair DNA damage and remove ROS prior to cell division. SIRT2 has also been reported to upregulate transcription of Foxo targets that allow cells to cope with ROS, such as p27kip and manganese superoxide dismutase [39]. In addition over expression of SIRT2 is capable of inducing transcription of the pro-apoptotic factor Bim [40]. This scenario is quite reminiscent of p53 [30].
CBP/p300
The HATs CBP and p300 have two distinct activities on Foxo transcription. Acetylation of Foxo by CBP/p300 results in a decrease in Foxo transcriptional activity. CBP is able to acetylate Foxo at Lys242, 245 and 262 [9]. These residues are all located in the DNA binding domain of Foxo, and in vitro experiments using mutants which mimic the acetylated state at these residues (by replacing the basic lysine residues with Alanines or Glutamines) have severely diminished DNA binding capacity in vitro [9]. This is a mechanism through which CBP/p300 can act to inhibit Foxo-dependent transcription (Fig. 2).
The second activity of CBP/p300 is to acetylate histones in a manner that allows Foxo increased access to promoter regions [41]. One example of this activity is during treatment of cells with ROS. Treatment with ROS has been shown to increases the association of CBP/p300 with Foxo. This interaction has been shown to be mediated by a redox sensitive disulfide bridge that covalently links Foxo to CBP/p300 through Cys477 on Foxo [42]. This could potentially lead to acetylation of Foxo and decreased levels of Foxo transcription, yet ROS are known to increase the expression levels of many Foxo regulated genes. The most probable explanation for this result is that CBP/p300 acetylation of histones favorably remodels chromatin while SIRT1/2 prevents acetylation of Foxo and allows it to bind DNA. Thus while CBP/p300 can act as repressors of Foxo through their protein acetyltransferase activity, they also act as coactivators, increasing Foxo transcription through their histone acetyltransferase activity. A structural representation of how CBP/p300 binds Foxo and acts as a coactivator to increase Foxo transcription has recently been solved [41].
UBIQUITINATION OF FOXO
Ubiquitination plays a key role in the regulation of Foxo proteins. Foxo, like many other proteins, is targeted for proteasomal degradation through polyubiquitination. In addition to this canonical role for ubiquitination in protein degradation, monoubiquitination also plays a role in Foxo response to ROS. This is achieved by controlling transcripttional activity and localization of Foxo. This suggests a complex role for ubiquitination in Foxo signaling. Monoubiquitination of Foxo leads to activation of its transcriptional activity. This is balanced by polyubiquitination which leads to degradation – essentially depression of Foxo transcription through degradation. Therefore, from the monoubiquitinated state, polyubiquitination can be used for long term inactivation of Foxo transcription or deubiquitination can occur, allowing for quick reactivation of Foxo if necessary (Fig. 2).
Monoubiquitination by MDM2
Exposure of cells to ROS results in monoubiquitination of Foxo at multiple sites. This monoubiquitination is lost upon treatment of cells with MDM2 siRNA suggesting this E3 ligase directly ubiquitinates Foxo. Expression of Foxo-ubiquitin fusion constructs suggest that monoubiquitination of Foxo results in an increase in nuclear localization of Foxo as wellenhancement of Foxo transcriptional activity. While MDM2 likely acts as a monoubiquitinating E3 ligase for Foxo, this event probably primes Foxo for branching ubiquitination by SKP2. This is suggested by the observation that very high levels of MDM2 expression (or phosphorylation of Foxo by ERK), which increases MDM2-Foxo interaction, results in decreased levels of Foxo protein [43].
Mutant Foxo constructs and treatment with deacetylase inhibitors suggests that the lysine residues that are ubiquitinated are the same residues that are acetylated by CBP/p300 [44]. Under some circumstances a competition exists between the ubiquitin ligases and acetylases for these residues. This is potentially important as it suggests a posttranslationally modified state of Foxo where acetylases could interact with Foxo as cofactors (allowing optimal chromatin structure) without acetylating Foxo (which would inhibit its transcriptional activity). It is important to note that this state is independent of SIRT deacetylation.
Deubiquitination by USP7
Deubiquitination of Foxo is mediated by the deubiquitinating enzyme USP7. Foxo has been shown to interact with USP7 both through yeast two hybrid and immunoprecipitation experiments. This interaction is enhanced by increased levels of ROS with kinetics which are slower than the monoubiquitination of Foxo [44]. These observations suggest that Foxo exists in a dynamic equilibrium: Foxo monoubiquitination by MDM2 is induced by ROS, and this monoubiquitination is offset by USP7-dependent deubiquitination. Deubiquitinated Foxo is less transcriptionally active, thus USP7 acts as a negative regulator on Foxo transcription. While USP7 inhibits Foxo through deubiquitination it has no effect on protein levels of Foxo reinforcing the view that monoubiquitination of Foxo acts independent of protein degradation.
Polyubiquitination by SKP2
SKP2 physically interacts with Foxo in a phosphorylation-dependent manner. Phosphorylation of Foxo at Ser253 (preferentially phosphorylated by AKT) is required for Foxo SKP2 interaction and its subsequent ubiquitination [45]. SKP2 polyubiquitinates Foxo after binding and the ability of SKP2 to decrease Foxo protein levels is proteasome-dependent (it is abolished by MG132 treatment [46]). Elevated SKP2 levels are found in a wide variety of human cancers, and overexpression of this protein in mice leads to tumor formation. The ability to degrade Foxo (and thus eliminate its pro-apoptotic activity) may help explain the oncogenic properties of SKP2. This is also consistent with the oncogenic properties of PTEN deficiency. The loss of PTEN activity enhances AKT signaling and phosphorylation of Foxo at Ser253. This not only promotes nuclear exclusion of Foxo but polyubiquitination by SKP2 which leads to degradation of Foxo. This link between SKP2 and AKT phosphorylation also helps to explain the capacity of PTEN deficiency to induce tumorogenesis.
FOXO COFACTORS AND FOXO AS A COFACTOR
Foxo transcriptional activity can be affected by cofactors. These cofactors may have a general effect on Foxo transcriptional activity or they may confer Foxo transcriptional specificity. In addition, Foxo acts as a cofactor for a number of other transcription factors, affecting their transcriptional activity independently of Foxo’s own DNA binding activity. This increases the scope of Foxo activity beyond the already impressively large number of Foxo transcriptional processes.
β-Catenin Increases Foxo Transcriptional Activity
β-Catenin directly binds to Foxo and increases its transcriptional activity. ROS increase the interaction between β-catenin and Foxo [47]. This interaction may play a role in determining whether cells progress through the cell cycle or arrest. In the absence of ROS, β-catenin interacts with members of the T cell factor (TCF) family of transcription factors, which act to promote progression of the cell cycle. In Foxo Activity and Specificity contrast, increased Foxo transcription results in higher levels of cell cycle inhibitors such as p27kip. In this way the ROS dependent interaction between Foxo and β-catenin may act like a switch, allowing cells to pause and clear harmful ROS when Foxo and β-catenin interact. When oxidative stress subsides, β-catenin preferentially interacts with TCF, allowing cells to proceed through the cell cycle.
Foxo acts as a Corepressor of HIF1
As mentioned before, PTEN deficiency results in a lack of nuclear Foxo and is associated with tumorogenesis. PTEN-deficient tumors are known to be quite aggressive and highly vascular. This knowledge led to a study of the effects of PTEN on Hypoxia Inducible Factor 1 (HIF1) a key transcription factor controlling the expression of many angiogenic genes. PTEN alters HIF1 transcriptional activetion via Foxo’s cellular localization [48]. Even in PTEN deficient cells, if Foxo is localized to the nucleus, then HIF1 transactivation is inhibited. This is accomplished by Foxo complexing with HIF1 and interfering with HIFl’s interaction with its coactivator p300 [48]. This discovery adds to the complexity of Foxo’s anticancer properties. In addition to affecting cell cycle progression, clearing ROS, and pro-apoptotic capacity, Foxo also seems to act as a repressor of the HIF1 transcription factor and neovascularization.
Foxo is a Corepressor of the Androgen Receptor
Much like its effect on HIF-1, Foxo is capable of repressing the transcriptional activity of the Androgen Receptor (AR). This inhibition is not dependent on the transcriptional capacity of Foxo but simply on its nuclear localization. Transcriptionally inactive Foxo (mutant in its DNA binding domain) inhibits AR as robustly as the wild type protein [49]. Much like HIF-1, Foxo and the AR form a complex on AR promoter regions. HDAC3 is also a part of this complex and seems to play an important role in Foxo’s ability to inhibit AR transcription. Foxo inhibition of the AR prevents both adrenergic and non-adrenergic activation of theAR[49].
Foxo Acts as a Coactivator of Ets-1
There are 15 consensus phosphorylation sites on Foxo which are predicted to be regulated by members of the Mitogen Activated Protein Kinase (MAPK) family. Nine of the serine residues are capable of being phosphorylated in vivo by ERK and of these, five can also be phosphorylated by p38. Mutational analysis has revealed that phosphorylation at these sites can influence Foxo interaction with another transcription factor, Ets-1. Foxo interaction with Ets-1 leads to an increase in Ets-1 transcriptional activity as assessed by Ets-1 activity on the Flk-1 promoter, which promotes angiogenesis. Foxo has no activity at the Flk-1 promoter region but Ser to Ala mutations at the putative ERK/p38 sites that disrupt the Foxo-Ets-1 interaction greatly diminish Ets-1 interaction with Flk-1 [50]. This suggests that Foxo can act as a cofactor for other transcription factors indicating an even broader role of Foxo in transcriptional regulation. As discussed previously, phosphorylation of Foxo by ERK decreases Foxo stability potentially acting as a negative regulator of this signaling pathway.
FOXO SIGNALING AS A POTENTIAL THERAPEUTIC TARGET FOR DISEASE
As of yet, the only clinical use of drugs that target the Foxo signaling pathway is in cancer. Inactivation of Foxo through aberrant PTEN, AKT, IKK or ERK signaling have all been shown to promote tumorogenesis. A number of chemotherapeutic drugs have been shown to mediate their activity at least in part through restoring Foxo activity to cells, inducing Foxo pro-apoptotic target genes such as Bim. For example, Paclitaxel is known to increase Foxo activity through repression of AKT and induction of JNK signaling which cooperatively lead to nuclear localization and activation of Foxo [1]. While no drugs that function through the Foxo signaling pathway are currently available for treating diseases other than cancer, some research has suggested that under certain circumstances promoting Foxo signaling may be used in a pro-survival manner as well. The marine sponge compound Psammaplysene A has been shown to be neuroprotective both in vitro and in vivo against neurotoxic insults [2] in a Foxo depentent manner. This reinforces the observation that induction of Foxo can have varying activity based on the cellular context of Foxo activation. This raises the intriguing possibility of developing drugs in the future which act on Foxo to modulate its transcriptional profile. Current drug strategies take advantage of the fact that Foxo, through the integration of various posttranslational modifications, is able to activate pro-survival or pro-death genes depending on the state of cell. If we fully understood the signaling pathways that mediate Foxo gene expression choice, we could develop more potent drugs that not only activate Foxo, but ensure that it is transcribing only those genes which will give the greatest benefit in a specific disease state.
Acknowledgments
The Kalb Laboratory is supported by the United States Public Health Service (NS052325, NS060754 and NS064232).
References
- 1.Yang JY, Hung MC. A new fork for clinical application: targeting forkhead transcription factors in cancer. Clin Cancer Res. 2009;15:752–7. doi: 10.1158/1078-0432.CCR-08-0124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mojsilovic-Petrovic J, Nedelsky N, Boccitto M, et al. FOXO3a is broadly neuroprotective in vitro and in vivo against insults implicated in motor neuron diseases. J Neurosci. 2009;29:8236–47. doi: 10.1523/JNEUROSCI.1805-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barthelemy C, Henderson CE, Pettmann B. Foxo3a induces motoneuron death through the Fas pathway in cooperation with JNK. BMC Neurosci. 2004;5:48. doi: 10.1186/1471-2202-5-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brownawell AM, Kops GJ, Macara IG, Burgering BM. Inhibition of nuclear import by protein kinase B (Akt) regulates the subcellular distribution and activity of the forkhead transcription factor AFX. Molecular and cellular biology. 2001;21:3534–46. doi: 10.1128/MCB.21.10.3534-3546.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Biggs WH, 3rd, Meisenhelder J, Hunter T, Cavenee WK, Arden KC. Protein kinase B/Akt-mediated phosphorylation promotes nuclear exclusion of the winged helix transcription factor FKHR1. Proc Natl Acad Sci USA. 1999;96:7421–6. doi: 10.1073/pnas.96.13.7421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brunet A, Bonni A, Zigmond MJ, et al. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857–68. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- 7.Brunet A, Park J, Tran H, Hu LS, Hemmings BA, Greenberg ME. Protein kinase SGK mediates survival signals by phosphorylating the forkhead transcription factor FKHRL1 (FOXO3a) Mole Cel Biol. 2001;21:952–65. doi: 10.1128/MCB.21.3.952-965.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jacobs FM, van der Heide LP, Wijchers PJ, Burbach JP, Hoekman MF, Smidt MP. FoxO6, a novel member of the FoxO class of transcription factors with distinct shuttling dynamics. J Biol Chem. 2003;278:35959–67. doi: 10.1074/jbc.M302804200. [DOI] [PubMed] [Google Scholar]
- 9.Matsuzaki H, Daitoku H, Hatta M, Aoyama H, Yoshimochi K, Fukamizu A. Acetylation of Foxol alters its DNA-binding ability and sensitivity to phosphorylation. Proc Natl Acad Sci USA. 2005;102:11278–83. doi: 10.1073/pnas.0502738102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koul D. PTEN signaling pathways in glioblastoma. Cancer Biol Ther. 2008;7:1321–5. doi: 10.4161/cbt.7.9.6954. [DOI] [PubMed] [Google Scholar]
- 11.Majumder PK, Sellers WR. Akt-regulated pathways in prostate cancer. Oncogene. 2005;24:7465–74. doi: 10.1038/sj.onc.1209096. [DOI] [PubMed] [Google Scholar]
- 12.Dillon RL, White DE, Muller WJ. The phosphatidyl inositol 3-kinase signaling network: implications for human breast cancer. Oncogene. 2007;26:1338–45. doi: 10.1038/sj.onc.1210202. [DOI] [PubMed] [Google Scholar]
- 13.Reagan-Shaw S, Ahmad N. RNA interference-mediated depletion of phosphoinositide 3-kinase activates forkhead box class O transcription factors and induces cell cycle arrest and apoptosis in breast carcinoma cells. Cancer Res. 2006;66:1062–9. doi: 10.1158/0008-5472.CAN-05-1018. [DOI] [PubMed] [Google Scholar]
- 14.Knippschild U, Gocht A, Wolff S, Huber N, Lohler J, Stoter M. The casein kinase 1 family: participation in multiple cellular processes in eukaryotes. Cell Signal. 2005;17:675–89. doi: 10.1016/j.cellsig.2004.12.011. [DOI] [PubMed] [Google Scholar]
- 15.Rena G, Woods YL, Prescott AR, et al. Two novel phosphorylation sites on FKHR that are critical for its nuclear exclusion. EMBO J. 2002;21:2263–71. doi: 10.1093/emboj/21.9.2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Park J, Song WJ, Chung KC. Function and regulation of DyrklA: towards understanding Down syndrome. Cell Mol Life Sci. 2009;66:3235–40. doi: 10.1007/s00018-009-0123-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Woods YL, Rena G, Morrice N, et al. The kinase DYRK1A phosphorylates the transcription factor FKHR at Ser329 in vitro, a novel in vivo phosphorylation site. Biochem J. 2001;355:597–607. doi: 10.1042/bj3550597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ghosh S, Karin M. Missing pieces in the NF-kappaB puzzle. Cell. 2002;109(Suppl):S81–96. doi: 10.1016/s0092-8674(02)00703-1. [DOI] [PubMed] [Google Scholar]
- 19.Hu MC, Lee DF, Xia W, et al. IkappaB kinase promotes tumorigenesis through inhibition of forkhead FOXO3a. Cell. 2004;117:225–37. doi: 10.1016/s0092-8674(04)00302-2. [DOI] [PubMed] [Google Scholar]
- 20.Torii S, Nakayama K, Yamamoto T, Nishida E. Regulatory mechanisms and function of ERK MAP kinases. J Biochem. 2004;136:557–61. doi: 10.1093/jb/mvh159. [DOI] [PubMed] [Google Scholar]
- 21.Yang JY, Zong CS, Xia W, et al. ERK promotes tumorigenesis by inhibiting FOXO3a via MDM2-mediated degradation. Nat Cell Biol. 2008;10:138–48. doi: 10.1038/ncb1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Essers MA, Weijzen S, de Vries-Smits AM, et al. FOXO transcription factor activation by oxidative stress mediated by the small GTPase Ral and JNK. EMBO J. 2004;23:4802–12. doi: 10.1038/sj.emboj.7600476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yuan Z, Becker EB, Merlo P, et al. Activation of FOXO1 by Cdkl in cycling cells and postmitotic neurons. Science. 2008;319:1665–8. doi: 10.1126/science.1152337. [DOI] [PubMed] [Google Scholar]
- 24.Liu P, Kao TP, Huang H. CDK1 promotes cell proliferation and survival via phosphorylation and inhibition of FOXO1 transcription factor. Oncogene. 2008;27:4733–44. doi: 10.1038/onc.2008.104. [DOI] [PubMed] [Google Scholar]
- 25.Radu M, Chernoff J. The DeMSTification of mammalian Ste20 kinases. Curr Biol. 2009;19:R421–5. doi: 10.1016/j.cub.2009.04.022. [DOI] [PubMed] [Google Scholar]
- 26.Jang SW, Yang SJ, Srinivasan S, Ye K. Akt phosphorylates MstI and prevents its proteolytic activation, blocking FOXO3 phosphorylation and nuclear translocation. J Biol Chem. 2007;282:30836–44. doi: 10.1074/jbc.M704542200. [DOI] [PubMed] [Google Scholar]
- 27.Yuan Z, Lehtinen MK, Merlo P, Villen J, Gygi S, Bonni A. Regulation of neuronal cell death by MSTI-FOXO1 signaling. J Biol Chem. 2009;284:11285–92. doi: 10.1074/jbc.M900461200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ramaswamy S, Nakamura N, Sansal I, Bergeron L, Sellers WR. A novel mechanism of gene regulation and tumor suppression by the transcription factor FKHR. Cancer Cell. 2002;2:81–91. doi: 10.1016/s1535-6108(02)00086-7. [DOI] [PubMed] [Google Scholar]
- 29.Dijkers PF, Medema RH, Lammers JW, Koenderman L, Coffer PJ. Expression of the pro-apoptotic Bcl-2 family member Bim is regulated by the forkhead transcription factor FKHR-L1. Curr Biol. 2000;10:1201–4. doi: 10.1016/s0960-9822(00)00728-4. [DOI] [PubMed] [Google Scholar]
- 30.Stiewe T. The p53 family in differentiation and tumorigenesis. Nat Rev Cancer. 2007;7:165–8. doi: 10.1038/nrc2072. [DOI] [PubMed] [Google Scholar]
- 31.Greer EL, Oskoui PR, Banko MR, et al. The energy sensor AMP-activated protein kinase directly regulates the mammalian FOXO3 transcription factor. J Biol Chem. 2007;282:30107–19. doi: 10.1074/jbc.M705325200. [DOI] [PubMed] [Google Scholar]
- 32.Choi CH, Zimon A, Usheva A. Metabolic stress regulates basic transcription through acetyl-coenzyme A. Cell Mol Life Sci. 2005;62:625–8. doi: 10.1007/s00018-005-4516-6. [DOI] [PubMed] [Google Scholar]
- 33.Brunet A, Sweeney LB, Sturgill JF, et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004;303:2011–5. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- 34.Giannakou ME, Partridge L. The interaction between FOXO and SIRT1: tipping the balance towards survival. Trends Cell Biol. 2004;14:408–12. doi: 10.1016/j.tcb.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 35.Motta MC, Divecha N, Lemieux M, et al. Mammalian SIRT1 represses forkhead transcription factors. Cell. 2004;116:551–63. doi: 10.1016/s0092-8674(04)00126-6. [DOI] [PubMed] [Google Scholar]
- 36.van der Horst A, Tertoolen LG, de Vries-Smits LM, Frye RA, Medema RH, Burgering BM. FOXO4 is acetylated upon peroxide stress and deacetylated by the longevity protein hSir2(SIRTl) J Biol Chem. 2004;279:28873–9. doi: 10.1074/jbc.M401138200. [DOI] [PubMed] [Google Scholar]
- 37.Jung-Hynes B, Nihal M, Zhong W, Ahmad N. Role of sirtuin histone deacetylase SIRT1 in prostate cancer. A target for prostate cancer management via its inhibition? J Biol Chem. 2009;284:3823–32. doi: 10.1074/jbc.M807869200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang Y, Hou H, Haller EM, Nicosia SV, Bai W. Suppression of FOXO1 activity by FHL2 through SIRT1-mediated deacetylation. EMBO J. 2005;24:1021–32. doi: 10.1038/sj.emboj.7600570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Daitoku H, Hatta M, Matsuzaki H, et al. Silent information regulator 2 potentiates Foxol-mediated transcription through its deacetylase activity. Proc Natl Acad Sci USA. 2004;101:10042–7. doi: 10.1073/pnas.0400593101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang F, Nguyen M, Qin FX, Tong Q. SIRT2 deacetylates FOXO3a in response to oxidative stress and caloric restriction. Aging Cell. 2007;6:505–14. doi: 10.1111/j.1474-9726.2007.00304.x. [DOI] [PubMed] [Google Scholar]
- 41.Wang F, Marshall CB, Li GY, Yamamoto K, Mak TW, Ikura M. Synergistic Interplay between Promoter Recognition and CBP/p300 Coactivator Recruitment by FOXO3a. ACS Chem Biol. 2009;4:1017–27. doi: 10.1021/cb900190u. [DOI] [PubMed] [Google Scholar]
- 42.Dansen TB, Smits LM, van Triest MH, et al. Redox-sensitive cysteines bridge p300/CBP-mediated acetylation and FoxO4 activity. Nat Chem Biol. 2009;5:664–72. doi: 10.1038/nchembio.194. [DOI] [PubMed] [Google Scholar]
- 43.Brenkman AB, de Keizer PL, van den Broek NJ, Jochemsen AG, Burgering BM. Mdm2 induces mono-ubiquitination of FOXO4. PLoS One. 2008;3:e2819. doi: 10.1371/journal.pone.0002819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.van der Horst A, de Vries-Smits AM, Brenkman AB, et al. FOXO4 transcriptional activity is regulated by monoubiquitination and USP7/HAUSP. Nat Cell Biol. 2006;8(10):1064–73. doi: 10.1038/ncb1469. [DOI] [PubMed] [Google Scholar]
- 45.Matsuzaki H, Daitoku H, Hatta M, Tanaka K, Fukamizu A. Insulin-induced phosphorylation of FKHR (Foxo1) targets to proteasomal degradation. Proc Natl Acad Sci USA. 2003;100:11285–90. doi: 10.1073/pnas.1934283100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huang H, Regan KM, Wang F, et al. Skp2 inhibits FOXO1 in tumor suppression through ubiquitin-mediated degradation. Proc Natl Acad Sci USA. 2005;102:1649–54. doi: 10.1073/pnas.0406789102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Essers MA, de Vries-Smits LM, Barker N, Polderman PE, Burgering BM, Korswagen HC. Functional interaction between beta-catenin and FOXO in oxidative stress signaling. Science. 2005;308:1181–4. doi: 10.1126/science.1109083. [DOI] [PubMed] [Google Scholar]
- 48.Emerling BM, Weinberg F, Liu JL, Mak TW, Chandel NS. PTEN regulates p300-dependent hypoxia-inducible factor 1 transcriptional activity through Forkhead transcription factor 3a (FOXO3a) Proc Natl Acad Sci USA. 2008;105:2622–7. doi: 10.1073/pnas.0706790105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu P, Li S, Gan L, Kao TP, Huang H. A transcription-independent function of FOXO1 in inhibition of androgen-independent activation of the androgen receptor in prostate cancer cells. Cancer Res. 2008;68:10290–9. doi: 10.1158/0008-5472.CAN-08-2038. [DOI] [PubMed] [Google Scholar]
- 50.Asada S, Daitoku H, Matsuzaki H, et al. Mitogen-activated protein kinases, Erk and p38, phosphorylate and regulate Foxol. Cell Signal. 2007;19:519–27. doi: 10.1016/j.cellsig.2006.08.015. [DOI] [PubMed] [Google Scholar]