

Abstract
Background
Excessive gestational weight gain (GWG) increases risk of large for gestational age neonates and subsequent tracking of excess weight throughout the life course for both mother and child. Although the physiological mechanisms underlying these associations are incomplete, the insulin-like growth factor (IGF) axis has garnered attention for its role in fetal growth and development. Our purpose was to characterize the IGF axis protein expression patterns in mother–infant dyads in respect of excessive GWG.
Methods
We obtained fasting serum samples and corresponding cord blood from eight controls (ADHERE group: ie, those who gained in accordance with 2009 Institute of Medicine GWG recommendations) and 13 exceeders (EXCEED group: ie, those who exceeded Institute of Medicine GWG recommendations). At study completion, we examined protein expression of IGF-I, IGF-II, IGF binding protein (IGFBP)-1, IGFBP-3, IGFBP-4, and hormone concentrations in both maternal and cord blood.
Results
Between-group comparisons were made and revealed elevated maternal leptin (P ≤ 0.05) concentrations in gravidas who exceeded recommendations. There was a significantly higher number of obese women in the EXCEED group (P < 0.05). After adjustment, maternal leptin levels were positively correlated with maternal homeostasis model of assessment for insulin resistance score and excessive GWG (P ≤ 0.01). However, serum IGFBP-3 expression in the EXCEED mothers was greater than that in the ADHERE group (P ≤ 0.05).
Conclusion
These findings provide preliminary evidence suggesting that small deviations in IGFBP-regulated IGF bioavailability arising from excessive GWG/positive energy balance may affect adipocyte differentiation through subclinical insulin resistance.
Keywords: gestational weight gain, insulin-like growth factors, insulin-like growth factor binding protein-3, pregnancy, insulin sensitivity
Introduction
Excessive gestational weight gain (GWG), defined as that above the 2009 Institute of Medicine (IOM) recommendations,1 is associated with a greater likelihood of having a large for gestational age neonate2 with increased adiposity and insulin resistance.3 Similarly, excess GWG is also implicated in adverse maternal outcomes, including gestational diabetes,4 hypertension, and preeclampsia,5 as well as greater postpartum weight retention (PPWR).6 The insulin-like growth factor (IGF) axis plays a vital role in growth and development and regulates cellular differentiation, hypertrophy, and glycemic homeostasis.7 IGFs elicit their intracellular effects by interacting with the IGF type 1 receptor (IGF-1R) or IGF type 2 receptor (IGF-2R) and insulin–IGF receptor hybrids.8 Initial reports identified IGF binding protein (IGFBP)-3 as an insulin-antagonizing peptide, binding with high affinity to free IGF(s) and governing bioactivity. Recently, IGFBP-3 has been shown to act independent of the traditional receptors via nuclear translocation and is thereby capable of directly interrupting transcriptional regulation.8 In respect of excessive GWG, IGFBP-3 has been shown to inhibit adipocyte differentiation9 and induce insulin resistance in vivo and in vitro.10 Furthermore, IGF axis protein expression is compromised in pregnancies complicated by obesity.11 However, the influence that excessive GWG has on IGF and IGFBP expression during human pregnancy is unknown. Understanding differences in weight accretion during critical periods of maternal–fetal growth and development may lead to more efficacious obesity management strategies, as maternal insulin sensitivity and PPWR are known contributors to the intergenerational obesity cycle.12
Consequently, the IGF system may be a candidate pathway involved in maternal substrate storage and nutrient flux through the placenta during pregnancy where excessive weight is gained. Positive energy balance and aberrant hormone responses may contribute to alterations in maternal–fetal body composition. For instance, fetal adipogenesis peaks in late gestation,13 with more than 90% of fetal body fat accrued in the third trimester.14 In obese women, bioactive IGF-I is directly correlated with IGFBP-3 concentrations in blood,15 and IGF-I stimulates the differentiation of preadipocytes through modulation of the IGFBPs.16 At term, the pregnancy-associated rise in maternal IGFBP-3 serum level correlates positively with birth weight,17,18 whereas the increase in free IGF-I during pregnancy is thought to be mediated by IGFBP-3 proteolysis.19 Traditionally, IGFBP-1 acts to inhibit IGF function and is inversely related to insulin levels. Instances of chronic weight gain over time (ie, obesity) display low serum concentrations of IGFBP-1 that may regulate in vivo IGF-I bioavailability based on fuel supply.15 Lastly, although the exact function of IGFBP-4 is not yet understood, it may play a role in stem cell differentiation into muscle and fat and be modified in both pregnancy and weight gain.20 Thus, we aimed to examine protein expression of regulatory components of the IGF system in controls (ADHERE group: ie, those who gained in accordance with IOM GWG recommendations) and mothers and their term neonates who exceeded IOM recommendations (EXCEED group: ie, those who exceeded IOM GWG recommendations). Specifically, we determined whether IGF-I, IGF-II, and IGFBP-1, -3, and -4 expression differed between groups of mother-infant dyads. Given that mothers who gain excessively during pregnancy generally give birth to larger infants, we hypothesized that (1) the growth-enhancing components of the IGF system would be expressed to a greater extent, and (2) the growth-inhibiting proteins downregulated in pregnancies with excessive GWG
Methods and procedures
Participants
Patients were recruited from the Ottawa Hospital General Campus (a tertiary care center), Maternal Fetal Medicine, and Obstetrics clinics after the study was approved by the Ottawa Hospital Research Ethics Board. Informed consent was obtained from all participants. Women who smoked and those with type 1, type 2, or gestational diabetes or any other medical complication such as hypertension, cardiac disease, vascular disease, autoimmune disease, and thrombophilias and a past history of fetal loss, fetal growth restriction, or hypertensive diseases of pregnancy were excluded from either group. Women carrying fetuses with congenital or chromosomal anomalies were also excluded. All women were classified as glucose tolerant following their prenatal glucose challenge test, according to Canadian guidelines.
Sample collection
Fasting blood samples were obtained via peripheral venipuncture from 21 gravidas (eight who met and 13 who exceeded IOM GWG recommendations relative to prepregnancy body mass index [BMI]) immediately prior to term elective cesarean section, to avoid any potential influence of labor. Corresponding venous serum was obtained from the umbilical cord following removal of the placenta as clinically indicated. The placenta was then weighed to the nearest gram using a calibrated electronic scale (Olympic Smart Scale; Natus Medical Inc, Seattle, WA, USA). Following blood draws, the samples remained at room temperature for 30 minutes to allow for clotting, and were subsequently spun at 1,700 × g for 15 minutes at 4°C. The supernatant was removed aliquoted into separate cryovials, and frozen at −80°C for batch analysis. Maternal and fetal clinical variables were collected through a chart review upon study completion.
Determination of IGF-II via Western blot
The expression of IGF-II in human circulation was determined as previously described by Qiu et al.21 Briefly, aliquots of 0.5 μL of sera were diluted with 1X SDS nonreducing sample buffer (62.5 mMTris-HCl, pH 6.8, 2% sodium dodecyl sulfate [SDS], 10% glycerol, 0.01% Bromophenol Blue), and subjected to electrophoresis with 10% tricine SDS-polyacrylamide gel electrophoresis (PAGE). The separated proteins were blotted on to a nitrocellulose membrane, treated with an antibody extender solution (Pierce, Rockford, IL, USA), blocked with 5% dehydrated nonfat milk in Tris-buffered saline (pH 7 with 0.3% Tween 20), and subsequently probed with mouse anti-IGF-II(1–67) monoclonal antibody (clone S1F2; Upstate Biotechnology, Lake Placid, NY, USA) and horseradish peroxidase (HRP)-conjugated antimouse immunoglobulin G. Bands of IGF-II variants were visualized with enhanced chemiluminescence, and their relative contents quantified using densitometry with Alpha Ease FCTM software (Alpha Innotech, San Leandro, CA,USA). This assay can detect a quantity of recombinant human IGF-II as low as 6.25 pg.21
Determination of IGF-I and IGFBP-4 via Western blot
The presence of IGF-I and IGFBP-4 in human and cord serum was determined as described previously, with the following modifications. Aliquots of serum (0.5 μL) were resolved by 15% glycine SDS-PAGE under nonreducing conditions and electrotransferred to nitrocellulose membranes. The membranes were blocked for 1 hour in 5% milk, immunoblotted with highly specific antibodies for IGF-I (#ab9572; Abcam, CA, USA) and IGFBP-4 (#sc6005; Santa Cruz Biotech, CA, USA), and quantified as described previously.
Determination of IGFBP-1 and -3 via Western ligand blot
IGFBPs were determined using aliquots of 0.5 μL of sera subjected to electrophoresis, transfer, antibody extend solution treatment, and blocking agents as described previously, for the determination of IGF-I and -II. The membranes were subsequently probed with biotinylated IGF-II (GroPep Ltd, Adelaide, SA, Australia) (50 ng/mL prepared with Tris-buffered saline and Tween 20) at 4°C overnight, followed by antibody highly specific to IGFBP-1 (#sc55474; Santa Cruz Biotech) and IGFBP-3 (#sc135947; Santa Cruz Biotech) and finally by streptavidin-HRP (GE Healthcare, Buckinghamshire, UK) for 1 hour at room temperature. Bands of IGFBPs were visualized after being independently probed on separate days with highly specific antibodies targeting solely the protein of interest, with enhanced chemiluminescence, and their relative contents were quantified via densitometry with Alpha Ease FCTM software as described previously.
Biochemical analyses of glucose, insulin, and leptin
Glucose concentrations were measured by oxidase enzymatic methodology using the Cholestech LDX analyzer as described by the manufacturer (Hayward, CA, USA). Circulating levels of insulin and leptin were analyzed in duplicate by enzyme-linked immunosorbent assay using Luminex xMAP technology for the human metabolic hormone magnetic bead panel assay as described by the manufacturer (Millipore Corporation, Billerica, MA, USA). The intra- and interassay coefficients of variation were 3% and 6% for insulin and 3% and 4% for leptin.
Computation of outcome variables
GWG was calculated from anthropometric values obtained from the medical record using the difference of directly measured maternal weight at last prenatal visit minus directly measured prepregnancy weight obtained by the patient’s general practitioner. Percentage (%) exceeding GWG was calculated as the quotient of total GWG divided by the upper limit of recommended gain using the 2009 IOM guidelines relative to pregravid BMI, then multiplied by 100.1 This allowed us to categorize women into ADHERE (≤100% recommended GWG) or EXCEED (>100% recommended GWG) groups independent of pregravid BMI, and to assess the effect of relative excessive GWG based on evidence-informed guidelines for prepregnancy BMI. Homeostasis model of assessment for insulin resistance (HOMA-IR) was calculated as fasting glucose (mmol/L) × fasting insulin (pmol/L)/22.5 and subsequently log-transformed.22 Fetal–placental weight ratio was calculated as infant birth weight (grams [g])/placenta weight (g).
Statistical analysis
SPSS Statistics software version 19 was used for outcome analysis (IBM Corporation, Armonk, NY, USA). Protein band optical densities were log-transformed and presented as the mean ± standard deviation percentage above internal control. The internal control used was a pooled sample consisting of equal volumes of maternal and fetal sera taken from each subject in the study. All other outcome variables are presented as the mean ± standard deviation unless otherwise specified. A nonparametric two-tailed independent samples Kruskal–Wallis test was used to make conservative comparisons between groups for all patient characteristics as well as maternal–fetal levels of glucose, insulin, and leptin. Spearman correlations were used to examine relationships between maternal and fetal characteristics and outcomes of interest. Significance was set to P ≤ 0.05. Adjustments for multiple comparisons were made if necessary and presented within.
Results
Patient characteristics
Eight women who met and 13 who exceeded 2009 IOM GWG recommendations, of comparable age and height, were studied. To assess relative GWG, the mean percentage of women exceeding GWG guidelines based on prepregnancy BMI differed between the groups, as designed. Mean prepregnancy BMI and weight were greater in the EXCEED group, as expected, given the narrower range of acceptable gain as pregravid BMI increases (Table 1). In the ADHERE group, seven had pregravid BMIs classified as normal weight, whereas one was classified as obese. In the EXCEED group, four and nine women were categorized as normal weight and obese, respectively. However, birth weight, gestational age at delivery, prenatal glucose screen, placenta weight, and fetal–placental weight ratio did not differ between groups.
Table 1.
Patient characteristics
| Patient characteristics | GWG ADHERE | GWG EXCEED |
|---|---|---|
| Age (y) | 33.88 (6.6) | 32.31 (5.0) |
| Height (m) | 1.62 (0.9) | 1.63 (0.7) |
| Pregravid weight (kg) | 64.07 (20.0) | 86.92 (22.3)* |
| BMI (kg/m2) | 24.51 (8.7) | 32.75 (9.8)* |
| Parity | 2.38 (0.7) | 2.08 (0.4) |
| Gestational age (wk) | 38.38 (1.1) | 38.77 (0.9) |
| Prenatal GCT (mmol/L) | 6.36 (1.9) | 6.39 (1.3) |
| HOMA-IR (log) | 0.22 (0.1) | 0.52 (0.3) |
| Absolute GWG (kg) | 11.89 (3.6) | 14.97 (4.5) |
| % exceed IOM upper limit | 78.21 (18.0) | 136.82 (33.6)* |
| Infant birth weight (g) | 3,216.25 (505.8) | 3,547.77 (310.1) |
| Placenta weight (g) | 731.00 (157.9) | 742.83 (94.7) |
| Fetal–placental weight ratio | 4.47 (0.5) | 4.87 (0.6) |
Notes: Data presented mean (standard deviation).
Denotes P ≤ 0.05.
Abbreviations: BMI, body mass index; GCT, glucose challenge test; GWG, gestational weight gain; HOMA-IR, homeostasis model of assessment for insulin resistance; % exceed IOM, Institute of Medicine upper limit of GWG based on prepregnancy BMI.
IGF and IGFBP
Maternal protein expression of IGF-I, IGF-II, IGFBP-1, and IGFBP-4 and cord blood protein expression of IGF-I, IGF-II, IGFBP-1, IGFBP-3, and IGFBP-4 were similar between groups (P > 0.05). Protein expression of IGFBP-3 was increased in maternal serum from the EXCEED group when compared with the ADHERE group (P ≤ 0.05) (Figure 1).
Figure 1.
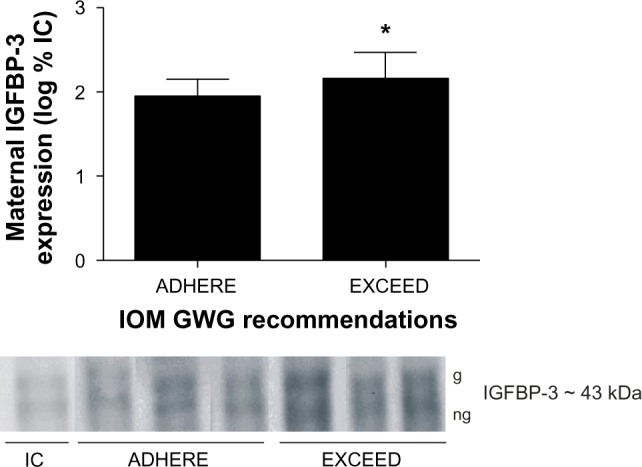
Excessive gestational weight gain augments maternal IGFBP-3 expression. Representative serum expression patterns of glycosylated (g) and nonglycosylated (ng) maternal IGFBP-3 isoforms of patients who adhere to (ADHERE) or exceed (EXCEED) the 2009 IOM GWG guidelines.
Note: *Denotes P ≤ 0.05.
Abbreviations: IC, internal control; GWG, gestational weight gain; IGFBP-3, insulin-like growth factor binding protein-3; IOM, Institute of Medicine.
Glucose, insulin, and leptin
Maternal glucose, insulin, and cord concentrations of insulin, glucose, and leptin did not differ between groups (Table 2). However, maternal leptin levels were elevated in the EXCEED group (Figure 2) and displayed a strong direct relationship with excess GWG and HOMA-IR (Figure 3 and Table 3). Following Bonferroni adjustment, nonsignificant trends (ie, where P < 0.05 prior to adjustment and P = 0.01–0.05 postadjustment) were observed between excessive GWG and infant birth weight, maternal IGFBP-4 expression, and HOMA-IR (Table 3).
Table 2.
Maternal and fetal outcome variables for mothers who adhere to (ADHERE) and exceed (EXCEED) GWG recommendations
| Maternal endpoints | GWG ADHERE | GWG EXCEED |
|---|---|---|
| IGF-I exp (log % IC) | 2.32 (0.1) | 2.24 (0.1) |
| IGF-II exp (log % IC) | 3.03 (0.3) | 2.95 (0.2) |
| IGFBP-1 exp (log % IC) | 2.09 (0.1) | 2.1 (0.1) |
| IGFBP-3 exp (log % IC) | 1.95 (0.2) | 2.16 (0.3)* |
| IGFBP-4 exp (log % IC) | 2.22 (0.1) | 2.08 (0.1) |
| Maternal insulin (log pg/mL) | 2.67 (0.1) | 2.81 (0.3) |
| Glucose (mmol/L) | 3.68 (0.5) | 4.10 (0.4) |
| Leptin (pg/mL) | 16,659.50 (11,300.0) |
29,560.33 (16,119.8)* |
| Fetal endpoints | ||
| IGF-I exp (log % IC) | 1.87 (0.2) | 1.67 (0.3) |
| IGF-II exp (log % IC) | 1.92 (0.1) | 1.92 (0.0) |
| IGFBP-1 exp (log % IC) | 1.95 (0.1) | 2.02 (0.1) |
| IGFBP-3 exp (log % IC) | 2.23 (0.2) | 2.19 (0.1) |
| IGFBP-4 exp (log % IC) | 2.11 (0.2) | 2.04 (0.1) |
| Cord insulin (log pg/mL) | 2.61 (0.3) | 2.83 (0.2) |
| Glucose (mmol/L) | 3.56 | 3.01 (0.1) |
| Leptin (pg/mL) | 182,70.71 (12319.1) |
25,837.75 (18,038.3) |
Notes: Data presented mean (standard deviation).
Denotes P ≤ 0.05.
Abbreviations: IGF, insulin-like growth factor; exp, protein expression relative to IC; GWG, gestational weight gain; IC, internal control; IGFBP, insulin-like growth factor binding protein.
Figure 2.
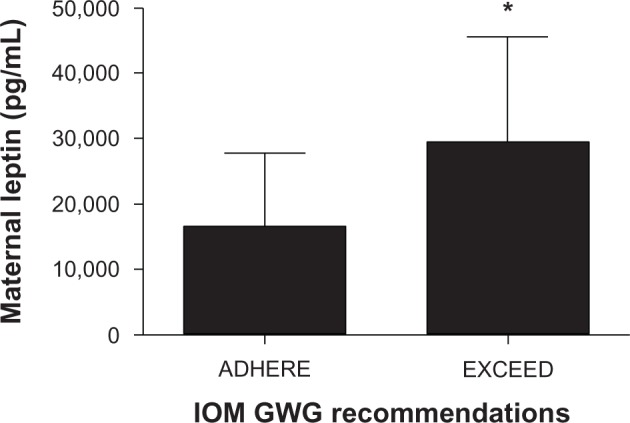
Maternal leptin level increase in women who exceed 2009 GWG recommendations.
Notes: n = 8 ADHERE group; n = 9 EXCEED group. *Denotes P ≤ 0.05.
Abbreviations: GWG, gestational weight gain; IOM, Institute of Medicine.
Figure 3.
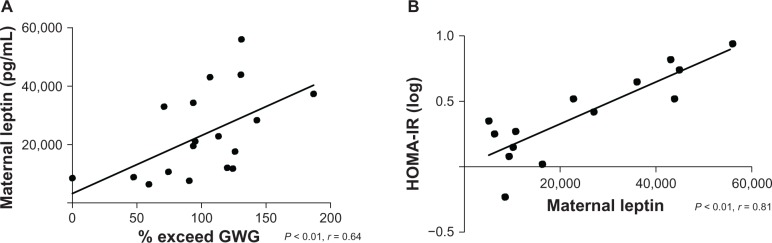
Maternal leptin levels positively correlate with (A) excessive GWG and (B) HOMA-IR.
Notes: Leptin (n = 20); % exceed upper limit of gestational weight gain (GWG) target recommendation based on prepregnancy body mass index; HOMA-IR (n = 14), homeostasis model of assessment for insulin resistance; Bonferroni adjustment for multiple comparisons P ≤ 0.01.
Table 3.
Spearman correlations between maternal and fetal outcomes
| Spearman correlations | % exceed GWG | Maternal leptin | HOMA-IR | Maternal IGFBP-4 expression | Birth weight |
|---|---|---|---|---|---|
| % exceed GWG | *0.64 | ***0.58 | ***0.44 | **0.47 | |
| Maternal leptin (pg/mL) | *0.81 | −0.21 | 0.33 | ||
| HOMA-IR (log) | −0.31 | 0.14 | |||
| Maternal IGFBP-4 expression (log % IC) | −0.20 | ||||
| Birth weight (g) | |||||
Notes: Significant correlations following Bonferroni adjustment for multiple comparisons
P ≤ 0.01 (ie, 5/0.05 = 0.01). Significant trends were identified when P < 0.05 prior to adjustment and P = 0.01–0.05 postadjustment. Therefore,
P = 0.02
P ≤ 0.05 for trend post-Bonferroni adjustment.
Abbreviations: HOMA-IR, homeostasis model of assessment for insulin resistance; IC, internal control; IGFBP-4, insulin-like growth factor binding protein-4; % exceed GWG, percent exceed upper limit for gestational weight gain based on prepregnancy body mass index.
Discussion
The present study illustrates a preliminary examination of the IGF axis in otherwise healthy mothers and newborns who adhere to and exceed 2009 IOM GWG guidelines. The fact that participants suffered no medical or obstetrical complications (ie, no gestational diabetes mellitus [GDM] or hypertension) provided us with a unique opportunity to examine the influence of excessive GWG without the potential effects of the commonly associated complications or comorbidities. This investigation reveals a small, albeit significant, difference in the IGF axis in the EXCEED group versus the ADHERE group: specifically, augmented expression of IGFBP-3 in maternal blood from normoglycemic pregnancies where GWG is excessive. Given our design, we are unable to assess causality. However, the metabolic dysregulation of maternal growth-controlling factors in excessive gainers aligns with previous observations, demonstrating that IGFBP-3 induces insulin resistance in adipocytes10 via inhibition of adipocyte differentiation,9 thus modifying metabolic homeostasis.
Although induced during adipocyte differentiation, IGFBP-3 can paradoxically inhibit this process.23 As adipogenesis proceeds, increasing adipose tissue IGFBP-3 levels may signal a feedback mechanism, limiting further differentiation of preadipocytes.9 In fact, the dual regulatory role of IGFBP-3 in differentiation has been identified in other cell types, including myoblasts,24 keratinocytes,25 and chondrocytes.26 As such, it is possible that augmented protein expression of maternal IGFBP-3 may be, in part, due to a feedback response to excessive weight gain, whereby IGFBP-3 sequesters free IGFs, preventing further differentiation of adipocytes and antagonizing IGF-induced glucose uptake and insulin-like activity. Previous investigations have demonstrated that IGFBP-310,27 inhibits insulin-mediated glucose uptake in adipocytes, and have suggested that the increase in free IGF-I during pregnancy is mediated by IGFBP-3 proteolysis.19 In fact, Chan et al,9 using 3T3-L1 adipocytes, demonstrated that IGFBP-3 inhibits the peroxisome proliferator activated receptor-γ-dependent process of adipocyte differentiation by inhibiting the dimerization of retinoid X receptor-α and peroxisome proliferator activated receptor-γ. Taken together, these findings suggest that IGFBP-3 has the potential to disrupt glycemic control and substrate uptake in adipose tissue. For instance, Kim et al10 convincingly demonstrate that IGFBP-3 induces insulin resistance in adipocytes in vitro and in vivo. Furthermore, Nguyen et al28 show that mice overexpressing human IGFBP-3 in vivo have delayed insulin clearance and reduced glucose-stimulated insulin secretion in pancreatic islets acting by both IGF-dependent and IGF-independent mechanisms. Although several physiological factors are suspected to regulate IGFBP-3-induced alteration of glycemic control, we cannot preclude the possibility that augmented IGFBP-3 with excessive weight gain and obesity may preferentially elicit a hypertrophic response in existing adipocytes at the expense of attenuated adipogenesis. This may predispose women to preferentially store substrate in existing adipocytes, a fuel partitioning strategy that is metabolically disadvantageous when adipocyte proliferation rates are low.29,30 If excessive GWG with obesity compromises functional components of the IGF axis, as demonstrated in the present investigation, this suggests that the IGF axis may be implicated in maternal fat accretion, leptin metabolism, and insulin resistance.
It is known that serum levels of leptin correlate with total body fat and insulin resistance31 in pregnant32 and nonpregnant women.31–33 In a group of healthy pregnant women in the first trimester,34 as well as those with gestational diabetes,35 serum leptin positively relates with insulin resistance, a finding we substantiate at term in pregnancies with excessive GWG and obesity. Given that serum leptin concentrations decrease following delivery, it has been argued36 that factors other than fat mass alone cause increased leptin production throughout pregnancy, including placental contributions. However, Eriksson et al37 showed that serum leptin was elevated in gestational week 8 when increases in fat mass are minimal and when the placenta is not fully established, providing support that increased leptin levels are not only due to increased fat mass but influenced by other unknown factors. These findings parallel with our observation in women who gain excessively, as leptin levels have been reported to be commensurate with adipose stores in pregnant women at term.32,37 Although we acknowledge the possibility that the observed differences in maternal leptin at the end of pregnancy may simply reflect between-group differences in pregravid BMI, Shaarawy and el-Mallah,38 comparing nonpregnant and pregnant women matched for pregravid BMI, determined that elevated serum leptin was directly associated with maternal adiposity of pregnancy. Furthermore, in observations from a randomized controlled trial with and without GWG restriction in BMI-matched obese pregnant women, Wolff et al39 showed that when patients in the intervention group successfully limited food intake and restricted GWG, a significant 20% reduction in both insulin and leptin was observed. Lastly, in our ADHERE group, only a single woman was categorized as obese. Collectively, these results suggest that appropriate GWG may attenuate adipose accretion and the commensurate leptin response, whereas excessive GWG may exacerbate maternal fat accretion, thus raising leptin levels.
Further, we present a strong direct relationship between maternal leptin levels and HOMA-IR, supporting previous work identifying pregnancy as both an insulin- and leptin-resistant state,32 partly mediated by increasing adiposity during pregnancy.37 Indeed, Eriksson et al37 showed that total body fat percentage was significantly correlated with HOMA-IR and with serum leptin before and during pregnancy, whereas leptin correlated with HOMA-IR, identifying a complex interactive role of maternal leptin, adiposity, and insulin sensitivity. Although we cannot make conclusive inferences about the composition of excessive GWG based on the results of our study, previous work suggests that high maternal fat content predominates and stimulates fetal growth, as evidenced by data showing that maternal total body fat40 and excessive GWG2 positively relate to birth weight, and that neonates born to mothers who gain in excess have higher body fat.41 Furthermore, the pregnancy-enhancing effect on the relationship between maternal body fatness and insulin resistance37 may represent a physiological mechanism predisposing pregnant women to preferentially store body fat as weight is gained. It is now widely accepted that maternal body size affects fetal growth and metabolic homeostasis.3,42 Our findings, in addition to others’ findings,43 suggest that maternal body size and weight gain are important factors determining the amount of dietary energy required by the developing child. Excessive GWG with obesity may present a mechanism by which offspring size and body composition are regulated via alterations in the IGF axis in response to energy availability in the maternal milieu. Although we were unable to assess maternal or fetal body composition, we cannot exclude the hypothesis that GWG-induced perturbations in IGF axis function influence maternal–fetal body composition – a phenomenon thoroughly documented in mother–infant dyads who gain excessively and exacerbated as pregravid BMI increases, leading to greater relative fat mass accretion.6 Therefore, future longitudinal investigations should examine the influence of physical activity energy expenditure and dietary intake on IGF axis dynamics and maternal–fetal body composition to confirm our findings in a larger sample.
The limitations of our study include the cross-sectional design, relatively small sample size, and inability to control for maternal dietary intake and physical activity patterns. However, we present a novel finding identified with validated and highly specific methodology for the outcomes of interest, while controlling for the acute effects of postprandial metabolism, as all women were fasted for at least 12 hours. Through this design, we also attempted to control for diurnal variation in IGF metabolism, given that all blood was drawn at the same time of day (ie, the morning upon arrival to hospital admittance and fasting). Further, our study population was homogenous in nature, as all women were normoglycemic as defined by clinical standards. In an attempt to avoid a confounding effect of mode of delivery on cord blood IGF profile, all women in our study underwent elective cesarean section. Lastly, we used the most recent 2009 evidence-based recommendations for GWG as outlined by the IOM.1
Conclusion
Overall, this study provides further support for the clinical value and use of the 2009 IOM GWG recommendations in primary care, as women and providers can track and monitor weight status throughout pregnancy to reduce not only the likelihood of impairing maternal–fetal metabolic signals but also the incidence of fetal overgrowth.2 Specifically, if altering weight prepregnancy is not a feasible option, being aware that GWG is a modifiable risk factor may motivate women to gain within the limits in order to optimize maternal–fetal health, chiefly PPWR and infant birth weight, known contributors to the intergenerational obesity cycle. Our results align with epidemiological data suggesting that excessive GWG and obesity influence maternal growth signals during pregnancy. Although it appears that IGFBP-3 attempts to sequester free IGFs and prevent undesired increases in IGF bioactivity, the potential induction of impaired glycemic control renders excessive gain harmful. Therefore, this study suggests that small deviations in IGFBP-regulated IGF bioavailability arising from excessive weight gain and obesity may influence adipocyte differentiation and glycemic control.
Acknowledgments
We are indebted to the Ottawa Hospital General Campus Labour and Delivery Team for their assistance with patient recruitment, and to Dr Nina Fowler for her assistance with laboratory assays. This work was supported by the University of Ottawa, Faculty of Health Sciences/Children’s Hospital of Eastern Ontario Research Partnership Grant awarded to KBA and ZMF and a W Garfield Weston Foundation Award in Obesity and Reproductive Health given to AG and KBA. The Ministry of Research and Innovation–Early Researcher Award (KBA) provided graduate funding for ZMF, who was also a recipient of an Ontario Graduate Scholarship from the Ontario Ministry of Training, Colleges, and Universities.
Footnotes
Author contributions
ZMF conceived the study, recruited the patients, collected the samples, ran the experiments, analyzed the data, and led manuscript drafting. QQ supervised the experiments and assisted with data analysis. AG assisted with study design, helped interpret the data, and critically reviewed the manuscript. KBA assisted with study design, data interpretation, and critical review of the manuscript. All authors drafted and/or revised, then approved the final manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Institute of Medicine . Weight gain during pregnancy: reexamining the guidelines. Washington, DC: The National Academies Press; 2009. [PubMed] [Google Scholar]
- 2.Ferraro ZM, Barrowman N, Prud’homme D, et al. Excessive gestational weight gain predicts large for gestational age neonates independent of maternal body mass index. J Matern Fetal Neonatal Med. 2012;25:538–542. doi: 10.3109/14767058.2011.638953. [DOI] [PubMed] [Google Scholar]
- 3.Catalano PM, Presley L, Minium J, Hauguel-de MS. Fetuses of obese mothers develop insulin resistance in utero. Diabetes Care. 2009;32:1076–1080. doi: 10.2337/dc08-2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hedderson MM, Gunderson EP, Ferrara A. Gestational weight gain and risk of gestational diabetes mellitus. Obstet Gynecol. 2010;115:597–604. doi: 10.1097/AOG.0b013e3181cfce4f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fortner RT, Pekow P, Solomon CG, Markenson G, Chasan-Taber L. Prepregnancy body mass index, gestational weight gain, and risk of hypertensive pregnancy among Latina women. Am J Obstet Gynecol. 2009;200:167. doi: 10.1016/j.ajog.2008.08.021. [DOI] [PubMed] [Google Scholar]
- 6.Butte NF, Ellis KJ, Wong WW, Hopkinson JM, Smith EO. Composition of gestational weight gain impacts maternal fat retention and infant birth weight. Am J Obstet Gynecol. 2003;189:1423–1432. doi: 10.1067/s0002-9378(03)00596-9. [DOI] [PubMed] [Google Scholar]
- 7.Juul A. Serum levels of insulin-like growth factor I and its binding proteins in health and disease. Growth Horm IGF Res. 2003;13:113–170. doi: 10.1016/s1096-6374(03)00038-8. [DOI] [PubMed] [Google Scholar]
- 8.Jogie-Brahim S, Feldman D, Oh Y. Unraveling insulin-like growth factor binding protein-3 actions in human disease. Endocr Rev. 2009;30:417–437. doi: 10.1210/er.2008-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chan SS, Schedlich LJ, Twigg SM, Baxter RC. Inhibition of adipocyte differentiation by insulin-like growth factor-binding protein-3. Am J Physiol Endocrinol Metab. 2009;296:E654–E663. doi: 10.1152/ajpendo.90846.2008. [DOI] [PubMed] [Google Scholar]
- 10.Kim HS, Ali O, Shim M, et al. Insulin-like growth factor binding protein-3 induces insulin resistance in adipocytes in vitro and in rats in vivo. Pediatr Res. 2007;61:159–164. doi: 10.1203/pdr.0b013e31802d8a30. [DOI] [PubMed] [Google Scholar]
- 11.Ferraro ZM, Qiu Q, Gruslin A, Adamo KB. Characterization of the insulin-like growth factor axis in term pregnancies complicated by maternal obesity. Hum Reprod. 2012;27:2467–2475. doi: 10.1093/humrep/des189. [DOI] [PubMed] [Google Scholar]
- 12.Oken E. Maternal and child obesity: the causal link. Obstet Gynecol Clin North Am. 2009;36:361–377. doi: 10.1016/j.ogc.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 13.Mandrup S, Lane MD. Regulating adipogenesis. J Biol Chem. 1997;272:5367–5370. doi: 10.1074/jbc.272.9.5367. [DOI] [PubMed] [Google Scholar]
- 14.Widdowson E. Changes in body proportions and composition during growth. In: Davis J, Dobbing J, editors. Scientific Foundations of Pediatrics. London, UK: Heinemann; 1974. pp. 153–163. [Google Scholar]
- 15.Frystyk J, Brick DJ, Gerweck AV, Utz AL, Miller KK. Bioactive insulin-like growth factor-I in obesity. J Clin Endocrinol Metab. 2009;94:3093–3097. doi: 10.1210/jc.2009-0614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boney CM, Moats-Staats BM, Stiles AD, D’Ercole AJ. Expression of insulin-like growth factor-I (IGF-I) and IGF-binding proteins during adipogenesis. Endocrinology. 1994;135:1863–1868. doi: 10.1210/endo.135.5.7525256. [DOI] [PubMed] [Google Scholar]
- 17.Giudice LC, de Zegher F, Gargosky SE, et al. Insulin-like growth factors and their binding proteins in the term and preterm human fetus and neonate with normal and extremes of intrauterine growth. J Clin Endocrinol Metab. 1995;80:1548–1555. doi: 10.1210/jcem.80.5.7538146. [DOI] [PubMed] [Google Scholar]
- 18.Osorio M, Torres J, Moya F, et al. Insulin-like growth factors (IGFs) and IGF binding proteins-1, -2, and -3 in newborn serum: relationships to fetoplacental growth at term. Early Hum Dev. 1996;46:15–26. doi: 10.1016/0378-3782(96)01737-9. [DOI] [PubMed] [Google Scholar]
- 19.Hasegawa T, Hasegawa Y, Takada M, Tsuchiya Y. The free form of insulin-like growth factor I increases in circulation during normal human pregnancy. J Clin Endocrinol Metab. 1995;80:3284–3286. doi: 10.1210/jcem.80.11.7593439. [DOI] [PubMed] [Google Scholar]
- 20.Du M, Yan X, Tong JF, Zhao J, Zhu MJ. Maternal obesity, inflammation, and fetal skeletal muscle development. Biol Reprod. 2010;82:4–12. doi: 10.1095/biolreprod.109.077099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Qiu Q, Jiang JY, Bell M, Tsang BK, Gruslin A. Activation of endoproteolytic processing of insulin-like growth factor-II in fetal, early postnatal, and pregnant rats and persistence of circulating levels in postnatal life. Endocrinology. 2007;148:4803–4811. doi: 10.1210/en.2007-0535. [DOI] [PubMed] [Google Scholar]
- 22.Vogeser M, Konig D, Frey I, Predel HG, Parhofer KG, Berg A. Fasting serum insulin and the homeostasis model of insulin resistance (HOMA-IR) in the monitoring of lifestyle interventions in obese persons. Clin Biochem. 2007;40:964–968. doi: 10.1016/j.clinbiochem.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 23.Wabitsch M, Heinze E, Debatin KM, Blum WF. IGF-I- and IGFBP-3-expression in cultured human preadipocytes and adipocytes. Horm Metab Res. 2000;32:555–559. doi: 10.1055/s-2007-978685. [DOI] [PubMed] [Google Scholar]
- 24.Foulstone EJ, Savage PB, Crown AL, Holly JM, Stewart CE. Role of insulin-like growth factor binding protein-3 (IGFBP-3) in the differentiation of primary human adult skeletal myoblasts. J Cell Physiol. 2003;195:70–79. doi: 10.1002/jcp.10227. [DOI] [PubMed] [Google Scholar]
- 25.Edmondson SR, Thumiger SP, Kaur P, et al. Insulin-like growth factor binding protein-3 (IGFBP-3) localizes to and modulates proliferative epidermal keratinocytes in vivo. Br J Dermatol. 2005;152:225–230. doi: 10.1111/j.1365-2133.2004.06350.x. [DOI] [PubMed] [Google Scholar]
- 26.Spagnoli A, Hwa V, Horton WA, et al. Antiproliferative effects of insulin-like growth factor-binding protein-3 in mesenchymal chondrogenic cell line RCJ3.1C5.18. relationship to differentiation stage. J Biol Chem. 2001;276:5533–5540. doi: 10.1074/jbc.M005088200. [DOI] [PubMed] [Google Scholar]
- 27.Chan SS, Twigg SM, Firth SM, Baxter RC. Insulin-like growth factor binding protein-3 leads to insulin resistance in adipocytes. J Clin Endocrinol Metab. 2005;90:6588–6595. doi: 10.1210/jc.2005-0595. [DOI] [PubMed] [Google Scholar]
- 28.Nguyen KH, Yao XH, Moulik S, Mishra S, Nyomba BL. Human IGF binding protein-3 overexpression impairs glucose regulation in mice via an inhibition of insulin secretion. Endocrinology. 2011;152:2184–2196. doi: 10.1210/en.2010-1324. [DOI] [PubMed] [Google Scholar]
- 29.Arner E, Westermark PO, Spalding KL, et al. Adipocyte turnover: relevance to human adipose tissue morphology. Diabetes. 2010;59:105–109. doi: 10.2337/db09-0942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lonn M, Mehlig K, Bengtsson C, Lissner L. Adipocyte size predicts incidence of type 2 diabetes in women. FASEB J. 2010;24:326–331. doi: 10.1096/fj.09-133058. [DOI] [PubMed] [Google Scholar]
- 31.Silha JV, Nyomba BL, Leslie WD, Murphy LJ. Ethnicity, insulin resistance, and inflammatory adipokines in women at high and low risk for vascular disease. Diabetes Care. 2007;30:286–291. doi: 10.2337/dc06-1073. [DOI] [PubMed] [Google Scholar]
- 32.Highman TJ, Friedman JE, Huston LP, Wong WW, Catalano PM. Longitudinal changes in maternal serum leptin concentrations, body composition, and resting metabolic rate in pregnancy. Am J Obstet Gynecol. 1998;178:1010–1015. doi: 10.1016/s0002-9378(98)70540-x. [DOI] [PubMed] [Google Scholar]
- 33.Considine RV. Human leptin: an adipocyte hormone with weight-regulatory and endocrine functions. Semin Vasc Med. 2005;5:15–24. doi: 10.1055/s-2005-871738. [DOI] [PubMed] [Google Scholar]
- 34.Mastorakos G, Valsamakis G, Papatheodorou DC, et al. The role of adipocytokines in insulin resistance in normal pregnancy: visfatin concentrations in early pregnancy predict insulin sensitivity. Clin Chem. 2007;53:1477–1483. doi: 10.1373/clinchem.2006.084731. [DOI] [PubMed] [Google Scholar]
- 35.McLachlan KA, O’Neal D, Jenkins A, Alford FP. Do adiponectin, TNFalpha, leptin and CRP relate to insulin resistance in pregnancy? Studies in women with and without gestational diabetes, during and after pregnancy. Diabetes Metab Res Rev. 2006;22:131–138. doi: 10.1002/dmrr.591. [DOI] [PubMed] [Google Scholar]
- 36.Bajoria R, Sooranna SR, Ward BS, Chatterjee R. Prospective function of placental leptin at maternal-fetal interface. Placenta. 2002;23:103–115. doi: 10.1053/plac.2001.0769. [DOI] [PubMed] [Google Scholar]
- 37.Eriksson B, Lof M, Olausson H, Forsum E. Body fat, insulin resistance, energy expenditure and serum concentrations of leptin, adiponectin and resistin before, during and after pregnancy in healthy Swedish women. Br J Nutr. 2010;103:50–57. doi: 10.1017/S0007114509991371. [DOI] [PubMed] [Google Scholar]
- 38.Shaarawy M, el-Mallah SY. Leptin and gestational weight gain: relation of maternal and cord blood leptin to birth weight. J Soc Gynecol Investig. 1999;6:70–73. doi: 10.1016/s1071-5576(99)00003-9. [DOI] [PubMed] [Google Scholar]
- 39.Wolff S, Legarth J, Vangsgaard K, Toubro S, Astrup A. A randomized trial of the effects of dietary counseling on gestational weight gain and glucose metabolism in obese pregnant women. Int J Obes (Lond) 2008;32:495–501. doi: 10.1038/sj.ijo.0803710. [DOI] [PubMed] [Google Scholar]
- 40.Forsum E, Lof M, Olausson H, Olhager E. Maternal body composition in relation to infant birth weight and subcutaneous adipose tissue. Br J Nutr. 2006;96:408–414. doi: 10.1079/bjn20061828. [DOI] [PubMed] [Google Scholar]
- 41.Hull HR, Thornton JC, Ji Y, et al. Higher infant body fat with excessive gestational weight gain in overweight women. Am J Obstet Gynecol. 2011;205:211–217. doi: 10.1016/j.ajog.2011.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Catalano PM, Thomas A, Huston-Presley L, Amini SB. Increased fetal adiposity: a very sensitive marker of abnormal in utero development. Am J Obstet Gynecol. 2003;189:1698–1704. doi: 10.1016/s0002-9378(03)00828-7. [DOI] [PubMed] [Google Scholar]
- 43.King JC. Physiology of pregnancy and nutrient metabolism. Am J Clin Nutr. 2000;71:1218S–1225S. doi: 10.1093/ajcn/71.5.1218s. [DOI] [PubMed] [Google Scholar]