

Background: RecN is an SMC (structural maintenance of chromosomes) family protein that is required for DNA double-strand breaks (DSBs) repair.
Results: We identified a RecA mutant that is deficient in interacting with RecN.
Conclusion: A functional interaction between RecN and RecA is required for assembly of RecN at the sites of DSBs.
Significance: RecN is critical for protecting the structural integrity of chromosomes during DSBs repair.
Keywords: ATPases, DNA Damage, DNA Damage Response, DNA Recombination, DNA Repair, DNA-Protein Interaction, DNA Double-strand Breaks
Abstract
Escherichia coli RecN is an SMC (structural maintenance of chromosomes) family protein that is required for DNA double-strand break (DSB) repair. Previous studies show that GFP-RecN forms nucleoid-associated foci in response to DNA damage, but the mechanism by which RecN is recruited to the nucleoid is unknown. Here, we show that the assembly of GFP-RecN foci on the nucleoid in response to DNA damage involves a functional interaction between RecN and RecA. A novel RecA allele identified in this work, recAQ300R, is proficient in SOS induction and repair of UV-induced DNA damage, but is deficient in repair of mitomycin C (MMC)-induced DNA damage. Cells carrying recAQ300R fail to recruit RecN to DSBs and accumulate fragmented chromosomes after exposure to MMC. The ATPase-deficient RecNK35A binds and forms foci at MMC-induced DSBs, but is not released from the MMC-induced DNA lesions, resulting in a defect in homologous recombination-dependent DSB repair. These data suggest that RecN plays a crucial role in homologous recombination-dependent DSB repair and that it is required upstream of RecA-mediated strand exchange.
Introduction
DNA double-strand breaks (DSBs)3 are serious genomic lesions that are potentially lethal at the cellular level. DSBs are caused by exogenous agents such as ionizing radiation, chemical mutagens, reactive oxygen species, and replicative stress (i.e. collapsed replication forks) (1, 2). In bacteria, homologous recombination (HR) plays a major role in repairing DSBs. HR enzymes and pathways have been extensively characterized in Escherichia coli, and E. coli HR is a paradigm for understanding HR-related processes in all organisms (3–5).
In E. coli, the repair of DSBs is initiated by RecBCD, which generates 3′ single-stranded DNA (ssDNA) tails at DSB sites via its helicase and nuclease activities; the ssDNA tails are then substrates for homologous pairing by RecA protein (4, 6). The RecF pathway is involved in the daughter strand gap repair in wild-type cells (3). RecF also provides an alternative pathway for HR-dependent DSB repair in recBC mutants when two additional nucleases, ExoI and SbcCD, are inactivated (7, 8). However, recent studies suggest that the RecFOR pathway may play a crucial role in DSB repair in bacterial species other than E. coli (9–13). In both pathways, RecA is loaded onto the ssDNA tail to form a nucleoprotein filament at the DSB sites. The RecA strand exchange activity generates recombination intermediates via its strand exchange activity (4, 14), which are then processed either by the Holliday junction resolvase, RuvABC, or by the RecG helicase to produce mature products (15–17). In addition, RecA is essential for the induction of the SOS response (18). RecA assembled on ssDNA stimulates self-cleavage of the LexA repressor, which in turn induces downstream SOS genes (19–21).
Structural maintenance of chromosomes (SMC) proteins are ubiquitous proteins that maintain and modulate chromosome structure in prokaryotic and eukaryotic cells (22, 23). E. coli RecN is a highly conserved, DNA damage-inducible SMC-like protein in bacteria that has two SOS boxes in its promoter region. Therefore, the expression of recN is tightly regulated by the LexA repressor (24–26). GFP-RecN forms nucleoid-associated foci in response to DNA damage and forms aggregates in the cytoplasm (27). RecN aggregates are then degraded by the ClpXP protease (27, 28), which is required for efficient recovery from DNA damage. E. coli recN mutants are highly sensitive to ionizing radiation, bleomycin, and the DNA cross-linking agent mitomycin C (MMC), but not to UV irradiation (29, 30). Mutants of recN are defective in conjugational recombination in recBC sbcBC strains (24), suggesting that RecN plays a role in the RecF pathway. However, RecN is also required for RecBCD-dependent repair of DSBs (31–33). Thus, RecN plays a specific role in the repair of DNA DSBs, and its role is not limited to a single branch or subpathway of HR.
In this study, we examine the mechanism by which RecN is recruited to the nucleoid in response to DNA damage. We show that a functional interaction between RecN and RecA is required for assembly of RecN foci at MMC-induced DSBs; conversely, conditions that abrogate or disrupt a stable RecN-RecA interaction lead to chromosome fragmentation and loss of cell viability in cells exposed to MMC. The RecN ATPase is not required for formation of RecN-DSB foci, but is required for release of RecN from DSBs and completion of RecA/HR-dependent DSB repair. These data demonstrate that the SMC-like protein RecN plays a crucial role in promoting RecA-dependent DSB repair.
EXPERIMENTAL PROCEDURES
Media and General Methods
Standard methods for E. coli genetics and recombinant DNA techniques were as described by Miller (34) and Sambrook et al. (35). Ampicillin (50 μg/ml), tetracycline (10 μg/ml), chloramphenicol (100 μg/ml), and kanamycin (30 μg/ml) were used where indicated. Mitomycin C (2.5 mg/ml) was dissolved in 10 mm Tris-HCl (pH 8.5) buffer. Sensitivity to UV damage was measured as described previously (36). To measure sensitivity to MMC, cultures were grown in LB broth to an A650 of ∼0.4, serially diluted, spotted onto LB medium containing the indicated concentration of MMC, and incubated at 37 °C.
Bacterial Strains and Plasmids
Strains used in this study were isogenic with BW25141 (37) except for strains with PBAD-I-SceI. Wild-type strains and deletion mutants were provided by the National BioResource Project (NBRP) (38). The strains carrying the PBAD-I-SceI were a gift from S. M. Rosenberg (39). The strains carrying the inducible fluorescent repressor gene (araC PBAD-lacI-ecfp) and the lacO array were described previously (40). A fragment containing the SOS promoter and open reading frame of recN was cloned into the low copy plasmid pSCH19, generating pRecN (27). RecN was tagged with an enhanced GFP cassette at its NH2 terminus to generate pSG101. Arabinose-inducible pBAD GFP-recN (pTF271) was constructed as described previously (27). recNK35A was generated from pUC19-recN by site-directed PCR mutagenesis, and it was substituted for wild-type recN in pSG101 to generate pSG105. The structures of recombinant plasmids were confirmed by DNA sequencing.
Isolation of recA Mutant
A library of recA mutants was generated by carrying out PCR-mediated random mutagenesis, as described previously (36). The resultant recA mutant clones were transformed into a recA strain. The transformants were resuspended in M9 salts, plated on LB plates containing ampicillin, and then irradiated with UV (20 J/m2). After overnight incubation, UV-resistant colonies were replica-plated on LB plates containing MMC (1 μg/ml). Clones that grew very poorly or did not grow at all on the MMC plates were selected.
SOS Induction
SOS induction was assessed by measuring the degradation of LexA as described previously (36). Log phase cultures were treated with MMC (0.5 μg/ml), and aliquots were taken at multiple time points for immunoblot analysis with anti-LexA (BioAcademia), anti-RecA (BioAcademia), or anti-RecN (26). LexA resynthesis was inhibited by adding chloramphenicol (100 μg/ml) to the cultures 10 min before adding MMC.
Fluorescence Microscopy and Localization Analysis of GFP-RecN
Exponentially growing cultures were treated with 0.5 μg/ml MMC at 37 °C. Cells were fixed with methanol, stained with 1 μg/ml DAPI (4′, 6′-diamidino-2-phenylindole), and spread (1–2 μl) on a cover glass. GFP fluorescence was not affected by prior fixation of the sample. Fluorescence microscopy was performed on a Zeiss Axioplan2 (41). Scale bars of 5 or 10 μm are shown in the figures. Images of both nucleoids (blue as a color signal) and GFP-RecN foci (green as a color signal) were merged on identical cells. In the merged image, GFP-RecN foci localized on the nucleoid appeared as a light blue color, whereas GFP-RecN foci in the cytoplasm appeared as a green color. The localization of GFP-RecN foci was determined based on these visual criteria, and more than 150 individual cells were scored for each strain.
To observe the ori1 loci (15 kbp distance from oriC) on chromosomes, we constructed wild-type and ΔrecN strains carrying both a lacO array inserted into the ori1 locus and an inducible fluorescent repressor gene (araC PBAD-lacI-ecfp) (40, 42, 43). Cells were grown at 37 °C for 90 min in LB medium containing 0.2% arabinose and 1 μg/ml MMC and then analyzed by fluorescence microscopy.
Effect of I-SceI-induced DSBs on the Localization of RecN
To detect DSB-induced RecN foci, ΔrecA strains carrying the PBADI-SceI cassette in the chromosome and a single I-SceI recognition site at the codA21 locus in the F′ episome (39) were transformed with pSG101 (gfp-recN) and with pRecA or pRecAQ300R. When cultures had reached early log phase, I-SceI was induced by the addition of 0.2% arabinose (w/v). One hour after the addition of arabinose, aliquots were taken and examined under the microscope.
RESULTS
RecA Is Required for the Formation of Nucleoid-associated GFP-RecN Foci in MMC-treated Cells
When DNA is damaged or replication is inhibited, ssDNA-bound RecA becomes conformationally active and promotes cleavage of the LexA repressor, which results in the induction of SOS genes including recN (21, 44). Previously, we showed that GFP-RecN formed foci on nucleoids after DNA damage (27). This implied that RecN could be recruited to the nucleoid at a step after RecA is loaded onto damaged DNA. To specifically examine this sequence of events, we measured GFP-RecN foci in a ΔrecA strain. Wild-type recN is a part of the SOS regulon, and its expression is completely dependent on activated RecA. Therefore, this experiment was performed in cells that expressed GFP-recN under control of the inducible PBAD promoter. Fig. 1A shows that arabinose-inducible GFP-recN fully complements the MMC sensitivity of ΔrecN, when cells are grown in the presence of arabinose, but not when cells are grown in the presence of glucose. Furthermore, GFP-RecN foci form in the cytoplasm of both MMC-treated and untreated wild-type cells and ΔrecA cells, whereas nucleoid-associated GFP-RecN foci form in wild-type MMC-treated cells but are absent in ΔrecA MMC-treated cells (Fig. 1, B and C). These results suggest that RecA is required to recruit GFP-RecN to DNA damage sites in MMC-treated cells.
FIGURE 1.
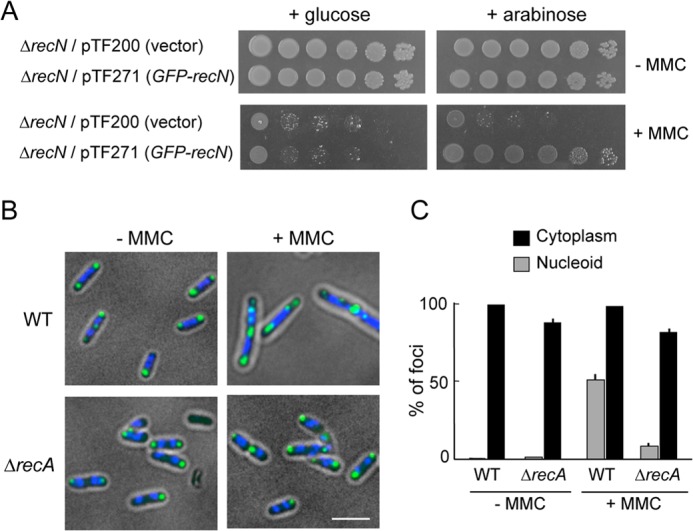
RecN foci in wild-type and ΔrecA cells with or without DNA damage. A, ΔrecN cells carrying either an arabinose-inducible GFP-recN gene (pTF271) or a pBAD vector (pTF200) were diluted and spotted onto LB plates with or without MMC (0.5 μg/ml) in the presence of either glucose or arabinose. B, the subcellular localization of GFP-RecN foci in response to MMC-induced damage. Wild-type or ΔrecA cells carrying pTF271 were exposed to MMC (0.5 μg/ml) followed by the addition of arabinose (0.05%, w/v) to induce GFP-RecN. The panels show GFP/DAPI-merged images of cells 30 min after the addition of arabinose. Scale bar indicates 2.5 μm. C, quantitative analysis of GFP-RecN foci. For cells incubated with or without MMC, ∼150 cells were examined. The results represent the average of at least three independent measurements. Error bars indicate S.D.
Isolation of a recA Mutant That Mimics the Phenotype of ΔrecN
Although RecA plays the central role in recombinational repair and is the master inducer of the SOS pathway, it is unclear what functions of RecA are required to recruit RecN to the nucleoid in MMC-treated cells. The phenotype of ΔrecA or SOS-deficient lexA3 cells (45) differs from the phenotype of ΔrecN cells; the former are hypersensitive to MMC and UV, whereas the latter are hypersensitive to MMC but insensitive to UV (Fig. 2A). Therefore, a library of recA mutants was generated and screened for mutants that provide resistance to UV but confer sensitivity to MMC. Three candidate mutants were isolated from ∼2,000 clones. All three mutants carry an arginine substitution at the highly conserved C-terminal Gln-300 of RecA (Fig. 2B). Fig. 2C shows that this allele, recAQ300R, fully complements the UV sensitivity of ΔrecA cells, but does not complement their MMC sensitivity. In the wild-type strain, MMC-induced DNA damage leads to proteolytic cleavage of LexA, the repressor of the SOS regulon, and induces the SOS response (Fig. 2D). Similarly, recAQ300R is capable of inducing expression of the SOS regulon and specific proteolytic cleavage of LexA in MMC-treated cells (Fig. 2D). Thus, recAQ300R is proficient in the DNA damage-induced SOS response, and its phenotype is similar to the phenotype of ΔrecN.
FIGURE 2.
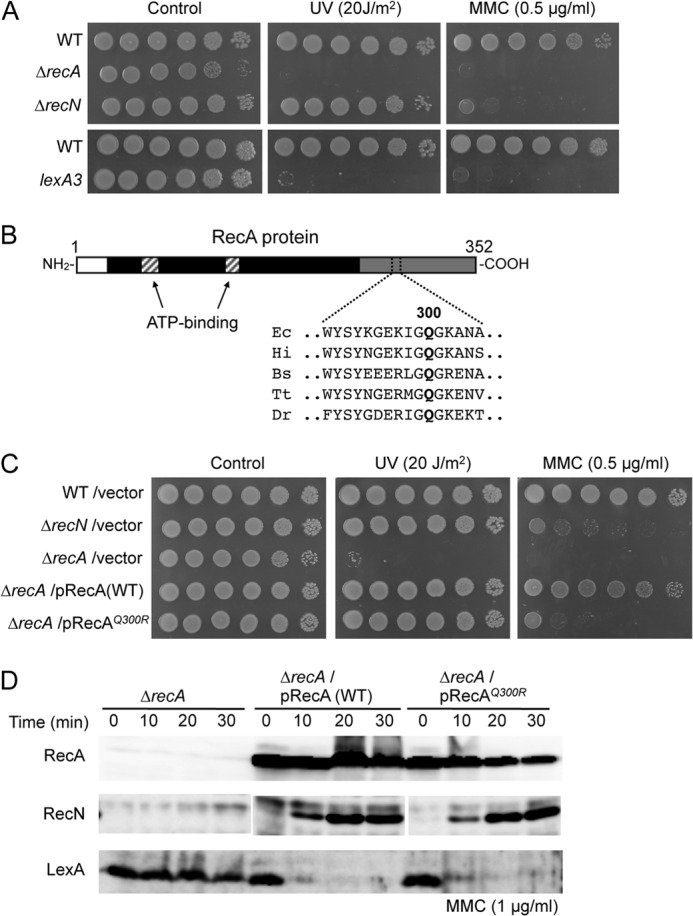
Effect of recAQ300R on DNA repair and SOS response. A, sensitivity of cells to MMC and UV irradiation. 10-fold serial dilutions of the indicated strains were spotted on LB plates. DNA damage was induced by either MMC or UV irradiation. B, Q300 is conserved in bacterial RecA orthologs. A map of the E. coli RecA region between amino acids 290 and 305 is shown. Ec, E. coli; Hi, H. influenzae; Bs, B.subtilis; Tt, Thermus thermophilus; Dr, D. radiodurans. C, sensitivity of recAQ300R cells to MMC and UV irradiation was examined as in A. D, RecAQ300R is proficient in SOS induction. Protein extracts from cells treated with MMC for the indicated times were prepared and analyzed by Western blot using anti-RecA, anti-RecN, or anti-LexA antibodies. For the LexA degradation assay, chloramphenicol (100 μg/ml) was added at time 0 to inhibit resynthesis of LexA protein.
Nucleoid Fragmentation in MMC-treated ΔrecN and recAQ300R Cells
To explore these results further, ΔrecA, recAQ300R, ΔrecN, and ΔruvB cells were stained with DAPI and examined by fluorescence and phase-contrast microscopy for genome integrity and cell morphology. Under conditions of exponential growth in the absence of MMC, all cells had a normal morphology, with two centrally located nucleoids per cell (Fig. 3A). Wild-type cells treated with MMC for 90 min became highly filamented with elongated, evenly spaced nucleoids (Fig. 3A). This morphology is typical of SOS-activated cells (46). By contrast, a large fraction of MMC-treated SOS-defective ΔrecA were anucleate, and filamentous cells were hardly detected (Fig. 3A). RuvABC resolvasome branch-migrates and resolves Holliday junctions, and inactivation of any of the three Ruv functions blocks resolution of recombinational repair intermediates (3). Therefore, RuvB plays a role in the later steps of HR. MMC-treated ruvB mutants formed both filamentous and anucleate cells. As reported previously (47), the nucleoids of filamentous ruvB cells were centrally located and little to no DNA migrated to cell poles, which is in contrast to the morphology of filamentous wild-type cells (i.e. well partitioned nucleoids). This indicates that the accumulation of intermediates of HR-mediated DSB repair results in chromosome nondisjunction and the production of anucleate cells. MMC-treated ΔrecN and recAQ300R cells were as filamentous as wild-type cells, but had an abnormal morphology characterized by multiple, short, diffuse nucleoids (Fig. 3A). In wild-type cells, the number of nucleoids per cell was largely unaffected by exposure to MMC, whereas the number of nucleoids per cell increased when ΔrecN and recAQ300R cells were exposed to MMC (Fig. 3B). Furthermore, abnormal nucleoid morphology was not generally observed in UV-irradiated ΔrecN and recAQ300R cells (supplemental Fig. S1). These results support the conclusion that recAQ300R is a phenocopy of ΔrecN. Our interpretation of this result is that RecN is dysfunctional in the recAQ300R mutant.
FIGURE 3.

Morphological changes in MMC-treated wild-type, ΔrecN, and recA cells. Exponentially growing cells were fixed and stained with DAPI and analyzed by fluorescence microscopy. A, the panels show DAPI images of cells incubated for 90 min in the presence or absence of MMC (1 μg/ml). Nucleoids are visualized as a light blue color. B, quantitative analysis of nucleoids. For cells with or without MMC-induced DNA damage, >200 cells were examined. The results represent the average of at least three independent measurements. Error bars indicate S.D. C, localization of a LacI-ECFP to the nucleoid. The wild-type and ΔrecN strains carry an ectopic tandem array of lacO at ori1 (15 kb counterclockwise of oriC). The panels show merged images of LacI-ECFP (light blue) and nucleoids (dark blue). White arrows indicate nucleoids that fail to bind LacI-ECFP, and by implication, oriC-lacking nucleoids. D, quantification of cells lacking ori1 foci. Wild-type and ΔrecN cells were treated with MMC for 90 min and examined by fluorescence microscopy. For cells with or without MMC-induced damage, >200 cells were examined.
We hypothesized that the abnormal nucleoid morphology of MMC-treated ΔrecN cells might reflect the presence of unrepaired DSBs and chromosome fragmentation. Therefore, a fluorescence-based method was used to visualize chromosome fragments. For this purpose, oriC was labeled indirectly, via LacI-ECFP (enhanced cyan fluorescent protein) bound to an ectopic tandem array of Lac repressor-binding sites (240 × lacO) at the ori1 locus (15 kb counterclockwise of oriC) (40). LacI-ECFP was expressed from the chromosomally integrated gene under the control of the PBAD promoter. Wild-type and ΔrecN cells were treated or not with MMC and visualized using fluorescence microscopy. The results revealed 2–4 ori1 foci per cell in the majority of wild-type and ΔrecN cells in the absence of MMC. In these cells, all nucleoids contained at least one ori1 focus (Fig. 3C). When treated with MMC, the number of ori1 foci per nucleoid increased significantly in wild-type and ΔrecN cells, and the foci were distributed throughout the elongated nucleoid (Fig. 3C). Notably, >15% of ΔrecN cells carried nucleoids lacking ori1 foci, whereas such nucleoids were infrequent in wild-type cells (<1.4%) (Fig. 3D). These results demonstrate the presence of aberrant nucleoids lacking oriC in MMC-treated ΔrecN cells, which likely represent subchromosomal fragments.
RecAQ300R Is Defective in Recruiting RecN to Nucleoids in MMC-treated Cells
The results described above suggest that RecAQ300R does not recruit RecN to the nucleoid, under conditions where wild-type RecA does so (i.e. in MMC-treated wild-type cells). To explore this further, GFP-RecN foci were quantified in MMC-treated ΔrecA ΔrecN cells expressing SOS-inducible GFP-recN and either wild-type recA or recAQ300R. After exposure to MMC for 60 min, >90% of wild-type cells contained nucleoid-associated GFP-RecN foci (Fig. 4, A and B). By contrast, <5% of cells expressing recAQ300R had nucleoid-associated GFP-RecN foci, whereas the number and fraction of cells with cytoplasmic GFP-RecN foci was higher in cells expressing recAQ300R than that in cells expressing wild-type recA (Fig. 4, A and B). These results indicate that RecA is required for the formation of MMC-induced, nucleoid-associated RecN foci and that RecAQ300R has a specific defect in this function/role.
FIGURE 4.
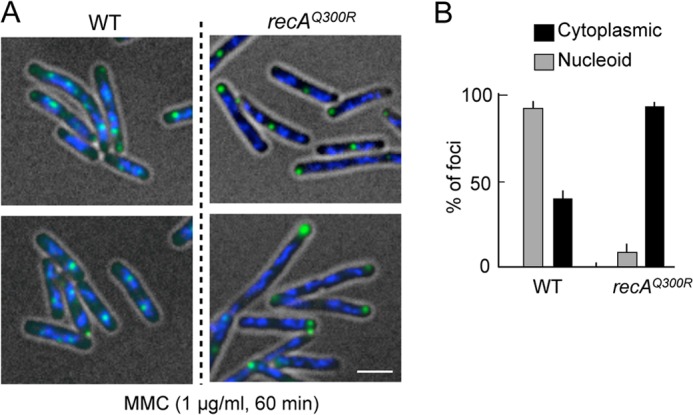
Nucleoid-associated RecN foci in response to MMC-induced DNA damage. A, MMC damage-induced RecN foci in recAQ300R cells. Cells carrying the SOS-inducible GFP-recN (pSG101) were exposed to MMC for 60 min. The panels show GFP/DAPI-merged images of cells. Scale bar indicates 2.5 μm. B, quantitative analysis of GFP-RecN foci. For cells with MMC damage, >150 cells were examined. The results represent the average of at least three independent measurements. Error bars indicate S.D.
RecA Is Required to Recruit RecN to sites of DSBs
To examine the recruitment of RecN to a unique DSB site in RecA-proficient cells, I-SceI was used to introduce a site-specific DSB into a strain that carries the PBADI-SceI cassette on the chromosome and a single I-SceI recognition site on the F′ episome (39). Appropriately engineered cells were transformed with a plasmid expressing GFP-recN from its native SOS-inducible promoter, grown to early log phase and exposed to arabinose to induce I-SceI. Control cells were grown in medium lacking arabinose. VspI endonuclease digestion resulted in a 1.8-kb fragment containing the I-SceI cleavage site. I-SceI digestion produced two fragments, one of which with a size of 1.2 kb hybridized to the site 1 probe (Fig. 5A). The kinetics of DSB formation was monitored by Southern blot analysis of VspI-digested DNA isolated from samples taken at different times after the addition of 0.2% arabinose or glucose to the culture. A 1.2-kb fragment was not detected when cells were maintained in glucose-containing medium, whereas it was detected within 30 min in wild-type cells proficient for RecBCD after the addition of arabinose (Fig. 5, A and B). The intensity of the 1.2-kb fragments increased with time, reaching a maximum intensity ∼1 h after the addition of arabinose. Similar results were obtained when the site 2 probe was used to detect the I-SceI cleavage site (Fig. 5A and supplemental Fig. S2). One possible explanation for the kinetics of DSB formation is that I-SceI digestion is not synchronous in the entire population, and the breaks may be repaired very efficiently. Thus, the only breaks generated just before samples were taken might be detected by Southern blotting. This is consistent with previous studies using chromosomally integrated I-SceI site, where DSB products are readily detected in wild-type cells even after 1 h of I-SceI induction (48).
FIGURE 5.

RecA is required for the assembly of RecN at the sites of DSBs. A, Southern blot of the flanking region of the I-SceI site before and after induction of I-SceI. Exponentially growing cells were cultured, and either arabinose (Ara) was added to induce I-SceI or glucose (Glu) was added as a control. Cells were taken at the indicated times. The DNA digested with VspI was analyzed on a 1% agarose gel and detected by Southern analysis using site 1 probe. The top panel illustrates the DNA sequences flanking the I-SceI cleavage site in the codA21::miniTn7Kan locus on the F′ episome. The location of VspI cut sites and the sizes of the DNA fragments after VspI digestion are shown. Site 1 and site 2 regions were used for Southern blot analysis. B, quantitation of Southern blot analysis. I-SceI break refers to the levels of the 1.2-kb fragment resulting from DSB formation. C, GFP-RecN foci at a unique I-SceI-induced nascent DSB. The panels show GFP/DAPI-merged images of cells with or without a single I-SceI recognition site. recAΔ cells expressing SOS-inducible GFP-recN and either wild-type recA or recAQ300R were incubated for 1 h in the presence of arabinose to induce I-SceI. Scale bar indicates 2.5 μm. D, quantitative analysis of GFP-RecN foci. The results represent the average of at least three independent measurements. Error bars indicate S.D.
Fig. 5C shows that nucleoid-associated GFP-RecN foci were detected in cells that expressed I-SceI and carried an F′ episome with an I-SceI cleavage site. By contrast, GFP-RecN foci were not observed when the same cells were grown in glucose-containing medium (to repress I-SceI) or if the cells did not carry an I-SceI-sensitive F′ episome (Fig. 5C). Furthermore, the number of nucleoid-associated GFP-RecN foci was much lower in recAQ300R mutant cells (<1%) than in cells expressing wild-type recA (18%) (Fig. 5D). These results indicate that, in wild-type cells, a single I-SceI-induced DSB induces an SOS response and promotes the formation of nucleoid-associated GFP-RecN foci in a RecA-dependent manner.
RecN ATPase Activity Is Required for Release from Growth Arrest in Cells with DNA Damage
RecN has a typical SMC family protein domain structure, including an extensive, centrally located coiled-coil domain and globular N- and C-terminal domains with Walker A and Walker B nucleotide-binding motifs, respectively (49). A previous study showed that substitution of Lys-35 with alanine in the Walker A motif resulted in a complete loss of RecN DNA repair activity in vivo (50). Biochemical characterization of Deinococcus radiodurans RecN showed that RecNK67A (an lysine-to-alanine substitution at position 67, which corresponds to E. coli RecN Lys-35) abolished ATPase activity, but did not impair ATP binding in vitro (49). Fig. 6A shows that expression of recNK35A in a ΔrecN background conferred sensitivity to MMC that was equivalent to that of ΔrecN. The overproduction of RecNK35A rendered wild-type cells sensitive to MMC (Fig. 6A), demonstrating that recNK35A is a dominant-negative allele of recN. GFP-RecNK35A formed nucleoid-associated foci in >80% of MMC-treated cells, and these foci failed to form in cells expressing recAQ300R (Fig. 6B). This result indicates that the ATPase activity of RecN is not required for formation of nucleoid-associated RecN foci. However, ΔrecN cells expressing wild-type GFP-recN resumed normal cell growth, and nucleoid-associated GFP-RecN foci dissociated after exposure to MMC was terminated (Fig. 6C). By contrast, ΔrecN cells expressing GFP-recNK35A became highly filamented, acquired fragmented nucleoid structures, and retained GFP-RecNK35A foci for 2 h after exposure to MMC was terminated (Fig. 6C). These results demonstrate that ATPase-defective RecNK35A is recruited to sites of DNA damage, but may not be properly released because of the defects in HR-mediated repair of MMC-induced DSBs.
FIGURE 6.

RecNK35A is deficient in HR-mediated recovery after exposure to MMC. A, sensitivity of cells to MMC. The indicated strains were grown in LB. Cells were diluted and spotted onto LB with or without MMC (0.5 μg/ml). B, subcellular localization of GFP-RecNK35A. The panels show GFP/DAPI images of ΔrecN cells containing SOS-inducible GFP-recNK35A after 30 min of incubation in the presence of MMC. Quantitative analysis of GFP-RecNK35A foci is shown to the right. The results represent the average of at least three independent measurements. C, wild-type and recNK35A cells were treated with MMC for 10 min and then transferred to MMC-free medium (t = 0). At the indicated time points, cells were analyzed for the presence of RecN foci. The right panels show the GFP/DAPI images of cells at the indicated time after transfer to MMC-free media. Scale bar indicates 2.5 μm. Error bars indicate S.D.
DISCUSSION
Previous studies demonstrate that RecN protein forms both nucleoid-associated and cytoplasmic foci in cells exposed to DSB-inducing agents and that cytoplasmic RecN aggregates are degraded by the ClpXP protease (27). Here, we demonstrate that RecN is recruited to nucleoids in a RecA-dependent manner. We characterize a novel recA allele, recAQ300R, which promotes expression of SOS-inducible genes but does not promote formation of nucleoid-associated RecN foci. RecN accumulates at a unique I-SceI-induced DSB in wild-type recA cells but not in recAQ300R cells. Thus, we conclude that RecA plays an essential role in DNA damage-induced expression of recN and the assembly of RecN foci at the sites of DSBs. ATPase-deficient recNK35A mutants are proficient in forming nucleoid-associated foci at DSBs, but fail to resume growth after release from MMC-induced cell-cycle arrest. This results in highly filamented cells with nucleoid-associated GFP-RecNK35A foci and fragmented nucleoid structures. One possible explanation for the presence of persistent foci associated with damaged DNA in recNK35A cells is that RecNK35A is recruited to DSBs, but is not released from DSB sites because it lacks ATPase activity; under such conditions, mutant RecNK35A DNA damage foci persist and accumulate, which interferes with RecA-mediated synaptic steps in the HR pathway.
This study also reveals that ΔrecN and recAQ300R cells are hypersensitive to MMC but not to UV. A previous study showed that cells expressing recAΔC17 (a deletion mutant lacking residues 336–352) are hypersensitive to MMC but not to UV (51). Here, we confirm that recAΔC17 cells are sensitive to MMC, although they are less sensitive than ΔrecN and recAQ300R cells (supplemental Fig. S3). We found that nucleoid-associated GFP-RecN foci form normally in MMC-treated recAΔC17 cells (supplemental Fig. S3), indicating that the defects in the response to MMC in recAQ300R cells are not a result of a dysfunctional RecA C-terminal domain. However, it still remains possible that the C-terminal region of RecA plays a role in modulating RecN function at a later step in the repair/response to MMC-induced DSBs.
Previous studies suggest that the SOS response plays a critical role in DSB repair in E. coli but not in Bacillus subtilis (52). Indeed, unlike in E. coli, the expression of B. subtilis RecN appears to be SOS-independent (53), and GFP-B. subtilis RecN foci associate with DSBs before RecA is recruited to the DNA lesion (10). By contrast, E. coli recN is typical of SOS-regulated genes in that its expression is tightly repressed in unstressed cells. This suggests that E. coli RecN participates in HR repair of DSBs after RecA senses DNA damage. Consistent with this, the present study indicates that RecA actively recruits RecN to DSBs. These results may reflect species-specific attributes of E. coli and B. subtilis HR pathways. The purified B. subtilis RecN (and also D. radiodurans RecN) binds to DNA and has DNA-stimulated ATPase activity in vitro (54, 55). Unfortunately, it has been difficult to purify E. coli RecN because it is relatively insoluble and highly susceptible to degradation (data not shown). A recent study showed that Haemophilus influenzae RecN can be purified to near homogeneity and is fully functional in E. coli (50). Purified Haemophilus influenzae RecN does not bind DNA, and DNA had no significant effect on Haemophilus influenzae RecN ATPase activity in vitro, which contrasts with the activity of B. subtilis RecN. This observation supports our conclusion that E. coli RecN is recruited to DSBs through its interaction with RecA. Thus, the difference in the DNA binding specificities of RecN orthologs may explain their different affinities for their respective bacterial nucleoids. However, our data do not exclude the possibility that E. coli RecN has DNA binding activity. It is also conceivable that RecA facilitates the binding and/or retention of RecN on damaged DNA.
SMC proteins are highly conserved ATPases whose role in higher order chromosome organization and dynamics is conserved from bacteria to humans. DSBs are one of the most cytotoxic forms of DNA damage, and therefore, the repair of DSBs is crucial for cell survival and for maintaining the integrity of the genome. In this study, we provide evidence that the recruitment of RecN to DSBs requires interaction with RecA. Any defect in the interaction results in chromosomal fragmentation, such as that observed in cells exposed to the DSB-inducing agent MMC. Based on these results and implications, we propose a mechanism by which RecN promotes RecA-dependent DSB repair. The initial presynaptic step of the DNA strand exchange reaction is formation of a RecA-ssDNA nucleoprotein filament. RecA-dependent recruitment of SMC-like RecN to DSBs follows, serving a scaffolding function to facilitate subsequent search by RecA for homologous templates in the segregated sister chromatids. Lastly, RecA mediates strand exchange. This model might be compatible with the recN studies in B. subtilis; here, we allow for the fact that RecN plays a role in an early step of DSB repair and that the mechanism by which RecN is recruited to DSBs differs.
In future studies, it will be interesting to investigate how RecN SMC complexes actually promote RecA-dependent DSB repair. Therefore, novel integrated biochemical and structural approaches to examine this and other questions concerning the roles of RecN and RecA will be required. The results of such studies should advance our understanding of the mechanism of DSB repair in prokaryotic and eukaryotic cells.
Acknowledgments
We are grateful to S. M. Rosenberg for providing the E. coli strains. We thank T. Kado for technical assistance. We thank the members of the Hishida laboratory for stimulating discussions.
This work was supported by grants-in-aid for scientific research on priority areas from the Ministry of Education, Culture, Sports, Science and Technology of Japan (to T. H.).

This article contains supplemental Figs. S1–S3.
- DSB
- DNA double-strand break
- HR
- homologous recombination
- MMC
- mitomycin C
- SMC
- structural maintenance of chromosomes
- ECFP
- enhanced cyan fluorescent protein.
REFERENCES
- 1. Cox M. M., Goodman M. F., Kreuzer K. N., Sherratt D. J., Sandler S. J., Marians K. J. (2000) The importance of repairing stalled replication forks. Nature 404, 37–41 [DOI] [PubMed] [Google Scholar]
- 2. Kuzminov A. (2001) DNA replication meets genetic exchange: chromosomal damage and its repair by homologous recombination. Proc. Natl. Acad. Sci. U.S.A. 98, 8461–8468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kuzminov A. (1999) Recombinational repair of DNA damage in Escherichia coli and bacteriophage λ. Microbiol. Mol. Biol. Rev. 63, 751–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kowalczykowski S. C. (2000) Initiation of genetic recombination and recombination-dependent replication. Trends Biochem. Sci. 25, 156–165 [DOI] [PubMed] [Google Scholar]
- 5. Cox M. M. (2001) Historical overview: searching for replication help in all of the rec places. Proc. Natl. Acad. Sci. U.S.A. 98, 8173–8180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Singleton M. R., Dillingham M. S., Gaudier M., Kowalczykowski S. C., Wigley D. B. (2004) Crystal structure of RecBCD enzyme reveals a machine for processing DNA breaks. Nature 432, 187–193 [DOI] [PubMed] [Google Scholar]
- 7. Lloyd R. G., Low K. B. (1996) Escherichia coli and Salmonella: Cellular and Molecular Biology, 2nd Ed., pp. 2236–2255, American Society for Microbiology Press, Washington, D.C. [Google Scholar]
- 8. Amundsen S. K., Smith G. R. (2003) Interchangeable parts of the Escherichia coli recombination machinery. Cell 112, 741–744 [DOI] [PubMed] [Google Scholar]
- 9. Rocha E. P., Cornet E., Michel B. (2005) Comparative and evolutionary analysis of the bacterial homologous recombination systems. PLoS Genet. 1, e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sanchez H., Kidane D., Castillo Cozar M., Graumann P. L., Alonso J. C. (2006) Recruitment of Bacillus subtilis RecN to DNA double-strand breaks in the absence of DNA end processing. J. Bacteriol. 188, 353–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cao Z., Mueller C. W., Julin D. A. (2010) Analysis of the recJ gene and protein from Deinococcus radiodurans. DNA Repair 9, 66–75 [DOI] [PubMed] [Google Scholar]
- 12. Bentchikou E., Servant P., Coste G., Sommer S. (2010) A major role of the RecFOR pathway in DNA double-strand-break repair through ESDSA in Deinococcus radiodurans. PLoS Genet. 6, e1000774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cox M. M., Keck J. L., Battista J. R. (2010) Rising from the ashes: DNA repair in Deinococcus radiodurans. PLoS Genet. 6, e1000815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lusetti S. L., Cox M. M. (2002) The bacterial RecA protein and the recombinational DNA repair of stalled replication forks. Annu. Rev. Biochem. 71, 71–100 [DOI] [PubMed] [Google Scholar]
- 15. Lloyd R. G., Sharples G. J. (1993) Dissociation of synthetic Holliday junctions by E. coli RecG protein. EMBO J. 12, 17–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shinagawa H., Iwasaki H. (1996) Processing the Holliday junction in homologous recombination. Trends in biochemical sciences 21, 107–111 [PubMed] [Google Scholar]
- 17. Sharples G. J., Ingleston S. M., Lloyd R. G. (1999) Holliday junction processing in bacteria: insights from the evolutionary conservation of RuvABC, RecG, and RusA. J. Bacteriol. 181, 5543–5550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Little J. W., Mount D. W. (1982) The SOS regulatory system of Escherichia coli. Cell 29, 11–22 [DOI] [PubMed] [Google Scholar]
- 19. Little J. W. (1991) Mechanism of specific LexA cleavage: autodigestion and the role of RecA coprotease. Biochimie 73, 411–421 [DOI] [PubMed] [Google Scholar]
- 20. Luo Y., Pfuetzner R. A., Mosimann S., Paetzel M., Frey E. A., Cherney M., Kim B., Little J. W., Strynadka N. C. (2001) Crystal structure of LexA: a conformational switch for regulation of self-cleavage. Cell 106, 585–594 [DOI] [PubMed] [Google Scholar]
- 21. Courcelle J., Khodursky A., Peter B., Brown P. O., Hanawalt P. C. (2001) Comparative gene expression profiles following UV exposure in wild-type and SOS-deficient Escherichia coli. Genetics 158, 41–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Graumann P. L. (2001) SMC proteins in bacteria: condensation motors for chromosome segregation? Biochimie 83, 53–59 [DOI] [PubMed] [Google Scholar]
- 23. Hirano T. (2006) At the heart of the chromosome: SMC proteins in action. Nat. Rev. Mol. Cell Biol. 7, 311–322 [DOI] [PubMed] [Google Scholar]
- 24. Lloyd R. G., Picksley S. M., Prescott C. (1983) Inducible expression of a gene specific to the RecF pathway for recombination in Escherichia coli K12. Mol. Gen. Genet. 190, 162–167 [DOI] [PubMed] [Google Scholar]
- 25. Finch P. W., Chambers P., Emmerson P. T. (1985) Identification of the Escherichia coli recN gene product as a major SOS protein. J. Bacteriol. 164, 653–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rostas K., Morton S. J., Picksley S. M., Lloyd R. G. (1987) Nucleotide sequence and LexA regulation of the Escherichia coli recN gene. Nucleic Acids Res. 15, 5041–5049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nagashima K., Kubota Y., Shibata T., Sakaguchi C., Shinagawa H., Hishida T. (2006) Degradation of Escherichia coli RecN aggregates by ClpXP protease and its implications for DNA damage tolerance. J. Biol. Chem. 281, 30941–30946 [DOI] [PubMed] [Google Scholar]
- 28. Neher S. B., Villén J., Oakes E. C., Bakalarski C. E., Sauer R. T., Gygi S. P., Baker T. A. (2006) Proteomic profiling of ClpXP substrates after DNA damage reveals extensive instability within SOS regulon. Mol. Cell 22, 193–204 [DOI] [PubMed] [Google Scholar]
- 29. Sargentini N. J., Smith K. C. (1983) Characterization of an Escherichia coli mutant (radB101) sensitive to γ and UV radiation, and methyl methanesulfonate. Radiat. Res. 93, 461–478 [PubMed] [Google Scholar]
- 30. Kosa J. L., Zdraveski Z. Z., Currier S., Marinus M. G., Essigmann J. M. (2004) RecN and RecG are required for Escherichia coli survival of Bleomycin-induced damage. Mutat. Res. 554, 149–157 [DOI] [PubMed] [Google Scholar]
- 31. Picksley S. M., Attfield P. V., Lloyd R. G. (1984) Repair of DNA double-strand breaks in Escherichia coli K12 requires a functional recN product. Mol. Gen. Genet. 195, 267–274 [DOI] [PubMed] [Google Scholar]
- 32. Lloyd R. G., Buckman C., Benson F. E. (1987) Genetic analysis of conjugational recombination in Escherichia coli K12 strains deficient in RecBCD enzyme. J. Gen. Microbiol. 133, 2531–2538 [DOI] [PubMed] [Google Scholar]
- 33. Wang T. C., Smith K. C. (1988) Different effects of recJ and recN mutations on the postreplication repair of UV-damaged DNA in Escherichia coli K-12. J. Bacteriol. 170, 2555–2559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Miller J. H. (1992) A Short Course in Bacterial Genetics: A Laboratory Manual and Handbook for Escherichia coli and Related Bacteria, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 35. Sambrook J., Fritsch E. F., Maniatis T. (1989) Molecular Cloning: A Laboratory Manual, 2nd Ed., Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 36. Shibata T., Hishida T., Kubota Y., Han Y. W., Iwasaki H., Shinagawa H. (2005) Functional overlap between RecA and MgsA (RarA) in the rescue of stalled replication forks in Escherichia coli. Genes Cells 10, 181–191 [DOI] [PubMed] [Google Scholar]
- 37. Datsenko K. A., Wanner B. L. (2000) One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U.S.A. 97, 6640–6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Baba T., Ara T., Hasegawa M., Takai Y., Okumura Y., Baba M., Datsenko K. A., Tomita M., Wanner B. L., Mori H. (2006) Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2, 2006.0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ponder R. G., Fonville N. C., Rosenberg S. M. (2005) A switch from high-fidelity to error-prone DNA double-strand break repair underlies stress-induced mutation. Mol. Cell 19, 791–804 [DOI] [PubMed] [Google Scholar]
- 40. Nozaki S., Niki H., Ogawa T. (2009) Replication initiator DnaA of Escherichia coli changes its assembly form on the replication origin during the cell cycle. J. Bacteriol. 191, 4807–4814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hishida T., Han Y. W., Shibata T., Kubota Y., Ishino Y., Iwasaki H., Shinagawa H. (2004) Role of the Escherichia coli RecQ DNA helicase in SOS signaling and genome stabilization at stalled replication forks. Genes Dev. 18, 1886–1897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lau I. F., Filipe S. R., Søballe B., Økstad O. A., Barre F. X., Sherratt D. J. (2003) Spatial and temporal organization of replicating Escherichia coli chromosomes. Mol. Microbiol. 49, 731–743 [DOI] [PubMed] [Google Scholar]
- 43. Hatano T., Yamaichi Y., Niki H. (2007) Oscillating focus of SopA associated with filamentous structure guides partitioning of F plasmid. Mol. Microbiol. 64, 1198–1213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Humayun M. Z. (1998) SOS and Mayday: multiple inducible mutagenic pathways in Escherichia coli. Mol. Microbiol. 30, 905–910 [DOI] [PubMed] [Google Scholar]
- 45. Little J. W., Harper J. E. (1979) Identification of the lexA gene product of Escherichia coli K-12. Proc. Natl. Acad. Sci. U.S.A. 76, 6147–6151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Friedberg E. C., Aguilera A., Gellert M., Hanawalt P. C., Hays J. B., Lehmann A. R., Lindahl T., Lowndes N., Sarasin A., Wood R. D. (2006) DNA repair: from molecular mechanism to human disease. DNA Repair 5, 986–996 [DOI] [PubMed] [Google Scholar]
- 47. Ishioka K., Fukuoh A., Iwasaki H., Nakata A., Shinagawa H. (1998) Abortive recombination in Escherichia coli ruv mutants blocks chromosome partitioning. Genes Cells 3, 209–220 [DOI] [PubMed] [Google Scholar]
- 48. Meddows T. R., Savory A. P., Lloyd R. G. (2004) RecG helicase promotes DNA double-strand break repair. Mol. Microbiol. 52, 119–132 [DOI] [PubMed] [Google Scholar]
- 49. Pellegrino S., Radzimanowski J., de Sanctis D., Boeri Erba E., McSweeney S., Timmins J. (2012) Structural and functional characterization of an SMC-like protein RecN: new insights into double-strand break repair. Structure 20, 2076–2089 [DOI] [PubMed] [Google Scholar]
- 50. Grove J. I., Wood S. R., Briggs G. S., Oldham N. J., Lloyd R. G. (2009) A soluble RecN homologue provides means for biochemical and genetic analysis of DNA double-strand break repair in Escherichia coli. DNA Repair 8, 1434–1443 [DOI] [PubMed] [Google Scholar]
- 51. Lusetti S. L., Wood E. A., Fleming C. D., Modica M. J., Korth J., Abbott L., Dwyer D. W., Roca A. I., Inman R. B., Cox M. M. (2003) C-terminal deletions of the Escherichia coli RecA protein. Characterization of in vivo and in vitro effects. J. Biol. Chem. 278, 16372–16380 [DOI] [PubMed] [Google Scholar]
- 52. Simmons L. A., Goranov A. I., Kobayashi H., Davies B. W., Yuan D. S., Grossman A. D., Walker G. C. (2009) Comparison of responses to double-strand breaks between Escherichia coli and Bacillus subtilis reveals different requirements for SOS induction. J. Bacteriol. 191, 1152–1161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Au N., Kuester-Schoeck E., Mandava V., Bothwell L. E., Canny S. P., Chachu K., Colavito S. A., Fuller S. N., Groban E. S., Hensley L. A., O'Brien T. C., Shah A., Tierney J. T., Tomm L. L., O'Gara T. M., Goranov A. I., Grossman A. D., Lovett C. M. (2005) Genetic composition of the Bacillus subtilis SOS system. J. Bacteriol. 187, 7655–7666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sanchez H., Alonso J. C. (2005) Bacillus subtilis RecN binds and protects 3′-single-stranded DNA extensions in the presence of ATP. Nucleic Acids Res. 33, 2343–2350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Reyes E. D., Patidar P. L., Uranga L. A., Bortoletto A. S., Lusetti S. L. (2010) RecN is a cohesin-like protein that stimulates intermolecular DNA interactions in vitro. J. Biol. Chem. 285, 16521–16529 [DOI] [PMC free article] [PubMed] [Google Scholar]