

Background: Afadin is an important regulator of cell-cell adhesion.
Results: PLEKHA7 binds to afadin and this binding is required for the proper formation of adherens junction (AJ).
Conclusion: PLEKHA7 plays a cooperative role with nectin and afadin in the proper formation of AJ in epithelial cells.
Significance: The result indicates a novel regulatory mechanism for the formation of cell-cell adhesion.
Keywords: Adherens Junction, Cadherins, Cell Adhesion, Epithelial Cell, Protein-Protein Interactions, PLEKHA7, Afadin, Nectin
Abstract
Adherens junction (AJ) is a specialized cell-cell junction structure that plays a role in mechanically connecting adjacent cells to resist strong contractile forces and to maintain tissue structure, particularly in the epithelium. AJ is mainly comprised of cell adhesion molecules cadherin and nectin and their associating cytoplasmic proteins including β-catenin, α-catenin, p120ctn, and afadin. Our series of studies have revealed that nectin first forms cell-cell adhesion and then recruits cadherin to form AJ. The recruitment of cadherin by nectin is mediated by the binding of α-catenin and p120ctn to afadin. Recent studies showed that PLEKHA7 binds to p120ctn, which is associated with E-cadherin, and maintains the integrity of AJ in epithelial cells. In this study, we showed that PLEKHA7 bound to afadin in addition to p120ctn and was recruited to the nectin-3α-based cell-cell adhesion site in a manner dependent on afadin, but not on p120ctn. The binding of PLEKHA7 to afadin was required for the proper formation of AJ, but not for the formation of tight junction, in EpH4 mouse mammary gland epithelial cells. These results indicate that PLEKHA7 plays a cooperative role with nectin and afadin in the proper formation of AJ in epithelial cells.
Introduction
Mono-layered columnar epithelial cells, which are simply referred here to as epithelial cells, in small intestine, for instance, form a sheet by adhering adjacent cells through cell-cell junctions, such as tight junction (TJ),2 adherens junction (AJ), and desmosome (1). In epithelial cells, TJ is localized at the most apical side of the cell-cell adhesion site, and AJ is formed just at the basal side of TJ. TJ is crucial for the barrier function that prevents the passage of soluble molecules through the gaps between cells (2) and is thought to be involved in the fence function that keeps the cell surface lipids at the basolateral region separated from those at the apical region (3), although the latter function remains controversial (4). AJ plays a role in mechanically connecting adjacent cells to resist strong contractile forces and to maintain tissue structure particularly in epithelial cells. In addition, AJ regulates the formation and maintenance of TJ and desmosome (5).
AJ is formed by cell adhesion molecules (CAMs) cadherin and nectin and their associating molecules and is undercoated with the actin cytoskeleton associated with myosin (6, 7). Cadherin is a key Ca2+-dependent CAM with a single transmembrane segment and comprises a family consisting of over 100 members. Cadherin directly binds β-catenin and p120ctn (8). β-Catenin in turn interacts with α-catenin, which binds many peripheral membrane proteins, such as vinculin, α-actinin, and EPLIN (9). Nectin is a Ca2+-independent Ig-like CAM with a single transmembrane segment and comprises a family consisting of four members: nectin-1, -2, -3, and -4 (10, 11). Nectin is associated with actin filaments (F-actin) through afadin, an F-actin-binding protein. Afadin has some splicing isoforms, and epithelial cells mainly express the longest one, l-afadin, hereafter referred to as afadin. Our series of studies have revealed that nectin first forms cell-cell adhesion and then recruits the cadherin-catenin complex to the nectin-based cell-cell adhesion site at least through the direct binding of afadin to α-catenin and the indirect binding of afadin to p120ctn to form AJ (6, 10–14).
Nectin is also involved in the formation of TJ in addition to AJ (15). The major CAMs at TJ are claudin, occludin, and junctional adhesion molecule. Claudin is a Ca2+-independent CAM with four transmembrane segments and comprises a family consisting of ∼24 members in humans and mice (3, 16). Claudin forms TJ strands to seal the apposed plasma membranes of adjacent cells. The structure of occludin is similar to that of claudin, but its function has not yet been established. In addition to these CAMs, several peripheral membrane proteins, such as ZO-1, -2, and -3, are localized at TJ and interact with claudin, occludin, and junctional adhesion molecule to link these CAMs to F-actin. During and/or after the formation of AJ, nectin recruits first junctional adhesion molecule and then claudin and occludin to the apical side of AJ, resulting in the formation of TJ. It remains unknown how the trans-interactions of nectin lead to the recruitment of the TJ components, but afadin and ZO-1 and the interaction of these two proteins are necessary for the recruitment of CAMs to TJ (15).
A recent study showed that PLEKHA7, a protein originally identified as a member of pleckstrin homology (PH) domain containing, family A (17, 18), binds to both p120ctn and Nezha (19). p120ctn binds to the juxtamembrane region of E-cadherin, whereas Nezha binds to the minus end of microtubules. Thus, PLEKHA7 connects the microtubule network to AJ through p120ctn and Nezha in human colon carcinoma Caco2 cells. E-cadherin, β-catenin, α-catenin, p120ctn, and EPLIN are localized not only at AJ but also at the basal side of AJ along the lateral plasma membranes of attached epithelial cells (9). In contrast, PLEKHA7 is strictly localized at AJ like the nectin-afadin system in Caco2 cells (19). It was more recently shown that PLEKHA7 binds to paracingulin, which is known to interact with ZO-1, and is localized at AJ in Madin-Darby canine kidney (MDCK) cells (20). However, paracingulin is localized not only at AJ but also at TJ (21). It was proposed that paracingulin is localized at AJ and TJ by binding to PLEKHA7 and ZO-1, respectively (20). Thus, it still remains unknown how PLEKHA7 is localized strictly at AJ in epithelial cells.
Because the nectin-afadin system is strictly localized at AJ in epithelial cells, we examined here whether PLEKHA7 is recruited to AJ through this system and found that PLEKHA7 bound to afadin in addition to p120ctn and was associated with nectin by binding to afadin. In addition, we found that this binding of PLEKHA7 to afadin was required for the proper formation of AJ, but not for the formation of TJ, in EpH4 mouse mammary gland epithelial cells. We show here this novel regulatory mechanism for the formation of AJ in epithelial cells.
EXPERIMENTAL PROCEDURES
Plasmid Constructions
A plasmid for HA-tagged PLEKHA7 (HA-PLEKHA7) was constructed by using MultiSite Gateway Pro system for two and three fragments (Invitrogen). The mouse cDNA for PLEKHA7, which corresponds to the longest one (1266 amino acids (aa)) among the transcripts predicted in the UCSC genome browser, was cloned from the kidney of C57/BL6 mice by RT-PCR and inserted into pDONR entry vector (Invitrogen). A cDNA for HA tag was amplified by PCR and inserted into pDONR entry vector. pCX4-puro-DEST was constructed by using a retroviral vector, pCX4 (22), which was modified with a drug-resistant gene against puromycin using Gateway RfA cassette (Invitrogen). pCX4-puro-HA-PLEKHA7 was obtained by using LR recombination reaction. The cDNAs for human p120ctn isoform 3AB (accession number AF062338) and mouse Nezha (accession number BC048787) were cloned from the human small intestine and the kidney of C57/BL6 mice, respectively, by RT-PCR and inserted into the pDONR entry vector. To construct the plasmids for the truncated mutants of PLEKHA7, the following fragments of PLEKHA7 were amplified by PCR and cloned into pDONR entry vectors: ΔWWPH (corresponding to 538–1266 aa), ΔCC (1–696 aa), WWPH (1–374 aa), WW (1–119 aa), and PH (120–374 aa). PLEKHA7-ΔAfBR (1–119 and 375–1266 aa), which lacks the afadin-binding region (AfBR), was constructed by QuikChange site-directed mutagenesis (Stratagene). The plasmid for EGFP-tagged rat afadin was constructed as described previously (14). The plasmids for the full-length and the following fragments of afadin N-terminally tagged with FLAG were constructed as described previously (15): N-PDZ (1–1100 aa), PDZ-C (1016–1829 aa), NN (2–500 aa), NC (501–1100 aa), RA (2–350 aa), FHA (351–500 aa), DIL (501–1020 aa), and PDZ (1001–1100 aa). For bacterial expression of GST-tagged AfBR of PLEKHA7, the N-terminal fragment of human p120ctn isoform 3AB (p120-N; 2–295 aa), and the C-terminal fragment of mouse Nezha (Nezha-C; 940–1252 aa), the cDNAs for each fragment were amplified by PCR and subcloned into pGEX-4T1 vector (GE Healthcare). The plasmids for shRNA expression were constructed using pSIREN-retroQ-GFP vector (Clontech) as described previously (23). The 21-base targeting sequence for mouse PLEKHA7 was 5′-AAGGAGAATAAAGATCAGCTA-3′, and that for mouse afadin was 5′-AAGACAATCCTGCTGTCTACT-3′. As a control, we used a nontargeting sequence, 5′-AAACAAGATGAAGAGCACCAA-3′. To construct the shRNA-resistant PLEKHA7 (sr-PLEKHA7), three bases in the shRNA target sequence in the HA-PLEKHA7 cDNA were mutated (5′-AAAGAAAACAAAGATCAGCTA-3′) by mutagenesis. DNA sequences of constructs were validated by sequencing.
Antibodies
A mouse mAb against afadin was prepared as described (24). A rat mAb against E-cadherin (ECCD2) was a kind gift from Dr. M. Takeichi (Center for Developmental Biology, RIKEN, Japan). The following rabbit polyclonal antibodies (pAbs) were purchased from commercial sources: anti-afadin (Sigma-Aldrich), anti-GFP (Medical and Biological Laboratories), anti-HA (Medical and Biological Laboratories), and anti-PLEKHA7 (HPA038610; Sigma-Aldrich). The following mouse mAbs were purchased from commercial sources: anti-actin (clone C4; Millipore), anti-FLAG (clone M2; Sigma-Aldrich), anti-HA (clone 16B12; Covance), anti-occludin (Zymed Laboratories Inc.), and anti-p120ctn (BD Bioscience). The following rat mAbs were purchased from commercial sources: anti-GFP (clone GF090R; Nacalai Tesque), anti-nectin-2 (clone 502-57; Medical and Biological Laboratories), anti-nectin-3 (clone 103-A1; Medical and Biological Laboratories), anti-occludin (Sanko Junyaku), and anti-ZO-1 (Millipore). Horseradish peroxidase-conjugated secondary Abs were purchased from GE Healthcare. Fluorophore-conjugated secondary Abs were purchased from Chemicon and Jackson ImmunoResearch.
Cells and Cell Culture
L cells stably expressing exogenous mouse nectin-3α or its mutant nectin-3α-ΔC, in which the C-terminal four amino acid residues of nectin-3α were deleted (nectin-3-L cells or nectin-3-ΔC-L cells, respectively), were prepared as described (25). EL cells were established by introducing the cDNA for E-cadherin into L cells (26). Nectin-3-L cells, nectin-3-ΔC-L cells, EL cells, human embryonic kidney HEK293E cells, and EpH4 mouse mammary gland epithelial cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum.
Transfection and Retrovirus Infection
Transfection of cells with plasmids or siRNA was performed using Lipofectamine 2000 or Lipofectamine RNAiMAX reagent (Invitrogen), respectively, according to the manufacturer's instructions. To generate the retroviral supernatant, HEK293E cells were co-transfected with pGP (Takara Bio), pE-Ampho (Takara Bio), and either pCX4-puro or pSIREN-retroQ-GFP plasmid. After a 48-h culture period, the culture medium was centrifuged, and the viral supernatant with 8 μg/ml polybrene was used for infection.
Knockdown Experiments
Stealth RNAi duplexes against mouse p120ctn (5′-GAAGCAGUGUGGACCUGCAUCGUUU-3′) and mouse afadin (5′-CCCAAGACAUAAACCUGGAGCUGUU-3′) were purchased from Invitrogen. As a control, we used stealth RNAi negative control medium GC duplex 2 (Invitrogen). To confirm the knockdown efficiency, the cells were washed with ice-cold PBS and lysed with a lysis buffer (20 mm Tris-HCl, pH 7.5, 150 mm NaCl, 5% glycerol, 0.5% Nonidet P-40, 1 mm MgCl2, 1 mm dithiothreitol, 1 mm Na3VO4, 10 mm sodium fluoride, 1 mm PMSF, and 10 μg/ml leupeptin). The lysates were added with 5× SDS sample buffer, boiled for 5 min, and subjected to SDS-PAGE, followed by Western blotting with appropriate Abs. For knockdown of PLEKHA7 in EpH4 cells, EpH4 cells were infected with the retrovirus encoding the shRNA sequence targeting PLEKHA7 for 24 h and cultured for 48 h. The cells were then plated on coverslips and further cultured for 24 h before fixation. In the rescue experiments, EpH4 cells were infected with the control retrovirus (mock) or the retrovirus coding for HA-sr-PLEKHA7-WT or HA-sr-PLEKHA7-ΔAfBR. Infected EpH4 cells were cultured for 24 h, washed, and selected by culturing with 2 μg/ml puromycin for 4–6 days. The selected EpH4 cells were infected with the shRNA retroviruses for 24 h and processed as described above.
Immunoprecipitation Assay
HEK293E cells were co-transfected with various combinations of plasmids. After incubation for 48 h, the cells were washed with ice-cold PBS and then lysed with the lysis buffer. The cell lysates were obtained by centrifugation at 20,000 × g for 15 min. The cell lysates were incubated with the rabbit anti-GFP pAb-conjugated protein A-Sepharose at 4 °C for 3 h. After the beads were extensively washed with the lysis buffer, the bound proteins were eluted by boiling the beads in SDS sample buffer. The samples were subjected to SDS-PAGE, followed by Western blotting with the rat anti-GFP, rat anti-HA, and mouse anti-FLAG mAbs.
GST Pulldown Assay
GST and GST-fused proteins were expressed in Escherichia coli, and the cells were sonicated in a sonication buffer (50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 0.1% Triton X-100, 1 mm dithiothreitol, 1 mm PMSF, and 10 μg/ml leupeptin). The homogenate was clarified by centrifugation, and the supernatant was then incubated with glutathione-Sepharose 4B (GE Healthcare) at 4 °C for 1 h to immobilize GST-fused proteins. After being washed with the lysis buffer, the beads were incubated with the cell lysates prepared from HEK293E cells exogenously expressing proteins of interest. After 3 h of incubation, the beads were extensively washed with the lysis buffer, and bound proteins were subjected to SDS-PAGE, followed by Western blotting with the appropriate Abs and Amido Black staining.
Immunofluorescence Microscopy
Cells seeded on coverslips were fixed with 1% paraformaldehyde in Hanks' balanced salt solution and then permeabilized with 0.2% Triton X-100 in PBS. After being blocked with 2% goat serum in PBS, the samples were incubated with the primary Abs, followed by incubation with appropriate fluorophore-conjugated secondary Abs. The samples were then washed three times with PBS and mounted in ProLong Gold mount gel (Invitrogen). Fluorescence signals were visualized with a confocal laser scanning microscope (LSM510; Zeiss). The fluorescence intensity of E-cadherin at the cell-cell adhesion site was quantified in randomly chosen fields by ImageJ software, and the average intensity value of each image was calculated. Statistical significance was assessed by Tukey's multiple comparisons test.
RESULTS
Requirement of Afadin for the Recruitment of PLEKHA7 to the Cell-Cell Adhesion Site in Epithelial Cells
We first examined whether afadin is required for the recruitment of PLEKHA7 to the cell-cell adhesion site in EpH4 cells that endogenously express PLEKHA7 (Fig. 1A). For this purpose, we knocked down afadin in EpH4 cells by using the retroviral shRNA vector and analyzed the localization of PLEKHA7. Infection of EpH4 cells with the retrovirus encoding the afadin shRNA significantly suppressed the expression of afadin but did not affect the expression of PLEKHA7 or E-cadherin (Fig. 1A). Because these retroviruses were engineered to simultaneously express shRNA and GFP, the infection of the retroviruses was confirmed by the expression of GFP. The immunofluorescence signals for afadin, PLEKHA7, E-cadherin, and p120ctn were all concentrated at the cell-cell adhesion site between the control retrovirus-infected cells (Fig. 1B, control). In contrast, the signals for all of these molecules were significantly decreased at the cell-cell adhesion site between the afadin shRNA retrovirus-infected cells as compared with that at the cell-cell adhesion site between uninfected GFP-negative cells (Fig. 1B, afadin KD). The reductions of the signals for PLEKHA7 and E-cadherin at the cell-cell adhesion sites by the afadin knockdown were not caused by the reductions of the total amounts of these proteins as judged by Western blotting (Fig. 1A). The reduction of the signal for E-cadherin was consistent with the earlier observations in MDCK cells (15, 27). These results indicate that afadin is required for the recruitment of PLEKHA7 as well as E-cadherin and p120ctn to the cell-cell adhesion site in EpH4 cells. However, it cannot be evaluated from these experiments whether afadin is involved in the recruitment of PLEKHA7 to the cell-cell adhesion site in a p120ctn-independent manner.
FIGURE 1.

Requirement of afadin for the recruitment of PLEKHA7 to the cell-cell adhesion site in EpH4 cells. A, suppression of afadin by shRNA. EpH4 cells infected with the control (Control) or afadin shRNA retrovirus (afadin KD) were cultured for 72 h. The cell lysates were subjected to Western blotting with the indicated Abs. IB, immunoblot. B, localization of PLEKHA7, E-cadherin, and p120ctn in the control and afadin KD cells. EpH4 cells infected with retrovirus coding for the control or afadin shRNA were cultured for 48 h and then replated on coverslips. Twenty-four hours after the replating, the cells were fixed and analyzed by immunostaining with the indicated Abs. Scale bars, 20 μm. The results shown are representative of three independent experiments.
Afadin-dependent and p120ctn-independent Recruitment of PLEKHA7 to the Nectin-3α-based Cell-Cell Adhesion Site in L Cells Stably Expressing Nectin-3α
We therefore next examined whether afadin recruits PLEKHA7 to the cell-cell adhesion site in a manner independent of p120ctn and E-cadherin by using L fibroblasts, because this cell line lacks classical cadherins (28). Afadin and the small amounts of nectin-1 and -2 were expressed in L cells (29). α-Catenin and β-catenin were down-regulated because of the lack of cadherin in this cell line (13). When L cells stably expressing exogenous nectin-3α (nectin-3-L cells) were cultured, the nectin-3α-based cell-cell adhesion was formed as described (Fig. 2) (30). The immunofluorescence signals for both nectin-3α and afadin were concentrated at the cell-cell adhesion site (Fig. 2, arrowheads). To examine the recruitment of PLEKHA7 to the nectin-3α-based cell-cell adhesion site, HA-PLEKHA7 was expressed in nectin-3-L cells in which endogenous PLEKHA7 was undetectable by Western blotting (data not shown). The signal for HA-PLEKHA7 was co-localized with the signals for nectin-3α and afadin (Fig. 2, arrowheads). However, the signal for p120ctn was not detected there (Fig. 2, arrows). These results indicate that PLEKHA7 is recruited to the nectin-3α-based cell-cell adhesion site presumably in a p120ctn-independent manner.
FIGURE 2.
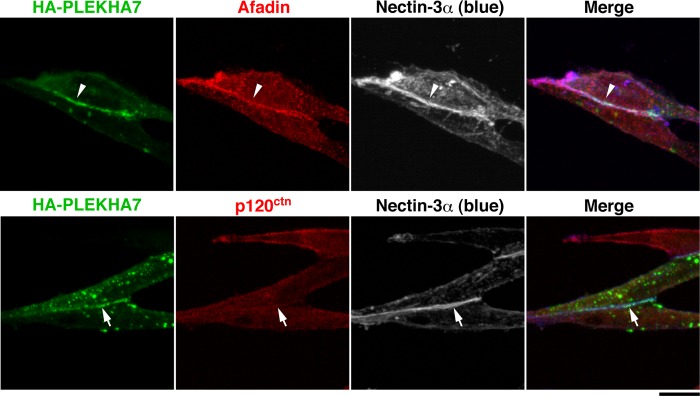
Recruitment of PLEKHA7 to the nectin-3α-based cell-cell adhesion site in L cells stably expressing nectin-3α. Nectin-3-L cells were transfected with HA-tagged PLEKHA7, cultured on coverslips for 24 h, and stained with various combinations of the indicated Abs. Scale bar, 10 μm. The results shown are representative of three independent experiments.
We further confirmed the p120ctn-independent recruitment of PLEKHA7 to the nectin-3α-based cell-cell adhesion site by knocking down p120ctn in nectin-3-L cells. When p120ctn was knocked down in nectin-3-L cells by using siRNA, the p120ctn protein was markedly reduced as estimated by Western blotting (Fig. 3Aa). In the p120ctn knockdown (KD)-nectin-3-L cells, the signals for nectin-3α, afadin, and HA-PLEKHA7 were concentrated at the cell-cell adhesion site to the extents similar to those in the control siRNA-transfected nectin-3-L cells and untransfected nectin-3-L cells (Fig. 3Ab, arrowheads; see also Fig. 2). In a quantitative analysis, the percentages of the nectin-3α-based cell-cell adhesion site at which afadin or HA-PLEKHA7 accumulated were not reduced in the p120ctn KD-nectin-3-L cells as compared with those in the control cells (Fig. 3Ac). These results further indicate that PLEKHA7 is recruited to the nectin-3α-based cell-cell adhesion site in a p120ctn-independent manner.
FIGURE 3.

Recruitment of PLEKHA7 to the nectin-3α-based cell-cell adhesion site in a p120ctn-independent and afadin-dependent manner. Aa, confirmation of the knockdown of p120ctn by Western blotting. Nectin-3-L cells were transfected with control siRNA (control) or p120ctn siRNA (p120ctn KD) and cultured for 48 h. The cell lysates were subjected to SDS-PAGE, followed by Western blotting with the indicated Abs. IB, immunoblot. Ab, localization of HA-PLEKHA7 in the control and p120ctn KD-nectin-3-L cells. Nectin-3-L cells were transfected with the control or p120ctn siRNA. After a 48-h culture, the cells were transfected with HA-PLEKHA7, incubated for 24 h, and stained with various combinations of the indicated Abs. Ac, quantitative analysis of Ab. The percentages of the nectin-3α-based cell-cell adhesion site at which the indicated proteins accumulated are shown as recruitment indices in the bar graph. The data are the means ± S.E. of three independent experiments. Ba, localization of HA-PLEKHA7 in nectin-3-ΔC-L cells. Nectin-3-ΔC-L cells were transfected with HA-PLEKHA7, cultured on coverslips for 24 h, and stained with various combinations of the indicated Abs. Bb, quantitative analysis of Ba. The percentages of the nectin-3α-based cell-cell adhesion site at which the indicated proteins accumulated are shown as the recruitment indices in the bar graph. The data are the means ± S.E. of three independent experiments. Scale bars, 10 μm. The results shown are representative of three independent experiments.
We then examined whether this recruitment of PLEKHA7 is dependent on afadin. For this purpose, we used nectin-3α-ΔC, a mutant of nectin-3α lacking the C-terminal four aa residues necessary for the binding to afadin (31, 32). When L cells stably expressing nectin-3α-ΔC (nectin-3-ΔC-L cells) were transfected with HA-PLEKHA7, the signal for nectin-3α-ΔC was concentrated at the cell-cell adhesion site as described previously (30), but the signal for afadin or HA-PLEKHA7 was not concentrated there (Fig. 3Ba, arrows). In a quantitative analysis, the percentages of the nectin-3α-based cell-cell adhesion site at which afadin or HA-PLEKHA7 accumulated were markedly reduced in the nectin-3-ΔC-L cells as compared with those in the nectin-3-L cells (Fig. 3Bb). These results suggest that the recruitment of PLEKHA7 to the nectin-3α-based cell-cell adhesion site is dependent on the binding of afadin to the cytoplasmic tail of nectin-3α. Taken together, it is likely that PLEKHA7 is recruited to the nectin-3α-based cell-cell adhesion site in a manner dependent on afadin but independent of p120ctn.
Afadin-independent Recruitment of PLEKHA7 to the E-cadherin-based Cell-Cell Adhesion Site in L Cells Stably Expressing E-cadherin
We then examined whether PLEKHA7 is recruited to the E-cadherin-based cell-cell adhesion site in an afadin-dependent or -independent manner. When HA-PLEKHA7 was expressed in L cells stably expressing E-cadherin (EL cells), it was recruited to the E-cadherin-based cell-cell adhesion site where the immunofluorescence signals for p120ctn and afadin were concentrated (Fig. 4A, arrowheads). When afadin was knocked down in EL cells by using siRNA, the afadin protein was markedly reduced as estimated by Western blotting (Fig. 4Ba). The signal for afadin was observed at the E-cadherin-based cell-cell adhesion site in the control cells (Fig. 4Bb, upper panels, arrowheads), but not in the afadin KD-EL cells (Fig. 4Bb, upper panels, arrows). Under these conditions, the signal for HA-PLEKHA7 was observed at the E-cadherin-based cell-cell adhesion site in the afadin KD-EL cells where the signal for afadin was not concentrated (Fig. 4Bb, upper panels, arrowheads). The signal for p120ctn was concentrated at the E-cadherin-based cell-cell adhesion site in both the control and afadin KD-EL cells (Fig. 4Bb, lower panels, arrowheads). In a quantitative analysis, the percentage of the E-cadherin-based cell-cell adhesion site at which afadin accumulated was clearly diminished in the afadin KD-EL cells as compared with that in the control EL cells, whereas HA-PLEKHA7 was still recruited to the E-cadherin-based cell-cell adhesion site in the afadin KD-EL cells with similar frequency in the control cells (Fig. 4Bc). These results indicate that PLEKHA7 is recruited to the E-cadherin-based cell-cell adhesion site in an afadin-independent manner. These results are apparently inconsistent with the results in Fig. 1 that the knockdown of afadin reduced the accumulations of the signals for E-cadherin and p120ctn at the cell-cell adhesion site in the afadin KD-EpH4 cells. The reason for this apparent inconsistency is discussed under “Discussion.”
FIGURE 4.

Afadin-independent recruitment of PLEKHA7 to the E-cadherin-based cell-cell adhesion site in L cells stably expressing E-cadherin. A, recruitment of PLEKHA7 to the E-cadherin-based cell-cell adhesion site in EL cells. EL cells were transfected with HA-PLEKHA7, cultured on coverslips for 24 h, and stained with the indicated Abs. B, afadin-independent recruitment of PLEKHA7 to the E-cadherin-based cell-cell adhesion site. Ba, confirmation of the knockdown of afadin by Western blotting. EL cells were transfected with the control or afadin siRNA and cultured for 48 h. The cell lysates were subjected to SDS-PAGE, followed by Western blotting with the indicated Abs. IB, immunoblot. Bb, recruitment of PLEKHA7 to the E-cadherin-based cell-cell adhesion site. EL cells were transfected with the control or afadin siRNA and cultured for 48 h. The cells were then transfected with HA-PLEKHA7, incubated for 24 h, and stained with various combinations of the indicated Abs. Bc, quantitative analysis of Bb. The percentages of the E-cadherin-based cell-cell adhesion site at which the indicated proteins accumulated are shown as the recruitment indices in the bar graph. The data are the means ± S.E. of three independent experiments. Scale bars, 10 μm. The results shown are representative of three independent experiments.
Binding of PLEKHA7 to Afadin
We next examined by the immunoprecipitation assay whether PLEKHA7 binds to afadin. When GFP-afadin and HA-PLEKHA7 were co-expressed in HEK293E cells and the cell lysates were immunoprecipitated with the anti-GFP pAb, HA-PLEKHA7 was co-immunoprecipitated with GFP-afadin (Fig. 5A). Under these conditions, endogenous p120ctn was not co-immunoprecipitated with GFP-afadin (data not shown).
FIGURE 5.

Binding of PLEKHA7 to afadin. A, co-immunoprecipitation of afadin with PLEKHA7. The cell lysates from HEK293E cells co-transfected with various combinations of the indicated plasmids were immunoprecipitated with the anti-GFP pAb. The samples were analyzed by Western blotting with the indicated Abs. B, the afadin-binding region of PLEKHA7. HEK293E cells were co-transfected with various deletion mutants of HA-PLEKHA7 and GFP-afadin. The cell lysates were immunoprecipitated with the anti-GFP pAb, and the samples were analyzed by Western blotting with the indicated Abs. Ba, co-immunoprecipitation of the N-terminal fragment (ΔCC), but not the C-terminal fragment (ΔWWPH), with GFP-afadin. Bb, co-immunoprecipitation of the PLEKHA7-PH fragment, but not of the WW fragment, with GFP-afadin. Bc, decrease of the binding of PLEKHA7-ΔAfBR to GFP-afadin. Bd, schematic diagram of the PLEKHA7 fragments and their binding to afadin. C, the PLEKHA7-binding regions of afadin. HEK293E cells were transfected with the FLAG-tagged afadin mutants, and the cell lysates were incubated with GST or GST-tagged PLEKHA7-AfBR (GST-AfBR) immobilized on glutathione-Sepharose. The samples were analyzed by Western blotting with the anti-FLAG mAb and Amido Black staining. Ca, binding of the N-terminal half of afadin (N-PDZ) to GST-AfBR. Cb, binding of both the NN and NC fragments of afadin to GST-AfBR. Cc, binding of the RA fragment of afadin to GST-AfBR. Cd, binding of the PDZ fragment of afadin to GST-AfBR. Ce, schematic diagram of the afadin fragments and binding to GST-AfBR of PLEKHA7. The results shown are representative of three independent experiments. IB, immunoblot; IP, immunoprecipitation.
To determine the afadin-binding region of PLEKHA7, a series of truncated mutants of PLEKHA7 shown in Fig. 5Bd were co-expressed with GFP-afadin in HEK293E cells, and GFP-afadin was immunoprecipitated with the anti-GFP pAb. In this assay, an N-terminal fragment (ΔCC), but not a C-terminal fragment (ΔWWPH), was co-immunoprecipitated with GFP-afadin (Fig. 5Ba). These results indicate that afadin binds the N-terminal region of PLEKHA7, which contains a WW domain and a PH domain. Therefore, we further examined whether the N-terminal fragment containing both the WW and PH domains and the fragments containing only the WW or PH domain bind to afadin. The fragment containing the WW and PH domains (WWPH) and the fragment containing only the PH domain (PH), but not the fragment containing only the WW domain (WW), were co-immunoprecipitated with GFP-afadin (Fig. 5Bb), indicating that the region containing the PH domain and its flanking residues of PLEKHA7 (120–374 aa) is responsible for the binding to afadin. Consistent with these results, the amount of a mutant of PLEKHA7 lacking this region, which was co-immunoprecipitated with GFP-afadin, was markedly reduced as compared with that of full-length PLEKHA7 co-immunoprecipitated with GFP-afadin (Fig. 5Bc). These results indicate that the region of PLEKHA7 (120–374 aa) is responsible for the binding to afadin. We hereafter refer to this region as the afadin-binding region (AfBR).
We next mapped the PLEKHA7-binding region of afadin by GST pulldown assay by using GST-PLEKHA7-AfBR (GST-AfBR). FLAG-tagged afadin and deletion mutants of afadin shown in Fig. 5Ce were expressed in HEK293E cells, and the lysates of these cells were incubated with GST-AfBR immobilized on glutathione-Sepharose. Full-length afadin (WT) and an N-terminal half of afadin (N-PDZ) were specifically co-precipitated with GST-AfBR, but not with GST (Fig. 5Ca). In contrast, a C-terminal half of afadin (PDZ-C) was not co-precipitated with GST-AfBR. When the N-PDZ fragment was divided into two fragments (NN and NC; see Fig. 5Ce) and each fragment was incubated with GST-AfBR, both the NN and NC fragments were co-precipitated with GST-AfBR (Fig. 5Cb). When the NN fragment was divided into two fragments (RA and FHA; see Fig. 5Ce) and each fragment was incubated with GST-AfBR, the RA fragment, but not the FHA fragment, was co-precipitated with GST-AfBR (Fig. 5Cc). When the NC fragment was divided into two fragments (DIL and PDZ; see Fig. 5Ce) and each fragment was incubated with GST-AfBR, the PDZ fragment, but not the DIL fragment, was co-precipitated with GST-AfBR (Fig. 5Cd). These results indicate that afadin contains at least two PLEKHA7-binding regions (2–350 aa and 1001–1100 aa).
Binding of PLEKHA7-ΔAfBR to p120ctn and Nezha
The afadin-binding region of PLEKHA7 is distinct from its p120ctn- and Nezha-binding regions (Fig. 5Bd). To exclude the possibility that the deletion of AfBR would affect the binding of PLEKHA7 to p120ctn and/or Nezha, we examined whether PLEKHA7-ΔAfBR binds to p120ctn and Nezha by GST pulldown assay. Because PLEKHA7 binds to an N-terminal region of p120ctn and a C-terminal region of Nezha (19), we used GST-tagged PLEKHA7-binding region of p120ctn (GST-p120-N) and that of Nezha (GST-Nezha-C). GST, GST-p120-N, or GST-Nezha-C was immobilized on beads and incubated with the lysates from HEK293E cells expressing HA-PLEKHA7-WT or -ΔAfBR. HA-PLEKHA7-WT was co-precipitated with both GST-p120-N and GST-Nezha-C, but not with GST, consistent with the previous observation that PLEKHA7-WT specifically bound to p120-N and Nezha-C (19) (Fig. 6). Under the same conditions, HA-PLEKHA7-ΔAfBR was also co-precipitated with both GST-p120-N and GST-Nezha-C (Fig. 6). These results indicate that the deletion of the afadin-binding region does not affect the binding of PLEKHA7 to p120ctn and Nezha.
FIGURE 6.
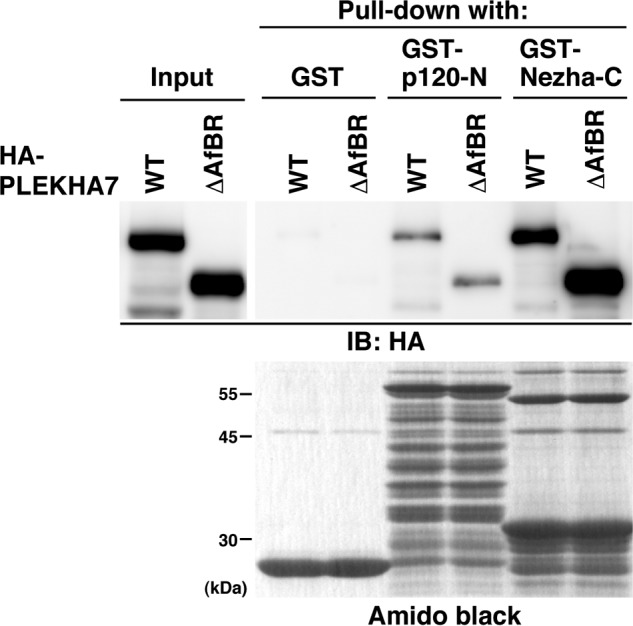
Binding of PLEKHA7-ΔAfBR to p120ctn and Nezha. The lysates from HEK293E cells expressing HA-PLEKHA7-WT or -ΔAfBR were incubated with GST, GST-p120-N, or GST-Nezha-C, which was immobilized on glutathione-Sepharose 4B. The cell lysates and precipitated proteins were subjected to SDS-PAGE, followed by Western blotting with the anti-HA Ab and Amido Black staining. The results shown are representative of three independent experiments. IB, immunoblot.
Requirement of the Binding of PLEKHA7 to Afadin for the Proper Formation of AJ, but Not for the Formation of TJ, in EpH4 Cells
We lastly examined whether the binding of PLEKHA7 to afadin is required for the formation of AJ and/or TJ. First, we knocked down PLEKHA7 in EpH4 cells by using retroviral shRNA vectors as in Fig. 1 and analyzed the junctional localizations of the AJ and TJ components. Infection of EpH4 cells with the retrovirus encoding the PLEKHA7 shRNA significantly suppressed the expression of PLEKHA7 without affecting those of afadin and E-cadherin (Fig. 7Aa). The signal for PLEKHA7 was concentrated at the cell-cell adhesion site between the control retrovirus-infected cells (Fig. 7Ab, control), but hardly observed at that between the PLEKHA7 shRNA retrovirus-infected cells (Fig. 7Ab, PLEKHA7 KD). In the control cells, the signals for nectin-2, afadin, E-cadherin, p120ctn, ZO-1, and occludin were all concentrated at the cell-cell adhesion site (Fig. 7B). In this cell line, nectin-2α and -2δ were expressed as judged by Western blotting (data not shown). When PLEKHA7 was knocked down by shRNA, the signals for E-cadherin and p120ctn at the cell-cell adhesion site were markedly decreased as compared with those in the control cells, although they still remained to small extents (Fig. 7B). The reduction of the accumulation of the signal for E-cadherin at the cell-cell adhesion sites by the PLEKHA7 knockdown was not caused by the reduction of the total amount of the E-cadherin protein (Fig. 7Aa). In contrast, the signal for afadin, nectin-2, occludin, or ZO-1 was not markedly changed (Fig. 7B). These results are consistent with the earlier observations in Caco2 cells (19) and indicate that PLEKHA7 is required for the proper formation of AJ, but not for the formation of TJ. The residual signals for E-cadherin and p120ctn at the cell-cell adhesion site in the PLEKHA7 KD cells might be caused by the recruitment of these molecules mediated by other linker molecules for the nectin-afadin and cadherin-catenin systems as described (10, 13).
FIGURE 7.

Requirement of the binding of PLEKHA7 to afadin for the proper formation of AJ, but not for the formation of TJ, in EpH4 cells. A, suppression of PLEKHA7 by shRNA. EpH4 cells infected with the control (control) or the PLEKHA7 shRNA retrovirus (PLEKHA7 KD) were cultured for 72 h. Expression of PLEKHA7 was analyzed by Western blotting (Aa) and immunostaining (Ab) with the indicated Abs. The cells infected with the retroviruses were marked by GFP fluorescence in Ab. IB, immunoblot. B, localization of the junctional proteins in the control and PLEKHA7 KD cells. EpH4 cells infected with retrovirus coding for the control or PLEKHA7 shRNA were cultured for 48 h and then replated on coverslips. Twenty-four hours after the replating, the cells were fixed and analyzed by immunostaining with the indicated Abs. Ca, restoration of the decrease of the signal for E-cadherin at the cell-cell adhesion site in PLEKHA7 KD cells by the re-expression of sr-PLEKHA7-WT, but not by the re-expression of sr-PLEKHA7-ΔAfBR. EpH4 cells were infected with the retrovirus encoding shRNA together with the retrovirus encoding HA-sr-PLEKHA7-WT or -ΔAfBR and then processed as described in B. Cb, quantitative analysis of the intensity of the signal for E-cadherin at the cell-cell adhesion site. The bar graph represents the relative fluorescence intensity of the signal for E-cadherin at the cell-cell adhesion site. The data are the means ± S.E. of 15 images from three independent experiments, with the mean fluorescence in the cells infected with control shRNA and mock retroviruses normalized to 1.0. *, p < 0.05 with Tukey's multiple comparisons test. Scale bars, 20 μm. The results shown are representative of three independent experiments.
We then examined whether the mutant of PLEKHA7 incapable of binding to afadin (PLEKHA7-ΔAfBR) does not rescue the formation of AJ in the PLEKHA7 KD cells under the conditions where full-length PLEKHA7 rescues it. To perform this rescue experiment, we constructed an shRNA-resistant PLEKHA7 (sr-PLEKHA7) cDNA bearing three silent mutations in the shRNA target sequence. When EpH4 cells were infected with the HA-sr-PLEKHA7-WT retrovirus and the PLEKHA7 shRNA retrovirus, HA-sr-PLEKHA7-WT was expressed in the GFP-positive PLEKHA7 shRNA-expressing cells, and the signal for HA-sr-PLEKHA7-WT was concentrated at the cell-cell adhesion site between GFP-positive cells (Fig. 7Ca). The signal for E-cadherin was significantly increased at the cell-cell adhesion site in these cells as compared with that at the cell-cell adhesion site in the PLEKHA7 KD cells infected with mock retrovirus (Fig. 7C, a and b). In contrast, the mutant of PLEKHA7 incapable of binding to afadin (HA-sr-PLEKHA7-ΔAfBR) was localized in a punctate pattern in the cytosol and at the cell-cell adhesion site; however, its accumulation at the cell-cell adhesion site was significantly weaker than that in the PLEKHA7 KD cells in which HA-sr-PLEKHA7-WT was expressed (Fig. 7Ca). The signal for E-cadherin was not significantly increased at the cell-cell adhesion site in these cells as compared with that at the cell-cell adhesion site in the PLEKHA7 KD cells infected with mock retrovirus (Fig. 7C, a and b). These results indicate that the mutant of PLEKHA7 incapable of binding to afadin does not rescue the proper formation of AJ in the PLEKHA7 KD cells under the conditions where full-length PLEKHA7 rescues it. Taken together, these results indicate that the binding of PLEKHA7 to afadin is required at least partly for the proper formation of AJ, but not for the formation of TJ.
DISCUSSION
It was previously shown that PLEKHA7 binds to p120ctn and is recruited to the E-cadherin-based cell-cell adhesion site in Caco2 cells (19). However, PLEKHA7 is more strictly localized at AJ, whereas E-cadherin and its associating proteins, such as β-catenin, p120ctn, and α-catenin, are localized not only at AJ but also at the basal side of AJ along the lateral plasma membranes of attached epithelial cells, such as MDCK, Caco2, DLD-1, and MCF-10A cells and small intestinal absorptive epithelial cells, although all of them are more highly concentrated at AJ than at the basal side of AJ along the lateral plasma membranes of these attached epithelial cells (9, 19, 33–36). We have shown here that: 1) PLEKHA7 binds to afadin in addition to p120ctn; 2) PLEKHA7 is recruited to the nectin-3α-based cell-cell adhesion site by binding to afadin in a p120ctn-independent manner; and 3) PLEKHA7 is recruited to the E-cadherin-based cell-cell adhesion site in an afadin-independent manner. Considering the previous result that nectin and afadin are strictly localized at AJ in epithelial cells (36), afadin is a likely candidate molecule that localizes PLEKHA7 strictly at AJ. However, the ability of PLEKHA7 to bind to afadin is not sufficient to explain its strict localization at AJ, because PLEKHA7 is able to bind to p120ctn, which is localized at both AJ and the basal side of AJ along the lateral plasma membranes of attached cells, and PLEKHA7 is recruited to the E-cadherin-based cell-cell adhesion site in an afadin-independent manner in EL cells (Fig. 4). In addition, another PLEKHA7-binding protein paracingulin is localized at both AJ and TJ (20). These three PLEKHA7-binding proteins all are localized at AJ, although p120ctn and paracingulin are distributed to other sites than AJ. Considering the previous result that E-cadherin, β-catenin, p120ctn, and α-catenin are more highly concentrated at AJ than at the basal side of AJ along the lateral plasma membranes of attached epithelial cells, such as MDCK and MCF-10A cells (33, 34), p120ctn concentrated at AJ would contribute to the strict localization of PLEKHA7 at AJ cooperatively with afadin. This likely cooperative role of afadin and p120ctn is supported by the results of the rescue experiment in which the mutant of PLEKHA7 incapable of binding to afadin did not rescue the proper formation of AJ, as estimated by E-cadherin accumulation at the cell-cell adhesion site, in the PLEKHA7 KD EpH4 cells under the conditions where full-length PLEKHA7 rescued it (Fig. 7C, a and b). In addition, the signal for this mutant of PLEKHA7 was observed at the cell-cell adhesion site, but its accumulation at the cell-cell adhesion site was significantly weaker as compared with wild-type PLEKHA7 (Fig. 7Ca). The weak localization of this mutant of PLEKHA7 at the cell-cell adhesion site was likely to be mediated by residual p120ctn, which bound to residual E-cadherin at AJ, but not by afadin, in the PLEKHA7 KD EpH4 cells. The deletion of the afadin-binding region of PLEKHA7 did not affect its binding to p120ctn (Fig. 6), and therefore PLEKHA7-ΔAfBR would be recruited to the cell-cell adhesion site where p120ctn is localized through its binding to p120ctn. Importantly, the depletion of afadin in EpH4 cells disrupted the accumulations of PLEKHA7, p120ctn, and E-cadherin at the cell-cell adhesion site (Fig. 1). This strongly supports the role for afadin in promoting the accumulations of these proteins at the cell-cell adhesion site. However, another possible mechanism in which an unidentified factor(s) is involved in the strict localization of PLEKHA7 at AJ in addition to afadin and p120ctn cannot be excluded. Further studies are needed to establish the mechanism that localizes PLEKHA7 strictly at AJ.
We have then shown here the role of the binding of PLEKHA7 to the nectin-afadin system. The binding of PLEKHA7 to afadin was necessary for the proper formation of AJ probably by promoting the recruitment of the cadherin-catenin complex to the nectin-based cell-cell adhesion site. Our previous series of studies have revealed that the nectin-afadin system first forms cell-cell adhesion and then recruits the cadherin-catenin complex to the nectin-based cell-cell adhesion site to form AJ (10). The association between the nectin-afadin and cadherin-catenin systems is mediated by afadin, α-catenin, and their binding proteins. Afadin binds to α-catenin directly (12, 13) and indirectly through afadin-binding proteins including LIM domain only 7, afadin dilute domain-interacting protein, and ponsin (10). In the PLEKHA7 KD-EpH4 cells, the immunofluorescence signals for E-cadherin and p120ctn at the cell-cell adhesion site were markedly reduced but still remained to small extents under the conditions where the signal for PLEKHA7 was mostly diminished (Fig. 7B). These residual signals for E-cadherin and p120ctn at the cell-cell adhesion site might be caused by the recruitment of these molecules mediated by these afadin-binding proteins other than PLEKHA7. Taken together, it is likely that PLEKHA7 regulates at least partly the association of the nectin-afadin and cadherin-catenin systems. However, the physical and functional associations of PLEKHA7 with other afadin-binding proteins remain unknown.
In contrast to the finding that the binding of PLEKHA7 to afadin is important to promote the accumulation of E-cadherin at cell-cell adhesion site in EpH4 cells (Fig. 7), the E-cadherin-based cell-cell adhesion was formed upon the knockdown of afadin in EL cells (Fig. 4). These apparently inconsistent results are probably attributed to the difference of the cell lines used in the experiments. Wild-type L cells, which lack cadherin, express nectin-1 and -2 to small extents but do not form cell aggregates in the cell aggregation assay (29). This result indicates that the amounts of nectins are not sufficient to form the nectin-based cell-cell adhesion in L cells. Despite this fact, when E-cadherin is exogenously expressed in L cells, these EL cells form the E-cadherin-based cell-cell adhesion under the condition where the endogenous nectin-afadin system would not work sufficiently, presumably because of the sufficient expression of E-cadherin. This is probably the reason why the E-cadherin-based cell-cell adhesion is formed in the afadin KD-EL cells. On the other hand, EpH4 cells endogenously express E-cadherin, nectin-2, occludin, p120ctn, afadin, and ZO-1. Under this more physiological condition, the uncoupling of PLEKHA7 to afadin resulted in the failure to promote the accumulation of E-cadherin at the cell-cell adhesion site. These results indicate the significance of the recruitment of PLEKHA7 to the cell-cell adhesion site through afadin for the proper formation of AJ in epithelial cells.
It is currently unknown at which stage PLEKHA7 is recruited to AJ by afadin and p120ctn and how it regulates the formation of AJ in epithelial cells. One possible mechanism is that when the nectin-afadin system first forms cell-cell adhesion, PLEKHA7 is recruited to this adhesion site through its binding to afadin and when the cadherin-catenin complex is recruited to the nectin-based cell-cell adhesion site where PLEKHA7 is localized, it is associated with PLEKHA7 through p120ctn, resulting in the formation of AJ. Another possible mechanism is that after the cell-cell adhesion is formed by both the nectin-afadin and cadherin-catenin systems, PLEKHA7 is recruited there cooperatively by afadin and p120ctn. PLEKHA7 recruited in this way then would promote the further accumulation of the cadherin-catenin complex at the cell-cell adhesion site to establish AJ properly. The expression of PLEKHA7 is detected mainly in epithelial tissues and cultured epithelial cells (19, 35), and the expression of PLEKHA7 in other nonepithelial cells is unknown. Therefore, the role for PLEKHA7 shown here in the proper formation of AJ may contribute to the formation of AJ specifically in epithelial cells. However, further studies are needed for our understanding of the precise molecular mechanisms for the formation of AJ in epithelial cells.
It was considered that the cadherin-based cell-cell adhesion is required for the formation of TJ (5), but recent studies have shown that the nectin-based cell-cell adhesion, but not the cadherin-based cell-cell adhesion, is necessary for the formation of TJ (37–42).3 Furthermore, we have shown that afadin is essential for the formation of TJ (15). We have shown here that the knockdown of PLEKHA7 reduces the accumulation of the cadherin-catenin complex at the cell-cell adhesion site but does not reduce the accumulation of TJ components, such as ZO-1 and occludin. These results are also consistent with the notion that the cadherin-catenin system is not absolutely required for the formation of TJ.
Acknowledgment
We thank Dr. M. Takeichi for generous gifts of reagents.
This work was supported by the Global Centers of Excellence Program “Global Center for Education and Research in Integrative Membrane Biology” and the Targeted Proteins Research Program from the Ministry of Education, Culture, Sports, Science and Technology, Japan, by grants-in-aid from the Japan Society for the Promotion of Science, by the Core Research for Evolutional Science and Technology from the Japan Science and Technology Agency, and by grants from the Naito Foundation, the Sagawa Foundation, and the Yasuda Medical Foundation.
T. Yamada, K. Kuramitsu, E. Rikitsu, S. Kurita, W. Ikeda, and Y. Takai, submitted for publication.
- TJ
- tight junction
- AJ
- adherens junction
- CAM
- cell adhesion molecule
- F-actin
- actin filament
- aa
- amino acid
- pAb
- polyclonal antibody
- KD
- knockdown
- AfBR
- afadin-binding region
- PH
- pleckstrin homology
- MDCK
- Madin-Darby canine kidney
- sr
- shRNA-resistant.
REFERENCES
- 1. Farquhar M. G., Palade G. E. (1963) Junctional complexes in various epithelia. J. Cell Biol. 17, 375–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Anderson J. M., Van Itallie C. M. (2009) Physiology and function of the tight junction. Cold Spring Harb. Perspect. Biol. 1, a002584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tsukita S., Yamazaki Y., Katsuno T., Tamura A. (2008) Tight junction-based epithelial microenvironment and cell proliferation. Oncogene 27, 6930–6938 [DOI] [PubMed] [Google Scholar]
- 4. Ikenouchi J., Suzuki M., Umeda K., Ikeda K., Taguchi R., Kobayashi T., Sato S. B., Kobayashi T., Stolz D. B., Umeda M. (2012) Lipid polarity is maintained in absence of tight junctions. J. Biol. Chem. 287, 9525–9533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gumbiner B., Stevenson B., Grimaldi A. (1988) The role of the cell adhesion molecule uvomorulin in the formation and maintenance of the epithelial junctional complex. J. Cell Biol. 107, 1575–1587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Takai Y., Irie K., Shimizu K., Sakisaka T., Ikeda W. (2003) Nectins and nectin-like molecules. Roles in cell adhesion, migration, and polarization. Cancer Sci. 94, 655–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Takeichi M. (1991) Cadherin cell adhesion receptors as a morphogenetic regulator. Science 251, 1451–1455 [DOI] [PubMed] [Google Scholar]
- 8. Takeichi M. (2007) The cadherin superfamily in neuronal connections and interactions. Nat. Rev. Neurosci. 8, 11–20 [DOI] [PubMed] [Google Scholar]
- 9. Abe K., Takeichi M. (2008) EPLIN mediates linkage of the cadherin catenin complex to F-actin and stabilizes the circumferential actin belt. Proc. Natl. Acad. Sci. U.S.A. 105, 13–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Takai Y., Ikeda W., Ogita H., Rikitake Y. (2008) The immunoglobulin-like cell adhesion molecule nectin and its associated protein afadin. Annu. Rev. Cell Dev. Biol. 24, 309–342 [DOI] [PubMed] [Google Scholar]
- 11. Takai Y., Miyoshi J., Ikeda W., Ogita H. (2008) Nectins and nectin-like molecules. Roles in contact inhibition of cell movement and proliferation. Nat. Rev. Mol. Cell Biol. 9, 603–615 [DOI] [PubMed] [Google Scholar]
- 12. Pokutta S., Drees F., Takai Y., Nelson W. J., Weis W. I. (2002) Biochemical and structural definition of the l-afadin- and actin-binding sites of α-catenin. J. Biol. Chem. 277, 18868–18874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tachibana K., Nakanishi H., Mandai K., Ozaki K., Ikeda W., Yamamoto Y., Nagafuchi A., Tsukita S., Takai Y. (2000) Two cell adhesion molecules, nectin and cadherin, interact through their cytoplasmic domain-associated proteins. J. Cell Biol. 150, 1161–1176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hoshino T., Sakisaka T., Baba T., Yamada T., Kimura T., Takai Y. (2005) Regulation of E-cadherin endocytosis by nectin through afadin, Rap1, and p120ctn. J. Biol. Chem. 280, 24095–24103 [DOI] [PubMed] [Google Scholar]
- 15. Ooshio T., Kobayashi R., Ikeda W., Miyata M., Fukumoto Y., Matsuzawa N., Ogita H., Takai Y. (2010) Involvement of the interaction of afadin with ZO-1 in the formation of tight junctions in Madin-Darby canine kidney cells. J. Biol. Chem. 285, 5003–5012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Van Itallie C. M., Anderson J. M. (2006) Claudins and epithelial paracellular transport. Annu. Rev. Physiol. 68, 403–429 [DOI] [PubMed] [Google Scholar]
- 17. Gerhard D. S., Wagner L., Feingold E. A., Shenmen C. M., Grouse L. H., Schuler G., Klein S. L., Old S., Rasooly R., Good P., Guyer M., Peck A. M., Derge J. G., Lipman D., Collins F. S., Jang W., Sherry S., Feolo M., Misquitta L., Lee E., Rotmistrovsky K., Greenhut S. F., Schaefer C. F., Buetow K., Bonner T. I., Haussler D., Kent J., Kiekhaus M., Furey T., Brent M., Prange C., Schreiber K., Shapiro N., Bhat N. K., Hopkins R. F., Hsie F., Driscoll T., Soares M. B., Casavant T. L., Scheetz T. E., Brown-stein M. J., Usdin T. B., Toshiyuki S., Carninci P., Piao Y., Dudekula D. B., Ko M. S., Kawakami K., Suzuki Y., Sugano S., Gruber C. E., Smith M. R., Simmons B., Moore T., Waterman R., Johnson S. L., Ruan Y., Wei C. L., Mathavan S., Gunaratne P. H., Wu J., Garcia A. M., Hulyk S. W., Fuh E., Yuan Y., Sneed A., Kowis C., Hodgson A., Muzny D. M., McPherson J., Gibbs R. A., Fahey J., Helton E., Ketteman M., Madan A., Rodrigues S., Sanchez A., Whiting M., Madari A., Young A. C., Wetherby K. D., Granite S. J., Kwong P. N., Brinkley C. P., Pearson R. L., Bouffard G. G., Blakesly R. W., Green E. D., Dickson M. C., Rodriguez A. C., Grimwood J., Schmutz J., Myers R. M., Butterfield Y. S., Griffith M., Griffith O. L., Krzywinski M. I., Liao N., Morin R., Palmquist D., Petrescu A. S., Skalska U., Smailus D. E., Stott J. M., Schnerch A., Schein J. E., Jones S. J., Holt R. A., Baross A., Marra M. A., Clifton S., Makowski K. A., Bosak S., Malek J. (2004) The status, quality, and expansion of the NIH full-length cDNA project. The Mammalian Gene Collection (MGC). Genome Res. 14, 2121–2127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Strausberg R. L., Feingold E. A., Grouse L. H., Derge J. G., Klausner R. D., Collins F. S., Wagner L., Shenmen C. M., Schuler G. D., Altschul S. F., Zeeberg B., Buetow K. H., Schaefer C. F., Bhat N. K., Hopkins R. F., Jordan H., Moore T., Max S. I., Wang J., Hsieh F., Diatchenko L., Marusina K., Farmer A. A., Rubin G. M., Hong L., Stapleton M., Soares M. B., Bonaldo M. F., Casavant T. L., Scheetz T. E., Brownstein M. J., Usdin T. B., Toshiyuki S., Carninci P., Prange C., Raha S. S., Loquellano N. A., Peters G. J., Abramson R. D., Mullahy S. J., Bosak S. A., McEwan P. J., McKernan K. J., Malek J. A., Gunaratne P. H., Richards S., Worley K. C., Hale S., Garcia A. M., Gay L. J., Hulyk S. W., Villalon D. K., Muzny D. M., Sodergren E. J., Lu X., Gibbs R. A., Fahey J., Helton E., Ketteman M., Madan A., Rodrigues S., Sanchez A., Whiting M., Young A. C., Shevchenko Y., Bouffard G. G., Blakesley R. W., Touchman J. W., Green E. D., Dickson M. C., Rodriguez A. C., Grimwood J., Schmutz J., Myers R. M., Butterfield Y. S., Krzywinski M. I., Skalska U., Smailus D. E., Schnerch A., Schein J. E., Jones S. J., Marra M. A. (2002) Generation and initial analysis of more than 15,000 full-length human and mouse cDNA sequences. Proc. Natl. Acad. Sci. U.S.A. 99, 16899–16903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Meng W., Mushika Y., Ichii T., Takeichi M. (2008) Anchorage of microtubule minus ends to adherens junctions regulates epithelial cell-cell contacts. Cell 135, 948–959 [DOI] [PubMed] [Google Scholar]
- 20. Pulimeno P., Paschoud S., Citi S. (2011) A role for ZO-1 and PLEKHA7 in recruiting paracingulin to tight and adherens junctions of epithelial cells. J. Biol. Chem. 286, 16743–16750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ohnishi H., Nakahara T., Furuse K., Sasaki H., Tsukita S., Furuse M. (2004) JACOP, a novel plaque protein localizing at the apical junctional complex with sequence similarity to cingulin. J. Biol. Chem. 279, 46014–46022 [DOI] [PubMed] [Google Scholar]
- 22. Akagi T., Sasai K., Hanafusa H. (2003) Refractory nature of normal human diploid fibroblasts with respect to oncogene-mediated transformation. Proc. Natl. Acad. Sci. U.S.A. 100, 13567–13572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sakisaka T., Baba T., Tanaka S., Izumi G., Yasumi M., Takai Y. (2004) Regulation of SNAREs by tomosyn and ROCK. Implication in extension and retraction of neurites. J. Cell Biol. 166, 17–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sakisaka T., Nakanishi H., Takahashi K., Mandai K., Miyahara M., Satoh A., Takaishi K., Takai Y. (1999) Different behavior of l-afadin and neurabin-II during the formation and destruction of cell-cell adherens junction. Oncogene 18, 1609–1617 [DOI] [PubMed] [Google Scholar]
- 25. Satoh-Horikawa K., Nakanishi H., Takahashi K., Miyahara M., Nishimura M., Tachibana K., Mizoguchi A., Takai Y. (2000) Nectin-3, a new member of immunoglobulin-like cell adhesion molecules that shows homophilic and heterophilic cell-cell adhesion activities. J. Biol. Chem. 275, 10291–10299 [DOI] [PubMed] [Google Scholar]
- 26. Nagafuchi A., Shirayoshi Y., Okazaki K., Yasuda K., Takeichi M. (1987) Transformation of cell adhesion properties by exogenously introduced E-cadherin cDNA. Nature 329, 341–343 [DOI] [PubMed] [Google Scholar]
- 27. Ooshio T., Fujita N., Yamada A., Sato T., Kitagawa Y., Okamoto R., Nakata S., Miki A., Irie K., Takai Y. (2007) Cooperative roles of Par-3 and afadin in the formation of adherens and tight junctions. J. Cell Sci. 120, 2352–2365 [DOI] [PubMed] [Google Scholar]
- 28. Nagafuchi A., Takeichi M. (1988) Cell binding function of E-cadherin is regulated by the cytoplasmic domain. EMBO J. 7, 3679–3684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Honda T., Shimizu K., Kawakatsu T., Yasumi M., Shingai T., Fukuhara A., Ozaki-Kuroda K., Irie K., Nakanishi H., Takai Y. (2003) Antagonistic and agonistic effects of an extracellular fragment of nectin on formation of E-cadherin-based cell-cell adhesion. Genes Cells 8, 51–63 [DOI] [PubMed] [Google Scholar]
- 30. Takekuni K., Ikeda W., Fujito T., Morimoto K., Takeuchi M., Monden M., Takai Y. (2003) Direct binding of cell polarity protein PAR-3 to cell-cell adhesion molecule nectin at neuroepithelial cells of developing mouse. J. Biol. Chem. 278, 5497–5500 [DOI] [PubMed] [Google Scholar]
- 31. Miyahara M., Nakanishi H., Takahashi K., Satoh-Horikawa K., Tachibana K., Takai Y. (2000) Interaction of nectin with afadin is necessary for its clustering at cell-cell contact sites but not for its cis dimerization or trans interaction. J. Biol. Chem. 275, 613–618 [DOI] [PubMed] [Google Scholar]
- 32. Takahashi K., Nakanishi H., Miyahara M., Mandai K., Satoh K., Satoh A., Nishioka H., Aoki J., Nomoto A., Mizoguchi A., Takai Y. (1999) Nectin/PRR. An immunoglobulin-like cell adhesion molecule recruited to cadherin-based adherens junctions through interaction with Afadin, a PDZ domain-containing protein. J. Cell Biol. 145, 539–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kinch M. S., Clark G. J., Der C. J., Burridge K. (1995) Tyrosine phosphorylation regulates the adhesions of ras-transformed breast epithelia. J. Cell Biol. 130, 461–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Näthke I. S., Hinck L., Swedlow J. R., Papkoff J., Nelson W. J. (1994) Defining interactions and distributions of cadherin and catenin complexes in polarized epithelial cells. J. Cell Biol. 125, 1341–1352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pulimeno P., Bauer C., Stutz J., Citi S. (2010) PLEKHA7 is an adherens junction protein with a tissue distribution and subcellular localization distinct from ZO-1 and E-cadherin. PLoS One 5, e12207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mandai K., Nakanishi H., Satoh A., Obaishi H., Wada M., Nishioka H., Itoh M., Mizoguchi A., Aoki T., Fujimoto T., Matsuda Y., Tsukita S., Takai Y. (1997) Afadin. A novel actin filament-binding protein with one PDZ domain localized at cadherin-based cell-to-cell adherens junction. J. Cell Biol. 139, 517–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Harris T. J., Peifer M. (2004) Adherens junction-dependent and -independent steps in the establishment of epithelial cell polarity in Drosophila. J. Cell Biol. 167, 135–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Okamoto R., Irie K., Yamada A., Katata T., Fukuhara A., Takai Y. (2005) Recruitment of E-cadherin associated with α- and β-catenins and p120ctn to the nectin-based cell-cell adhesion sites by the action of 12-O-tetradecanoylphorbol-13-acetate in MDCK cells. Genes Cells 10, 435–445 [DOI] [PubMed] [Google Scholar]
- 39. Capaldo C. T., Macara I. G. (2007) Depletion of E-cadherin disrupts establishment but not maintenance of cell junctions in Madin-Darby canine kidney epithelial cells. Mol. Biol. Cell 18, 189–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yamada A., Fujita N., Sato T., Okamoto R., Ooshio T., Hirota T., Morimoto K., Irie K., Takai Y. (2006) Requirement of nectin, but not cadherin, for formation of claudin-based tight junctions in annexin II-knockdown MDCK cells. Oncogene 25, 5085–5102 [DOI] [PubMed] [Google Scholar]
- 41. Kuramitsu K., Ikeda W., Inoue N., Tamaru Y., Takai Y. (2008) Novel role of nectin. Implication in the co-localization of JAM-A and claudin-1 at the same cell-cell adhesion membrane domain. Genes Cells 13, 797–805 [DOI] [PubMed] [Google Scholar]
- 42. Zhang L., Jouret F., Rinehart J., Sfakianos J., Mellman I., Lifton R. P., Young L. H., Caplan M. J. (2011) AMP-activated protein kinase (AMPK) activation and glycogen synthase kinase-3β (GSK-3β) inhibition induce Ca2+-independent deposition of tight junction components at the plasma membrane. J. Biol. Chem. 286, 16879–16890 [DOI] [PMC free article] [PubMed] [Google Scholar]