

Background: Abnormally up-regulated miR-29a is associated with several diseases, so miR-29a repression is important for homeostasis.
Results: miR-29a was repressed by androgen system and inversely inhibited AR expression by targeting IGF1 and CDC42/p85α-p53 pathways in mouse epididymis.
Conclusion: There is an androgen receptor-miR-29a regulatory circuitry in mouse epididymis.
Significance: The mutual repression between miR-29a and AR may be important for epididymal development and functions.
Keywords: Androgen, Androgen Receptor, Epididymis, Gene Regulation, MicroRNA
Abstract
MicroRNAs are involved in a number of cellular processes; thus, their deregulation is usually apt to the occurrence of diverse diseases. Previous studies indicate that abnormally up-regulated miR-29a is associated with several diseases, such as human acute myeloid leukemia and diabetes; therefore, the proper level of miR-29a is critical for homeostasis. Herein, we observed that miR-29a was repressed by androgen/androgen receptor signaling in mouse epididymis by targeting a conserved androgen response element located 8 kb upstream of miR-29b1a loci. It is well known that multiple regulatory programs often form a complicated network. Here, we found that miR-29a reversibly suppressed androgen receptor and its target genes by targeting IGF1 and p53 pathways. miR-29b1a-overexpressing transgenic mice displayed epididymis hypoplasia partially similar to the phenotype of those mice with an impaired androgen-androgen receptor signal system. Taken together, the results demonstrated that there is a regulatory circuitry between the androgen signaling pathway and miR-29a in mouse epididymis that may be vital for epididymal development and functions.
Introduction
MicroRNAs (miRNAs)3 are a class of endogenous, 20–23-nucleotide-long, small non-coding RNAs that serve as post-transcriptional regulators of gene expression (1). miRNAs are involved in a variety of cellular processes and in organ development through regulation of a large number of genes (2). The deregulation of miRNAs is consistently linked to distinct diseases (3–5). Therefore, the proper regulation of miRNAs in the physiological state is important.
Our and other previous studies reveal that miR-29a is up-regulated during postnatal development of many organs, such as heart, liver, lung, epididymis, aorta, brain, cornea, and skeletal muscle, and then maintained at a high level in the adult stage (6–11). However, an abnormally elevated miR-29a level is associated with several diseases. For example, miR-29a is increased in muscle, adipose tissue, and liver of diabetic rats, and the up-regulation of miR-29a causes insulin resistance in adipocytes (12). Recently, Han et al. (13) observed that a significant proportion of human acute myeloid leukemia exhibits enhanced miR-29a expression and suggested that miR-29a may also play a role in human myeloid leukemogenesis. In addition, miR-29a expression is greatly enhanced in patients with invasive breast carcinomas compared with non-invasive hyperplasias (14). All these data indicate that abnormal up-regulation of miR-29a is harmful; thus, miR-29a expression should be properly suppressed.
Indeed, previous studies reveal that miR-29a is suppressed by multiple factors, such as TGF-β/Smad3, c-Myc, Hedgehog, and NF-κB signaling pathways (15–18). In the male reproductive system, especially in epididymis, the androgen pathway plays very important regulatory roles in development, function, gene expression, and even miRNA expression. For example, miR-125b, miR-99a, and miR-100 are suppressed by androgen/AR in LNCaP prostate cancer cells (19). In addition, using chromatin immunoprecipitation followed by sequencing (ChIP-Seq), we discovered an AR-binding site (ARBS) located 8 kb upstream of miR-29b1a loci in mouse caput epididymis.4 All the evidence suggests that miR-29b1 and miR-29a may be involved in the androgen signaling pathway in the male reproductive system.
Epididymis, a highly androgen-sensitive organ responsible for sperm maturation and storage, was used here to test this possibility by using a castration model. Our results showed that miR-29a was repressed by androgen/AR in mouse epididymis. Disruption of the repression by overexpressing miR-29a in transgenic mice resulted in epididymis hypoplasia. Furthermore, elevated miR-29a expression can reversibly suppress AR expression by targeting IGF1 and CDC42/p85α-p53 pathways. Taken together, our results demonstrated a regulatory circuitry between androgen/AR signaling pathway and miR-29a that may be important for epididymal development and functions.
EXPERIMENTAL PROCEDURES
Animals and Cell Culture
Male C57BL/6 mice and male Sprague-Dawley rats were purchased from the Animal Center of the Chinese Academy of Sciences (Shanghai, China). Experiments were conducted according to a protocol approved by the Institute Animal Care Committee. The protocol conforms to internationally accepted guidelines for the humane care and use of laboratory animals. The approved permit number for this study is SYXK2007-0017.
Immortalized mouse epididymal cell line PC-1 from proximal caput epididymis was kindly provided by Dr. Orgebin-Crist (Department of Obstetrics and Gynecology, Vanderbilt University School of Medicine, Nashville, TN). PC-1 cells were grown at 33 °C with 5% CO2 in Iscove's modified Dulbecco's medium supplemented with 10% (v/v) fetal bovine serum (FBS), 1 mm sodium pyruvate, 0.1 mm nonessential amino acids, 4 mm glutamine, penicillin-streptomycin (25,000 units of penicillin G sodium, 25 mg of streptomycin sulfate), and 1 nm 5α-dihydrotestosterone (DHT) (20). The MCF-7 and HEK 293T cells were maintained in DMEM; LNCaP, RM-1, U-2 OS, and T24 cells were maintained in RPMI 1640 medium supplemented with 10% (v/v) FBS; MG-63 cells were maintained in minimum Eagle's medium supplemented with 10% (v/v) FBS; and RWPE-1 cells were maintained in keratinocyte serum-free medium. All of these cells were cultured at 37 °C with 5% CO2. All cell culture reagents were purchased from Invitrogen. For androgen deprivation, cells were plated in medium supplemented with 10% charcoal-stripped serum (Hyclone) 24 h before treatment with DHT. Ethanol was used as a vehicle. The IGF1 protein was purchased from ProSpec Co. (catalog number CYT-216).
Castration and Androgen Replacement
Castration and androgen replacement were conducted following a protocol described previously (21, 22). Briefly, for the mouse castration model, adult 10-week-old male C57BL/6 mice were castrated bilaterally under sodium pentobarbital anesthesia. The mice were divided into three groups (six mice per group), killed at different times after castration (0 and 2 days after the initial testosterone propionate (TP, 2.5 mg/kg of body weight) or sesame oil injections (7 days after castration)). For the rat castration model, 120-day-old normal male Sprague-Dawley rats were divided into nine groups (four to seven rats per group) and killed on days 1, 3, 5, and 7 after castration as well as 1, 3, 5, and 7 days after the initial TP injection. Androgen supplementation began on the 7th day after castration, and the rats were injected with TP (3 mg/kg of body weight) every 2 days. The epididymidis, prostate, and brain were excised and used for RNA extraction.
Northern Blotting
The RNA was extracted with TRIzol reagent (Invitrogen). Total RNA was resolved by a 15% denaturing polyacrylamide gel containing 8 m urea. The RNA was then transferred to GeneScreen Plus Hybridization Transfer Membrane (PerkinElmer Life Sciences). After baking at 80 °C for 0.5 h and then UV cross-linking, the membranes were stained by methylene blue to evaluate the transfer efficiency. The membranes were incubated in hybridization buffer (Toyobo, Osaka, Japan) at 45 °C for 30 min and then hybridized with specific γ-32P-labeled oligonucleotide probes (Sangon, Shanghai, China) complementary to each miRNA at 45 °C for 16 h. The membranes were washed twice with washing buffer (2× SSC with 0.1% SDS), and the signal was detected by a phosphorimaging scanner (Fujifilm 9000). The hybridized membranes were then rehybridized with specific γ-32P-labeled 5 S rRNA probes (Sangon), which were used as a loading control, at 50 °C for 2 h. The oligonucleotide probes for Northern blotting were as follows: miR-29a, 5′-TAACCGATTTCAGATGGTGCTA-3′; miR-29b, 5′-AACACTGATTTCAAATGGTGCTA-3′; 5 S rRNA, 5′-CGGTATTCCCAGGCGGTCT-3′.
ChIP, ChIP-PCR, ChIP-Quantitative PCR, Peak Finding, Motif Search, and Electrophoretic Mobility Shift Assay
The ChIP, ChIP-PCR, ChIP-quantitative PCR, peak finding, motif search, and electrophoretic mobility shift assay were conducted following a protocol described previously (21). The primers for ChIP-PCR and ChIP-quantitative PCR were: forward, 5′-TTGGTTCTCATCCGTCCTTTAT-3′; reverse, 5′-AGAATGGCTCATTCCCTCATAGA-3′.
Generation of Transgenic Mice
A precursor of mouse miR-29b1a was amplified with the following primers: forward, 5′-GTgcggccgcACGGACTTCACCTTCCCT-3′ (NotI); reverse, 5′-GAgagctcGTCAGCAATAACGTAGTCAG-3′ (SacI). The lowercase underlined nucleotides represent nucleotide sequences of the recognition site for the restriction enzymes. The 541-bp product was digested with restriction endonucleases NotI and SacI and then cloned into the expression vector pUBC containing a human ubiquitin C promoter, which was found to provide the most reliable expression across different cell types. After testing the efficacy of miR-29a and miR-29b overexpression in HEK 293T cells, the construct was linearized for pronuclear microinjection.
Real Time RT-PCR
Quantitative real time RT-PCR was performed to detect mRNA expression using Toyobo SYBR Green Real-time PCR Mix (Toyobo, Osaka, Japan) according to the manufacturer's protocol. Levels of mRNAs were normalized to Gapdh. The primers were as follows: mouse Ar, 5′-CTGGGAAGGGTCTACCCAC-3′ (forward) and 5′-GGTGCTATGTTAGCGGCCTC-3′ (reverse); mouse Adam7, 5′-GGTCATTGTGCTTGTCATGG-3′ (forward) and 5′-ACGGAGGATTAGCCCAGTCT-3′ (reverse); mouse Fkbp5, 5′-GTCCAAAGCCTCAGAGTCGTTC-3′ (forward) and 5′-AGCCTTTCTCATTGGCACTGTC-3′ (reverse); mouse Gldc, 5′-AAAAGTCCGCACAGTGAAAGGA-3′ (forward) and 5′-CCCCCAGGGCAAATGTTTA-3′ (reverse); mouse Gpx5, 5′-TATTGCGGTCTGACAATCCA-3′ (forward) and 5′-TTAAGGGGGAAGGAGGAAGA-3′ (reverse); mouse Igf1, 5′-CTGGACCAGAGACCCTTTGC-3′ (forward) and 5′-GGACGGGGACTTCTGAGTCTT-3′ (reverse); mouse Cdc42, 5′-AAAAGTGGGTGCCTGAGATAAC-3′ (forward) and 5′-GGCTCTTCTTCGGTTCTGGAG-3′ (reverse); mouse p85α, 5′-GCAGAGGGCTACCAGTACAGA-3′ (forward) and 5′-CTGAATCCAAGTGCCACTAAGG-3′ (reverse).
miRNAs, Small Interfering RNAs, and Transfection
RNA oligonucleotides were transfected into cells using RNAiMAX (Invitrogen) according to the manufacturer's protocol. miR-29a and miR-29b duplex (pre-miR-29a and pre-miR-29b) or miR-29a-specific inhibitor (anti-miR-29a) molecules and appropriate negative control molecules (pre-miR-negative control (pre-miR-NC) or anti-miR-negative control (anti-miR-NC)) were purchased from Ambion (Austin, TX). The following small interfering RNAs (siRNAs) were synthesized by GenePharma (Shanghai, China): Igf1 siRNA-1, 5′-CAGCCUUCCAACUCAAUUATT-3′; Igf1 siRNA-2, 5′-GUGACUUCUUGAAGAUAAATT-3′; p53 siRNA, 5′-GUACAUGUGUAAUAGCUCCTT-3′.
Western Blotting
Proteins were extracted from cells using radioimmune precipitation assay buffer (25 mm Tris, pH 7.4, 150 mm NaCl, 1% (v/v) Nonidet P-40, 1% (w/v) sodium deoxycholate, 0.1% (w/v) SDS). Each protein sample (30–50 μg) was separated on 10% SDS-polyacrylamide gels (for AR and p53) or 15% SDS-polyacrylamide gels (for IGF1) and then semidry blotted to polyvinylidene difluoride membranes (Amersham Biosciences). Immunodetection of proteins was carried out by standard procedures using rabbit polyclonal antibody to AR (Santa Cruz Biotechnology, Santa Cruz, CA; 1:500 dilution), IGF1 (Abcam, Cambridge, UK; 1:500 dilution), and p53 (Abcam; 1:500 dilution). The protein blots were probed with primary antibody and goat anti-rabbit secondary antibodies conjugated to horseradish peroxidase (HRP) (Sigma-Aldrich) and detected using ECL (GE Healthcare) reagents. β-Actin (Santa Cruz Biotechnology) was used as a loading control.
DNA Constructs and Mutagenesis
The 792-bp AR-binding region around the peak summit was amplified with primer 1 (5′-gaggtaccTCCCATGTCTTTGACTTGATTTTC-3′ (KpnI)) and primer 2 (5′-gaactcgagGCTCGCCCTTTGAGAAACATT-3′ (XhoI)) and subcloned into pGL3-Basic and pGL3-Promoter luciferase reporter vectors. The lowercase underlined nucleotides represent nucleotide sequences of the recognition site for the restriction enzymes. The ARE was mutated with primer 1, primer 2, primer 3 (5′-TTCAGGGCCTGAGTAGATAGATGT-3′), primer 4 (5′-TAAGAACATGGCTCAATTCTTTTTC-3′), and primer 5 (5′-TACTCAGGCCCTGAAtTAaAGCATaTTtTAAGAACATGGCTCA-3′). The lowercase, bold, underlined nucleotides represent mutated nucleotides. Briefly, the upstream ARBS fragment was amplified using primer 1 and primer 3, the downstream ARBS fragment was amplified using primer 2 and primer 4, and then the full-length mutated ARBS was amplified using primer 1, primer 2, and primer 5 with the upstream and downstream fragments as templates. Then the product was cut with KpnI and XhoI and ligated with pGL3-Promoter luciferase reporter vectors. HEK 293T cells were grown in 96-well plates, cotransfected with 150 ng of reporter constructs, 15 ng of pRL-TK (Renilla luciferase), and 15 ng of pcDNA3.1-Ar expression vector coding for full-length human AR using X-tremeGENE HP DNA Transfection Reagent (Roche Applied Science). After 12 h of transfection, cells were treated with 100 nm dihydrotestosterone or ethanol vehicle for 24 h. Luciferase activity was measured using the Dual-Luciferase reporter assay (Promega, Madison, WI) and a Mithras LB940 multimode microplate reader (Berthold, Bad Wildbad, Germany).
The 3′-untranslated regions (3′-UTRs) of human IGF1 mRNA were amplified using the primers 5′-CCGctcgagTCCATCTGTGGCATTTGTACC-3′ (XhoI) and 5′-GTAgcggccgcCCCTTGTGTCATCTTTGGCTCC-3′ (NotI). The lowercase underlined nucleotides represent nucleotide sequences of the recognition site for the restriction enzymes. After cutting with XhoI and NotI, the amplified products were subcloned downstream of the Renilla luciferase gene of the psiCHECKTM-2 plasmid (Promega). Site-directed mutagenesis was performed using the QuikChange site-directed mutagenesis kit (Stratagene, Santa Clara, CA). The primers for mutation of the target site of human IGF1 3′-UTR were 5′-GTAAAACTCTGTTTTTTAGTATAATGGGTAGATTTTGTAGTTTGTTATATGAAAGAGTC-3′ (forward) and 5′-GACTCTTTCATATAACAAACTACAAAATCTACCCATTATACTAAAAAACAGAGTTTTAC-3′ (reverse). The four nucleotides TGCT in “seed” sequences of the miR-29a potential target site were mutated to GTAG (underlined).
Luciferase Reporter Assay
HEK 293T cells were seeded to 24-well plates 24 h before transfection. HEK 293T cells were transiently transfected with 100 ng of psiCHECK-2 (Promega) vector containing the wild-type IGF1 3′-UTR (named IGF1-3′-UTR-WT) or IGF1 3′-UTR mutant (named as IGF1-3′-UTR-Mut) together with 30 nm pre-miR-29a or pre-miR-NC, respectively. Using the Dual-Luciferase assay kit (Promega), the luciferase activities were measured 24 h after transfection. The Renilla luciferase activity was normalized to firefly activity. Lipofectamine 2000 transfection reagent was used for cotransfection of RNA oligonucleotides and plasmid.
Hematoxylin and Eosin (H&E) Staining and Immunohistochemical Staining
H&E staining of the sections was carried out using standard protocols. Immunohistochemical staining was performed as described previously (22). Primary and secondary antibodies were diluted in PBS containing 10% normal goat serum. The 1:100-diluted anti-AR antiserum (Abcam, catalog number ab74272) was applied to the tissues overnight at 4 °C, and the 1:200-diluted biotin-conjugated goat anti-rabbit IgG was incubated for 1 h at room temperature. Expression was visualized using HRP substrate. As a negative control, serial sections were subjected to the same procedure with normal rabbit serum replacing the primary antibody. The expression was visualized using HRP substrate. The sections were mounted in 80% glycerol and examined with an Olympus BX-52 microscope.
In Situ Hybridization
In situ hybridization was performed as described previously (24). Briefly, the epididymal tissues were fixed in 4% paraformaldehyde and then embedded in paraffin. The embedded epididymidis was sectioned. The slides were dewaxed, then digested with proteinase K (500 ng/ml), hybridized with locked nucleic acid probes (Exiqon) specific for mmu-miR-29a, U6 snRNA, and scrambled oligonucleotides, which were labeled with digoxigenin. The temperature of hybridization was 65 °C. The expression of miR-29a or control small RNAs was visualized using alkaline phosphatase substrate.
TUNEL Assay
The TUNEL assay was performed according to the protocol of the in situ apoptosis detection kit (Takara Bio Inc., Shiga, Japan). In short, after deparaffinization and rehydration, the 5-μm paraplast tissue sections were incubated with 20 μg/ml proteinase K solution (Invitrogen/Molecular Probes, catalog number 25530049) for 15 min at 37 °C. Thereafter, sections were incubated with FITC-labeled terminal deoxynucleotidyltransferase. Then sections were rinsed in PBS and observed under a fluorescence microscope.
RESULTS
Androgen Negatively Regulates miR-29a Expression in Epididymis
Androgen acts through its receptor, AR, to control normal development of male reproductive organs and maintain male characteristics. Orchiectomy is considered as an ideal model to investigate androgen effects, for example, its regulation of gene expression. Therefore, herein a castration model was used to investigate the response of miR-29a to androgen manipulation. First, we found that the level of miR-29a was higher in the mouse epididymis at 9 days after orchiectomy (7 days after castration with sesame oil injection for 2 days) than in normal epididymis. The miR-29a level in the sesame oil injection group was also higher than in the TP injection group (Fig. 1A, left panel). These results strongly demonstrated that the miR-29a level was reversely related with the androgen level (Fig. 1A). Subsequently, similar phenomena were observed in rat epididymis by using a castration model (Fig. 1B). After castration, the ratio of epididymal cell types may be altered due to cell apoptosis occurring mainly in principle cells (25). To exclude the possibility that the altered ratios of cell types in the castrated epididymis caused the change of miR-29a, we investigated the miR-29a expression pattern in normal rat epididymis using in situ hybridization. The result showed that miR-29a was extensively expressed in epididymal epithelial cells (Fig. 1C). Thus, the change of miR-29a mainly caused the regulatory effects. Furthermore, we tested whether miR-29a was androgen-responsive in principal cells using an in vitro system. The PC-1 cell line, an immortalized epididymal principal cell line from mouse proximal caput, was used here. PC-1 cells were maintained in Iscove's modified Dulbecco's medium with different concentrations of DHT. Twenty-four hours later, miR-29a expression was determined using Northern blotting. The expression of miR-29a in PC-1 cells cultured without DHT was higher than that with DHT. Furthermore, the level of miR-29a displayed a dosage-dependent pattern (Fig. 1D), also further validating that the up-regulation of miR-29a in castrated epididymis was not the consequence of the alteration of cell type ratios but the alteration of the androgen level. Thus, miR-29a was an androgen-repressive miRNA in mouse and rat epididymis.
FIGURE 1.

Androgen negatively regulates miR-29a expression in mouse and rat epididymis. A, Northern blotting detection of miR-29a expression in mouse epididymis from normal and from castrated (Cas) mice supplemented at 7 days after castration with sesame oil or TP (2.5 mg/kg of body weight) for an additional 2 days. Each group included three mice. The right panel shows the concentrations of serum testosterone of every mouse. B, relative expression levels of miR-29a (Northern blotting) in the adult rat epididymis during androgen manipulation (n = 4–7). The right panel shows the average concentrations of serum testosterone of every group. C, hybridization showed that miR-29a was extensively expressed in adult rat epididymis. The probes were for scrambled oligonucleotides (a; negative control), U6 snRNA (b), and miR-29a (c–f). The blue signal represents positive signal. c–f represents initial segment, caput, corpus, and cauda, respectively. Scale bars, 20 μm. D, Northern blotting detection of miR-29a level in mouse PC-1 cells that were cultured without or with different concentrations (1 and 10 nm) of DHT for 24 h. -Fold change represents miR-29a relative to the non-stimulated state. E, Northern blotting detection of miR-29b in entire epididymis from normal and castrated mice supplemented at 7 days after castration with sesame oil or TP (2.5 mg/kg of body weight) for an additional 2 days. 5 S rRNA was used as an internal control.
miR-29b1, another member of the miR-29 family, is in close proximity to miR-29a on mouse chromosome 6 (26). Therefore, we detected the response of miR-29b to androgen. Similarly, miR-29b was also suppressed by androgen (Fig. 1E). In conclusion, the miR-29b1/a cluster was repressed by the androgen signaling system.
In contrast, Ribas et al. (27) found by miRNA microarray analysis that miR-29a may be one AR-induced miRNA in two human androgen-responsive prostate cancer cell lines, LNCaP and LAPC-4. To validate this result, LNCaP cells were plated in medium with 0–10 nm DHT, and 72 h later, the cells were harvested to detect the miR-29a level. The result showed that miR-29a was indeed induced by androgen in LNCaP cells (Fig. 2A), which differed from epididymal cells. Hereupon, we asked the question whether miR-29a was suppressed by androgen specifically in epididymis. To answer this question, we further investigated miR-29a expression in prostate and brain in castrated mouse. We found that the expression of miR-29a was repressed by androgen stimulation in mouse prostate tissue, whereas in brain, there was no apparent change (Fig. 2, B and C). Therefore, the repression of miR-29a by androgen was not epididymis-specific.
FIGURE 2.

Androgen response patterns of miR-29a in different tissues and cell lines. A, Northern blotting analysis of miR-29a dose response to DHT in the human prostate cancer cell line LNCaP treated with different concentrations of DHT for 72 h. B and C, Northern blotting detection of miR-29a expression in mouse prostate and brain from normal and castrated (Cas) mice supplemented at 7 days after castration with sesame oil or TP (2.5 mg/kg of body weight) for an additional 2 days. In B and C, each group included three mice. The concentration of serum testosterone of every mouse was the same as in Fig. 1A. D–I, Northern blotting analysis of miR-29a expression at 72 h in RM-1, RWPE-1, MCF-7, U-2 OS, MG-63, and T24 cells treated with different concentrations of DHT. 5 S rRNA was used as a loading control. -Fold change represents miR-29a relative to the non-stimulated state. J, Western blotting analysis of AR expression at 72 h in RM-1 cells treated with different concentration of DHT. β-Actin was used a loading control for Western blotting.
Next, we asked whether the response patterns of miR-29a were different between physiological and tumorigenic states. We then investigated the responses of miR-29a to androgen stimulation in mouse prostate cancer cell line RM-1 and normal human prostate epithelial cell line RWPE-1. We found that miR-29a was also suppressed by androgen in RM-1 cells and RWPE-1, although the change in RWPE-1 cells was weak (Fig. 2, D and E). Furthermore, we also tested the responses of miR-29a to androgen in several human AR-positive cancer cell lines, including MCF-7 (breast cancer), U-2 OS, and MG-63 (osteosarcoma), and T24 (bladder carcinoma). However, there were no apparent changes in these cell lines (Fig. 2, F–J). The RM-1 cell line is androgen-insensitive. Why does DHT also inhibit the expression of miR-29a in the androgen-insensitive RM-1 cell line? A previous study revealed that DHT treatment of the androgen-insensitive human prostate cancer cell line PC-3 resulted in up-regulation of p21CIP1 levels (28). Because the AR-mediated transactivation could be induced in a ligand-independent manner (29, 30), we deduced that miR-29a was suppressed in RM-1 cells possibly by a similar mechanism because AR also can be up-regulated by DHT stimulation in RM-1 cells (Fig. 2J). These results indicated that the repression of miR-29a by androgen was context-dependent.
Androgen Regulates miR-29b1a through AR Binding Upstream of miR-29b1a
Next, we investigated how androgen regulated miR-29a and miR-29b expression. It is well known that the effects of androgen are mediated through AR binding to ARE DNA (31, 32). Previously, by ChIP-Seq assay, we identified the binding sites of AR in mouse caput epididymis. These binding sites were associated with 63 miRNAs, including miR-29a (21). As shown in Fig. 3A, there was an AR-binding site enriched by AR 8 kb upstream of miR-29a and miR-29b1 precursor. We first confirmed this site was a bona fide AR-binding site by ChIP-PCR (Fig. 3B). Our results showed that the peak was androgen-dependent (Fig. 3C). Next, we performed bioinformatics analysis and found that the binding site contained two conserved AREs (Fig. 3D). Further experiments by EMSA showed that ARE1 but not ARE2 can be bound by AR (Fig. 3E). Mutation of ARE1 (Fig. 3D) abolished the binding activity (Fig. 3, F and G), indicating that androgen regulated miR-29b1a expression through AR binding to a conserved ARE upstream of miR-29b1a.
FIGURE 3.

Characterization of AR-binding sites associated with potential AR target gene miR-29b1a. A, screen shots of the UCSC Genome Browser mm9 showing AR-binding sites around miR-29b1a. The figure was generated by uploading one file containing unique ChIP-Seq sites in the BED format to the UCSC Genome Browser. The ChIP-Seq peak is shown at the top, and the AR target genes are shown at the bottom. B, validation of the ARBS associated with miR-29b1a by ChIP-PCR using anti-AR antibody. C, validation of the AR-binding sites associated with miR-29b1a by ChIP-quantitative PCR using anti-AR antibody. Enrichment -fold values were calculated using IgG enrichment as a control (mean ± S.D. (error bars), n = 3). ChIP samples were prepared from epididymidis of normal mice (Nor), mice castrated at 3 days (Cas), and mice castrated at 3 days and supplemented with TP for an additional 2 days. D, the ARBS included two potential conserved AREs; the wild-type and mutated sequences of the two AREs are listed. E–G, AR bound ARE1 but not ARE2 as demonstrated by EMSAs. E, AR bound ARE1 but not ARE2. The ARE of C3 gene served as a positive control. Control incubation performed in the absence of protein is presented in lane 2. In F and G, competitors are indicated above, and amounts of competitors used are given in molar excess. Competitors include unlabeled AREs, mutant AREs, and a consensus DNA-binding site for Sp1. H, ARBS identified by ChIP-Seq functions as an enhancer. The ARBSs with wild-type and mutated ARE1 were subcloned into the pGL3-Promoter plasmid. Plasmids were cotransfected with pcDNA3.1-Ar into HEK 293T cells, and luciferase activities were measured after treatment with ethanol or 100 nm DHT (mean ± S.D. (error bars), n = 3). Chr., chromosome; mARE, mouse ARE; hARE, human ARE; mut, mutant.
Then the ARBS was subcloned to pGL3-Basic luciferase reporter vectors. Compared with pGL3-Basic vector, the pGL3-Basic-ARBS did not show apparent increased luciferase activity with or without DHT stimulation (data not shown), indicating that ARBS may be an enhancer/silencer. Next, the ARBS was subcloned to pGL3-Promoter vector. As Fig. 3H shows, the luciferase activity was down-regulated after DHT stimulation compared with ethanol control, but the mutation of ARE1 abolished the repressive effect. These results indicated that the ARE1 was a bona fide repressive regulator of miR-29b1a expression.
Overexpression of miR-29b1a Leads to Smaller Epididymis
As described above, the androgen signaling pathway inhibited miR-29b1a expression in mouse epididymis. To further confirm this result in vivo and explore its biological function, we generated miR-29b1a transgenic mice by overexpressing a 523-nucleotide precursor of miR-29b1a under the control of the promoter of ubiquitin C (UBC) gene (Fig. 4A). The genotyping of the mice was performed by PCR on genomic DNA (data not shown). miR-29a and miR-29b expression levels were detected using Northern blotting. As anticipated, there was greater miR-29a and miR-29b expression in epididymis of transgenic mice compared with epididymis of littermates (Fig. 4B). Consequently, epididymis from transgenic mice displayed small size and weight (Fig. 4, C and D), demonstrating that miR-29b1a overexpression disturbed epididymal development. Although the transgenic mice displayed a smaller epididymis, the ratios of cell types showed no apparent change (Fig. 4E). There was no apparent difference in cell apoptosis in wild-type and transgenic mouse epididymis, but more apoptotic cells were observed in transgenic mouse testis (Fig. 4F).
FIGURE 4.

miR-29b1a-overexpressing transgenic mice showed smaller epididymidis. A, vector of miR-29b1apUBC transgene, which was controlled by the promoter of UBC gene. pA, polyadenylation. B, relative expression of miR-29a and miR-29b in epididymis from transgenic and littermate control mice at 7 weeks of age. C, macroscopic appearance of the epididymis from transgenic and littermate control mice at 7 weeks of age. D, ratio of testis weight to body weight is comparable in wild-type (WT) and miR-29b1a transgenic mice (mean ± S.E. (error bars), n = 4). E, H&E-stained sections of epididymis from wild-type and transgenic 10-week-old mice (magnification, ×400). F, TUNEL analysis of cell death in epididymis and testis from wild-type and transgenic 10-week-old mice. The arrows indicate apoptotic cells (magnification, ×100). Scale bars, 20 μm in E 200 μm in F. TG, transgenic; IS, initial segment; Cap, caput; Cor, corpus; Cau, cauda.
Feedback Inhibition of AR by miR-29a
It is well known that there is agenesis of the epididymis in Ar knock-out mice (33–35). We asked whether miR-29a disturbed epididymal development by inhibiting AR expression. To test this speculation, we detected Ar mRNA and protein levels in wild-type and transgenic mouse epididymis by using real time PCR and Western blotting, respectively. As expected, the AR level was significantly lower in transgenic mouse epididymis than in epididymis of their non-transgenic littermates (Fig. 5, A and B). Consistently, AR immunostaining showed a weak signal in transgenic mouse epididymis compared with wild type (Fig. 5C). Next, we tested this reverse suppression in vitro. Pre-miR-29a and scrambled RNA oligonucleotides were transfected into PC-1 cells. Forty-eight hours later, RNA and protein were extracted to detect the expression of AR. Compared with the scrambled RNA transfection group, the mRNA and protein levels of AR were decreased in the pre-miR-29a transfection group (Fig. 5, D and E). Consistently, we found that miR-29b can also suppress AR expression in PC-1 cells (Fig. 5F). Taken together, miR-29b1a suppresses the expression of AR in vitro and in vivo.
FIGURE 5.

miR-29a inhibited AR expression in mouse epididymis. A, real time PCR analysis of the Ar mRNA level in the epididymis from transgenic mice and littermate controls (mean ± S.E. (error bars), n = 3). B, Northern blotting and Western blotting detection of miR-29a and AR protein levels, respectively, in the epididymidis from three paired transgenic mice and littermate controls. tRNA staining was used as a loading control for Northern blotting, and β-actin was used as a loading control for Western blotting. C, AR expression in 10-week-old WT and transgenic mouse (TG) epididymis. AR was markedly reduced in all regions of epididymis in transgenic mice as shown by immunohistochemistry. IS, initial segment; Cap, caput; Cor, corpus; Cau, cauda. D, real time PCR analysis of Ar mRNA levels in PC-1 cells 48 h after miR-29a mimic transfection (mean ± S.E. (error bars), n = 3). E and F, Western blotting detection of the protein levels of AR in PC-1 cells 48 h after miR-29a and miR-29b transfection, respectively. β-Actin was used a loading control for Western blotting. G, Real time PCR analysis of mRNA levels of AR target genes Adam7, Fkbp5, Gldc, and Gpx5 in the epididymidis from transgenic mice and littermate controls (mean ± S.E. (error bars), n = 3).
Such being the case, miR-29a and miR-29b should also suppress the target genes of the androgen signaling pathway. Here, we detected four epididymal androgen-responsive genes, namely Adam7, Fkbp5, Gldc, and Gpx5 (36, 37). No miR-29a- and miR-29b-binding sites on the 3′-UTR of these four genes were predicted using the on-line software miRWalk, indicating that they should not be direct targets of miR-29a and miR-29b. These four genes were down-regulated in miR-29b1a transgenic mice at the mRNA level (Fig. 5G), demonstrating that miR-29a and miR-29b down-regulated AR expression and thereby suppressed the activity of androgen signaling in mouse epididymis.
miR-29a Represses AR Expression through IGF1 and p53 Pathways
Subsequently, possible miRNA target sites of mouse AR at 5′-UTR, coding region, and 3′-UTR were predicted using the on-line software miRWalk. miR-29a was not a candidate; thus, miR-29a probably regulates AR expression in an indirect way. We then tried to find which genes potentially mediated this regulation. A previous study revealed that the AR protein level is decreased in prostate from Igf1 knock-out mice (38). Therefore, we determined whether IGF1 can also activate AR expression in mouse epididymal cells. At a concentration of 1 ng/ml, IGF1 apparently increased the AR protein level (Fig. 6A) but slightly decreased the Ar mRNA level (Fig. 6B). Consistently, after IGF1 was down-regulated using specific siRNAs (Fig. 6C), the AR protein level was reduced (Fig. 6D), but Ar mRNA was slightly increased (Fig. 6E). It is possible that the IGF1 signaling pathway can weakly inactivate the transcription of Ar but increase the stability of AR protein. We then explored whether miR-29a suppressed IGF1 expression. Overexpression of miR-29a by transfection of pre-miR-29a into PC-1 cells suppressed the expression of Igf1 at both mRNA and protein levels (Fig. 6, F and G). Consistently, Igf1 mRNA and protein levels were down-regulated in the epididymis of the miR-29b1a transgenic mice (Fig. 6, H and I), although the IGF1 level in the circulation showed no apparent change (data not show). Our results, therefore, confirmed that miR-29a suppressed IGF1 expression both in vivo and in vitro.
FIGURE 6.

IGF1 mediated the suppression of AR by miR-29a. A and B, Western blotting and real time PCR detection of AR protein and mRNA levels in PC-1 cells treated with DHT (10 nm) and/or IGF1 protein (1 ng/ml) 48 h later, respectively (mean ± S.D. (error bars), n = 3). The basic medium was Iscove's modified Dulbecco's medium without FBS. C, real time PCR analysis of Igf1 mRNA levels in PC-1 cells transfected with siRNAs (si) against Igf1 (mean ± S.D. (error bars), n = 3). D, Western blotting analysis of AR and IGF1 protein levels in PC-1 cells transfected with siRNAs against Igf1. E, real time PCR detection of Ar mRNA levels in PC-1 cells transfected with mouse Igf1 siRNA 48 h later (mean ± S.D. (error bars), n = 3). F, real time PCR analysis of Igf1 mRNA levels in PC-1 cells transfected with miR-29a mimics or control mimics 48 h later (mean ± S.D. (error bars), n = 3). G, Western blotting analysis of AR and IGF1 protein levels in PC-1 cells transfected with miR-29a mimics or control mimics 48 h later. H and I, real time PCR and Western blotting analysis of Igf1 mRNA and protein levels in epididymis from transgenic mice and littermate controls, respectively (mean ± S.D. (error bars), n = 3). J, miR-29a targets human, mouse, and rat Igf1 3′-UTR. K, HEK 293T cells were transfected with reporters constructed by inserting either wild-type or mutant Igf1 3′-UTR downstream of the luciferase gene. Results are normalized to a scrambled RNA (mean ± S.D. (error bars), n = 3).
Furthermore, to determine whether IGF1 is a direct target of miR-29a, the Dual-Luciferase reporter assay was performed. First, IGF1 was predicted as a potentially conserved target of miR-29a using three on-line software programs: TargetScan, MiRanda, and PicTar (Fig. 6J). Then the wild-type or mutated 3′-UTR of Igf1 mRNA was cloned downstream of the Renilla luciferase gene and cotransfected with pre-miR-29a or scrambled oligonucleotides into HEK 293T cells. Luciferase activities were measured 24 h after transfection. Cells cotransfected with the reporter gene carrying wild-type Igf1 3′-UTR and pre-miR-29a exhibited an apparent reduction of the luciferase activities compared with cells transfected with the scrambled oligonucleotide (Fig. 6K, left). Next, four nucleotides (TGCT→GTAG) in seed sequences of the miR-29a potential target site were mutated. The reduction of luciferase activity of the reporter by miR-29a was abrogated by the mutations (Fig. 6K, right). These data indicated that the Igf1 3′-UTR was truly targeted by miR-29a. Consistent with this result, Hand et al. (39) also demonstrated recently that miR-29 a/b1 can target Igf1 in mouse liver. Taken together, IGF1 was a direct target of miR-29a, and it mediated the suppression of AR by miR-29a.
Additionally, Alimirah et al. (40) found that p53 can negatively regulate AR expression by binding to a p53 DNA-binding consensus sequence of AR promoter in human prostate cancer cells. Another study found that p53 can be induced by miR-29 through targeting CDC42 and p85α (41). Thus, we deduced that miR-29a may repress AR expression by inducing p53 expression in epididymis too. To verify this hypothesis, first, we determined whether miR-29a can also promote p53 expression in epididymal cells. As expected, CDC42 and p85α, which negatively regulated p53 expression (30), were down-regulated in PC-1 cells transfected with pre-miR-29a (Fig. 7A). Consequently, overexpression of miR-29a in PC-1 cells elevated the p53 protein level (Fig. 7B). Consistently, when miR-29a was inhibited, p53 protein decreased (Fig. 7C). As expected, Cdc42 and p85α mRNA levels decreased (Fig. 7D), whereas the p53 protein level increased in miR-29b1a transgenic mouse epididymis (Fig. 7E). Thus, miR-29a can also promote p53 expression in mouse epididymis. Second, we wanted to know whether p53 can also inhibit AR expression in mouse epididymis. PC-1 cells were transfected with specific siRNA against mouse p53, and 48 h later, the AR protein level was indeed up-regulated (Fig. 7F). Therefore, p53 can also serve as a mediator of the suppression of AR by miR-29a in mouse epididymis. In summary, miR-29a and AR were mutually inhibited and formed a regulatory circuitry to regulate epididymal development (Fig. 8).
FIGURE 7.

miR-29a suppressed AR expression by promoting p53 protein level. A, real time PCR analysis of Cdc42 and p85α mRNA levels in PC-1 cells transfected with miR-29a mimics or control mimics (mean ± S.E. (error bars), n = 3). B, Western blotting analysis of AR and p53 protein levels in PC-1 cells transfected with miR-29a mimics or control mimics. C, Western blotting analysis of AR and p53 protein levels in PC-1 cells transfected with miR-29a inhibitor or control inhibitor. D, real time PCR analysis of Cdc42 and p85α mRNA levels in epididymis from transgenic mice and littermate controls (mean ± S.E. (error bars), n = 3). E, Western blotting analysis of p53 protein levels in epididymis from transgenic mice and littermate controls. F, Western blotting analysis of AR and p53 protein levels in PC-1 cells transfected with siRNAs (si) against mouse p53.
FIGURE 8.
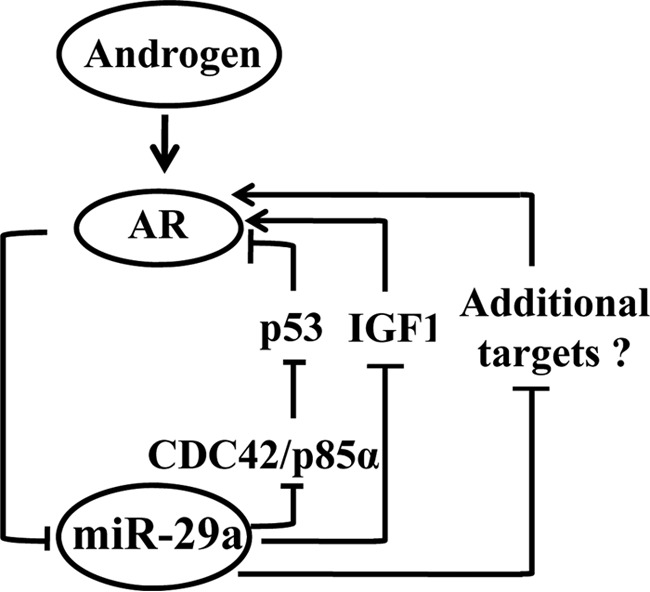
A model depicting androgen-AR-miR-29a regulation loop. The model depicts the role of the androgen, AR, and miR-29 regulatory circuit in mouse epididymis. AR directly suppresses miR-29a expression. Conversely, on one side, miR-29 miRNAs directly suppresses CDC42 and p85α, resulting in the stabilization of p53, and then p53 inhibits the transcription of AR; on the other side, miR-29a inhibits IGF1 expression, attenuating the promotion on AR expression by the IGF1 signaling pathway.
DISCUSSION
Androgen and its receptor regulate the biogenesis of several miRNAs in different tissues or cell lines. For example, the expression of miR-21, miR-125b, miR-101, miR-148a, and miR-338 is modulated by androgen/AR in prostate cancer cells (27, 42–45). In addition, miR-122 and miR-216a are induced in female mouse liver and hepatocarcinogenesis by androgen (46, 47). The facts that androgen is critical for epididymal development and function and that miR-29a plays vital roles in epididymal development and function (3) suggest an androgen regulation of miR-29a in epididymis. Herein, we identified a bona fide ARE upstream of miR-29b1a and demonstrated that androgen negatively regulated miR-29a in epididymis. Our data first confirmed that miR-29a was an androgen-repressive miRNA in mouse and rat epididymis as well as in a mouse epididymal cell line and extended the current understanding of miRNA regulation by androgen under different physiological and pathological states. RM-1 is an androgen-insensitive cell line; however, miR-29a was also repressed in RM-1 cells by DHT. A previous study revealed that AR-mediated transactivation could be induced in a ligand-independent manner (29, 30). We deduced that miR-29a was suppressed in RM-1 cells possibly by a similar mechanism because AR also can be up-regulated by DHT stimulation in RM-1 cells (Fig. 2J).
As the mediator of androgen functions, AR expression is well regulated by some transcription factors such as the Wnt and NF-κB signaling pathways in prostate cancer cells (48, 49). Serving as post-transcriptional regulators, miRNAs can also regulate AR. Previous studies have reported that several miRNAs, including miR-488*, let-7c, and miR-331-3p, down-regulated AR (23, 50, 51). Our study demonstrated that miR-29a and miR-29b regulated AR expression in mouse epididymis. Furthermore, all the previous studies demonstrated that miRNAs inhibited AR at the cell level. Our studies provide the first in vivo evidence that miRNAs regulate AR at the organism level. Moreover, although androgen is critical for epididymal development and functions, how AR is regulated in epididymis was unclear up to now. This study has shed new light on our understanding of AR regulation in epididymis.
Androgen repressed miR-29a, and miR-29a down-regulated AR. Thus, there is a regulatory circuitry between miR-29a and AR. The regulatory circuitry fine-tuned epididymal development and functions. Once the regulatory circuitry was disrupted, epididymis disorders occurred. Our data showed an elevated level of miR-29a expression accompanied by androgen withdrawal after castration. It has been reported that miR-29a can induce cell apoptosis by targeting multiple genes (34–36). Thus, the castration-mediated miR-29a elevation might contribute to apoptosis of epididymal epithelial cells. Additionally, elevated miR-29a suppressed AR expression and exacerbated the effects of castration. Therefore, both castration and miR-29a overexpression mimicked the AR knock-out-mediated epididymis agenesis.
Moreover, we further characterized the miR-29 and AR regulatory circuitry in epididymis by identifying IGF1 and p53 as critical mediators of the regulatory circuitry. As in the prostate (38), IGF1 enhanced AR expression and was directly suppressed by miR-29a in mouse epididymis. Therefore, the suppression of miR-29a by AR might be mediated or at least partially mediated by IGF1. Also, p53 was shown to bind to the promoter region of the Ar gene to inhibit Ar (40). miR-29a has been shown to elevate p53 by down-regulation of CDC42 and p85α (41). Herein, we demonstrated in epididymis that miR-29a directly down-regulated CDC42 and p85α to promote p53 and hence inhibited AR expression.
Acknowledgment
We thank Jin-Mei Chen for mouse handling. We thank Zimei Ni for construction of castration mouse model. We thank Aihua Liu for technical assistance with immunohistochemical staining.
This work was supported by National Natural Science Foundation of China Grants 31201113 and 30930053, Chinese Academy of Sciences Knowledge Innovation Program Grant KSCX2-EW-R-07, and National Basic Research Program of China (2014CB943103).
S. Hu, G. Yao, X. Guan, Z. Ni, W. Ma, E. Wilson, F. French, Q. Liu, and Y. Zhang, unpublished data.
- miRNA
- microRNA
- DHT
- 5α-dihydrotestosterone
- TP
- testosterone propionate
- NC
- negative control
- AR
- androgen receptor
- ARE
- androgen response element
- ChIP-Seq
- chromatin immunoprecipitation followed by sequencing
- ARBS
- AR-binding site
- UBC
- ubiquitin C
- Adam7
- disintegrin and metalloproteinase domain-containing protein 7
- Fkbp5
- FK506-binding protein 5
- Gldc
- glycine decarboxylase
- Gpx5
- glutathione peroxidase 5.
REFERENCES
- 1. Bartel D. P. (2009) MicroRNAs: target recognition and regulatory functions. Cell 136, 215–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kasinski A. L., Slack F. J. (2011) Epigenetics and genetics. MicroRNAs en route to the clinic: progress in validating and targeting microRNAs for cancer therapy. Nat. Rev. Cancer 11, 849–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Alvarez-Garcia I., Miska E. A. (2005) MicroRNA functions in animal development and human disease. Development 132, 4653–4662 [DOI] [PubMed] [Google Scholar]
- 4. Hagen J. W., Lai E. C. (2008) MicroRNA control of cell-cell signaling during development and disease. Cell Cycle 7, 2327–2332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Thum T., Mayr M. (2012) Review focus on the role of microRNA in cardiovascular biology and disease. Cardiovasc. Res. 93, 543–544 [DOI] [PubMed] [Google Scholar]
- 6. Ma W., Xie S., Ni M., Huang X., Hu S., Liu Q., Liu A., Zhang J., Zhang Y. (2012) MicroRNA-29a inhibited epididymal epithelial cell proliferation by targeting nuclear autoantigenic sperm protein (NASP). J. Biol. Chem. 287, 10189–10199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Williams A. E., Moschos S. A., Perry M. M., Barnes P. J., Lindsay M. A. (2007) Maternally imprinted microRNAs are differentially expressed during mouse and human lung development. Dev. Dyn. 236, 572–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ott C. E., Grünhagen J., Jäger M., Horbelt D., Schwill S., Kallenbach K., Guo G., Manke T., Knaus P., Mundlos S., Robinson P. N. (2011) MicroRNAs differentially expressed in postnatal aortic development downregulate elastin via 3′ UTR and coding-sequence binding sites. PLoS One 6, e16250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Podolska A., Kaczkowski B., Kamp Busk P., Søkilde R., Litman T., Fredholm M., Cirera S. (2011) MicroRNA expression profiling of the porcine developing brain. PLoS One 6, e14494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Li Y., Piatigorsky J. (2009) Targeted deletion of Dicer disrupts lens morphogenesis, corneal epithelium stratification, and whole eye development. Dev. Dyn. 238, 2388–2400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Huang T. H., Zhu M. J., Li X. Y., Zhao S. H. (2008) Discovery of porcine microRNAs and profiling from skeletal muscle tissues during development. PLoS One 3, e3225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hulsmans M., De Keyzer D., Holvoet P. (2011) MicroRNAs regulating oxidative stress and inflammation in relation to obesity and atherosclerosis. FASEB J. 25, 2515–2527 [DOI] [PubMed] [Google Scholar]
- 13. Han Y. C., Park C. Y., Bhagat G., Zhang J., Wang Y., Fan J. B., Liu M., Zou Y., Weissman I. L., Gu H. (2010) microRNA-29a induces aberrant self-renewal capacity in hematopoietic progenitors, biased myeloid development, and acute myeloid leukemia. J. Exp. Med. 207, 475–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gebeshuber C. A., Zatloukal K., Martinez J. (2009) miR-29a suppresses tristetraprolin, which is a regulator of epithelial polarity and metastasis. EMBO Rep. 10, 400–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mott J. L., Kurita S., Cazanave S. C., Bronk S. F., Werneburg N. W., Fernandez-Zapico M. E. (2010) Transcriptional suppression of mir-29b-1/mir-29a promoter by c-Myc, hedgehog, and NF-κB. J. Cell. Biochem. 110, 1155–1164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Qin W., Chung A. C., Huang X. R., Meng X. M., Hui D. S., Yu C. M., Sung J. J., Lan H. Y. (2011) TGF-β/Smad3 signaling promotes renal fibrosis by inhibiting miR-29. J. Am. Soc. Nephrol 22, 1462–1474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang B., Komers R., Carew R., Winbanks C. E., Xu B., Herman-Edelstein M., Koh P., Thomas M., Jandeleit-Dahm K., Gregorevic P., Cooper M. E., Kantharidis P. (2012) Suppression of microRNA-29 expression by TGF-β1 promotes collagen expression and renal fibrosis. J. Am. Soc. Nephrol. 23, 252–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Winbanks C. E., Wang B., Beyer C., Koh P., White L., Kantharidis P., Gregorevic P. (2011) TGF-β regulates miR-206 and miR-29 to control myogenic differentiation through regulation of HDAC4. J. Biol. Chem. 286, 13805–13814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sun D., Lee Y. S., Malhotra A., Kim H. K., Matecic M., Evans C., Jensen R. V., Moskaluk C. A., Dutta A. (2011) miR-99 family of microRNAs suppresses the expression of prostate-specific antigen and prostate cancer cell proliferation. Cancer Res. 71, 1313–1324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Araki Y., Suzuki K., Matusik R. J., Obinata M., Orgebin-Crist M. C. (2002) Immortalized epididymal cell lines from transgenic mice overexpressing temperature-sensitive simian virus 40 large T-antigen gene. J. Androl. 23, 854–869 [PubMed] [Google Scholar]
- 21. Hu S., Yao G., Guan X., Ni Z., Ma W., Wilson E. M., French F. S., Liu Q., Zhang Y. (2010) Research resource: genome-wide mapping of in vivo androgen receptor binding sites in mouse epididymis. Mol. Endocrinol. 24, 2392–2405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhu C. F., Liu Q., Zhang L., Yuan H. X., Zhen W., Zhang J. S., Chen Z. J., Hall S. H., French F. S., Zhang Y. L. (2007) RNase9, an androgen-dependent member of the RNase A family, is specifically expressed in the rat epididymis. Biol. Reprod. 76, 63–73 [DOI] [PubMed] [Google Scholar]
- 23. Epis M. R., Giles K. M., Barker A., Kendrick T. S., Leedman P. J. (2009) miR-331-3p regulates ERBB-2 expression and androgen receptor signaling in prostate cancer. J. Biol. Chem. 284, 24696–24704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gupta A., Mo Y. Y. (2011) Detection of microRNAs in cultured cells and paraffin-embedded tissue specimens by in situ hybridization. Methods Mol. Biol. 676, 73–83 [DOI] [PubMed] [Google Scholar]
- 25. Fan X., Robaire B. (1998) Orchidectomy induces a wave of apoptotic cell death in the epididymis. Endocrinology 139, 2128–2136 [DOI] [PubMed] [Google Scholar]
- 26. Wang H., Garzon R., Sun H., Ladner K. J., Singh R., Dahlman J., Cheng A., Hall B. M., Qualman S. J., Chandler D. S., Croce C. M., Guttridge D. C. (2008) NF-κB-YY1-miR-29 regulatory circuitry in skeletal myogenesis and rhabdomyosarcoma. Cancer Cell 14, 369–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ribas J., Ni X., Haffner M., Wentzel E. A., Salmasi A. H., Chowdhury W. H., Kudrolli T. A., Yegnasubramanian S., Luo J., Rodriguez R., Mendell J. T., Lupold S. E. (2009) miR-21: an androgen receptor-regulated microRNA that promotes hormone-dependent and hormone-independent prostate cancer growth. Cancer Res. 69, 7165–7169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Alimirah F., Chen J., Basrawala Z., Xin H., Choubey D. (2006) DU-145 and PC-3 human prostate cancer cell lines express androgen receptor: implications for the androgen receptor functions and regulation. FEBS Lett. 580, 2294–2300 [DOI] [PubMed] [Google Scholar]
- 29. Culig Z., Hobisch A., Cronauer M. V., Radmayr C., Trapman J., Hittmair A., Bartsch G., Klocker H. (1995) Androgen receptor activation in prostatic tumor cell lines by insulin-like growth factor-I, keratinocyte growth factor and epidermal growth factor. Eur. Urol. 27, 45–47 [DOI] [PubMed] [Google Scholar]
- 30. Hobisch A., Eder I. E., Putz T., Horninger W., Bartsch G., Klocker H., Culig Z. (1998) Interleukin-6 regulates prostate-specific protein expression in prostate carcinoma cells by activation of the androgen receptor. Cancer Res. 58, 4640–4645 [PubMed] [Google Scholar]
- 31. Heinlein C. A., Chang C. (2002) Androgen receptor (AR) coregulators: an overview. Endocr. Rev. 23, 175–200 [DOI] [PubMed] [Google Scholar]
- 32. Heemers H. V., Tindall D. J. (2007) Androgen receptor (AR) coregulators: a diversity of functions converging on and regulating the AR transcriptional complex. Endocr. Rev. 28, 778–808 [DOI] [PubMed] [Google Scholar]
- 33. Yeh S., Tsai M. Y., Xu Q., Mu X. M., Lardy H., Huang K. E., Lin H., Yeh S. D., Altuwaijri S., Zhou X., Xing L., Boyce B. F., Hung M. C., Zhang S., Gan L., Chang C. (2002) Generation and characterization of androgen receptor knockout (ARKO) mice: an in vivo model for the study of androgen functions in selective tissues. Proc. Natl. Acad. Sci. U.S.A. 99, 13498–13503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. O'Hara L., Welsh M., Saunders P. T., Smith L. B. (2011) Androgen receptor expression in the caput epididymal epithelium is essential for development of the initial segment and epididymal spermatozoa transit. Endocrinology 152, 718–729 [DOI] [PubMed] [Google Scholar]
- 35. Krutskikh A., De Gendt K., Sharp V., Verhoeven G., Poutanen M., Huhtaniemi I. (2011) Targeted inactivation of the androgen receptor gene in murine proximal epididymis causes epithelial hypotrophy and obstructive azoospermia. Endocrinology 152, 689–696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chauvin T. R., Griswold M. D. (2004) Androgen-regulated genes in the murine epididymis. Biol. Reprod. 71, 560–569 [DOI] [PubMed] [Google Scholar]
- 37. Lareyre J. J., Claessens F., Rombauts W., Dufaure J. P., Drevet J. R. (1997) Characterization of an androgen response element within the promoter of the epididymis-specific murine glutathione peroxidase 5 gene. Mol. Cell. Endocrinol. 129, 33–46 [DOI] [PubMed] [Google Scholar]
- 38. Svensson J., Kindblom J., Shao R., Movérare-Skrtic S., Lagerquist M. K., Andersson N., Sjögren K., Venken K., Vanderschueren D., Jansson J. O., Isaksson O., Ohlsson C. (2008) Liver-derived IGF1 enhances the androgenic response in prostate. J. Endocrinol. 199, 489–497 [DOI] [PubMed] [Google Scholar]
- 39. Hand N. J., Horner A. M., Master Z. R., Boateng L. A., LeGuen C., Uvaydova M., Friedman J. R. (2012) MicroRNA profiling identifies miR-29 as a regulator of disease-associated pathways in experimental biliary atresia. J. Pediatr. Gastroenterol. Nutr. 54, 186–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Alimirah F., Panchanathan R., Chen J., Zhang X., Ho S. M., Choubey D. (2007) Expression of androgen receptor is negatively regulated by p53. Neoplasia 9, 1152–1159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Park S. Y., Lee J. H., Ha M., Nam J. W., Kim V. N. (2009) miR-29 miRNAs activate p53 by targeting p85α and CDC42. Nat. Struct. Mol. Biol. 16, 23–29 [DOI] [PubMed] [Google Scholar]
- 42. Shi X. B., Xue L., Yang J., Ma A. H., Zhao J., Xu M., Tepper C. G., Evans C. P., Kung H. J., deVere White R. W. (2007) An androgen-regulated miRNA suppresses Bak1 expression and induces androgen-independent growth of prostate cancer cells. Proc. Natl. Acad. Sci. U.S.A. 104, 19983–19988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cao P., Deng Z., Wan M., Huang W., Cramer S. D., Xu J., Lei M., Sui G. (2010) MicroRNA-101 negatively regulates Ezh2 and its expression is modulated by androgen receptor and HIF-1α/HIF-1β. Mol. Cancer 9, 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Murata T., Takayama K., Katayama S., Urano T., Horie-Inoue K., Ikeda K., Takahashi S., Kawazu C., Hasegawa A., Ouchi Y., Homma Y., Hayashizaki Y., Inoue S. (2010) miR-148a is an androgen-responsive microRNA that promotes LNCaP prostate cell growth by repressing its target CAND1 expression. Prostate Cancer Prostatic Dis. 13, 356–361 [DOI] [PubMed] [Google Scholar]
- 45. Ambs S., Prueitt R. L., Yi M., Hudson R. S., Howe T. M., Petrocca F., Wallace T. A., Liu C. G., Volinia S., Calin G. A., Yfantis H. G., Stephens R. M., Croce C. M. (2008) Genomic profiling of microRNA and messenger RNA reveals deregulated microRNA expression in prostate cancer. Cancer Res. 68, 6162–6170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Delić D., Grosser C., Dkhil M., Al-Quraishy S., Wunderlich F. (2010) Testosterone-induced upregulation of miRNAs in the female mouse liver. Steroids 75, 998–1004 [DOI] [PubMed] [Google Scholar]
- 47. Chen P. J., Yeh S. H., Liu W. H., Lin C. C., Huang H. C., Chen C. L., Chen D. S. (2012) Androgen pathway stimulates microRNA-216a transcription to suppress the tumor suppressor in lung cancer-1 gene in early hepatocarcinogenesis. Hepatology 56, 632–643 [DOI] [PubMed] [Google Scholar]
- 48. Yang X., Chen M. W., Terry S., Vacherot F., Bemis D. L., Capodice J., Kitajewski J., de la Taille A., Benson M. C., Guo Y., Buttyan R. (2006) Complex regulation of human androgen receptor expression by Wnt signaling in prostate cancer cells. Oncogene 25, 3436–3444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhang L., Altuwaijri S., Deng F., Chen L., Lal P., Bhanot U. K., Korets R., Wenske S., Lilja H. G., Chang C., Scher H. I., Gerald W. L. (2009) NF-κB regulates androgen receptor expression and prostate cancer growth. Am. J. Pathol. 175, 489–499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sikand K., Slaibi J. E., Singh R., Slane S. D., Shukla G. C. (2011) miR 488* inhibits androgen receptor expression in prostate carcinoma cells. Int. J. Cancer 129, 810–819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Nadiminty N., Tummala R., Lou W., Zhu Y., Zhang J., Chen X., eVere White R. W., Kung H. J., Evans C. P., Gao A. C. (2012) MicroRNA let-7c suppresses androgen receptor expression and activity via regulation of Myc expression in prostate cancer cells. J. Biol. Chem. 287, 1527–1537 [DOI] [PMC free article] [PubMed] [Google Scholar]