

Background: The duration of S phase in Xenopus is controlled by the nucleus/cytoplasm ratio through unknown mechanisms.
Results: Modulation of protein phosphatase 2A (PP2A) levels controls origin firing at high nucleus/cytoplasm.
Conclusion: Depletion of PP2A and its B55α subunit at high nucleus/cytoplasm causes an extension of S phase.
Significance: This mechanism may influence cell cycle remodeling that occurs in early embryogenesis.
Keywords: Cell Cycle, Checkpoint Control, DNA Replication, PP2A, Xenopus, B55α, Checkpoint Kinase, Midblastula Transition, Nucleus/Cytoplasm Ratio
Abstract
The nucleus/cytoplasm (N/C) ratio controls S phase dynamics in many biological systems, most notably the abrupt remodeling of the cell cycle that occurs at the midblastula transition in early Xenopus laevis embryos. After an initial series of rapid cleavage cycles consisting only of S and M phases, a critical N/C ratio is reached, which causes a sharp increase in the length of S phase as the cell cycle is reconfigured to resemble somatic cell cycles. How the N/C ratio determines the length of S phase has been a longstanding problem in developmental biology. Using Xenopus egg extracts, we show that DNA replication at high N/C ratio is restricted by one or more limiting substances. We report here that the protein phosphatase PP2A, in conjunction with its B55α regulatory subunit, becomes limiting for replication origin firing at high N/C ratio, and this in turn leads to reduced origin activation and an increase in the time required to complete S phase. Increasing the levels of PP2A catalytic subunit or B55α experimentally restores rapid DNA synthesis at high N/C ratio. Inversely, reduction of PP2A or B55α levels sharply extends S phase even in low N/C extracts. These results identify PP2A-B55α as a link between DNA replication and N/C ratio in egg extracts and suggest a mechanism that may influence the onset of the midblastula transition in vivo.
Introduction
During early embryonic development in many animals, including Xenopus laevis, Drosophila melanogaster, and Caenorhabditis elegans, cell cycle duration is controlled by the nucleus/cytoplasm ratio of the proliferating blastomeres. In C. elegans, this effect is seen as early as the two-cell stage, where the smaller P1 blastomere requires more time to complete S phase than its larger sister cell, AB (1). Early development in X. laevis and D. melanogaster begins with a series of rapid cell divisions that quickly subdivide the fertilized egg into thousands of individual cells (2–4). These early cleavage divisions lack gap phases entirely, consisting instead of alternating rounds of S and M phases without cell growth (5). After a fixed number of such divisions, the cell cycle abruptly lengthens during the midblastula transition (MBT)2 (6). Here, the lengths of S phase and mitosis expand, gap phases are incorporated, and cell cycle checkpoints begin to exert their influence (7–10). Newport and Kirschner (6) have shown that the MBT in Xenopus is triggered when the rapidly dividing embryo crosses a threshold N/C ratio. Similarly, in Drosophila, the N/C ratio controls the onset of the MBT, a point shown clearly by the observation that haploid embryos take one cell cycle longer to reach the MBT than do their diploid counterparts (3). Although it is clear that the N/C ratio determines the duration of early embryonic cell cycles in many organisms, the means by which the embryo senses the N/C ratio has remained an important unsolved problem in developmental biology.
Recent work in Drosophila has begun to unravel the molecular mechanism for cell cycle remodeling at the MBT in this organism. Fly embryos undergo 13 rapid cleavage divisions, and then, at the 14th division, S phase is abruptly lengthened, a G2 phase is introduced, and zygotic transcription is robustly activated. Activation of transcription allows the embryo to target Twine, an ortholog of the Cdc25 mitotic inducer, for rapid destruction, and this appears to be the key event that triggers cell cycle remodeling at the MBT (11–13).
In contrast, the MBT in Xenopus embryos occurs independently of zygotic transcription (6). After 12 rounds of cleavage divisions, the MBT is reached during interphase of the 13th division, and the cell cycle here is initially characterized by an increase in the length of S phase (8). Previous work has suggested that the extension of S phase and subsequent cell cycle changes may be due to either a developmental activation of the replication checkpoint (14, 15), depletion of DNA replication factors (6, 15), or depletion of dNTPs (16). In the present study, we have sought to shed light on this process by establishing how the progression of DNA synthesis is influenced by the N/C ratio in vitro using extracts derived from Xenopus eggs.
One longstanding model for how the N/C ratio affects S phase in Xenopus suggests that a positively acting DNA replication factor is titrated from the cytoplasm by the exponentially increasing number of nuclei during the rapid cleavage divisions. Once the availability of this factor drops below a threshold, DNA replication would take longer to complete, and S phase would thereby be extended. Any model invoking a limiting factor must, however, be reconciled with more recent studies that have examined how replication initiation is regulated in Xenopus and how the N/C ratio influences this regulation (17–20). Work using egg extracts has shown that increased N/C ratio results in lengthening of replicon size, which reflects an expansion of the distance between active origins of replication. This indicates that the N/C ratio influences S phase duration by limiting the number of active origins. Furthermore, other work has shown that the ATR-based replication checkpoint, a signaling pathway that is activated during replication, negatively regulates origin firing. Importantly, it has also been shown that abrogation of ATR signaling uncouples the rate of origin firing from the N/C ratio, such that extracts lacking ATR activity replicate DNA quickly even when the N/C ratio is high. It therefore appears that the limiting factor for DNA replication at high N/C is not a component of the replication fork itself, but rather is likely to be a regulator of origin firing. However, a molecular basis for how the N/C ratio works through the ATR pathway to control the rate of origin firing was not previously known.
ATR is a protein kinase that controls progression through S phase by restricting origin firing in the presence of replication stress and, to a lesser extent, during unperturbed replication (19–22). The observation that loss of ATR activity prevents control of S phase by the N/C ratio suggests two simple possibilities for how the N/C ratio is sensed. First, it may be that at high N/C ratio, the ATR pathway is more efficiently activated by ongoing DNA replication. Alternatively, an activity that neutralizes the ATR-dependent suppression of origin firing may become limiting at high N/C ratio, and this would allow the ATR pathway to control origin firing more stringently. In this work, we present evidence that the second model is correct. We report that the protein phosphatase PP2A plays a key positive role in controlling the rate of origin firing and, furthermore, that the PP2A activity state is what allows the N/C ratio to limit origin firing. At high N/C ratio, PP2A activity becomes limiting due to titration of its key regulatory subunit, the B55α protein, and this drop in PP2A activity is the critical event that allows for the extension of S phase. These results provide a mechanistic basis for how the N/C ratio is sensed to regulate S phase duration during early development.
EXPERIMENTAL PROCEDURES
Xenopus Egg Extracts
Interphase egg extracts and sperm chromatin were prepared as described previously (23). Cytostatic factor-arrested extracts were prepared as described (24), except that aprotinin (G Biosciences) was used in place of chymostatin and pepstatin, and Versilube was omitted. For protein expression in cytostatic factor-arrested extracts, mRNA templates were incubated with extract for 2 h at 21 °C and then activated by the addition of CaCl2 to 600 μm. Chromatin was then added after a 13-min incubation. DNA synthesis was measured by supplementing the extracts with low levels of [α-32P]dATP (about 0.05% of endogenous dATP levels) and quantifying incorporation into DNA using agarose gel electrophoresis and autoradiography, as described previously (23). Fresh extracts were prepared for all experiments.
Isolation of Nuclei from Extracts
After progressing into S phase, extract samples were transferred to ice and then centrifuged at 700 × g for 6 min at 4 °C to form an upper layer of nuclei. To minimize cytoplasmic contamination, 80% of the underlying cytoplasm and pelleted debris was first removed with a needle and syringe. The remaining extract containing the nuclei was then suspended in 500 μl of ELB (250 mm sucrose, 2.5 mm MgCl2, 50 mm KCl, 10 mm HEPES, 1 mm DTT, 50 μg/ml cycloheximide) by gentle inversion. Extracts were underlain with 1 ml of ELB, 0.9 m sucrose, and nuclei were pelleted at 8,000 × g for 5 min. Nuclei were resuspended in ELB and centrifuged once more through ELB, 0.9 m sucrose as above.
In Vitro Transcription
Linear DNA templates were generated by PCR using the LinTempGenSet system (5 Prime). mRNA was produced using the mMessage mMachine kit (Ambion) according to the manufacturer's instructions.
DNA Fiber Labeling
Preparation of labeled DNA fibers was performed as described previously (25). Interphase egg extracts were prepared and supplemented with volume of 1 mm biotin-16-dUTP (bio-dUTP; Roche Applied Science) and 1 mm digoxigenin-11-dUTP (dig-dUTP; Roche Applied Science) at the indicated times. To assess replication fork density, dig-dUTP was added prior to the start of the reaction, whereas bio-dUTP was added 28–60 min later during early to mid-S phase. To assess fork velocity, bio-dUTP was added to extracts in early to mid-S phase, followed 10 min later by dig-dUTP. To ensure completion of replication of high N/C ratio samples, all reactions were subsequently diluted to a final concentration of 1,000 nuclei/μl with fresh extract containing both bio-dUTP and dig-dUTP and incubated until 120 min. This step allows for the complete labeling of input DNA, which is essential for accurate assessment of fork density. To measure fork density in the presence of aphidicolin, additional steps were necessary. After the initial labeling, nuclei were gently isolated by diluting in 20 volumes of a buffer (50 mm KCl, 5 mm MgCl2, 1 mm DTT, 50 mm HEPES-KOH, pH 7.6) and centrifuging at 1,000 × g for 2 min. Nuclei were then resuspended in fresh extract containing bio-dUTP and dig-dUTP but lacking aphidicolin. This manipulation allowed completion of replication even when large doses of aphidicolin were used (data not shown).
Digoxigenin-labeled fibers were detected using mouse anti-digoxin FITC antibody (Sigma) and then fixed in 4% paraformaldehyde. Consecutive incubations in AlexaFluor488-labeled secondary antibodies (goat anti-mouse, chicken anti-goat, goat anti-chicken, and rabbit anti-goat; Invitrogen) were used to amplify the signal. Concurrently, biotin-labeled fibers were detected using three layers of streptavidin AlexaFluor594 (Invitrogen) interspersed with two layers of rabbit biotinylated anti-streptavidin antibody (Rockland). Fibers were photographed using an Olympus BX-51 microscope with attached camera and SPOT software (Diagnostic Instruments). Fibers were measured using ImageJ software (National Institutes of Health) with a conversion ratio of 2 kb/μm (26). Fork density was determined by dividing the total number of replication forks by the total length of DNA measured, as described previously (19). The S.E. value of this measurement was determined by performing a bootstrap resampling procedure on the raw data using R statistical software (27). Statistical significance was assessed using bootstrap percentile confidence intervals for the difference between fork densities. To measure replication fork velocity, the lengths of biotin-labeled overhangs within fibers were measured and divided by the time interval between the bio-dUTP and dig-dUTP additions. Statistical significance was assessed with Student's t tests (two-tailed).
Immunodepletion
Immunodepletion of egg extracts was carried out using Protein G-Sepharose beads (GE Healthcare). For PP2A depletions, we used mouse ascites fluid recognizing the A subunit (6F9, Covance). Antisera against PP2A B subunits were a very generous gift from Dr. Satoru Mochida. The appropriate antibody or antiserum was bound to beads by tumbling for 1 h at room temperature and then washed in ELB. Extract was incubated with a 30% volume of beads for three rounds of 50 min each. Residual fluid was first removed from beads by centrifugation through a small slit cut into the bottom of the tube. Extract was recovered after each step by centrifugation in the same manner.
Immunoblotting
Antibodies against phosphorylated Chk1 (Ser-345) and PP2A C were purchased from Cell Signaling and Millipore, respectively. Chk1 (FL-476) and PP2A A (6G3) antibodies were purchased from Santa Cruz Biotechnology, Inc. Antibodies recognizing ORC2 were affinity-purified from rabbit antiserum raised against Xenopus ORC2, which was produced in bacteria. Affinity-purified antibodies specific for B55α were a gift from Dr. Satoru Mochida. Antibodies against Cdk2 and phosphorylated Cdk2 (Tyr-15) were purchased from Novus and Cell Applications, respectively. ImageJ software was used to quantify band intensities.
Reagents
His8-tagged Xenopus PP2A Cβ was produced in High Five cells as described previously (28) and purified by modified ethanol precipitation (29) and affinity chromatography with nickel-nitrilotriacetic acid resin. (Qiagen). GST-geminin was purified as described previously (30, 31). Recombinant human PP2A catalytic subunit (ΔL309) was purchased from Cayman Chemical. Caffeine (Sigma) was dissolved in 10 mm PIPES, pH 7.4, at 125 mm and used at a final concentration of 5 mm. Okadaic acid (Sigma) was dissolved in DMSO at a stock concentration of 500 μm and used at a final concentration of 0.5–1.5 μm. A stock solution of 40 μm roscovitine (EMD Millipore) was prepared in DMSO and used at a final concentration of 0.5 μm. Aphidicolin (Sigma) was dissolved in DMSO at a concentration of 30 mm and used at a final concentration of 0.5–30 μm.
RESULTS
S Phase Elongation in Xenopus Egg Extracts Containing High N/C Ratio
To investigate the mechanism for regulation of cell cycle duration by the N/C ratio, we sought to recapitulate in egg extracts the S phase lengthening that occurs at high N/C ratio in vivo. In this model system, extracts made from Xenopus eggs are combined with Xenopus sperm chromatin. Nuclei then form around this chromatin, and each nucleus conducts a single round of DNA replication. To model different points during early development, we varied the amount of chromatin added to produce different N/C ratios. We first measured DNA synthesis, via uptake of radiolabeled dATP, on a per nucleus basis in extracts across a range of N/C ratios (Fig. 1A). In extracts with low N/C ratios (1,000–2,000 nuclei/μl), which correspond to early, cleavage stage embryos, DNA synthesis was virtually complete by 50 min. However, extracts with intermediate nuclei concentration (5,000 nuclei/μl) were somewhat slower, and high N/C extracts (10,000 nuclei/μl), which reflect an MBT stage embryo, showed a severe reduction in DNA synthesis (∼40–80%, depending upon the batch of extract), in agreement with previous observations (7, 17). As a result of this reduction, the time required to complete S phase more than doubled at high N/C ratio (Fig. 1B). These data demonstrate that the rate of DNA synthesis, and thus the duration of S phase, is controlled by the N/C ratio in egg extracts. The delay in replication at high N/C ratio could not be explained by a failure to form nuclei, because nuclei formed reliably at all nuclei concentrations tested, and nuclei entered S phase in a nearly synchronous manner at both low and high N/C ratio (data not shown).
FIGURE 1.

High N/C ratio reduces DNA synthesis by reducing origin firing and slowing replication fork progression. A and B, interphase extracts were supplemented with [α-32P]dATP and either 1,000, 2,000, 5,000, or 10,000 nuclei/μl. Extracts were incubated at 21 °C, and DNA synthesis was monitored at the times indicated. The time scale of 32P incorporation varied somewhat between extracts, so the data shown are from individual experiments and are representative of multiple, independent experiments. C, to analyze replication fork density, bio-dUTP was added at 28 min to mark sites of ongoing replication. Total DNA was labeled with dig-dUTP. D, DNA fibers labeled as in C were prepared and stained as described under “Experimental Procedures.” The locations of replication forks at 28 min on two sample fibers are indicated by arrowheads. The bio-dUTP and dig-dUTP signals were photographed separately and merged at a slight vertical offset to facilitate inspection of each labeling pattern. E, a large number of fibers (at least 15 Mb of DNA) labeled as in C were analyzed for the presence of replication forks. Error bars, S.E. ****, significant at p < 0.0001. F, to determine fork velocity, bio-dUTP was added to extracts during early S phase and followed 10 min later with dig-dUTP to mark the extent of fork progression. G, sample fibers labeled as in F are shown. Digoxigenin-negative tracts used to determine fork progression are marked with brackets. H, fork progression, in nt/min, was determined for at least 260 forks at both low and high N/C ratios. Mean velocity ± S.E. is shown. ****, significant at p < 0.0001 (Student's t test). a.u., arbitrary units.
Replication Slowing Is Caused by both Reduced Origin Firing and Reduced Fork Progression
We reasoned that the drop in replication rate must be due to a decrease in the number of active replication forks (17), a decrease in the speed of those forks (19), or both. To address this question in our system, we used DNA fiber labeling, according to published procedures (19), to directly measure both the density of replication forks and the rate of fork progression in early S phase extracts in parallel experiments. To examine changes in replication fork density, the locations of replication forks during early S phase were marked with bio-dUTP (Fig. 1, C and D), and DNA fibers were prepared and analyzed as described under “Experimental Procedures.” At high N/C ratio, replication fork density was reduced up to 60% relative to low N/C ratio (Fig. 1E), indicating that reduced origin firing was contributing to the lower replication rate. To measure fork velocity, bio-dUTP was chased with dig-dUTP after 10 min to mark the progression of individual forks (Fig. 1, F and G). Although the velocities of individual replication forks within each sample varied considerably, quantification of a large number of forks revealed that the mean fork velocity at high N/C ratio was 26% lower than that of low N/C forks (321 nt/min versus 432 nt/min; Fig. 1H). Based on these data, we conclude that the major effect on replication at high N/C ratio is a reduction in origin firing (∼60%) and that the more modest reduction in fork velocity (∼26%) also plays a minor role.
Many proposed explanations for the cell cycle lengthening at the time of the MBT invoke the concept of a limiting factor that is required to support high levels of replication, and the depletion of this factor may explain the reduction in replication that we observe at high N/C ratio. If true, then resupplying this factor should remedy the reduction in replication. As a simple test of this concept, we asked whether the slow replication rate of high N/C ratio extracts could be reversed by adjusting the N/C ratio downward after S phase entry. Extracts containing 10,000 nuclei/μl were incubated for 30 min to allow for the formation of nuclei and entry into S phase. We then diluted these reactions with fresh extract to a final concentration of 2,000 nuclei/μl, thus resupplying any potentially limiting factor. After a short lag, the amount of DNA synthesis in these nuclei increased sharply (Fig. 2). This indicates that the rate of replication is not predetermined during very early S phase but rather that it depends on sufficient concentrations of one or more limiting substances. To test whether the limiting factor was functioning in the activation of additional origins, we pretreated the diluting extract with the CDK inhibitor roscovitine, which prevents subsequent origin firing. Once treated with roscovitine, the extract used for dilution lost nearly all of its ability to rescue the slow replication of high N/C ratio extracts (Fig. 2). This suggests that the limiting factor is primarily involved in the firing of new origins of replication.
FIGURE 2.
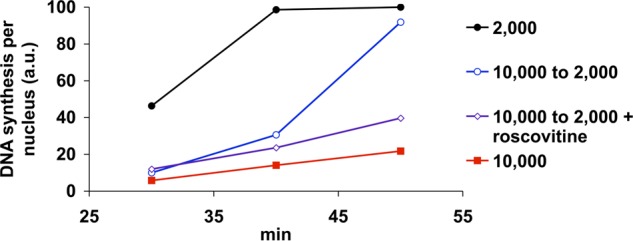
High N/C ratio extracts resupplied with fresh extract soon recover rapid replication rates. Extracts containing 10,000 nuclei/μl were incubated at room temperature for 30 min to allow for the formation of nuclei and entry into S phase. A subset of samples (open symbols) were then diluted to 2,000 nuclei/μl with 4 volumes of fresh extract in the presence or absence of 0.5 μm roscovitine, and DNA synthesis was measured. Alternatively, extracts prepared at either 10,000 or 2,000 nuclei were left undiluted (closed symbols) for comparison. The data shown are from a single experiment and are representative of multiple, independent experiments.
The Influence of Checkpoint Pathways at High N/C Ratios
The data presented thus far suggest that a replication initiation factor becomes limiting at high N/C ratio, and this in turn limits origin activation and thereby extends the length of S phase. Previous work has shown that ATR signaling is a major pathway that limits origin activation in Xenopus (see Introduction), and additional work in vivo suggests that developmental activation of the ATR-Chk1 pathway at some threshold level of N/C may be responsible for cell cycle lengthening at the MBT (14, 15). Therefore, we next analyzed the role of ATR signaling in modulation of replication by the N/C ratio. In mammalian somatic cells and other systems (32), ATR regulates origin firing through phosphorylation and activation of its effector kinase, Chk1. Activated Chk1 can phosphorylate Cdc25 and thereby target it for destruction. Loss of Cdc25 activity allows accumulation of inhibitory phosphorylation on CDK, and this in turn reduces origin firing, which is dependent on CDK. We therefore sought to determine whether the lengthening of S phase at high N/C ratios was a result of Chk1 activation and its effects on origin firing.
As observed both in vivo and previously in extracts (14, 33), Chk1 was phosphorylated at high levels at high N/C ratio (Fig. 3A). Rather than exhibiting an abrupt activation once a certain threshold level of nuclei was reached, however, the amount of Chk1 phosphorylation increased incrementally with the number of nuclei, even at very low N/C ratio. Chk1 phosphorylation was not dependent upon the N/C ratio per se but rather on the total amount of ongoing DNA synthesis. Reduction of replication initiation with geminin, an inhibitor of prereplication complex formation, led to a commensurate decrease in Chk1 activation at both low (Fig. 3, D and E) and high N/C ratios (Fig. 3, B and C). In these experiments, geminin did not directly interfere with checkpoint activation, because it was still possible to activate the checkpoint in the presence of geminin when aphidicolin was included (Fig. 3F). Taken together, these results suggest that the source of Chk1 activation at high N/C ratio is ordinary replication in many replicating nuclei.
FIGURE 3.

Chk1 phosphorylation in extracts is correlated with the total amount of ongoing replication. A, extracts were prepared across a range of nuclei concentrations and analyzed for Chk1 phosphorylation after 30 min. The asterisk marks an unknown nonspecific band. B, extracts were pretreated with varying amounts of recombinant geminin, and sperm chromatin was added to 10,000 nuclei/μl. Replication per nucleus was determined as in Fig. 1A. C, samples were prepared in parallel to those in B using the same batch of extract, and Chk1 phosphorylation was analyzed. D, extracts were pretreated with geminin at a range of concentrations, as in B, and then supplemented with 2,000 nuclei/μl. DNA synthesis was then measured. A control showing replication at 10,000 nuclei/μl was included for comparison. E, in a parallel experiment to D, Chk1 phosphorylation was analyzed in extracts pretreated with geminin and supplemented with 2,000 nuclei/μl. F, Chk1 activation in the presence of geminin. Extracts were pretreated with geminin and supplemented with 2,000 or 10,000 nuclei/μl. Chk1 phosphorylation was induced in geminin-treated extracts using 30 μm aphidicolin as indicated.
Therefore, if increased ATR-Chk1 signaling is to account for the change in replication behavior at high N/C, replicating nuclei would have to activate more Chk1 at high N/C than they do at low N/C. To look at this directly, we examined the efficiency of Chk1 activation at both low and high N/C ratios by normalizing the signal for both the number of nuclei and the number of replication forks contributing to it. Although high N/C extracts contained a greater amount of Chk1 on a per volume basis (Figs. 3A and 4A, top), the amount of Chk1 activation per nucleus was actually reduced at high N/C relative to low N/C (Fig. 4A, middle). However, because there are fewer replication forks contributing to this signal in high N/C nuclei (Figs. 1E and 4B), we also normalized this signal based upon the total number of replication forks in these extracts, which was calculated from the density of replication forks on DNA (Fig. 4B) and the concentration of nuclei in each sample. This gel shows that forks at high N/C ratio do not activate more Chk1 than forks at low N/C ratio (Fig. 4A, bottom), suggesting that high N/C ratio does not hyperstimulate ATR signaling. In contrast, slight inhibition of replication (Fig. 4, B and C) with a low dose of aphidicolin (4 μm) readily increased Chk1 activation per fork, demonstrating the effectiveness of this approach. These data indicate that Chk1 does not undergo any special activation at high N/C ratio, suggesting that it does not trigger the restriction of origin firing at high N/C ratio.
FIGURE 4.

ATR/Chk1 signaling efficiency is not affected by high N/C ratio. A, extracts were prepared with either 10,000 nuclei/μl, 1,000 nuclei/μl, or 1,000 nuclei/μl with low doses of aphidicolin. Chk1 phosphorylation was then analyzed at 30 min. Top, an equal volume of each extract was loaded, as in Fig. 3A. Middle, volumes of extract were loaded such that an equal number of nuclei were present in each lane. Bottom, volumes of extract were loaded such that an equal number of replication forks were present in each lane. The relative number of replication forks was determined by the fork density (B) and the concentration of nuclei in each sample. In the bottom two panels, fresh extract was added after application of SDS-PAGE sample buffer to equalize the total volume of extract in each lane. B, replication fork density was determined, as in Fig. 1C, in high or low N/C extracts or low N/C extracts treated with small doses of aphidicolin. To ensure that aphidicolin-treated samples replicated to completion, nuclei were transferred into fresh extracts after completion of the initial labeling, as described under “Experimental Procedures.” Error bars, S.E. Data marked with different letters are statistically different (p < 0.05). C, using a second set of samples, the replication fork velocity of the samples in B was determined in parallel (mean ± S.E.). Means with different letters are significantly different (Student's t test, p < 0.05). D, Cdk2 phosphorylation was analyzed after 30 min in low and high N/C extracts and in the presence or absence of aphidicolin. E, extracts with 1,000 or 10,000 nuclei/μl were treated with UCN01, and DNA synthesis was measured. F, extracts at 1,000 or 10,000 nuclei/μl were prepared in the presence or absence of 5 mm caffeine, and DNA synthesis was measured.
To confirm these findings, we examined the inhibitory phosphorylation of Cdk2, an important downstream target of Chk1 for control of origin firing. The data show no increase in Tyr-15 phosphorylation on Cdk2 at a high N/C ratio (Fig. 4D), again suggesting that Chk1 is not hyperstimulated. We also observed no increase in DNA synthesis when high N/C ratio extracts were treated with UCN01, a specific inhibitor of Chk1 activity, suggesting that Chk1 is not functionally responsible for controlling replication in our system (Fig. 4E). In contrast, upstream inhibition of ATR/ATM with the inhibitor caffeine did accelerate replication (Fig. 4F), suggesting that origin firing is controlled by an ATR-dependent, Chk1-independent mechanism. Consistent with our findings, other work has shown that depletion of Chk1 from egg extracts does not prevent ATR from inhibiting replication when ATR is hyperstimulated by replication stress (18). Based on these results, we conclude that, in Xenopus, ATR does not suppress origin firing via the canonical ATR-Chk1-Cdc25-CDK pathway and instead uses a novel pathway to accomplish this task. Furthermore, we also conclude that differences in the ATR activity state, as defined by phosphorylation of Chk1, cannot explain how origin firing is suppressed at high N/C ratios.
Control of DNA Replication by PP2A
Because a model where ATR is more efficiently activated at high N/C ratio does not appear to account for how the N/C ratio influences origin firing, we next considered that the limiting factor required for high replication rates might instead be involved in the suppression or attenuation of the ATR pathway. Previous work has implicated PP2A in suppression of an ATR and ATM-mediated checkpoint in response to DNA double strand breaks (34). In this pathway, PP2A acted in opposition to ATR/ATM to allow loading of the Cdc45 replication factor during replication initiation. We therefore determined whether a similar mechanism might respond to changes in the N/C ratio.
To investigate whether phosphatase activity was important for regulating replication rates at low or high N/C ratio, we tested whether treatment with okadaic acid, a potent inhibitor of PP2A (and, to a much lesser extent, PP1), would slow down the replication rate of low N/C ratio extracts. To do so, we first incubated sperm chromatin in extract at high N/C ratio to initiate S phase, as in Fig. 2. As before, reduction of the N/C ratio by dilution with fresh extract to low N/C ratio (1,000 nuclei/μl) quickly intensified DNA synthesis in these extracts (Fig. 5A). However, treatment of the extract with okadaic acid at the time of dilution prevented this increase almost entirely, suggesting that high phosphatase activity is required for the high replication rates that characterize low N/C ratio extracts.
FIGURE 5.
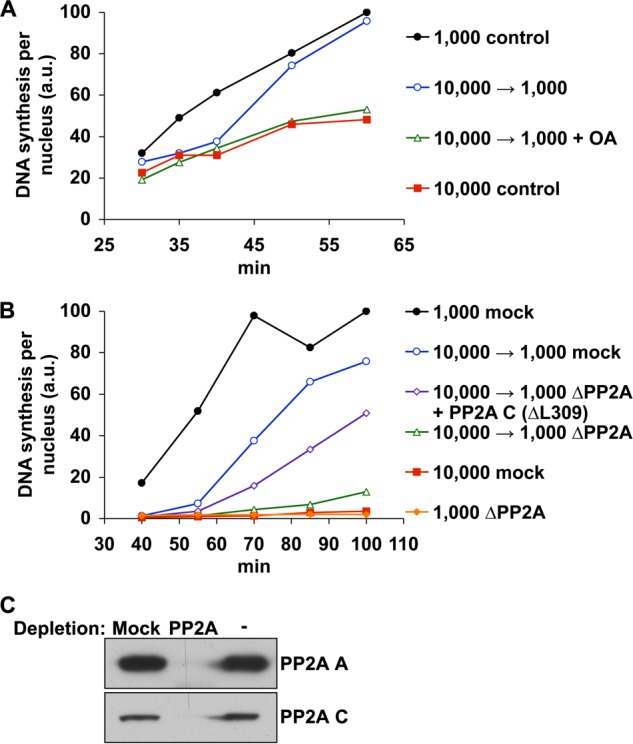
PP2A activity is essential for rapid S phase progression at low N/C ratio. A, extracts containing 10,000 nuclei/μl were allowed to assemble nuclei and progress into early S phase as in Fig. 2. At 30 min, these samples were diluted to 1,000 nuclei/μl with fresh extract (open symbols) and supplemented with DMSO or 0.5 μm okadaic acid (OA). Alternatively, extracts were left undiluted. DNA synthesis was then monitored as indicated. For comparison, DNA synthesis at low N/C ratio (not diluted) is also shown. B, mock-depleted extracts containing 10,000 nuclei/μl were allowed to initiate S phase as in A. After 40 min, extracts were diluted to 1,000 nuclei/μl using either mock-depleted extract, PP2A-depleted extract, or PP2A-depleted extract supplemented with recombinant human PP2A catalytic subunit (ΔL309). Alternatively, extract was left undiluted. Mock-depleted extract containing 1,000 nuclei/μl is shown for comparison. C, the effectiveness of PP2A depletion was monitored by immunoblot. The data shown are from single experiments and are representative of multiple, independent experiments. a.u., absorbance units.
To confirm that PP2A activity was responsible for this rescue effect, we repeated these experiments using extract immunodepleted of PP2A. Whereas mock-depleted extract readily restored a high rate of replication, PP2A-depleted extract lost nearly all of this capability (Fig. 5, B and C), demonstrating that PP2A is essential to regain rapid rates of DNA synthesis. This activity could be mostly restored, however, by the addition of recombinant human PP2A catalytic subunit (ΔL309; hPP2A C). These results demonstrate that PP2A plays a highly important role in controlling DNA synthesis, because every other component of the extract except for PP2A can be resupplied to high N/C ratio extracts without rescuing the replication rate.
PP2A Is Limiting for Replication at High N/C Ratio
If differences in PP2A can explain the reduction in replication at high N/C ratio, then the activity of PP2A would have to be rate-limiting under these conditions. Providing exogenous PP2A to these extracts should therefore counteract the reduction in DNA synthesis. We prepared high N/C extracts and supplemented them with either recombinant Xenopus PP2A catalytic subunit (rPP2A C) or buffer. We then monitored DNA synthesis to check for any effect on S phase progression. The addition of rPP2A C at a concentration of three times endogenous levels more than doubled the amount of DNA synthesis in high N/C ratio extracts (Fig. 6A), suggesting that its activity is functionally limiting for replication. In comparison, low N/C ratio extracts were much less sensitive to this effect, suggesting that a surfeit of PP2A activity was already present. Taken together with previous results, these data confirm that PP2A activity is a limiting factor for replication at high N/C ratio.
FIGURE 6.

PP2A is a limiting factor for S phase progression at high N/C ratio. A, extracts containing either 1,000 or 10,000 nuclei/μl were supplemented with either 478 nm Xenopus PP2A catalytic subunit or buffer, and DNA synthesis was measured. The data shown are from a single experiment and are representative of multiple, independent experiments. B and C, extracts were prepared as in A but were instead labeled with bio-dUTP and dig-dUTP and used to analyze replication fork density and velocity by fiber staining, as in Fig. 1, C and F. Error bars, S.E. ****, p < 0.0001; n.s., not significant (p > 0.05). D, mock-depleted extract, PP2A-depleted extract, or a mixture of PP2A-depleted (60%) and mock-depleted (40%) extract was supplemented with 1,000 nuclei/μl, and DNA synthesis was measured. E and F, mock-depleted extract or a mixture containing 60% PP2A-depleted and 40% mock-depleted extract was supplemented with 1,000 nuclei/μl and labeled with bio-dUTP and dig-UTP. Fork density and fork velocity were then determined at 55 min. a.u., absorbance units.
PP2A could potentially affect S phase progression by improving fork progression, activating additional origins, or both. To better understand how PP2A influences the replication rates in extracts, we used DNA fiber labeling to investigate replication dynamics in the presence and absence of rPP2A C. Although high N/C extracts again exhibited reduced origin firing relative to low N/C extracts, the addition of rPP2AC reversed this effect (Fig. 6B). The rate of fork progression, however, was not changed in these samples (Fig. 6C). These data indicate that PP2A controls the replication rate in extracts by influencing the rate of origin firing, not the rate of fork progression.
If the reduction in origin firing at high N/C ratio is caused by limiting amounts of PP2A, we would expect that partial depletion of PP2A would mimic these effects even at low N/C ratio. We therefore investigated the effect of loss of PP2A activity at low N/C ratio. Because complete loss of PP2A activity prevents nearly all replication (Fig. 6D) (35), we removed ∼60% of PP2A by immunodepletion and then analyzed fork density and velocity by DNA fiber analysis. Although the rates of fork progression in mock-depleted and 60% PP2A-depleted extracts were indistinguishable, the number of replication forks was sharply lower in PP2A-depleted extracts (Fig. 6, E and F). We conclude that the ability of low N/C ratio extracts to rapidly complete S phase depends upon extensive origin firing facilitated by high concentrations of PP2A.
Although PP2A activity is functionally limiting at high N/C ratio, this could be due to either an overall shortage of PP2A complexes, a failure to import sufficient PP2A complexes to the appropriate location within nuclei, or a separate regulatory mechanism that simply regulates nuclear PP2A complexes more strictly. In Xenopus eggs, PP2A exists in multiple forms. The 36-kDa catalytic C subunit makes a dimer with the 65-kDa A subunit to form the core enzyme, which can then form a trimer with one of many different regulatory B subunits to form the holoenzyme. The core and holoenzymes exist in a 1:1 ratio in the egg (35), and there is no detectable free C subunit. To test whether PP2A becomes physically limiting within nuclei at high N/C ratio, we analyzed nuclear isolates for A and C subunits. The amount of both A and C was reduced in high N/C ratio nuclei relative to low N/C ratio nuclei (Fig. 7, A and B), suggesting that increased N/C ratio results in a decrease in the amount of nuclear PP2A. This reduction in nuclear PP2A C could be rescued by the addition of recombinant catalytic subunit (Fig. 7D), a condition that also improves S phase progression (Fig. 6A). These results are consistent with the notion that the reduction in replication rate at high N/C ratio is caused by a failure to accumulate sufficient PP2A into nuclei, which could be caused by either a direct depletion of phosphatase components or altered nuclear transport dynamics. If a depletion mechanism is responsible, we would expect to see the amounts of remaining cytoplasmic PP2A begin to dwindle at high N/C ratio. However, the amount of PP2A C remaining in corresponding cytoplasmic isolates remained unchanged (Fig. 7C), suggesting that it is present in excess with regard to its nuclear functions. These data suggest that PP2A C, although present in excess in extracts, does not efficiently accumulate in nuclei at high N/C ratios.
FIGURE 7.

High N/C ratio nuclei contain lower amounts of PP2A. A and B, nuclei were isolated from extracts containing either 1,000 or 10,000 nuclei/μl and analyzed by immunoblot for PP2A components and ORC2. For 10,000 nuclei/μl, two different isolations were performed. In the first, an equal number of nuclei were isolated relative to the 1,000 nuclei/μl sample. In the second, nuclei were isolated from an equal volume of extract relative to the 1,000 nuclei/μl sample. A sample containing no nuclei is included to control for residual cytoplasmic protein. The results from two independent experiments are shown. C, after the removal of nuclei, cytoplasm from the samples in B was analyzed for PP2A C content. D, extracts were prepared with either 1,000 or 10,000 nuclei/μl and supplemented with either control buffer or 478 nm rPP2A C (Xenopus). An equal number of nuclei were isolated from each condition and blotted for the indicated proteins.
Specific Form of PP2A Involved in Replication
The substrate specificity and subcellular location of PP2A are imparted by the binding of one of a variety of regulatory B subunits. The functions of PP2A relevant to DNA replication are therefore likely to be mediated by one or a subset of these forms. Because the PP2A core enzyme is present at high concentrations in extracts (∼1% of total protein) and PP2A C appears to be present in excess with regard to nuclear functions, it is likely that only a specific form of PP2A becomes depleted at high N/C ratio. We therefore sought to identify which class of PP2A holoenzymes was involved in control of DNA replication. To test the importance of each B subunit, we immunodepleted known Xenopus B subunits from extracts and tested for replication defects. The depletion of PR70, B56γ, B56ϵ, or B55δ did not hinder DNA synthesis, even at a moderately high N/C ratio (6,000 nuclei/μl), a condition under which extracts should have elevated sensitivity to the depletion of a limiting factor (data not shown). However, extracts depleted using antibodies against B55α exhibited a sharp decrease in replication (over 70%), suggesting that the predominant form of PP2A involved in replication utilizes B55α regulatory subunits (Fig. 8, A and B). This is consistent with a recent report that found that inhibition of the B55 family of holoenzymes could compromise replication under conditions of artificially reduced PP2A activity (36).
FIGURE 8.

B55α levels control the amount of DNA synthesis at high N/C ratio. A, interphase extracts were immunodepleted of B55α or mock-depleted. Sperm chromatin was added to 6,000/μl, and DNA synthesis was measured. The data shown are from a single experiment and are representative of multiple, independent experiments. B, the extent of depletion was monitored by immunoblot. The asterisk marks an unknown nonspecific band. C, mock-depleted or B55α-depleted extracts were supplemented with 1,000 nuclei/μl. Nuclei were then isolated after 83 min and blotted for PP2A C and ORC2. D and E, cytostatic factor-arrested extracts were incubated with up to 100 or 133 ng/μl B55α mRNA for 2 h at room temperature, activated with CaCl2, and then supplemented with sperm chromatin to 10,000/μl. DNA synthesis was measured as indicated. A sample containing only 1,000 nuclei/μl is shown for comparison. The results from two independent experiments are shown. F, production of B55α was monitored by immunoblot. The asterisk marks an unknown nonspecific band. G, the B55α bands in F were quantified, and the relative intensities of each are shown. a.u., absorbance units.
Because the propensity of extracts for rapid DNA synthesis has been associated with the levels of nuclear PP2A C (Fig. 7), we next asked whether the nuclear accumulation of PP2A C was compromised in B55α-depleted extracts. Although PP2A C accumulated efficiently into nuclei of mock-depleted extracts, nuclei formed in B55α-depleted extracts did not contain any detectable PP2A C, even at later points in S phase (Fig. 8C). Nuclei were slightly smaller in B55α-depleted extract (data not shown), however, and this may contribute somewhat to this reduction in PP2A C. Nevertheless, the absence of PP2A C in these nuclei may explain the severe restriction of replication under these conditions (Fig. 8A).
If limitation of B55α is responsible for reduced PP2A function and DNA synthesis at high N/C ratio, then increasing its levels exogenously would be expected to counteract these effects. We were unable to produce sufficient quantities of functional, recombinant Xenopus B55α, despite testing many different expression systems and purification methods. Therefore, we decided to increase B55α levels by supplementing cytostatic factor-arrested extracts, which lack cycloheximide and are thus capable of translation, with B55α mRNA. Extracts were first preincubated with B55α mRNA (or water) to allow for protein synthesis and then activated with CaCl2. We then monitored DNA replication at high N/C ratio over the course of 2 h. Based on microscopic examination of nuclei, these extracts did not enter M phase during this interval. Measurements of DNA synthesis show that extracts incubated with B55α mRNA synthesized an amount of DNA at 60 min more than triple that of control samples (Fig. 8, D and E), indicating that B55α is functionally limiting for DNA replication at high N/C ratio. This potent effect occurred despite only a small increase in B55α protein levels (Fig. 8, F and G). Together with previous data, these results suggest that the essential function of PP2A during DNA replication is mediated primarily by holoenzymes containing B55α subunits. Furthermore, these results suggest that titration of this scarce form of the phosphatase is responsible for a significant lengthening of S phase when the N/C ratio exceeds a threshold.
DISCUSSION
Control of DNA Replication by the N/C Ratio through Titration of PP2A-B55α Complexes
In this study, we have examined how the length of S phase is controlled by the N/C ratio in Xenopus egg extracts. We report that high N/C extracts replicate DNA more slowly, and this is due in large part to a reduction in origin firing during early S phase (Fig. 1). In addition, and consistent with a previous report (19), we also observed a decrease in fork velocity at high N/C ratio. To pursue why high N/C extracts fire fewer replication origins during early S phase, we performed experiments that defined a limiting factor(s) for origin firing at high N/C (Fig. 2) and went on to identify the limiting factor as the PP2A-B55α holoenzyme. The addition of recombinant PP2A C to high N/C extract accelerates replication (Fig. 6), as does the addition of exogenous B55α, the regulatory subunit (Fig. 8). We also observed that reduction of PP2A in low N/C extracts reduces the rate of replication (Fig. 6). Furthermore, we have shown that the amount of PP2A C within nuclei becomes limiting as the N/C ratio increases (Fig. 7) and that the ability of PP2A to accumulate in nuclei is dependent on the B55α subunit (Fig. 8). All of the observed effects of PP2A on replication are at the level of origin activation, since modulation of PP2A activity in the extract produced a profound effect on replication fork density, a direct measure of origin activation, and had no effect on fork velocity (Fig. 6). Our data therefore suggest that origin firing is extremely sensitive to the amount of PP2A present in nuclei and, furthermore, that the amount of nuclear PP2A correlates directly with the N/C ratio. Taken together, these data suggest a simple model for how the N/C ratio regulates origin firing, and that is through titration of PP2A-B55α complexes. At low N/C ratio, there is a sufficient amount of PP2A-B55α within nuclei to support a high rate of origin firing. By contrast, at high N/C ratio, PP2A-B55α becomes limiting, the rate of origin activation drops, and S phase is thereby extended.
The Role of B55α in DNA Replication
A recent study has implicated B55 family members in the control of DNA replication (36), and our data both confirm and extend these observations through the demonstration that B55α is absolutely required for replication in egg extracts (Fig. 8A). Furthermore, we have shown that B55α is required for the nuclear accumulation of PP2A C. Interestingly, we have also shown that the addition of recombinant PP2A C alone to high N/C extract is sufficient to both stimulate replication (Fig. 6, A and B) and increase the amount of nuclear PP2A (Fig. 7D). How might exogenous PP2A C subunit alone produce these effects? It is important to consider the stoichiometry of PP2A in egg extracts; the A-C core enzyme is present at a 1:1 ratio with A-C-B holoenzymes, and there is no free C subunit present (35). The A-C core enzyme cannot accumulate in nuclei on its own (Fig. 8C); however, the 34-kDa C subunit is small enough to enter nuclei via passive diffusion. Thus, when C is added exogenously to extract, it is likely that some will accumulate in nuclei. Indeed, this is what was observed (Fig. 7D). Taken together, these data suggest that a major role of B55α in replication initiation is to allow nuclear accumulation of PP2A and that this function can be bypassed through the addition of exogenous PP2A C. B55α may also play additional roles (e.g. to mediate interaction between PP2A and its substrate(s) within nuclei), and further analysis will be required to determine if this is so.
The multifunctional nature of PP2A presents the possibility that it may also contribute to the progression of DNA synthesis in alternative ways, such as the promotion of nuclear assembly (37). Although nuclei formed successfully in PP2A-depleted extracts, they were slightly smaller than those in mock-depleted extracts (data not shown), as seen previously (35). Extracts depleted of B55α sometimes showed a similar phenotype (data not shown). Thus, PP2A may support rapid replication rates in part by facilitating the earlier assembly of nuclei. However, this would not explain how PP2A influences replication in nuclei-free extract systems (34); nor would it explain the requirement for PP2A in replication even after formation of nuclei is completed (Fig. 5, A and B). Furthermore, reduced nuclear size was not essential for the slower replication kinetics of high N/C extracts. We therefore believe that the effects of PP2A on replication are primarily mediated by another mechanism.
ATR-PP2A Conflict during DNA Replication
Taken together with previous reports (35, 36), the data presented here show clearly that PP2A-B55α is an important replication initiation factor. Previous studies have shown that PP2A counteracts the negative regulation of replication imposed by ATM/ATR during a DNA damage response (34), and we suggest that an ATR-PP2A tussle also exists during an unperturbed S phase. Importantly, we have shown that ATR signaling, as assessed by phosphorylation of its critical substrate Chk1, does not fluctuate as the N/C ratio changes (Fig. 4A). However, PP2A levels in the nucleus do fluctuate, and thus we envision that at low N/C ratio, PP2A can effectively neutralize ATR signaling to allow a high rate of origin firing. Our data also suggest that when the N/C ratio passes a threshold where nuclear PP2A becomes limiting, then ATR predominates, and origin firing is attenuated as an outcome. Although this simple model can readily explain all available data, the critical test can only come when the ATR/PP2A substrate for origin activation is identified. Data presented here and elsewhere (18) can exclude Chk1 as the ATR/PP2A substrate for replication control, and thus a high priority for future work will be to identify this factor or factors.
Potential Roles for S Phase Lengthening in Response to PP2A Titration in Vivo
The abrupt extension of S phase that marks the beginning of the MBT is a critical part of the cell cycle remodeling that occurs here and may be required for other developmental changes, such as the onset of transcription. In both Xenopus and Drosophila, rapid completion of S phase interferes with transcription, and premature lengthening or inhibition of DNA synthesis results in the early onset of transcription (3, 38, 39). Extension of S phase also appears to be responsible for the delay of the mitotic cycle, probably in a checkpoint-dependent manner (40). Thus, titration of PP2A-B55α provides a mechanism for the extension of S phase but probably plays a role in the occurrence of other MBT changes as well. Additionally, the potential involvement of PP2A-B55α in regulating ATR function may be important to the manifestation of cell cycle checkpoints near the MBT (7, 41). An abundance of PP2A-B55α in the early cleavage cycles, acting in opposition to ATR, may explain why early embryonic cycles are refractory to cell cycle pausing in response to DNA damage and other disruptions of S phase. Although we have shown that titration of PP2A activity results in substantial changes to replication in an in vitro system, subsequent evaluation of this mechanism in Xenopus embryos will be critical to determining the extent of its influence on developmental processes.
Acknowledgments
We thank Satoru Mochida for helpful discussions and for generously sharing many reagents used in this report. We are also grateful to Nicholas Rhind, Nicholas Willis, and Shankar Das for assistance with DNA fiber labeling experiments. We thank Irene Chiolo and Oscar Aparicio for comments on a draft of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grant GM067735 (to W. M. M.).
- MBT
- midblastula transition
- PP2A
- protein phosphatase 2A
- hPP2A C and rPP2A C
- human and Xenopus recombinant PP2A catalytic subunit, respectively
- N/C
- nucleus/cytoplasm
- dig-dUTP
- digoxigenin-11-dUTP
- bio-dUTP
- biotin-16-dUTP
- CDK
- cyclin-dependent kinase.
REFERENCES
- 1. Brauchle M., Baumer K., Gönczy P. (2003) Differential activation of the DNA replication checkpoint contributes to asynchrony of cell division in C. elegans embryos. Curr. Biol. 13, 819–827 [DOI] [PubMed] [Google Scholar]
- 2. Hara K., Tydeman P., Kirschner M. (1980) A cytoplasmic clock with the same period as the division cycle in Xenopus eggs. Proc. Natl. Acad. Sci. U.S.A. 77, 462–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Edgar B. A., Kiehle C. P., Schubiger G. (1986) Cell cycle control by the nucleo-cytoplasmic ratio in early Drosophila development. Cell 44, 365–372 [DOI] [PubMed] [Google Scholar]
- 4. Kane D. A., Kimmel C. B. (1993) The zebrafish midblastula transition. Development 119, 447–456 [DOI] [PubMed] [Google Scholar]
- 5. Graham C., Morgan R. (1966) Changes in the cell cycle during early amphibian development. Dev. Biol. 14, 439–460 [Google Scholar]
- 6. Newport J., Kirschner M. (1982) A major developmental transition in early Xenopus embryos. I. Characterization and timing of cellular changes at the midblastula stage. Cell 30, 675–686 [DOI] [PubMed] [Google Scholar]
- 7. Newport J., Dasso M. (1989) On the coupling between DNA replication and mitosis. J. Cell Sci. Suppl. 12, 149–160 [DOI] [PubMed] [Google Scholar]
- 8. Iwao Y., Uchida Y., Ueno S., Yoshizaki N., Masui Y. (2005) Midblastula transition (MBT) of the cell cycles in the yolk and pigment granule-free translucent blastomeres obtained from centrifuged Xenopus embryos. Dev. Growth Differ. 47, 283–294 [DOI] [PubMed] [Google Scholar]
- 9. Clute P., Masui Y. (1997) Microtubule dependence of chromosome cycles in Xenopus laevis blastomeres under the influence of a DNA synthesis inhibitor, aphidicolin. Dev. Biol. 185, 1–13 [DOI] [PubMed] [Google Scholar]
- 10. Finkielstein C. V., Lewellyn A. L., Maller J. L. (2001) The midblastula transition in Xenopus embryos activates multiple pathways to prevent apoptosis in response to DNA damage. Proc. Natl. Acad. Sci. U.S.A. 98, 1006–1011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Farrell J. A., O'Farrell P. H. (2013) Mechanism and regulation of cdc25/twine protein destruction in embryonic cell-cycle remodeling. Curr. Biol. 23, 118–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Di Talia S., She R., Blythe S. A., Lu X., Zhang Q. F., Wieschaus E. F. (2013) Posttranslational control of Cdc25 degradation terminates Drosophila's early cell-cycle program. Curr. Biol. 23, 127–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sung H. W., Spangenberg S., Vogt N., Grosshans J. (2013) Number of nuclear divisions in the Drosophila blastoderm controlled by onset of zygotic transcription. Curr. Biol. 23, 133–138 [DOI] [PubMed] [Google Scholar]
- 14. Shimuta K., Nakajo N., Uto K., Hayano Y., Okazaki K., Sagata N. (2002) Chk1 is activated transiently and targets Cdc25A for degradation at the Xenopus midblastula transition. EMBO J. 21, 3694–3703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sibon O. C., Stevenson V. A., Theurkauf W. E. (1997) DNA-replication checkpoint control at the Drosophila midblastula transition. Nature 388, 93–97 [DOI] [PubMed] [Google Scholar]
- 16. Vastag L., Jorgensen P., Peshkin L., Wei R., Rabinowitz J. D., Kirschner M. W. (2011) Remodeling of the metabolome during early frog development. PLoS One 6, e16881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Walter J., Newport J. W. (1997) Regulation of replicon size in Xenopus egg extracts. Science 275, 993–995 [DOI] [PubMed] [Google Scholar]
- 18. Luciani M. G., Oehlmann M., Blow J. J. (2004) Characterization of a novel ATR-dependent, Chk1-independent, intra-S-phase checkpoint that suppresses initiation of replication in Xenopus. J. Cell Sci. 117, 6019–6030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Marheineke K., Hyrien O. (2004) Control of replication origin density and firing time in Xenopus egg extracts. Role of a caffeine-sensitive, ATR-dependent checkpoint. J. Biol. Chem. 279, 28071–28081 [DOI] [PubMed] [Google Scholar]
- 20. Shechter D., Costanzo V., Gautier J. (2004) ATR and ATM regulate the timing of DNA replication origin firing. Nat. Cell Biol. 6, 648–655 [DOI] [PubMed] [Google Scholar]
- 21. Syljuåsen R. G., Sørensen C. S., Hansen L. T., Fugger K., Lundin C., Johansson F., Helleday T., Sehested M., Lukas J., Bartek J. (2005) Inhibition of human Chk1 causes increased initiation of DNA replication, phosphorylation of ATR targets, and DNA breakage. Mol. Cell Biol. 25, 3553–3562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cimprich K. A., Cortez D. (2008) ATR. An essential regulator of genome integrity. Nat. Rev. Mol. Cell Biol. 9, 616–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Walter J., Newport J. (1999) The use of Xenopus laevis interphase egg extracts to study genomic DNA replication. in Eukaryotic DNA Replication: A Practical Approach (Cotterill S., ed) pp. 201–222, Oxford University Press, Oxford, UK [Google Scholar]
- 24. Murray A. W. (1991) Cell cycle extracts. Methods Cell Biol. 36, 581–605 [PubMed] [Google Scholar]
- 25. Marheineke K., Goldar A., Krude T., Hyrien O. (2009) Use of DNA combing to study DNA replication in Xenopus and human cell-free systems. Methods Mol. Biol. 521, 575–603 [DOI] [PubMed] [Google Scholar]
- 26. Michalet X., Ekong R., Fougerousse F., Rousseaux S., Schurra C., Hornigold N., van Slegtenhorst M., Wolfe J., Povey S., Beckmann J. S., Bensimon A. (1997) Dynamic molecular combing. Stretching the whole human genome for high-resolution studies. Science 277, 1518–1523 [DOI] [PubMed] [Google Scholar]
- 27. R Development Core Team (2009) R. A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria [Google Scholar]
- 28. Ikehara T., Shinjo F., Ikehara S., Imamura S., Yasumoto T. (2006) Baculovirus expression, purification, and characterization of human protein phosphatase 2A catalytic subunits α and β. Protein Expr. Purif. 45, 150–156 [DOI] [PubMed] [Google Scholar]
- 29. Brandt H., Capulong Z. L., Lee E. Y. (1975) Purification and properties of rabbit liver phosphorylase phosphatase. J. Biol. Chem. 250, 8038–8044 [PubMed] [Google Scholar]
- 30. Michael W. M., Ott R., Fanning E., Newport J. (2000) Activation of the DNA replication checkpoint through RNA synthesis by primase. Science 289, 2133–2137 [DOI] [PubMed] [Google Scholar]
- 31. Stokes M. P., Van Hatten R., Lindsay H. D., Michael W. M. (2002) DNA replication is required for the checkpoint response to damaged DNA in Xenopus egg extracts. J. Cell Biol. 158, 863–872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Petermann E., Woodcock M., Helleday T. (2010) Chk1 promotes replication fork progression by controlling replication initiation. Proc. Natl. Acad. Sci. U.S.A. 107, 16090–16095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gotoh T., Kishimoto T., Sible J. C. (2011) Phosphorylation of Claspin is triggered by the nucleocytoplasmic ratio at the Xenopus laevis midblastula transition. Dev. Biol. 353, 302–308 [DOI] [PubMed] [Google Scholar]
- 34. Petersen P., Chou D. M., You Z., Hunter T., Walter J. C., Walter G. (2006) Protein phosphatase 2A antagonizes ATM and ATR in a Cdk2- and Cdc7-independent DNA damage checkpoint. Mol. Cell Biol. 26, 1997–2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lin X. H., Walter J., Scheidtmann K., Ohst K., Newport J., Walter G. (1998) Protein phosphatase 2A is required for the initiation of chromosomal DNA replication. Proc. Natl. Acad. Sci. U.S.A. 95, 14693–14698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Krasinska L., Domingo-Sananes M. R., Kapuy O., Parisis N., Harker B., Moorhead G., Rossignol M., Novák B., Fisher D. (2011) Protein phosphatase 2A controls the order and dynamics of cell-cycle transitions. Mol. Cell 44, 437–450 [DOI] [PubMed] [Google Scholar]
- 37. Schmitz M. H., Held M., Janssens V., Hutchins J. R., Hudecz O., Ivanova E., Goris J., Trinkle-Mulcahy L., Lamond A. I., Poser I., Hyman A. A., Mechtler K., Peters J.-M., Gerlich D. W. (2010) Live-cell imaging RNAi screen identifies PP2A-B55α and importin-β1 as key mitotic exit regulators in human cells. Nat. Cell Biol. 12, 886–893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shermoen A. W., O'Farrell P. H. (1991) Progression of the cell cycle through mitosis leads to abortion of nascent transcripts. Cell 67, 303–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kimelman D., Kirschner M., Scherson T. (1987) The events of the midblastula transition in Xenopus are regulated by changes in the cell cycle. Cell 48, 399–407 [DOI] [PubMed] [Google Scholar]
- 40. Takada S., Kwak S., Koppetsch B. S., Theurkauf W. E. (2007) grp (chk1) replication-checkpoint mutations and DNA damage trigger a Chk2-dependent block at the Drosophila midblastula transition. Development 134, 1737–1744 [DOI] [PubMed] [Google Scholar]
- 41. Anderson J. A., Lewellyn A. L., Maller J. L. (1997) Ionizing radiation induces apoptosis and elevates cyclin A1-Cdk2 activity before but not after the midblastula transition in Xenopus. Mol. Biol. Cell 8, 1195–1206 [DOI] [PMC free article] [PubMed] [Google Scholar]