

Background: The role of SNX26, a brain-enriched RhoGAP, in mature neurons remains unknown.
Results: SNX26 interacts with PSD-95 and regulates formation of dendritic spines.
Conclusion: SNX26 has a role in the activity-dependent structural change of dendritic spines in mature neurons.
Significance: Dynamic regulation of SNX26 plays a pivotal role in dendritic arborization of mature neurons.
Keywords: Actin, Cdc42, Dendrite, Neurons, Synapses, Synaptic Plasticity, PSD-95, RhoGAP, SNX26, Dendritic Spine
Abstract
SNX26, a brain-enriched RhoGAP, plays a key role in dendritic arborization during early neuronal development in the neocortex. In mature neurons, it is localized to dendritic spines, but little is known about its role in later stages of development. Our results show that SNX26 interacts with PSD-95 in dendritic spines of cultured hippocampal neurons, and as a GTPase-activating protein for Cdc42, it decreased the F-actin content in COS-7 cells and in dendritic spines of neurons. Overexpression of SNX26 resulted in a GTPase-activating protein activity-dependent decrease in total protrusions and spine density together with dramatic inhibition of filopodia-to-spine transformations. Such effects of SNX26 were largely rescued by a constitutively active mutant of Cdc42. Consistently, an shRNA-mediated knockdown of SNX26 significantly increased total protrusions and spine density, resulting in an increase in thin or stubby type spines at the expense of the mushroom spine type. Moreover, endogenous expression of SNX26 was shown to be bi-directionally modulated by neuronal activity. Therefore, we propose that in addition to its key role in neuronal development, SNX26 also has a role in the activity-dependent structural change of dendritic spines in mature neurons.
Introduction
Dendritic spines, highly specialized and micron-sized postsynaptic compartments extending from dendrites, are key mediators of excitatory synaptic transmission in the brain (1). During development, and in response to a variety of physiological stimuli affecting synaptic activity, marked changes in the number and shape of spines occur; such changes are thought to affect various processes such as learning and memory formation (2–4). Improper formation and/or morphological abnormalities of dendritic spines are associated with many neurological disorders, including mental retardation, schizophrenia, Alzheimer disease, autism spectrum disorders, and Down syndrome (5–8). During spinogenesis, thinly elongated highly motile protrusions (dendritic filopodia) that are rich in actin transform into dendritic spines, which, anatomically, are composed of a head connected by a neck to the dendrite (9, 10). A major component of both dendritic spines and filopodia is filamentous actin (F-actin), which can be dynamically remodeled (9, 11, 12). Dendritic spines can mature into stable mushroom-like shapes and can undergo remodeling in response to various forms of synaptic plasticity, including long term potentiation (LTP)3 and long term depression (LTD). LTP induces an increase in spinal density or size, and LTD results in shrinkage or retraction of spines in the forebrain (13, 14). In addition, it has been reported that drugs such as cytochalasins, which inhibit actin dynamics, can interfere with LTP (15).
The small GTPases of the Rho family, including Cdc42, Rac1, and RhoA, are prominent regulators of actin cytoskeleton reorganization (16). As such, these proteins control a variety of processes, including adhesion, migration, neuronal development, and morphogenesis. Cdc42 stimulates the formation of highly dynamic microspike-like structures called filopodia, whereas Rac1 stimulates the formation of membrane ruffles and lamellipodia. RhoA induction results in the formation of focal adhesions and stress fibers (16). Rho GTPases cycle in a tightly regulated manner between a GDP-bound inactive state and a GTP-bound active state (17). This cycling is regulated by the actions of three major classes of proteins as follows: guanine nucleotide exchange factors, guanine nucleotide dissociation inhibitors, and GTPase-activating proteins (GAPs). Among them, GAPs stimulate the relatively weak intrinsic GTP hydrolyzing activity of their substrate GTPases, thereby inactivating them. It has been reported that numerous guanine nucleotide exchange factors that activate Rho GTPases are involved in dendritic spine regulation; however, there are few reports on the role of GAPs in inactivating Rho GTPases (18).
Sorting nexin 26 (SNX26; also known as TC10 and Cdc42 GTPase-activating protein or neurite outgrowth multiadaptor-GAP) is a brain-enriched RhoGAP with multiple domains and was initially reported to be involved in insulin-stimulating glucose transporter 4 translocation in fat cells (19). It includes an N-terminal Phox homology (PX) domain followed by an Src homology 3 (SH3) domain and a RhoGAP domain, and it has repeated proline-rich sequences at the C terminus. Its expression is reported to be specific in mRNA levels of the brain and testis (19). It is predominant in postsynaptic density fractions and shows an age-dependent decrease in expression. SNX26 can be phosphorylated by the tyrosine kinase Fyn; furthermore, nerve growth factor (NGF)-stimulated neurite outgrowth can be suppressed by SNX26 overexpression and enhanced by a GAP-defective mutant form of SNX26 in PC12 cells (20). Recently, only depletion of SNX26 caused a decrease in dendritic outgrowth and branching with neither SNX26 overexpression nor down-regulation having an effect on axonal morphogenesis in primary cultured cortical neurons (21). It has been also reported that SNX26 regulates dendritic arborization and cortical thickness through the control of Cdc42 and cofilin activity during late cortical development in genetically modified mice (22).
Although SNX26 is expressed at high levels during early postnatal development, its expression is maintained in mature neurons; moreover, it is concentrated in the postsynaptic density region and is localized especially to dendritic spines, raising the possibility that SNX26 might also affect synaptic plasticity in mature neurons, but its roles in mature neurons remain mostly unreported.
Here, we report that SNX26 interacts with PSD-95 in dendritic spines. We also reveal that SNX26 decreases cytosolic F-actin content in COS-7 cells as well as in dendritic spines of neurons. In addition, both overexpression and knockdown of SNX26 were found to alter markedly the number and morphology of dendritic spines, especially inhibiting the conversion of filopodia-to-spines in a GAP activity-dependent manner. Neuronal activity was observed to modulate bi-directionally the expression of SNX26. Based on these results, we suggest that coordinated dynamic regulation of SNX26 may play a pivotal role in dendritic arborization of developing neurons and in the structural change of mature neurons.
EXPERIMENTAL PROCEDURES
Plasmid Construction, Antibody, and Reagent
SNX26 constructs originated from human and mouse were kindly provided by Dr. Tadashi Yamamoto (University of Tokyo, Tokyo, Japan). DNAs corresponding to the regions of human and mouse SNX26 or rat PSD-95 were amplified by polymerase chain reaction and subcloned into the indicated expression vectors enhanced GFP and HA. To visualize F-actin structures, we have performed in vitro annealing for Lifeact, a 17-amino acid peptide, known to be localized at F-actin structures without interfering with actin dynamics (23) and subcloned into mCherry vector (generously provided from Dr. Roger Y. Tsien, University of California at San Diego). The fidelity of all constructs was verified by sequencing. The following antibodies were used: GFP (Abcam, Cambridge, UK); tubulin-β (Abcam); SNX26 (Abcam); PSD-95 (SYSY, Göttingen, Germany); Cdc42 and RhoA (Santa Cruz Biotechnology); Rac1 (EMD Millipore, Billerica, MA), and anti-HA (Covance, Princeton, NJ). Anti-SNX26 mouse antibody was provided by Dr. Tadashi Yamamoto (University of Tokyo). HRP-conjugated secondary antibodies were obtained from Jackson ImmunoResearch (West Grove, PA). Alexa 488-conjugated donkey anti-goat antibody, Texas Red-conjugated goat anti-mouse antibody, and Texas Red-X phalloidin (TxRed-phalloidin) were from Molecular Probes (Eugene, OR), and all other reagents were from Sigma.
Cell Culture, Transfection, and Preparation for Quantification of F-actin Content
Experiments were performed in accordance with the guidelines set forth by the Seoul National University Council Directive for the proper care and use of laboratory animals.
COS-7 cells (Korean Cell Line Bank, Seoul, South Korea) and HEK293T cells (ATCC, Manassas, VA) were grown in Dulbecco's modified Eagle's medium (Invitrogen) supplemented with 10% fetal bovine serum (Hyclone, Logan, UT) at 37 °C in 5% CO2. Transfection was carried out using Lipofectamine 2000 reagent (Invitrogen), and cells were grown for 24 h. For preparation of the quantification of F-actin contents, cells were fixed in 4% paraformaldehyde, 4% sucrose, phosphate-buffered saline (PBS) for 15 min, washed two times for 5 min in PBS, and permeabilized for 5 min in 0.25% Triton X-100/PBS. They were blocked for 30 min in 10% bovine serum albumin (BSA)/PBS at 37 °C and incubated with TxRed-phalloidin, 3% BSA/PBS for 30 min at 37 °C. For neuron cultures, primary rat hippocampal neurons were prepared as described (24). Briefly, hippocampi were dissected from embryonic day 18 Sprague-Dawley fetal rats, dissociated with papain, and triturated with a polished half-pore Pasteur pipette. Cells (2.5 × 105) in minimum Eagle's medium supplemented with 0.6% glucose, 1 mm pyruvate, 2 mm l-glutamine, 10% fetal bovine serum and antibiotics were plated on poly-d-lysine-coated glass coverslips in a 60-mm Petri dish. Four hours after plating, the medium was replaced with Neurobasal (Invitrogen) supplemented with 2% B-27 and 0.5 mm l-glutamine. The neurons were transfected at indicated days in vitro (DIV) using the calcium phosphate method (24). Briefly, a total of 6 μg of cDNA and 9.3 μl of 2 m CaCl2 were mixed in distilled water to a total volume of 75 μl, and the same volume of 2× borate-buffered saline was added. The neuron culture medium was completely replaced by transfection medium (minimum Eagle's medium, 1 mm pyruvate, 0.6% glucose, 19 mm glutamine, and 19 mm HEPES, pH 7.65), and the cDNA mixture was added to the neurons, which were then incubated in a 5% CO2 incubator for 60 min. They were washed with transfection medium, pH 7.35, and then returned to the original culture medium.
Microscopy, Image Analysis, and Quantification of F-actin Content
Images were acquired with a Zeiss Axiovert 200 M inverted microscope (Carl Zeiss, Oberkochen, Germany) equipped with a ×40 oil-immersion objective lens, N.A. 1.0, using an ORCA-R2 CCD camera (Hamamatsu Photonics, Hamamatsu, Japan) driven by MetaMorph imaging software (Molecular Devices, Sunnyvale, CA) with a GFP- or a monomeric red fluorescent protein-optimized filter set (Omega Optical, Brattleboro, VT). For quantification of F-actin content, we have manually selected transfected cells showing moderate levels of GFP expression (300–600 arbitrary fluorescence units) and excluded the cells showing either low or high levels of GFP expression. Analysis and quantification of data were performed with MetaMorph software, and statistical analyses were carried out with PASW Statistics 18 (IBM, Armonk, NY). Data are presented as means ± S.E. For multiple conditions, we compared means by analysis of variance (ANOVA) followed by Tukey's honestly significant difference (HSD) post hoc test to reveal statistically different groups.
Image Analysis and Quantification for Dendritic Spines and Filopodia
Digital images for morphometric analysis of dendritic spines were obtained with a Nikon A1 confocal microscope (Nikon Instruments, Tokyo, Japan) equipped with a CFI Plan Apochromat ×60 oil-immersion objective lens, N.A. 1.4 at a resolution 512 × 512 using digital zooming. For analysis, well branched pyramidal or multipolar neurons were randomly selected. One to three secondary dendrites in each neuron were chosen. To determine the number of dendritic spines and filopodia, spines were defined as dendritic protrusions of <5 μm in length with heads and filopodia as dendritic protrusions of <10 μm in length without apparent heads. Spine head was defined as the tip structure that should be at least two times thicker than the spine neck. Spines can be morphologically divided into three types as follows: thin, mushroom-shaped, and stubby spines (25). To distinguish spine types, the thin type was defined as spines having a long thin neck and a small bulbous head; the mushroomed-shaped was defined as having a neck and a large head, and the stubby type was defined as ones devoid of a neck. The density and types of spines from a single dendrite were grouped and averaged. Analysis and quantification of data were performed with MetaMorph software, and statistical analyses were carried out with PASW Statistics 18. Data are presented as means ± S.E. For multiple conditions, we compared means by ANOVA followed by Tukey's HSD post hoc test. Statistical differences between two conditions were determined using Student's t test.
Co-immunoprecipitation and Immunoblotting
To detect SNX26 binding to PSD-95 in vivo, HEK293T cells were transfected using Lipofectamine 2000 (Invitrogen). The cells were washed twice with cold PBS and extracted at 4 °C for 1 h in lysis buffer (10 mm Tris-HCl, pH 7.4, 5 mm EDTA, 150 mm NaCl, 10% glycerol, 1% Triton X-100, 1 mm sodium orthovanadate, 1 mm PMSF, 10 mm leupeptin, 1.5 mm pepstatin, and 1 mm aprotinin). They were then clarified by centrifugation at 15,000 × g for 15 min, and protein concentrations were determined with a Bradford protein assay reagent kit (Bio-Rad). Samples containing 1 mg of total protein were immunoprecipitated for 4 h with anti-GFP antibody, followed by an additional 2 h of incubation at 4 °C with protein A-Sepharose beads (GE Healthcare). The beads were extensively washed with lysis buffer and subjected to SDS-PAGE and transferred to a PVDF membrane (Bio-Rad). The membrane was blocked with 5% skim milk/TBS-T for 1 h, washed, and probed with primary antibody for 2 h at room temperature. After extensive washing in TBS-T, the membrane was incubated with HRP-conjugated secondary antibody (Jackson ImmunoResearch). Proteins were visualized with ECL reagent (GE Healthcare).
RNA Interference
The small hairpin RNA (shRNA) for RNA interference against both rat and mouse SNX26, mRNA was designed on the basis of the mouse SNX26 cDNA sequence (GenBankTM accession number NM_178252.2), targeting to the region of nucleotide 1149–1169. Complementary oligonucleotides were synthesized separately, with the addition of a BamHI site at the 5′ end and an EcoRI site at the 3′ end. The forward targeting sequence of shRNA for SNX26 (shRNA-SNX26) was 5′-GAGGCTTCGGCATGAGTTTGA-3′. The forward targeting sequence of scrambled shRNA for SNX26 (shRNA-scr) was 5′-GACTGTGGTCGATGGCAGTTA-3′. The annealed cDNA fragments were cloned into the BamHI-EcoRI sites of the vector pSIREN-DNR-DsRed-Express (BD Biosciences). The efficiency of shRNAs was tested on protein levels of GFP-tagged mouse SNX26 (GFP-mSNX26) in HEK293T cells as described previously (21). It has been reported that this sequence is also effective in cultured rat cortical neurons (21). For evading RNA interference, silence mutations within the shRNA-SNX26 targeting sequence (T1155G and C1156A) in GFP-mSNX26 were generated using the QuikChange® site-directed mutagenesis kit (Stratagene, Austin, TX). The fidelity of all constructs was verified by sequencing.
RESULTS
SNX26 Expression Is Developmentally Regulated in Postnatal Brain and Neuron Culture
SNX26 mRNA was expressed predominantly in the brain and testis and concentrated in the cortex and hippocampus (19, 20). The SNX26 protein levels in the brain of postnatal mice are reported to be negatively regulated during postnatal development (20). In this study, we investigated the SNX26 protein distribution in different tissues and confirmed the developmental expression pattern of SNX26 in the postnatal mouse brain. Consistent with the results in previous reports (19, 20), SNX26 protein is highly expressed in brain, particularly in the cortex and hippocampus (Fig. 1A); moreover, expression of SNX26 is negatively regulated during postnatal development (Fig. 1B).
FIGURE 1.

Expression patterns of SNX26 protein in various tissues and during development of postnatal brain and neuron culture. A, tissue distribution of SNX26 protein from 2-week-old mice. Arrow points to the size of SNX26, ∼150 kDa. B, Western blot analysis of postnatal mouse brains. Postnatal brains were lysed, evenly loaded (100 μg) in SDS-polyacrylamide gels, and immunoblotted with a specific anti-SNX26 mouse antibody. SNX26 expression gradually decreases during maturation. C, developmental changes of SNX26 protein expression in cultured rat hippocampal neurons. Cultured primary hippocampal neurons were prepared at the indicated DIV, lysed, and evenly loaded (75 μg) in SDS-polyacrylamide gels. Western blot analysis was performed using a specific anti-SNX26 antibody. SNX26 expression rises during DIV 2–7 and then decreased, and after DIV 16 the levels persisted. D, developmental changes of SNX26, Cdc42, Rac1, and RhoA protein expression in cultured rat cortical neurons. Cultured primary cortical neurons were prepared at the indicated DIV, lysed, and evenly loaded (75 μg) in SDS-polyacrylamide gels. Western blot analysis was performed using each specific antibody. The developmental expression pattern of Cdc42, but not of Rac1 or RhoA, are similar to that of SNX26. βTub, β-tubulin.
We also investigated the expression of SNX26 protein in primary cultured hippocampal neurons in vitro by Western blotting. The SNX26 expression levels gradually increased from DIV 2 to 7 and then decreased, and after DIV 16, the levels approached a steady state (Fig. 1, C and D). When we compared protein expression patterns of representative members of the Rho protein family (Cdc42, Rac1, and RhoA) with that of SNX26 in cultured cortical neurons (Fig. 1D), the developmental expression pattern of Cdc42, but not of Rac1 or RhoA, was similar to that of SNX26. The results suggest that SNX26 may function in relation to Cdc42, which is consistent with recent findings that SNX26 acts as a GAP toward Cdc42 in the control of neurite outgrowth (20, 21).
SNX26 Reduces Cytosolic F-actin Content via its GAP Activity
Because SNX26 exhibits GAP activity toward Cdc42, we tested whether SNX26 expression modulates the actin cytoskeleton. COS-7 cells were transfected with GFP, GFP-SNX26 full-length (FL), or its various deletion mutants, and then the cells were stained with TxRed-phalloidin for F-actin detection. In cells expressing GFP-SNX26 or its RhoGAP domain, F-actin content decreased, by ∼60%, from that in GFP-transfected control cells and in surrounding nontransfected cells (Fig. 2A). Consistently, typical F-actin structures such as stress fiber and the cortical actin had largely disappeared, and some amorphous actin aggregates were observed in central areas. Cells expressing other deletion mutants lacking RhoGAP, such as ΔRhoGAP, PRD, PX, or SH3, show no apparent disappearance of F-actin compared with control (Fig. 2).
FIGURE 2.

SNX26 decreases the F-actin content via its RhoGAP domain. A, representative images for SNX26-induced reduction in F-actin content. COS-7 cells expressing GFP or the GFP-tagged indicated deletion mutants of SNX26 were fixed and stained with TxRed-phalloidin to visualize F-actin. Scale bar, 10 μm. B, arbitrary fluorescence unit (A.F.U.) of TxRed intensity in each group was measured. Over 40 cells were analyzed for each group. Data are presented as means ± S.E. of three independent experiments. Tf, transfected cells; unTf, untransfected cells. ***, p < 0.001 (Student's t test). C, in each group, TxRed intensity of transfected cells was normalized to that of untransfected cells. Either SNX26 full-length or only the RhoGAP domain containing mutants decrease F-actin content. ***, p < 0.001 (ANOVA and Tukey's HSD post hoc test). CTL, control.
A GAP-defective mutant of SNX26 FL (Arg-350 to Ile-350; R350I) or its RhoGAP domain (RI-RhoGAP) (20) failed to have an effect on the alteration of F-actin content in COS-7 cells (Fig. 3, A and B), indicating that the GAP activity of SNX26 is responsible for its effect on F-actin content. Not only in COS-7 cells but also in the dendritic spines of cultured hippocampal neurons, the SNX26 FL or RhoGAP domain induced a marked reduction of F-actin content, which was fully cancelled by RI-RhoGAP (Fig. 3, C–F). These results suggest a direct involvement of the GAP activity of SNX26 in actin cytoskeleton remodeling.
FIGURE 3.

GAP activity of SNX26 is required to decrease the F-actin content in COS-7 cells as well as in dendritic spines of neurons. A, COS-7 cells expressing GFP, GFP-SNX26, GFP-RhoGAP, GFP-RI-RhoGAP, or GFP-R350I were fixed and stained with TxRed-phalloidin to visualize F-actin. Scale bar, 10 μm. B, normalized intensity ratio (transfected/untransfected (Tf/unTf)) of cells expressing SNX26 or RhoGAP was significantly lower than that of cells expressing GFP (CTL, control), GFP-RI-RhoGAP (GAP activity-deficient mutant of RhoGAP domain), or R350I (GAP activity-deficient mutant of SNX26 FL). Data are presented as means ± S.E. of three independent experiments in which >40 transfected cells were examined for each group. ***, p < 0.001 (ANOVA and Tukey's HSD post hoc test). C, representative images of dendritic spines of neurons. Cultured hippocampal neurons were transfected at DIV 15 with cyan fluorescent protein and with GFP or GFP-SNX26 FL, fixed at DIV 16, and then stained with TxRed-phalloidin. Scale bar, 2 μm. D, TxRed intensities in dendritic spines and neighboring dendritic shafts were compared. Over 30 transfected individual spines and dendrites were examined for each group. Data are presented as means ± S.E. of three independent experiments. The normalized intensity ratio (spine/shaft) of cells expressing SNX26 FL was much lower than that of cells expressing control (CTL). ***, p < 0.001 (ANOVA and Tukey's HSD post hoc test). E, representative images of dendritic spines of neurons. Cultured hippocampal neurons were transfected at DIV 15 with GFP, GFP-RhoGAP, or GFP-RI-RhoGAP, fixed at DIV 16, and then stained with TxRed-phalloidin. Scale bar, 2 μm. F, TxRed intensities in dendritic spines and neighboring dendritic shafts were compared. Over 30 transfected individual spines and dendrites were examined for each group. Data are presented as means ± S.E. of three independent experiments. The normalized intensity ratio (spine/shaft) of cells expressing RhoGAP was much lower than that of cells expressing control or RI-RhoGAP. ***, p < 0.001 (ANOVA and Tukey's HSD post hoc test).
SNX26 Interacts with PSD-95 via Its PX and RhoGAP Domains and Co-localizes to Dendritic Spines in Cultured Hippocampal Neurons
PSD-95, a PDZ domain scaffold protein that is located in dendritic spines of excitatory neurons, has prominent roles in both synaptic development and plasticity (26, 27). It is crucial for tethering a variety of other synaptic proteins, such as N-methyl-d-aspartate (NMDA) receptors, into the postsynaptic density region of spines (28). Because SNX26 is reported to be abundant in the postsynaptic density fraction and is localized to dendritic spines (20), we examined whether SNX26 interacts with PSD-95. HEK293T cells were co-transfected either with HA-tagged FL or deletion mutants of SNX26 or with GFP-tagged FL or deletion mutants of PSD-95 and were subsequently immunoprecipitated with specific antibodies. Because PSD-95 has an SH3 domain in its middle, an interaction between the SH3 domain of PSD-95 and the proline-rich domain (PRD) of SNX26 was expected (Fig. 4, A and B); however, the PX and RhoGAP domains of SNX26 bound to PSD-95 (Fig. 4C). We further investigated those interactions and determined that SNX26 FL and PX domain interacts either with PSD-95 FL or with its ΔSH3 mutant, whereas the RhoGAP domain only interacted with PSD-95 FL (Fig. 4, D and E). There was no interaction between SNX26-PRD and PSD-95 (Fig. 4E). Immunoprecipitation assays with brain lysates showed that SNX26 bound to PSD-95 in rat brains (Fig. 4F). When overexpressed in cultured hippocampal neurons, SNX26 co-localized with PSD-95 at dendritic spines (Fig. 4G); in addition, endogenous SNX26 partially co-localized with PSD-95 (Fig. 4H). Thus, SNX26 in dendritic spines interacts with PSD-95 and may have a role in the regulation of dendritic spines.
FIGURE 4.

SNX26 interacts with PSD-95 through PX and RhoGAP domains. A, schematic representation of SNX26 full-length and various deletion mutants used in the experiments. Abbreviation used is as follows: RhoGAP, Rho GTPase-activating protein. B, schematic representation of full-length PSD-95 and deletion mutants used in the experiments. C–E, HEK293T cells were co-transfected with GFP, GFP-SNX26, or GFP-tagged deletion mutants and HA-PSD-95 (C) or co-transfected with GFP, PSD-95-GFP, or GFP-tagged deletion mutants and HA-SNX26 (D). Twenty four hours after transfection, the cells were lysed, immunoprecipitated with anti-GFP antibody, and immunoblotted with anti-HA antibody. IP, immunoprecipitation; IB, immunoblot. Interaction of SNX26 with PSD-95 was further investigated to pin down binding domains. E, HEK293T cells were co-transfected with each construct indicated, lysed, and immunoprecipitated with anti-GFP antibody followed by immunoblotting with anti-HA antibody. F, SNX26 endogenously interacts with PSD-95 in rat brains. Brain lysates were immunoprecipitated with an anti-PSD-95 antibody or normal rabbit serum (NRS), and immunoblotted with an anti-SNX26 antibody. G, neurons were co-transfected at DIV 13 with PSD-95-GFP and mCherry-SNX26 and fixed at DIV 14. PSD-95-GFP and mCherry-SNX26 are co-localized at spines. Scale bar, 15 μm. H, endogenous SNX26 partially co-localizes with PSD-95 at dendritic spines. Cultured hippocampal neurons at DIV 14 were fixed and immunostained with either a specific SNX26 or a specific PSD-95 antibody, followed by either Alexa 488-conjugated donkey anti-goat (SNX26) or Texas Red-conjugated anti-mouse antibody (PSD-95). Arrowheads indicate dendritic spines where two proteins were co-localized. Scale bar, 7 μm.
SNX26 Affects Dendritic Spine Formation
The number of dendritic spines increased during 10–15 DIV and reached a plateau at later stages. To investigate the effects of SNX26 on dendritic spine formation, cultured hippocampal neurons at DIV 13 were co-transfected with GFP or GFP-SNX26 and with mCherry empty vector (23) and then fixed at DIV 14. SNX26 overexpression induced an ∼38.2% decrease in total dendritic protrusions. We defined spines as dendritic protrusions of <5 μm in length with heads, and filopodia as dendritic protrusions of <10 μm in length without apparent heads (see under “Experimental Procedures”). When we classified dendritic protrusions into those two groups, despite the overall decrease in protrusion density, we found that spine density was markedly decreased, whereas filopodia density was dramatically increased compared with that in the control group (Fig. 5). Because dendritic filopodia are considered spine precursors, these results indicate that overexpression of SNX26 inhibits the transformation from dendritic filopodia to dendritic spines.
FIGURE 5.

Overexpression of SNX26 alters dendritic spine formation. A, cultured hippocampal neurons were co-transfected at DIV 13 with GFP-SNX26 and mCherry and fixed at DIV 14. Asterisks point to some of representative filopodia. Scale bar, 15 μm. B–E, quantification of the number of total protrusions, spines, and filopodia with or without expression of SNX26. Spines were defined as dendritic protrusions of <5 μm in length with heads and filopodia as dendritic protrusions of <10 μm in length without heads. Data were collected from 24 to 36 dendrites of 16 to 24 neurons for each group in three independent experiments. ***, p < 0.001 (Student's t test); CTL, control.
To identify which domain(s) of SNX26 may be responsible for the observed effects, neurons were co-transfected with mCherry and various deletion mutants of SNX26. Among the mutants, only the RhoGAP domain showed effects similar to those of SNX26 FL (Fig. 6), indicating that the RhoGAP domain is responsible for the effects of SNX26 on dendritic spine formation. This was confirmed by results from a GAP-defective mutant of SNX26 (R350I), which showed a comparable level of dendritic spine development to that in the control group (Fig. 7).
FIGURE 6.

Effects of SNX26 domains on dendritic spine formation. A, cultured hippocampal neurons were co-transfected at DIV 12 with the indicated GFP-tagged SNX26 domains and mCherry and fixed at DIV 14. Scale bar, 15 μm. B–E, quantification of the number of total protrusions, spines, and filopodia with expression of each domain of SNX26. Spines were defined as dendritic protrusions of <5 μm in length with heads and filopodia as dendritic protrusions of <10 μm in length without heads. Data were collected from 22 to 36 dendrites of 14–24 neurons for each group in three independent experiments. ***, p < 0.001; *, p < 0.01 (ANOVA and Tukey's HSD post hoc test). CTL, control.
FIGURE 7.
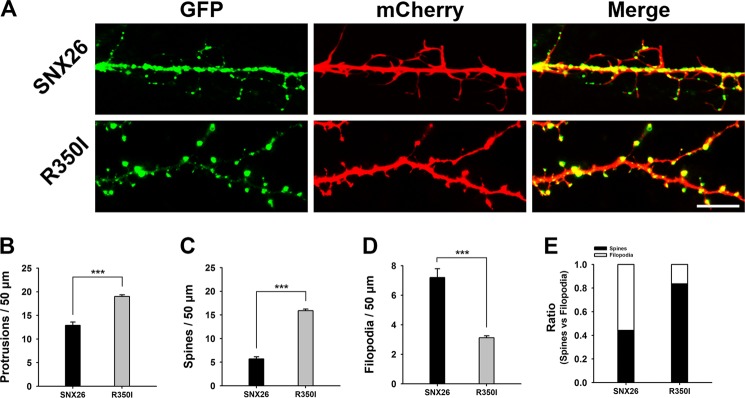
GAP activity of SNX26 is attributed to its effect on dendritic spine formation. A, cultured hippocampal neurons were co-transfected at DIV 13 with GFP-SNX26 or GFP-R350I and mCherry and fixed at DIV 14. Scale bar, 15 μm. B–E, quantification of the number of total protrusions, spines, and filopodia with expression of SNX26 or R350I. Spines were defined as dendritic protrusions of <5 μm in length with heads and filopodia as dendritic protrusions of <10 μm in length without heads. Data were collected from 20 to 24 dendrites of 15–16 neurons for each group in three independent experiments. ***, p < 0.001 (Student's t test).
To determine whether the spine-inhibiting effect of SNX26 is mediated by SNX26 acting as a GAP for Cdc42, neurons were co-transfected with GFP or the GFP-tagged constitutive active form of Cdc42 (Cdc42CA) and mCherry-SNX26. Cdc42CA largely rescued the SNX26-induced defects in spine formation (Fig. 8), suggesting that SNX26 can act as a GAP for Cdc42 and can regulate spine formation in neurons.
FIGURE 8.

Inhibitory effect of SNX26 on spine formation can be largely abrogated by constitutively active Cdc42 co-expression. A, cultured hippocampal neurons were co-transfected at DIV 13 with GFP or GFP-Cdc42CA and mCherry-SNX26 and fixed at DIV 14. Scale bar, 20 μm. B–E, quantification of the number of total protrusions, spines, and filopodia with expression of SNX26 or SNX26 with Cdc42CA. Spines were defined as dendritic protrusions of <5 μm in length with heads and filopodia as dendritic protrusions of <10 μm in length without heads. Data were collected from 18 to 26 dendrites of 10–18 neurons for each group in three independent experiments. ***, p < 0.001; *, p < 0.01 (Student's t test).
SNX26 Depletion Alters the Number and Morphology of Dendritic Spines in Neurons
To assess further the physiological effects of endogenous SNX26 on spine formation, we generated small hairpin RNA (shRNA) for mouse SNX26. Suppression of SNX26 expression by shRNA-SNX26, but not by scrambled shRNA-SNX26 (shRNA-scr), was confirmed by performing immunofluorescence and Western blot analyses of transfected HEK293T cells (Fig. 9A) and was further confirmed by immunofluorescence staining of SNX26 in hippocampal neurons (Fig. 9B). We also generated the GFP fusion construct resistant to shRNA-SNX26 (GFP-mSNX26-res) and confirmed its resistancy to shRNA-SNX26 (Fig. 9A).
FIGURE 9.

Knockdown of endogenous SNX26 by shRNA causes alterations in dendritic spine formation, which can be partially rescued by resistant mouse SNX26. A, HEK293T cells were co-transfected with GFP-mSNX26 or GFP-mSNX26-res and shRNA-scr or shRNA-SNX26, respectively. The knockdown efficiency of shRNA-SNX26 was confirmed by immunostaining or Western blotting with anti-GFP antibody. Although the expression of GFP-mSNX26 was successfully reduced by shRNA-SNX26, that of GFP-mSNX26-res was not affected by shRNA-SNX26. The term, res, indicates silent mutation that is resistant to shRNA-SNX26. Scale bar, 10 μm; βTub, β-tubulin. B, endogenous expression of SNX26 in cultured hippocampal neurons was reduced by shRNA-SNX26. Hippocampal neurons were transfected with shRNA-scr or shRNA-SNX26 at DIV 8, fixed at DIV 13, and immunostained with anti-SNX26 antibody, followed by Alexa 488-conjugated donkey anti-goat antibody. Alexa 488 channel shows endogenous levels of SNX26. dsRed channel shows the neurons transfected with shRNA-scr or shRNA-SNX26. Arrowheads indicated the soma of neurons transfected with shRNAs. The neuron expressing shRNA-SNX26 showed the reduced levels of SNX26 expression at soma compared with nontransfected neurons (arrows). Scale bar, 10 μm. C–H, cultured hippocampal neurons were transfected at DIV 8 with shRNA-scr or shRNA-SNX26 with GFP or shRNA-SNX26 with GFP-mSNX26-res and fixed at DIV 13. Quantification of the number of total protrusions, spines, and filopodia with expression of each group is shown. Spines were defined as dendritic protrusions of <5 μm in length with heads and filopodia as dendritic protrusions of <10 μm in length without heads. Data were collected from 22 to 42 dendrites of 12–24 neurons for each group in three independent experiments. *, p < 0.01 (ANOVA and Tukey's HDS post hoc test); ***, p < 0.001 (Student's t test). G, representative images of spine morphologies from the dendrite in the neuron expressing shRNA-scr or shRNA-SNX26 with GFP or shRNA-SNX26 with GFP-mSNX26-res, respectively. Scale bar, 15 μm. G, pie graphs illustrating the changes in the portion of different morphological types of spine protrusions in the neurons expressing shRNA-scr or shRNA-SNX26 with GFP or shRNA-SNX26 with GFP-mSNX26-res.
In cultured hippocampal neurons, spines emerged during DIV 9 and 10, increased in abundance during DIV 11–15, and reached a plateau at later stages (data not shown). To investigate the effect of shRNA-SNX26 on dendritic spine formation, neurons were transfected at DIV 8 with shRNA-scr or shRNA-SNX26 and GFP or with shRNA-SNX26 and GFP-mSNX26-res and were then fixed at DIV 13. When endogenous SNX26 was knocked down, the densities of protrusions and spines, but not that of filopodia, were significantly increased (Fig. 9,C–F). These effects were successfully rescued by co-expression of shRNA-SNX26 and GFP-mSNX26-res (Fig. 9, C–F). Although with co-expression the spine density was overly decreased, and filopodia density was increased, we believe that these effects were from overexpression of GFP-mSNX26-res, consistent with SNX26 overexpression phenotype observed in Fig. 5.
The predominant effect of SNX26 depletion in neurons was on dendritic spine morphology. The absence of SNX26 resulted in a decrease in the proportion of mushroom-shaped spines and a concomitant increase in the proportions of thin and stubby spines (Fig. 9, G and H). Again, these were largely rescued by co-expression of shRNA-SNX26 and GFP-mSNX26-res (Fig. 9, G and H).
Consistently, Cdc42CA but not RacCA expression resulted in an increase in the number of both thin and stubby spines and a reduction in the number of mushroom-shaped spines (Fig. 10F). As expected, a Cdc42 dominant negative form (Cdc42DN) induced a similar dendritic morphological phenotype as SNX26 expression (Figs. 5 and 10). Taken together, these results suggest that SNX26, by acting as a GAP toward Cdc42, regulates dendritic spine formation as well as spine morphogenesis.
FIGURE 10.

Cdc42CA overexpression resulted in an increase in the number of both thin and stubby spines and a reduction in the number of mushroom-shaped spines, although Cdc42DN overexpression is similar to SNX26 overexpression. A, representative images of spine morphologies from the dendrite in the neuron expressing GFP, GFP-tagged constitutively active Cdc42 (Cdc42CA), or Rac1 (RacCA), or dominant negative Cdc42 (Cdc42DN), respectively. Neurons at DIV 13 were transfected with indicated GFP or GFP-tagged constitutively active constructs and incubated 6 h after transfection, and neurons were fixed and imaged. Neurons showing mild GFP signals ranging from 150 and 450 arbitrary fluorescence units (A.F.U.) were imaged and analyzed. For GFP-Cdc42DN, neurons were transfected at DIV 12, fixed at DIV 14, and imaged. Scale bar, 7 μm. B–E, quantification of the number of total protrusions, spines, and filopodia with expression of the indicated constructs. Spines were defined as dendritic protrusions of <5 μm in length with heads and filopodia as dendritic protrusions of <10 μm in length without heads. Data were collected from 18 to 26 dendrites of 8–12 neurons for each group in the three independent experiments. ***, p < 0.001; *, p < 0.05, ANOVA and Tukey's HSD post hoc test. F, pie graphs illustrating the changes in the portion of different morphological types of spine protrusions in the neurons expressing GFP, GFP-Cdc42CA, GFP-RacCA, or GFP-Cdc42CA.
SNX26 Expression Is Activity-dependently Altered in Mature Neurons
A variety of synaptic activities can induce changes in the shape and number of dendritic spines in mature neurons (13, 14, 29). Because overexpression or depletion of SNX26 can have profound effects on dendritic spine development, we speculated on whether neuronal activity can affect SNX26 expression. Picrotoxin-mediated inhibition of GABAA inhibitory tone was reported to increase spontaneous synaptic activity in cultured hippocampal neurons, whereas tetrodotoxin-mediated inhibition of Na+ channels induced the opposite effect (30). We found that SNX26 levels significantly increased or decreased in response to picrotoxin or tetrodotoxin treatment, respectively (picrotoxin, 0.39 ± 0.05; tetrodotoxin, 1.19 ± 0.03; Fig. 11, A and B).
FIGURE 11.

Neuronal activity and inactivity bi-directionally modulate the endogenous expression levels of SNX26 in mature neurons. A and B, cultured hippocampal neurons at DIV 18 were treated with either 100 μm picrotoxin or 1 μm tetrodotoxin for 24 h and lysed with lysis buffer. Each sample was evenly loaded (100 μg) in SDS-polyacrylamide gels, and Western blot analysis was performed with anti-SNX26 antibody. The intensity ratio of SNX26/β-tubulin (βTub) indicates that picrotoxin and tetrodotoxin treatments induce an ∼60% decrease and an ∼20% increase in the endogenous levels of SNX26, respectively. **, p < 0.01 (ANOVA and Tukey's HSD post hoc test). CTL, control. C, schematic diagram shows the protocol for induction of chem-LTP or chem-LTD by glycine or NMDA treatment, respectively. D and E, induction of synaptic plasticity alters SNX26 levels in cultured hippocampal neurons. After induction of chem-LTP and chem-LTD, neurons at DIV 19 were lysed and immunoblotted with a specific SNX26 antibody. The intensity ratio of SNX26/β-tubulin indicates that LTP and LTD induce an ∼60% decrease and an ∼25% increase in the endogenous levels of SNX26, respectively. **, p < 0.01 (ANOVA and Tukey's HSD post hoc test).
Consistently, chemically induced synaptic plasticity with NMDA and/or glycine (named chem-LTD and chem-LTP hereafter) (20) treatment also resulted in significant alterations in SNX26 expression levels. Induction of chem-LTP significantly reduced SNX26 expression, whereas chem-LTD induction increased it (chem-LTP, 0.44 ± 0.05; chem-LTD, 1.24 ± 0.11, Fig. 11, D and E). These results suggest that SNX26 expression levels are bi-directionally regulated, depending on neuronal activity.
DISCUSSION
Recent studies reveal that SNX26 functions as a Cdc42-specific GAP that can regulate dendrite growth of cortical neurons and neocortical dendritic complexity during early neuronal development (21, 22). Those authors reported increases in total dendritic length and branch numbers following SNX26 overexpression in cultured rat cortical neurons. In contrast, knockdown of endogenous SNX26 inhibited the increases in total dendritic length and branch numbers. Those results were expected because the effects of SNX26 were reversed by co-expressing an shRNA-resistant SNX26 in cortical neurons during early developmental stages, both in vitro and in vivo. In support of those results, Rosario et al. (22) have reported that SNX26 depletion results in oversimplification of cortical dendritic complexity, hyperactivation of Cdc42, and cofilin phosphorylation. They also reported a significant decrease in neocortical thickness in various brain regions, including the parietal, somatosensory, and auditory cortices, a decrease that is associated with developmental defects in SNX26-deficient mice. Taken together, these results indicate that SNX26 is involved in the regulation of dendritic branching and neuronal complexity in the developing brain, despite its expression in mature neurons and its localization in dendritic spines; however, its roles in the later stages of neuronal development have not been described (21).
Dendritic spines can undergo dynamic changes in size, shape, and number depending on neuronal activity, and such changes are closely associated with various neurological diseases, such as mental retardation, schizophrenia, Alzheimer disease, Down syndrome, and autism spectrum disorder. Therefore, identifying factors that regulate dendritic spine formation and morphology and that result in spine dysfunction is essential to describing in more detail the physiology and pathophysiology of the nervous system.
A previous study reported that the suppression of NGF-induced neurite outgrowth by SNX26, and its GAP-defective mutant R350I, promotes neurite outgrowth in PC12 cells (20). In contrast, others reported that SNX26 has significant GAP activity in vitro but no detectable GAP activity in vivo (19). Despite the inconsistency in the GAP activity of SNX26 in vivo, our results show that overexpression of SNX26 causes a significant reduction in cytosolic F-actin content in COS-7 cells and neuron spines (Fig. 3). Moreover, RI-RhoGAP, a defective GAP mutant of SNX26-RhoGAP, failed to induce these changes, indicating that modulation of the actin cytoskeleton by SNX26 depends on its GAP activity.
Neurons in early developmental stages have abundant filopodia, the precursors of dendritic spines, and as spine maturation progresses, neurons undergo transformation of filopodia-to-spines. Our results show that overexpression of SNX26 inhibits this transformation. A recent study reported that cofilin, an actin-severing protein, is a downstream effector involved in actin regulation by SNX26 (22). Cofilin breaks and dismantles actin filaments, an action that stimulates actin dynamics by increasing the pool of actin subunits available for recycling. That study also suggested that activation of Cdc42 by SNX26 depletion may induce filopodia formation and concomitantly reduce actin dynamics through cofilin inhibition. Our results show that SNX26 expression remains low during the later stages of development, and SNX26 overexpression during those stages inhibited transformation of filopodia-to-spines. Therefore, we find it probable that SNX26 plays a role in the initiation of dynamic filopodia during early developmental stages, but the conversion of filopodia into stable dendritic spines during later stages requires inhibition of SNX26.
To date, the only reported regulator of the GAP activity of SNX26 is Fyn (20), which phosphorylates SNX26 on the 406 tyrosine residue and inhibits its GAP activity. Moreover, Fyn has key roles in synapse development and synaptic plasticity (31, 32) and has been associated with various higher brain functions such as LTP, spatial memory (33), and contextual fear memory (34, 35). Fyn-knock-out mice exhibit decreased spine density and aberrant spine morphology over time (36, 37). Moreover, Fyn phosphorylated protein-tyrosine phosphatase receptor T, a receptor-type protein-tyrosine phosphatase, which increased the hemophilic interactions of protein-tyrosine phosphatase receptor T and inhibited the interaction between protein-tyrosine phosphatase receptor T and neuroligin, leading to negative regulation of spine and synapse formation (38). Taken together, these results indicate that SNX26 may interact with PSD-95 and Fyn, thereby contributing to elaborate spine formation of developing neurons as well as to precise spine regulation in response to synaptic activity of mature neurons by controlling the actin cytoskeleton.
Knockdown of SNX26 by shRNA increases the number of spines, but with a reduction in the proportion of mushroom-shaped spines and an increase in the proportion of thin and stubby spines (Fig. 9H). Stubby and thin spines are considered premature spines that can mature into mushroom-shaped spines (39); thus, our results indicate that SNX26 knockdown can increase premature spine density via hyperactivation of Cdc42. Similar results were obtained with alteration of Cdc42CA expression (Fig. 10). Thus, SNX26 depletion results in sustained activation of Cdc42 leading to an increase in spine number and a change in spine morphology.
Our investigations into the effects of SNX26 domains on spine formation indicate a relationship between SNX26 and spine formation based on molecular interactions. For example, the PX domain promotes spine formation without an alteration in the number of filopodia (Fig. 6). In contrast, the SNX26 PRD domain decreases the number of spines. Our results indicate that PX seems to interact mainly with PSD-95; therefore, an interaction between SNX26 and PSD-95 may be endogenously interrupted by the overexpression of the PX domain, and consequently, the proper function of endogenous SNX26 is perturbed, finally leading to an increase in spine number. The PRD domain has multiple PXXP motifs, which interact with the SH3 domain-containing proteins, and is responsible for SNX26 FL aggregation (Figs. 2 and 4H), leading to a decrease in spine number by perturbing the actions of signaling proteins in spines.
Intriguingly, SNX26 shares significant redundancy with RICS (PX-RICS/ARHGAP32/Grit/p200RhoGAP/p250RhoGAP/GC-GAP), including redundancy in domain structures, binding partners, and function. They are both mainly expressed in the same brain regions and concentrated in the postsynaptic density region. Structurally, RICS is similar to SNX26 and has an N-terminal PX domain, an SH3 domain followed by a RhoGAP domain, and has multiple proline-rich sequences in the C terminus. The RhoGAP domain of RICS is very similar to that of SNX26, but there are inconsistent reports on the GAP activity of RICS toward small GTPases (40–43). One recent study showed that RICS exhibits GAP activity toward RhoA and is involved in NMDA receptor-mediated RhoA activation, leading to morphological plasticity of the spines (41). Both SNX26 and RICS are reported to bind the TrkA receptor (43, 44) and PSD-95 (40), findings that are supported by our current results. Furthermore, SNX26 and RICS are known to be regulated by tyrosine phosphorylation (20, 45). Nevertheless, knock-outs of RICS do not have any phenotypes at all (46), whereas knock-outs of SNX26 produce marked alterations in dendritic arborization (22). However, a recent study showed that a knockdown of RICS reduced the dendritic complexity of layer 2/3 neurons (47), a phenotype that is similar to that of SNX26 knock-out mice (22). Taken together, these results suggest that SNX26 and RICS may have different downstream effectors and upstream regulators but that suggestion requires further study.
Neuronal activity has profound effects on the structural and functional plasticity of the nervous system (2, 3, 13, 14). In this study, we observed that sub-chronic treatment of picrotoxin resulted in a significant increase in SNX26 expression, whereas that of tetrodotoxin induced the opposite effect (Fig. 11, A and B). Considering the marked effects of SNX26 on both neuronal morphology and dendritic spinogenesis, our results suggest that the expression level of SNX26 may be an important controller, through which neuronal activity can regulate neuronal structural plasticity. This possibility is supported by our observation that induction of chem-LTD significantly increased SNX26 expression, whereas that of chem-LTP had the opposite effect (Fig. 11, D and E).
We suggest that SNX26, acting as a Cdc42-specific GAP, interacts with PSD-95 and other upstream/downstream signaling proteins and has an important role in dendritic morphogenesis. Furthermore, our results raise the possibility that the expression (and possibly the activity) of SNX26 might be controlled by synaptic activity leading to changes in neuronal morphology not only during early neuronal development but also during synaptic plasticity in mature neurons.
Acknowledgments
We thank Boyoon Lee and Sang-Eun Lee for technical assistance. Confocal microscopy data for this study were acquired and analyzed in the Biomedical Imaging Center at Seoul National University College of Medicine.
This work was supported by grants from Basic Research Promotion Fund Grant 313-2008-2-C00690 (to S. C.) funded by the National Research Foundation of Korea, Brain Research Center of the 21st Century Frontier Research Program Grant M103KV010009-06K2201-00910 (to S. C.), and Basic Science Research Program Grant NRF-2011-355-C00121 (to Y. K.) funded by the Ministry of Education and Science and Technology, Republic of Korea.
- LTP
- long term potentiation
- GAP
- GTPase-activating protein
- ANOVA
- analysis of variance
- HSD
- honestly significant difference
- SH
- Src homology
- LTD
- long term depression
- PX
- Phox homology
- TxRed-phalloidin
- TxRed-phalloidin
- DIV
- day in vitro
- FL
- full-length
- PRD
- proline-rich domain.
REFERENCES
- 1. Arellano J. I., Espinosa A., Fairén A., Yuste R., DeFelipe J. (2007) Nonsynaptic dendritic spines in neocortex. Neuroscience 145, 464–469 [DOI] [PubMed] [Google Scholar]
- 2. Engert F., Bonhoeffer T. (1999) Dendritic spine changes associated with hippocampal long-term synaptic plasticity. Nature 399, 66–70 [DOI] [PubMed] [Google Scholar]
- 3. Maletic-Savatic M., Malinow R., Svoboda K. (1999) Rapid dendritic morphogenesis in CA1 hippocampal dendrites induced by synaptic activity. Science 283, 1923–1927 [DOI] [PubMed] [Google Scholar]
- 4. Yuste R., Majewska A. (2001) On the function of dendritic spines. Neuroscientist 7, 387–395 [DOI] [PubMed] [Google Scholar]
- 5. Kaufmann W. E., Moser H. W. (2000) Dendritic anomalies in disorders associated with mental retardation. Cereb Cortex 10, 981–991 [DOI] [PubMed] [Google Scholar]
- 6. Stephan K. E., Friston K. J., Frith C. D. (2009) Dysconnection in schizophrenia: from abnormal synaptic plasticity to failures of self-monitoring. Schizophr. Bull. 35, 509–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Walsh D. M., Klyubin I., Fadeeva J. V., Rowan M. J., Selkoe D. J. (2002) Amyloid-β oligomers: their production, toxicity and therapeutic inhibition. Biochem. Soc. Trans. 30, 552–557 [DOI] [PubMed] [Google Scholar]
- 8. Südhof T. C. (2008) Neuroligins and neurexins link synaptic function to cognitive disease. Nature 455, 903–911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fiala J. C., Feinberg M., Popov V., Harris K. M. (1998) Synaptogenesis via dendritic filopodia in developing hippocampal area CA1. J. Neurosci. 18, 8900–8911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Portera-Cailliau C., Pan D. T., Yuste R. (2003) Activity-regulated dynamic behavior of early dendritic protrusions: evidence for different types of dendritic filopodia. J. Neurosci. 23, 7129–7142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ethell I. M., Pasquale E. B. (2005) Molecular mechanisms of dendritic spine development and remodeling. Prog. Neurobiol. 75, 161–205 [DOI] [PubMed] [Google Scholar]
- 12. Matus A. (2000) Actin-based plasticity in dendritic spines. Science 290, 754–758 [DOI] [PubMed] [Google Scholar]
- 13. Matsuzaki M., Honkura N., Ellis-Davies G. C., Kasai H. (2004) Structural basis of long-term potentiation in single dendritic spines. Nature 429, 761–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhou Q., Homma K. J., Poo M. M. (2004) Shrinkage of dendritic spines associated with long-term depression of hippocampal synapses. Neuron 44, 749–757 [DOI] [PubMed] [Google Scholar]
- 15. Krucker T., Siggins G. R., Halpain S. (2000) Dynamic actin filaments are required for stable long-term potentiation (LTP) in area CA1 of the hippocampus. Proc. Natl. Acad. Sci. U.S.A. 97, 6856–6861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hall A. (1998) Rho GTPases and the actin cytoskeleton. Science 279, 509–514 [DOI] [PubMed] [Google Scholar]
- 17. Heasman S. J., Ridley A. J. (2008) Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat. Rev. Mol. Cell Biol. 9, 690–701 [DOI] [PubMed] [Google Scholar]
- 18. Tolias K. F., Duman J. G., Um K. (2011) Control of synapse development and plasticity by Rho GTPase regulatory proteins. Prog. Neurobiol. 94, 133–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chiang S. H., Hwang J., Legendre M., Zhang M., Kimura A., Saltiel A. R. (2003) TCGAP, a multidomain Rho GTPase-activating protein involved in insulin-stimulated glucose transport. EMBO J. 22, 2679–2691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liu H., Nakazawa T., Tezuka T., Yamamoto T. (2006) Physical and functional interaction of Fyn tyrosine kinase with a brain-enriched Rho GTPase-activating protein TCGAP. J. Biol. Chem. 281, 23611–23619 [DOI] [PubMed] [Google Scholar]
- 21. Shen P. C., Xu D. F., Liu J. W., Li K., Lin M., Wang H. T., Wang R., Zheng J. (2011) TC10β/CDC42 GTPase activating protein is required for the growth of cortical neuron dendrites. Neuroscience 199, 589–597 [DOI] [PubMed] [Google Scholar]
- 22. Rosário M., Schuster S., Jüttner R., Parthasarathy S., Tarabykin V., Birchmeier W. (2012) Neocortical dendritic complexity is controlled during development by NOMA-GAP-dependent inhibition of Cdc42 and activation of cofilin. Genes Dev. 26, 1743–1757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Riedl J., Crevenna A. H., Kessenbrock K., Yu J. H., Neukirchen D., Bista M., Bradke F., Jenne D., Holak T. A., Werb Z., Sixt M., Wedlich-Soldner R. (2008) Lifeact: a versatile marker to visualize F-actin. Nature methods 5, 605–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chang S., De Camilli P. (2001) Glutamate regulates actin-based motility in axonal filopodia. Nat. Neurosci. 4, 787–793 [DOI] [PubMed] [Google Scholar]
- 25. Peters A., Kaiserman-Abramof I. R. (1970) The small pyramidal neuron of the rat cerebral cortex. The perikaryon, dendrites and spines. A. J. Anat. 127, 321–355 [DOI] [PubMed] [Google Scholar]
- 26. El-Husseini A. E., Schnell E., Chetkovich D. M., Nicoll R. A., Bredt D. S. (2000) PSD-95 involvement in maturation of excitatory synapses. Science 290, 1364–1368 [PubMed] [Google Scholar]
- 27. Marrs G. S., Green S. H., Dailey M. E. (2001) Rapid formation and remodeling of postsynaptic densities in developing dendrites. Nat. Neurosci. 4, 1006–1013 [DOI] [PubMed] [Google Scholar]
- 28. Kennedy M. B. (2000) Signal-processing machines at the postsynaptic density. Science 290, 750–754 [DOI] [PubMed] [Google Scholar]
- 29. Okamoto K., Nagai T., Miyawaki A., Hayashi Y. (2004) Rapid and persistent modulation of actin dynamics regulates postsynaptic reorganization underlying bidirectional plasticity. Nat. Neurosci. 7, 1104–1112 [DOI] [PubMed] [Google Scholar]
- 30. Papa M., Segal M. (1996) Morphological plasticity in dendritic spines of cultured hippocampal neurons. Neuroscience 71, 1005–1011 [DOI] [PubMed] [Google Scholar]
- 31. Maness P. F. (1992) Nonreceptor protein tyrosine kinases associated with neuronal development. Dev. Neurosci. 14, 257–270 [DOI] [PubMed] [Google Scholar]
- 32. Grant S. G., Silva A. J. (1994) Targeting learning. Trends Neurosci. 17, 71–75 [DOI] [PubMed] [Google Scholar]
- 33. Kitazawa H., Yagi T., Miyakawa T., Niki H., Kawai N. (1998) Abnormal synaptic transmission in the olfactory bulb of Fyn-kinase-deficient mice. J. Neurophysiol. 79, 137–142 [DOI] [PubMed] [Google Scholar]
- 34. Huerta P. T., Scearce K. A., Farris S. M., Empson R. M., Prusky G. T. (1996) Preservation of spatial learning in fyn tyrosine kinase knockout mice. Neuroreport 7, 1685–1689 [DOI] [PubMed] [Google Scholar]
- 35. Isosaka T., Hattori K., Kida S., Kohno T., Nakazawa T., Yamamoto T., Yagi T., Yuasa S. (2008) Activation of Fyn tyrosine kinase in the mouse dorsal hippocampus is essential for contextual fear conditioning. Eur. J. Neurosci. 28, 973–981 [DOI] [PubMed] [Google Scholar]
- 36. Morita A., Yamashita N., Sasaki Y., Uchida Y., Nakajima O., Nakamura F., Yagi T., Taniguchi M., Usui H., Katoh-Semba R., Takei K., Goshima Y. (2006) Regulation of dendritic branching and spine maturation by semaphorin3A-Fyn signaling. J. Neurosci. 26, 2971–2980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Babus L. W., Little E. M., Keenoy K. E., Minami S. S., Chen E., Song J. M., Caviness J., Koo S. Y., Pak D. T., Rebeck G. W., Turner R. S., Hoe H. S. (2011) Decreased dendritic spine density and abnormal spine morphology in Fyn knockout mice. Brain Res. 1415, 96–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lim S. H., Kwon S. K., Lee M. K., Moon J., Jeong D. G., Park E., Kim S. J., Park B. C., Lee S. C., Ryu S. E., Yu D. Y., Chung B. H., Kim E., Myung P. K., Lee J. R. (2009) Synapse formation regulated by protein-tyrosine phosphatase receptor T through interaction with cell adhesion molecules and Fyn. EMBO J. 28, 3564–3578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Miller M., Peters A. (1981) Maturation of rat visual cortex. II. A combined Golgi-electron microscope study of pyramidal neurons. J. Comp. Neurol. 203, 555–573 [DOI] [PubMed] [Google Scholar]
- 40. Okabe T., Nakamura T., Nishimura Y. N., Kohu K., Ohwada S., Morishita Y., Akiyama T. (2003) RICS, a novel GTPase-activating protein for Cdc42 and Rac1, is involved in the β-catenin-N-cadherin and N-methyl-d-aspartate receptor signaling. J. Biol. Chem. 278, 9920–9927 [DOI] [PubMed] [Google Scholar]
- 41. Nakazawa T., Kuriu T., Tezuka T., Umemori H., Okabe S., Yamamoto T. (2008) Regulation of dendritic spine morphology by an NMDA receptor-associated Rho GTPase-activating protein, p250GAP. J. Neurochem. 105, 1384–1393 [DOI] [PubMed] [Google Scholar]
- 42. Zhao C., Ma H., Bossy-Wetzel E., Lipton S. A., Zhang Z., Feng G. S. (2003) GC-GAP, a Rho family GTPase-activating protein that interacts with signaling adapters Gab1 and Gab2. J. Biol. Chem. 278, 34641–34653 [DOI] [PubMed] [Google Scholar]
- 43. Nakamura T., Komiya M., Sone K., Hirose E., Gotoh N., Morii H., Ohta Y., Mori N. (2002) Grit, a GTPase-activating protein for the Rho family, regulates neurite extension through association with the TrkA receptor and N-Shc and CrkL/Crk adapter molecules. Mol. Cell. Biol. 22, 8721–8734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rosário M., Franke R., Bednarski C., Birchmeier W. (2007) The neurite outgrowth multiadaptor RhoGAP, NOMA-GAP, regulates neurite extension through SHP2 and Cdc42. J. Cell Biol. 178, 503–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Taniguchi S., Liu H., Nakazawa T., Yokoyama K., Tezuka T., Yamamoto T. (2003) p250GAP, a neural RhoGAP protein, is associated with and phosphorylated by Fyn. Biochem. Biophys. Res. Commun. 306, 151–155 [DOI] [PubMed] [Google Scholar]
- 46. Nasu-Nishimura Y., Hayashi T., Ohishi T., Okabe T., Ohwada S., Hasegawa Y., Senda T., Toyoshima C., Nakamura T., Akiyama T. (2006) Role of the Rho GTPase-activating protein RICS in neurite outgrowth. Genes Cells 11, 607–614 [DOI] [PubMed] [Google Scholar]
- 47. Long H., Zhu X., Yang P., Gao Q., Chen Y., Ma L. (2013) Myo9b and RICS modulate dendritic morphology of cortical neurons. Cereb. Cortex 23, 71–79 [DOI] [PubMed] [Google Scholar]