

Background: All known cilia-related proteins regulate Hedgehog signaling through their role in ciliogenesis.
Results: The centrosomal protein DZIP1 interacts with and sequesters GLI3 transcription factor in the cytoplasm and also regulates ciliogenesis.
Conclusion: DZIP1 is the first known cilia-related protein that regulates Hedgehog signaling through a dual mechanism.
Significance: Understanding how DZIP1 regulates Hedgehog signaling provides new insights into the molecular mechanism of Hedgehog signal transduction.
Keywords: Cilia, Hedgehog, Protein-Protein Interactions, Signaling, Transcription Factors
Abstract
The primary cilium is required for Hedgehog signaling. So far, all known ciliogenic proteins regulate Hedgehog signaling through their role in ciliogenesis. Here we show that the mouse DZIP1 regulates Hedgehog signaling through two mechanisms. First, DZIP1 interacts with GLI3, a transcriptional regulator for Hedgehog signaling, and prevents GLI3 from entering the nucleus. Second, DZIP1 is required for ciliogenesis. We show that DZIP1 colocalizes and interacts with CEP164, a protein localizing at appendages of the mother centrioles, and IFT88, a component of the intraflagellar transport (IFT) machinery. Functionally, both CEP164 and Ninein appendage proteins fail to localize to ciliary appendages in Dzip1 mutant cells; IFT components are not recruited to the basal body of cilia. Importantly, the accumulation of GLI3 in the nucleus is independent of loss of primary cilia in Dzip1 mutant cells. Therefore, DZIP1 is the first known ciliogenic protein that regulates Hedgehog signaling through a dual mechanism and that biochemically links IFT machinery with Hedgehog pathway components.
Introduction
The family of secreted Hedgehog (Hh) molecules plays an important role in the development of almost all embryonic tissues in vertebrates. An inappropriate activation of the Hh pathway is also associated with several types of human cancer (1). Thus, a thorough understanding of the molecular mechanism of Hh signal transduction is essential not only for the elucidation of the molecular mechanism of the embryonic developmental process but also for the prevention and treatment of Hh-related cancers.
The molecular mechanism of Hh signal transduction has been extensively studied. In vertebrates, Hh ligands bind to the cell surface receptor, Patched (PTCH) (2, 3). This binding prevents PTCH from inhibiting another transmembrane protein known as Smoothened (SMO), which then transduces signals to activate the GLI transcription factors downstream. Of the three GLI family members, GLI2 and GLI3 act as transcriptional repressors (GLI2Rep and GLI3Rep) in the absence of Hh signaling, because the C-terminal activation domains of the full-length proteins (GLI2FL and GLI3FL) are proteolytically processed (4, 5). Hh signaling inhibits this proteolytic processing and converts the latent full-length proteins into activators, which subsequently activate the downstream transcriptional targets. One of these targets is Gli1, which further enforces the Hh pathway activation, whereas Ptch is also a target to establish a negative feedback regulation of the pathway.
One of the recent advances in Hh signal transduction is the discovery that vertebrate Hh signaling occurs in primary cilia (6). Consistent with this, the Hh pathway core components, such as PTCH, SMO, GLI2 and GLI3 proteins, SUFU, and KIF7, localize to the cilia (7–11). Primary cilia, also called non-motile cilia, are solitary microtubule-based organelles that protrude from the cell surface and serve as a site for transducing extracellular signals. Defects in cilia structure are associated with a diverse array of developmental abnormalities, collectively termed “ciliopathies,” including polycystic kidney disease, respiratory and visual disorders, hydrocephalus, obesity, and mental retardation (12).
Primary cilia assemble during the G1 phase from the mother centrioles. The mother centriole is characterized by unique distal and subdistal appendages made of specific proteins, including Odf2, Ninein, and CEP164 (13–16). The mother centriole serves as a basal body and a docking station for ciliary microtubules and intraflagellar transport (IFT)2 machinery during primary cilia elongation. IFT machinery is also essential for the maintenance of primary cilia. There are two IFT protein complexes, IFT-A and IFT-B. IFT-B, together with a kinesin motor, is responsible for anterograde trafficking of protein complexes and vesicles in the cilia, whereas IFT-A, together with a dynein motor, is responsible for retrograde trafficking (17, 18). Increasingly, more ciliary proteins are found to be involved in Hh signaling. These proteins include motor proteins, components of the IFT-A and IFT-B complexes, centrosomal proteins, small GTPases (Rab and Arf or Arl), etc. (6). Among them, only KIF7, a kinesin motor and core component of the Hh cytoplasmic signaling complex, and MIM, a protein enriched in the basal body, have been shown to associate with GLI proteins physically (7, 19–21), although the in vivo role of MIM in Hh signaling and ciliogenesis needs to be further determined (22). However, none of these proteins have thus far been biochemically shown to bridge Hh pathway components and IFT machinery. The effect of these proteins on Hh signaling also appears to be only through their regulation of ciliogenesis or protein transport in the cilia.
In this study, we show that a mutation in the Dzip1 gene, the mouse homolog of the zebrafish Iguana (Igu) (23, 24), results in phenotypes consistent with loss of Hh signaling. DZIP1 regulates Hh signaling through two mechanisms. First, DZIP1 binds to GLI3 but not to GLI2 or GLI1, thus preventing the GLI3 protein from entering the nucleus. Consistent with this, the DZIP1 mutation leads to accumulation of GLI3 in the nucleus. Second, DZIP1 plays an essential role in ciliogenesis. We show that DZIP1 specifically accumulates at the distal appendage of the mother centrioles. In DZIP1 mutant cells, primary cilia are not formed; Ninein and Cep164, two ciliary appendage proteins, fail to localize to the appendages of the mother centrioles. In addition, IFT machinery is not recruited to the basal body. More interestingly, DZIP1 can interact with both CEP164 and IFT88. Thus, DZIP1 is necessary not only for recruiting IFT machinery to the distal appendages of the mother centrioles but also for bridging IFT88 to the cargo that contains GLI3. The sequestration of GLI3 in the cytoplasm and the role of ciliogenesis by DZIP1 are two independent functions. Therefore, DZIP1 is the first known protein that regulates Hh signaling through this dual mechanism and biochemically connects Hh signaling components with IFT machinery.
EXPERIMENTAL PROCEDURES
Mouse Strains and the Generation of the Dzip1 Mutant Allele
A PAC clone containing mouse Dzip1 genomic DNA sequences (Geneservices, Inc.) was used to create a Dzip1 targeting construct. The construct was engineered by replacing the first two exons of the Dzip1 gene with a neomycin cassette flanked by loxP sites in reverse orientation relative to the Dzip1 gene (Fig. 1A). The linearized construct was electroporated into W4 ES cells, and targeted ES cell clones were identified by digestion of genomic DNA with EcoRI, followed by a Southern blot analysis of ES cell DNA using 5′- and 3′-probes (Fig. 1B). Two Dzip1-targeted ES cell clones were injected into C57BL/6 blastocysts to generate chimeric founders, which were then bred with C57BL/6 to establish F1 heterozygotes. The Dzip1 heterozygotes were maintained in a 129/SVE and C57BL/6 mix background. The neomycin cassette was removed by crossing the Dzip1 mutant mice with actin-cre mice. PCR analysis was used for routine genotyping with the following primers: forward primer P1 (5′-ATC GAC GTG GAC AAG GTT GC-3′) and reverse primer P2 (5′-GCC AGC CTG CTT GGT GAG CAG C-3′) for the wild type allele, which produced a 282-bp fragment and forward primer P3 (5′-CAA GTG TAA CCT TAC TGT GGA GT-3′) and reverse primer P4 (5′-AGG TCC CTC GAC CTG CAG CCC AAG-3′) for the targeted Dzip1 allele, which produced a 260-bp fragment.
FIGURE 1.

Inactivation of DZIP1 disrupts Hedgehog signaling in mouse embryos. A, the gene-targeting strategy used to create a mouse Dzip1 mutant allele. Open rectangles, exons; lines, introns. Restriction enzymes are BamHI (B), EcoRI (RI), and NheI (Nh). B, Southern blot analysis shows a representative mutant and wild type (WT) ES cell clones. C, Western blots of WT and Dzip1 mutant embryos with anti-DZIP1 antibody and anti-α-tubulin antibody. Note that a protein smaller than WT DZIP1 was detected in the mutant embryos. D, the morphology of WT and Dzip1 mutant embryos at E9.5. The mutant embryos are underdeveloped (somites are indicated by an arrow). E, immunofluorescent staining (Foxa2, Hb9, Nkx2.2, Nkx6.1, and Pax6) and in situ hybridization of WT and Dzip1 mutant neural tubes (Ptch1 and Gli1). The ventral transcription factors examined are not expressed or diminished, whereas the dorsal marker Pax6 expression is expanded throughout the neural tube. Ptch1 and Gli1 expression is significantly reduced in the mutant neural tube. Scale bar, 100 μm. F, a Western blot showing that levels of GLI2FL are increased and GLI3 processing is reduced in the Dzip1 mutant embryos. Shown in the graph is the ratio of GLI3FL to GLI3Rep.
Cell Lines and Cell Culture
Wild type and mutant Dzip1 mouse embryonic fibroblasts (MEFs) were generated from E9.5 mouse embryos. IFT88 mutant MEFs were derived from E10.5 mouse embryos (25). The MEFs were cultured in DMEM supplemented with heat-inactivated 10% FBS, penicillin, and streptomycin until cells were immortalized. HEK293 cells and human RPE-1 cells were cultured in the same medium. C3H10T1/2 cells were also grown in the same medium supplemented with ZnSO4 (0.1 μg/ml) and β-mercaptoethanol (0.1 μm).
cDNA and shRNA Constructs and Cloning
Mouse and human full-length Dzip1 cDNAs were purchased from Open Biosystems and cloned into the pCMV-3xFLAG vector (Sigma) by restriction enzyme digestion and/or PCR. pLNCX-FLAGHA-Dzip1 was created by cloning FLAGHA-Dzip1 into the pLNCX retroviral vector (Clontech). The Dzip1 mutant constructs were created by the same method. Human GLI3NT (aa 1–478), GLI3ΔZF (aa 478–645 deleted), GLI3CT (aa 645–1596), and GLI3CTΔPDD (aa 860–1596) were cloned into the pRK expression vector by the same method. The scramble and hDZIP1 shRNA set were purchased from Open Biosystems (RHS4533). Two of the five constructs (TRCN0000146638 and TRCN0000155187) from the hDZIP1 shRNA set were able to knock down endogenous hDZIP1 in RPE-1 cells to undetectable levels (data not shown). Data presented in this study were obtained using one of the two shRNA constructs.
Tissue Immunohistochemistry and in Situ Hybridization
For immunohistochemistry, mouse embryos at 9.5 days postcoitus (E9.5) were dissected, fixed in 4% paraformaldehyde plus PBS for 20–30 min at 4 °C, equilibrated in 30% sucrose plus PBS overnight at 4 °C, and embedded in OCT. The frozen embryos were transversely cryosectioned at the forelimb areas (10 μm). Tissue sections were immunostained using antibodies against Foxa2 (concentrated), Nkx2.2, Hb9, Isl1/2, Nkx6.1, and Pax6 (Developmental Study Hybridoma Bank) as described (26). The secondary antibodies (Jackson ImmunoResearch) were Cy3-conjugated donkey anti-mouse IgG. In situ hybridization of embryonic sections with Ptch1 or Gli1 digoxigenin-labeled probes was performed as described (26).
Cell Immunofluorescence and Microscopy
For GLI3 protein nuclear translocation, cells were incubated with a culture medium containing 50 nm leptomycin B (LMB) for 6 h prior to immunofluorescence. For cell ciliation studies, cells were plated on coverslips coated with poly-d-lysine overnight and then incubated with DMEM supplemented with 0.1% FBS and penicillin/streptomycin for 1 day to arrest the cells. For centrosome staining, cells were fixed 2–3 min in −20 °C 100% methanol. For the cytoplasmic and cilia staining, cells were fixed for 15 min in 4% paraformaldehyde plus PBS. After washing with PBS, the cells were incubated in PBS with 0.2% Triton X-100 for 15 min, followed by a wash with PBS. The cells were then incubated with primary antibodies in PBS, 1% BSA, 4% donkey serum for 1 h at room temperature. The cells were washed with PBS and then incubated with secondary antibodies in the same solution for 1 h at room temperature. After three washes with PBS, the coverslips were mounted to glass slides with Vectashield mounting fluid with DAPI (Vector Labs). The staining was visualized using a Zeiss Axiovert fluorescent microscope.
Antibodies
Antibodies to DZIP1, GLI2, IFT88, and GFP were generated by Covance, Inc. Rabbits were immunized with an insoluble His-tagged mouse DZIP1 fragment (aa 1–552), a GLI2 fragment (aa 605–1460), a soluble GST-IFT88 (aa 693–824), or an insoluble GST-GFP purified from bacteria. The DZIP1 and GFP antibodies were used at a 1:1000 dilution for Western blotting. The IFT88 antibody was used at a 1:1000 dilution for cell staining. Other antibodies used for cell staining included FLAG M2 mAb (1:1000), acetylated tubulin (1:2000), γ-tubulin (1:2000) (Sigma), CP110 (1:200) (Bethyl Laboratories, A301-343A), GLI3 crude serum (1:1000) (5), ODF2 (Abcam, ab43840, 1:200), Ninein (1:5000) (27), CEP97 (1:500) (28), CEP164 (1:200) (Sigma, SAB3500022), IFT57 (1:1000), and IFT140 (1:1000) (29). Secondary antibodies Dylight 488-conjugated donkey anti-rabbit IgG and Cy3-conjugated donkey anti-mouse IgG were purchased from Jackson Immunoresearch, Inc.
Immunoprecipitations and Immunoblots
E9.5 mouse embryos or cells grown in dishes were washed once in PBS and lysed in radioimmune precipitation buffer buffer (50 mm Tris-HCl (pH 7.4), 150 mm NaCl, 1 mm EDTA, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS, protease inhibitors) for 10 min on ice. After being cleared by centrifugation, the lysates were mixed with protein loading buffer, resolved in SDS-polyacrylamide gel, and transferred to a nitrocellulose membrane, followed by Western blotting as described (5). For immunoprecipitation, the lysates were incubated with the indicated antibodies for 2 h on ice and subsequently with 15 μl of protein G-conjugated agarose beads with rotation at 4 °C for 1 h. The beads were washed with radioimmune precipitation buffer, and proteins were eluted with protein loading buffer and subjected to Western blotting.
RESULTS
A Dzip1 Mutation Impairs Hh Signaling in Embryos
Genetic evidence shows that the zebrafish Igu gene product acts downstream of Smo to modulate both Gli activator and repressor function (23, 24). To determine whether DZIP1, the mouse homolog of Igu, is involved in Hh signaling, we mutated the Dzip1 gene by deleting the first two exons to create the Dzip1ΔE1-2 allele (Fig. 1, A and B). Only about one-third of the homozygous Dzip1 mutant mice (7 of 22) died around E9.5, whereas the rest did not show noticeable phenotypes and survived to adulthood. The mutant embryos that showed abnormal phenotypes were smaller than their wild type littermates. In particular, the neural tube and somites were underdeveloped (Fig. 1D). The incomplete penetration of the mutant phenotype could have resulted from the possibly partial inactivation of the Dzip1 gene, because a truncated DZIP1 protein was still expressed in the mutant (Fig. 1C).
To determine the possible initiation codon for the mutant DZIP1 protein, we inspected the DZIP1 amino acid sequence and found that any of the four methionine residues at positions 193, 229, 258, and 285 could serve as an initiation codon. To determine which residue is most likely to be the initiation codon, four expression constructs, each starting from one of these methionines, were created and transfected into HEK293 cells. Western blot analysis indicated that the size of the mutant DZIP1 protein was similar to that of the DZIP1 protein initiated at residue 258 (data not shown). Therefore, the DZIP1 mutant protein most likely lacks the N-terminal 257 residues (DZIP1Δ257), which includes the zinc finger domain (aa 185–230).
To determine whether the Dzip1 mutant phenotypes are caused by dysfunctional Hh signaling, we examined the neural tube patterning of the affected embryos. Several transcription factors that are normally expressed in the developing ventral neural tube and whose expression is induced by Sonic Hedgehog (Shh) (30) were missing in the Dzip1 mutant. These included Foxa2, a floor plate marker; Hb9, a motor neuron marker; Nkx2.2, a marker for V3 progenitors; and Nkx6.1, a protein normally expressed in the V1–V3 domains. In contrast, Pax6 expression, normally restricted by Shh signaling, expanded throughout the entire ventral neural tube (Fig. 1E).
To determine if the Dzip1 mutation directly affects Hh signaling, we examined the expression of Gli1 and Ptch1 RNA, transcriptional targets of Hh signaling. In situ hybridization showed that the expression of both Gli1 and Ptch1 was significantly reduced in the mutant neural tube (Fig. 1E), indicating that Hh signaling was impaired in the Dzip1 mutant.
To investigate whether the mutant phenotypes are related to GLI2 and GLI3 function, we examined GLI2 and GLI3 protein levels in the mutant embryos by immunoblotting with GLI2 and GLI3 antibodies. The results showed that levels of both of the full-length GLI2 and GLI3 proteins, GLI2FL and GLI3FL, were increased, whereas GLI3 repressor (GLI3Rep) levels were significantly reduced (Fig. 1F). Taken together, these results indicate that the Dzip1 mutation impairs both GLI2 activator function and GLI3 processing.
DZIP1 Interacts with and Sequesters GLI3 in the Cytoplasm
To explore the potential mechanism by which DZIP1 regulates GLI protein activity, we first determined whether DZIP1 interacts with the three GLI proteins. A FLAG-tagged DZIP1 was coexpressed with each of GLI1, GLI2, and GLI3 in HEK293 cells. GLI proteins were precipitated with Sepharose beads conjugated with a GLI-binding oligonucleotide, and the precipitates were then immunoblotted with FLAG antibody (4). The results showed that DZIP1 interacted with GLI3 but not GLI1 or GLI2 (Fig. 2A). The interaction between endogenous DZIP1 and GLI3 was also detected by coimmunoprecipitation using wild type MEFs. Interestingly, the interaction between endogenous GLI3 and the mutant DZIP1 was significantly reduced, indicating that the DZIP1 N-terminal region is necessary for high affinity binding to GLI3 (Fig. 2B). In addition, human DZIP1 was able to interact with hGLI3 (Fig. 2C), indicating that DZIP1 and GLI3 interaction is conserved in mice and humans.
FIGURE 2.

DZIP1 interacts with GLI3 in the cell. A, DZIP1 interacts with GLI3, but not GLI1 and GLI2. FLAG-DZIP1 was coexpressed with GLI1, GLI2, or GLI3 in HEK293 cells, as indicated. The protein lysates made from the cells were subjected to immunoblotting with the indicated antibodies (left and right panels) or precipitation with Gli-binding or control oligonucleotide-conjugated beads followed by immunoblotting with FLAG antibody. B, endogenous DZIP1 interacts with GLI3. Both WT and Dzip1 mutant MEFs were used for coimmunoprecipitation analysis, as indicated. IP, immunoprecipitation; PI, preimmune serum. C, coimmunoprecipitation analysis shows that the human DZIP1 also interacted with hGLI3 protein when the two proteins were coexpressed in HEK293 cells. D, the various FLAG-DZIP1 mutant constructs were coexpressed with GLI3 in HEK293 cells. The protein lysates from the cells were subjected to immunoblotting with either FLAG or GLI3 antibodies (middle or bottom panels) or pull-down with Gli-binding oligonucleotides followed by immunoblotting with FLAG antibody (top panels). The specific DZIP1 proteins are indicated by asterisks. E and F, GLI3 PDD domain is necessary for the interaction of GLI3 with DZIP1. The proteins shown at the top were coexpressed in HEK293 cells. The protein lysates from the cells were subjected to coimmunoprecipitation (C, top panels), GST pull-down (D, top), or immunoblotting with the indicated antibodies (middle and bottom panels). G and H, diagrams summarize the results from A–E. ZF, zinc finger; CC, coiled coil.
To map the regions in DZIP1 that interact with GLI3, GLI3 was coexpressed with various Dzip1 mutant constructs in HEK293 cells. The interaction between GLI3 and the DZIP1 mutant proteins was examined as described above. The results revealed that two C-terminally truncated DZIP1 proteins, DZIP1(1–350) and DZIP1(1–437), which mimic two Igu mutations (23, 24) and lack the coiled coil domain, were still able to bind to GLI3, whereas DZIP1 C-terminal fragments containing the coiled coil domain were not (Fig. 2, D and G). Thus, the coiled coil domain is not required for GLI3 binding. Conversely, coimmunoprecipitation using protein lysates made from cells co-expressing FLAG-DZIP1 and various GLI3 mutants showed that both the N-terminal and C-terminal regions of GLI3 bind to DZIP1. The binding site in the C terminus is located in the processing determinant domain (PDD) region, which determines the extent of GLI3 processing (31), because the GLI3 C terminus lacking the PDD failed to bind DZIP1 (Fig. 2, E (lane 8) and H). The binding of GLI3PDD to DZIP1 was specific, because glutathione S-transferase (GST) fused with GLI2 PDD did not bind to DZIP1 (Fig. 2F). Taken together, the results indicate that the N-terminal region of DZIP1 and the GLI3 PDD domain are essential for the two proteins' interaction. This is consistent with the fact that binding of the DZIP1 mutant protein to GLI3 in Dzip1 mutant embryos is significantly reduced (Fig. 2B).
Because DZIP1 interacts with GLI3, we next investigated whether DZIP1 regulates GLI3 subcellular localization. When FLAG-DZIP1 was expressed in C3H10T1/2 Hh-responsive cells, the DZIP1 protein localized exclusively to the cytoplasm (Fig. 3, B panels). DZIP1 does not shuttle between the nucleus and cytoplasm, because treatment of cells with LMB, an inhibitor for nuclear protein export, did not affect DZIP1 cytoplasmic localization (Fig. 3F′). On the other hand, although GLI3 was predominantly found in the cytoplasm of the C3H10T1/2 cells, LMB treatment caused the GLI3 protein to translocate to the nucleus (Fig. 3, A panels and E panels). This observation indicates that GLI3 shuttles between the nucleus and cytoplasm. However, when coexpressed with FLAG-DZIP1, GLI3 was retained in the cytoplasm even upon LMB treatment (Fig. 3, F panels). However, upon coexpression of DZIP1(1–350) and DZIP1(1–437), which localize to the cytoplasm and both the nucleus and cytoplasm (Fig. 3, C panels and D panels), respectively, GLI3 protein was not retained in the cytoplasm following LMB treatment (Fig. 3, G panels and H panels). Because both the DZIP1(1–350) and DZIP1(1–437) mutant proteins can bind GLI3, these results indicate that it is the DZIP1 C terminus that prevents GLI3 from entering the nucleus by tethering GLI3 in the cytoplasm.
FIGURE 3.

Dzip1 prevents Gli3 from its translocation into the nucleus. A–H, C3H10T1/2 cells were transfected with the expression constructs indicated above the panels. After being treated without (A–D) or with (E–H) LMB, the cells were stained for GLI3 (green), FLAG-DZIP1 proteins (red), or both and DNA (DAPI) (blue). I, diagrams show the Dzip1 constructs. ZF, zinc finger; CC, coiled coil.
If DZIP1 is required for GLI3 to be retained in the cytoplasm, one would predict that overexpressed GLI3 in Dzip1 mutant MEFs would localize to the nucleus. To test this prediction, both wild type and Dzip1 mutant MEFs were transfected with a GLI3 expression construct, and the GLI3 subcellular localization was determined by immunofluorescence with a GLI3 antibody. As shown in Fig. 4, A, A′, and E, GLI3 localized to the cytoplasm in most transfected wild type MEFs (>80%). In contrast, GLI3 was found in the nucleus in most transfected Dzip1 mutant MEFs (nearly 70%) (Fig. 4, B, B′, and E). This nuclear localization could be partially reversed by coexpression of exogenous DZIP1 in the mutant cells (reduced to about 40%, Fig. 4, D, D′, and E). The partial rescue of Gli3 cytoplasmic localization is probably due to the variation in the levels of DZIP1 expression.
FIGURE 4.
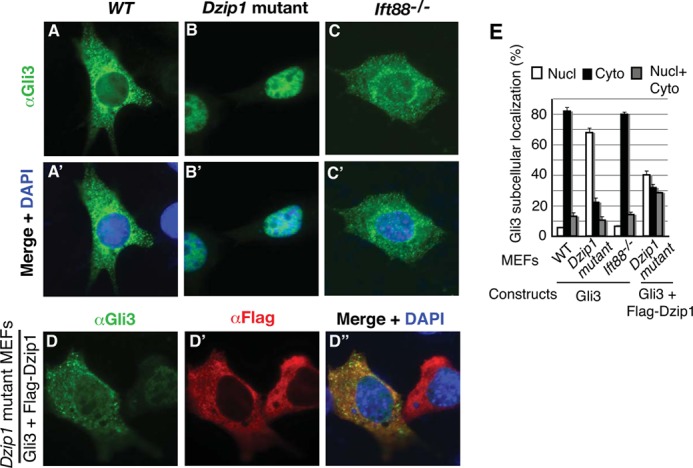
DZIP1 is required for GLI3 cytoplasmic localization. A–D, WT, Dzip1, and Ift88 mutant MEFs were transfected with GLI3 (A–C) or both GLI3 and FLAG-DZIP1 (D) expression constructs. The cells were stained for GLI3 (green), FLAG-DZIP1 proteins (red), or both and DNA (DAPI) (blue). E, the percentage of transfected cells that show GLI3 subcellular localization. Unpaired t test results: p = 0.2191 between WT and Ift88 mutant (not significant) and p < 0.0001 between WT and Dzip1 mutant or between Dzip1 mutant and Dzip1 cotransfected cells (highly significant). Nucl, nucleus; Cyto, cytoplasm; Nucl + Cyto, both nucleus and cytoplasm. More than 50 cells were counted.
Because Dzip1 mutant cells lack primary cilia (see below), we wanted to know whether the nuclear localization of GLI3 resulted from this or from a loss of the ability of DZIP1 to sequester GLI3 in the cytoplasm. To address this question, GLI3 subcellular localization was also examined in transfected Ift88 mutant MEFs, which lack cilia (11). The results showed that, as that in wild type cells, overexpressed GLI3 in Ift88 mutant cells was predominantly found in the cytoplasm (about 80%) (Fig. 4, C, C′, and E). Therefore, the nuclear localization of GLI3 in the Dzip1 mutant cells is due to the loss of DZIP1 function, not the lack of cilia. Together, these results indicate that the ability of DZIP1 to sequester GLI3 in the cytoplasm is independent of its function in ciliogenesis (see below).
DZIP1 Localizes to the Basal Body and Is Required for Ciliogenesis
The fact that Hh signaling and GLI3 processing are impaired in Dzip1 mutants suggests that DZIP1 is required for both GLI2 and GLI3 activator and repressor functions. This is reminiscent of mouse embryos exhibiting cilia defects (6). Thus, we asked whether primary cilia are present in Dzip1 mutant MEFs. Immunofluorescent staining for acetylated tubulin, which marks the axoneme of primary cilia, showed that primary cilia were present in most wild type MEFs but not in the mutant MEFs, except for a small dotted staining (Fig. 5A). Thus, DZIP1 is required for cilia formation.
FIGURE 5.
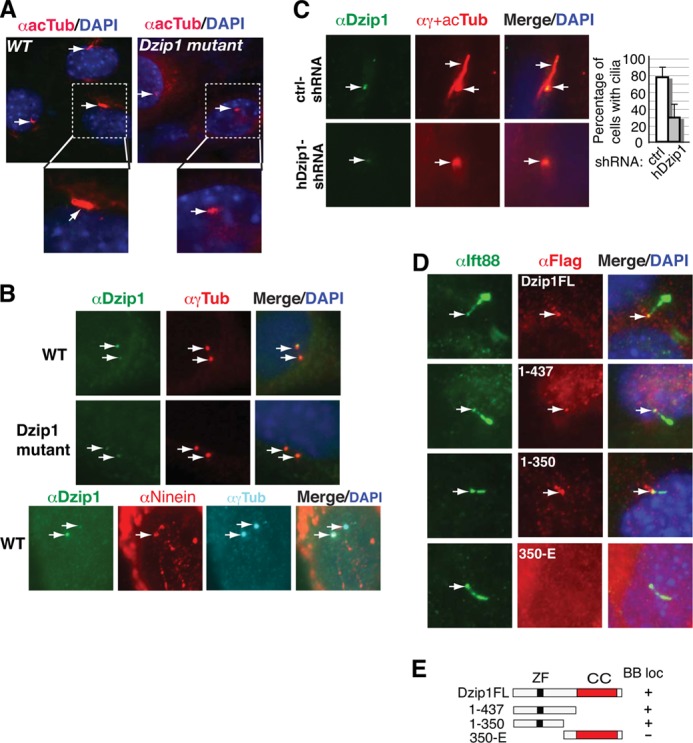
DZIP1 localizes at the basal body of cilia and is required for cilia formation. A, Dzip1 mutant cells lack cilia. WT and Dzip1 mutant MEFs were stained with acetylated α-tubulin antibody (red) for cilia and DAPI for the nucleus (blue). B, endogenous DZIP1, but not the Dzip1 mutant protein, accumulates at the mother centrioles. WT MEFs were co-stained with antibodies, as indicated. Note that the strong DZIP1 staining overlapped with that of Ninein. C, human DZIP1 is also required for ciliogenesis. Human RPE-1 cells were transduced with lentivirus carrying control shRNA or hDZIP1 shRNA expression constructs and subsequently subjected to immunostaining antibodies, as indicated. The percentage of cells with cilia is shown by a graph (more than 100 cells were counted, p < 0.0001). D, the N terminus is required for the localization of DZIP1 at the basal body. WT MEFs were transfected with the indicated FLAG-DZIP1 constructs. The transfected cells were co-stained for FLAG-DZIP1 proteins (red), IFT88, which localizes at the basal body and axoneme of cilia, and the nucleus (DAPI) (blue). E, diagrams showing the constructs used in D. Arrows, basal bodies. ZF, zinc finger; CC, coiled coil; BB loc, basal body-localized.
The involvement of DZIP1 in ciliogenesis prompted us to determine whether DZIP1 localizes to the cilia. To this end, wild type MEFs were co-immunostained for DZIP1 and γ-tubulin, a marker for centrioles. The results showed that DZIP1 localized to two centrioles asymmetrically. The weaker DZIP1-immunoreactive signal completely overlapped with one centriole, whereas the stronger signal only partially overlapped with the other centriole (Fig. 5B, top panels). Interestingly, only the weaker DZIP1-immunoreactive signals were detected in the centrioles of Dzip1ΔE1–2/ΔE1–2 MEFs (Fig. 5B, middle panels), indicating that the Dzip1 mutation abolishes DZIP1 accumulation to one of the centrioles. To determine whether the DZIP1 accumulation occurred at the mother or daughter centrioles, wild type MEFs were co-immunostained for DZIP1, Ninein (a subdistal appendage marker for the mother centrioles) (13), and γ-tubulin. The results showed that the stronger DZIP1 staining overlapped with that of Ninein (Fig. 5B, bottom panels), indicating that DZIP1 accumulates at the basal body and may be a part of the ciliary appendage structure. The role of DZIP1 in ciliogenesis is also conserved in human cells, because knockdown of hDZIP1 expression by shRNA significantly reduced cilia number in RPE-1 cells (Fig. 5C).
To determine whether overexpressed DZIP1 was able to recapitulate the basal body localization of endogenous DZIP1, wild type MEFs were transfected with FLAG-DZIP1. The cells were then immunostained for FLAG-DZIP1 and IFT88, an IFT component that localizes to the basal bodies and accumulates in the tips of the primary cilia in quiescent cells (32). As expected, the DZIP1 full-length protein, DZIP1FL, was enriched in the base of the cilia and colocalized with IFT88 at the basal body in addition to its diffuse localization in the cytoplasm (Fig. 5D, top panels). To define the region responsible for DZIP1 basal body localization, the same experiments were performed using several DZIP1 mutant constructs. DZIP1(1–437) and DZIP1(1–350) remained localized to the basal body (Fig. 5D, middle panels). However, DZIP1–350-E, which contains the coiled coil domain, failed to localize to the basal body (Fig. 5D, bottom panels). These results are consistent with the observation that the DZIP1ΔE1–2 mutant protein, which presumably lacks the N-terminal 257 residues, fails to localize to the basal body (Fig. 5, B (middle panels) and E). Taken together, these observations indicate that the accumulation of DZIP1 at the basal body is dependent on the DZIP1 N-terminal region but not the DZIP1 coiled coil domain.
DZIP1 Is Required for the Localization of CEP164 and Ninein to the Appendages of the Mother Centrioles
To understand the molecular mechanism underlying the absence of primary cilia in Dzip1 mutant cells, we examined the localization of several centrosomal and ciliary proteins in the wild type and mutant MEFs. Coimmunostaining of cells with antibodies against γ-tubulin and CP110 or CEP97, two proteins localizing to the distal end of the centrioles (28, 33), showed that the Dzip1 mutation did not affect the localization of either, suggesting that the distal centriole structure of centrioles still forms in Dzip1 mutant cells. CEP164, however, was not detected in the mutant cells at the distal appendage of the mother centrioles (Fig. 6A), where it normally localizes (16). This absence of CEP164 in Dzip1 mutant cells could not have been due to a change in expression, because the levels of CEP164 expression in the mutant cells were comparable with those in wild type cells (data not shown). It also cannot simply be a consequence of the loss of cilia in the Dzip1 mutant cells, because CEP164 still localized to the distal appendage of the mother centrioles in the Ift88 mutant MEFs (Fig. 6B).
FIGURE 6.

DZIP1 is required for the localization of CEP164 and Ninein at the primary cilia appendages. A, the Dzip1 mutation disrupts the localization of CEP164 and Ninein at the cilia appendages. WT and Dzip1 mutant MEFs were stained for the indicated proteins (green), γ-tubulin (a centriolar marker) (red), and DNA (DAPI) (blue). Arrows, centrioles. B, the localization of CEP164 to the cilia distal appendage is not disrupted in Ift88 mutant cells. Ift88 mutant MEFs were stained as in A. C, DZIP1 colocalizes with CEP164 and partially with Ninein. WT MEFs were transfected with FLAG-DZIP1 and then stained with FLAG and CEP164 or Ninein antibodies and DAPI. Arrows, centriolar staining of the proteins. D and E, coimmunoprecipitation shows that DZIP1 interacts with CEP164. HEK293 cells transiently transfected with the indicated expression constructs (D) or NIH3T3 cells stably transfected with FLAGHA-DZIP1 (E) were lysed. The resulting protein lysates were subjected to immunoblotting (two bottom panels) or immunoprecipitation (IP) followed by immunoblotting (IB) with the indicated antibodies (top panels). Note that only the DZIP1, and not DZIP1Δ257, was able to interact with CEP164.
We also examined whether the Dzip1 mutation affects the localization of Ninein. In the quiescent wild type cells, Ninein staining usually showed three dots in the mother centrioles, one in the proximal end and two in the subdistal appendages, and one dot in the daughter centrioles (Fig. 6A) (13, 34). However, in Dzip1 mutant cells, only a single dotted signal in the mother centrioles was detected and presumably localized to the proximal end of the mother centrioles (Fig. 6A). The absence of Ninein at the mother centriole appendages is not simply due to the lack of primary cilia in Dzip1 mutant cells, because Ninein was still found at the appendages in Ift88 mutant cells (Fig. 6B). Unlike that of Ninein, the localization of ODF2 to the appendages of the mother centrioles (34) in the mutant cells was unaffected (Fig. 6A). Taken together, these data indicate that DZIP1 is required for CEP164 and Ninein to localize to the distal and subdistal appendages of the mother centrioles, respectively.
To gain insight into the mechanism by which CEP164 and Ninein are absent in the distal and subdistal appendages, we asked whether DZIP1 co-localizes with these two proteins. Wild type MEFs were transfected with a FLAG-DZIP1 expression construct, and the transfected cells were subjected to co-immunostaining with antibodies against FLAG and CEP164 or Ninein. The results showed that FLAG-DZIP1 exactly colocalized with CEP164 and partially overlapped with Ninein (Fig. 6C), indicating that DZIP1 also localizes to the distal appendage of the mother centrioles.
The colocalization of DZIP1 with CEP164 prompted us to determine whether DZIP1 interacts with CEP164. FLAG-DZIP1 or FLAG-DZIP1Δ257 and GFP-tagged CEP164 (GFP-Cep164) were coexpressed in HEK293 cells, and the cell lysates were subjected to coimmunoprecipitation. The results showed that FLAG-DZIP1, but not FLAG-DZIP1Δ257, could be coimmunoprecipitated with GFP-CEP164 only when the two proteins were coexpressed and not when the two proteins were expressed separately (Fig. 6D). A similar experiment did not show any interaction between DZIP1 and Ninein (data not shown). To rule out the possibility that the interaction between DZIP1 and CEP164 was due to a high level of protein expression, NIH3T3 cells that stably expressed only modest levels of FLAG- and HA-tagged DZIP1 (FLAGHA-DZIP1) were used to repeat the same experiments. The results confirmed the interaction between DZIP1 and CEP164 (Fig. 6E). From these data, we conclude that DZIP1 and CEP164 interact with each other in the cell and that DZIP1 is required for both CEP164 and Ninein to localize to the appendages of the mother centrioles.
DZIP1 Is Required for the Recruitment of IFT Machinery to the Mother Centrioles
IFT machinery is recruited to the basal body during cilia elongation. To determine whether loss of Dzip1 affects this recruitment to the mother centrioles, we examined the centriolar localization of three IFT proteins: IFT88, IFT57, and IFT140. IFT88 accumulated in the basal body and the tips of the cilia in wild type cells but was not detected in the mother centrioles of the mutant cells. Similarly, both IFT57 and IFT140 were recruited to the mother centrioles in the wild type cells but not in the mutant cells (Fig. 7A). Because IFT88 and IFT57 are components of the IFT-B complex, whereas IFT140 is a component of the IFT-A complex, these data indicate that Dzip1 mutation disrupts recruitment of both IFT complexes to the mother centrioles.
FIGURE 7.
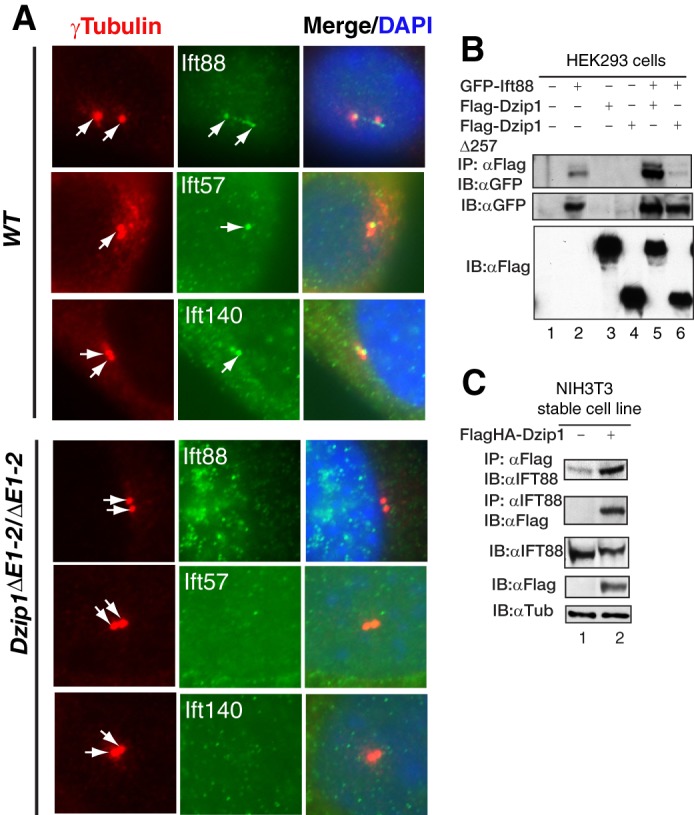
DZIP1 is required for the recruitment of IFT proteins to the basal body of cilia. A, WT and Dzip1 mutant MEFs were stained for γ-tubulin, a centrosome marker, and the indicated IFT proteins. None of the IFT proteins examined were detected in the centrioles of the mutant cells. B and C, DZIP1 interacts with IFT88. HEK293 cells transiently transfected with the indicated expression constructs or NIH3T3 cells stably transfected with FLAGHA-DZIP1 were lysed. The resulting protein lysates were subjected to immunoblotting (bottom panels) or immunoprecipitation (IP) followed by immunoblotting (IB) with the indicated antibodies (top panels). Note that only the DZIP1, and not DZIP1Δ257, was able to interact with IFT88.
The fact that both DZIP1 and IFT88 localize to the mother centrioles raises the possibility that they may interact with each other. To test this, HEK293 cells were transfected with GFP-IFT88 and FLAG-DZIP1 or FLAG-DZIP1Δ257 expression constructs. Coimmunoprecipitation analysis showed that FLAG-DZIP1, but not FLAG-DZIP1Δ257, was able to interact with GFP-IFT88 (Fig. 7B). This interaction was confirmed in NIH3T3 cells that stably express modest levels of FLAGHA-DZIP1 (Fig. 7C), suggesting that the DZIP1 and IFT88 interaction is unlikely to be due to the overexpression. Therefore, DZIP1 recruits IFT components to the base of the cilia through its interaction with IFT88.
DISCUSSION
In the present study, we investigate the role and mechanism of DZIP1 in Hh signaling in mice. The deletion of the first two exons of Dzip1 compromises Hh signaling during embryogenesis. DZIP1 regulates Hh signaling through two mechanisms. First, DZIP1 directly binds the GLI3 protein and prevents GLI3 from entering the nucleus. Second, DZIP1 is required for activation of GLI proteins through a cilia-dependent mechanism. Therefore, DZIP1 is the first known ciliogenic protein to regulate Hh signaling through this dual mechanism.
Dzip1 mutant embryos that show severe developmental defects have dorsalized neural tubes and underdeveloped somites, indicating that GLI activator function is impaired. Consistent with this, the expression of Ptch1 and Gli1 is dramatically reduced. However, levels of both GLI2FL and GLI3FL are increased, whereas the GLI3 repressor level is reduced (Fig. 1, D–F). Thus, like its zebrafish homolog Igu (23, 24) and many other known cilia-related proteins (6), DZIP1 is required for the functions of both GLI2 and GLI3 activators and repressors. However, unlike many known ciliary proteins, DZIP1 has also been implicated in the regulation of embryonic stem cells and germ cells (35). Thus, DZIP1 may have a broader function than the other known ciliary proteins.
The Dzip1 mutant phenotypes reported here are only partially penetrant. The embryos with the most severe phenotypes, about a third of the mutant embryos, died around E9.5, whereas the rest of the animals were born and grew to maturity without any noticeable phenotypes. The phenotypes appear to correlate with the presence of the primary cilia, because those without noticeable phenotypes had normal cilia (data not shown), whereas those with severe phenotypes lacked cilia (Fig. 5A). The partial penetrance of the Dzip1 mutant phenotypes could be related to the possibility that the mutant allele reported in this study might be hypomorphic, because a truncated protein is expressed in the mutant (Fig. 1C). It could also be attributed to potentially functional redundancy of different members of the DZIP1 family, such as DZIP1L. Further studies are necessary to determine the cause of the partial penetrance of the DZIP1 mutant phenotypes.
Most GLI3FL proteins localize to the cytoplasm, and the protein shuttles between the nucleus and cytoplasm (31, 36, 37). One protein that has been reported to regulate the GLI3 localization is SUFU, a negative regulator of GLI proteins that can retain GLI proteins in the cytoplasm when both proteins are overexpressed (38–40). DZIP1 also regulates GLI3 cytoplasmic localization, because it too can prevent GLI3 from entering the nucleus. GLI3 overexpressed in Dzip1 mutant MEFs is mostly found in the nucleus (Fig. 4, B and E), indicating that DZIP1 alone is sufficient to retain GLI3 in the cytoplasm. Because two of the DZIP1 mutants that are C-terminally truncated retain the ability to bind to GLI3 but are unable to keep Gli3 in the cytoplasm (Figs. 2D and 3, G and H), this binding alone is not sufficient to sequester the GLI3 in the cytoplasm. Thus, DZIP1 probably retains GLI3 in the cytoplasm by binding GLI3 to its N-terminal region, whereas its C-terminal region, which contains the coiled coil domain, may anchor DZIP1 and its binding proteins in the cytoplasm.
DZIP1 appears to regulate the cytoplasmic localization of GLI3 only, because it does not interact with GLI1 or GLI2 (Fig. 2, A and B). Because GLI3 is a weak activator, and GLI2 and GLI1 are strong transcription activators, the decrease in GLI3FL activators in the nucleus resulting from DZIP1 binding should lead to an overall increase in GLI activating activity. However, in Dzip1 mutant cells, increased nuclear localization of GLI3FL protein would consequently lead to an overall decrease in Gli activating function, because there are more weak GLI3FL activators competing against the stronger GLI1 and GLI2 activators for the Gli binding sites. Therefore, DZIP1 serves as a positive regulator for Hh signaling through its regulation of GLI3 subcellular localization.
Primary cilia are required for Hh signaling. Mutations in proteins involved in ciliogenesis are associated with ciliopathies and affect Hh signaling. There are an increasing number of proteins identified to regulate ciliogenesis. The cilia proteins that are known to affect Hh signaling include the kinesin and dynein motor proteins, components of the IFT-A and -B complexes, small GTPases, centriolar proteins, etc. (6). Recent studies have shown that zebrafish with either mutations in the Igu gene (the zebrafish Dzip1 homolog) or Igu morpholino knockdown lacks primary cilia (41–43). However, the underlying mechanism of the cilia defect is unknown. In the present study, we show that mouse Dzip1 mutant cells lack primary cilia (Fig. 5A), like zebrafish Igu mutants or morphants, and provide a mechanistic insight into how Dzip1 mutation may disrupt ciliogenesis. First, DZIP1 accumulates at the mother centrioles (Fig. 5, B–D) and colocalizes and interacts with CEP164 (Fig. 6, C–E), a distal appendage marker (16). This localization is dependent on the N-terminal region of the DZIP1 protein (Fig. 5D). Consistent with this localization, CEP164 and Ninein, two basal body appendage proteins (13), are absent at the appendages of the mother centrioles in Dzip1 mutant cells, whereas Ninein localization in the proximal end of the mother centrioles is unaffected (Fig. 6A). The failure of CEP164 and Ninein to localize to the appendages of the mother centrioles is not simply because of the lack of primary cilia in Dzip1 mutant cells, because the localization of these two proteins at the appendages is not affected in Ift88 mutant cells, which also lack primary cilia (Fig. 6, A and B). Therefore, DZIP1 is specifically required for the proper localization of CEP164 and Ninein at the appendages of the mother centrioles. Second, the components of both the IFT-A and IFT-B complexes fail to localize to the basal body of the primary cilia in Dzip1 mutant cells (Fig. 7A). This is probably due to a failed interaction between the Dzip1 mutant protein and IFT88 (Fig. 7, B and C). These results suggest that the recruitment of IFT machinery to the basal body, which is essential for cilia elongation, is through the interaction of IFT88 with DZIP1. These data may explain why cilia are not formed in Dzip1 mutant cells.
Hh signaling is generally thought to occur in primary cilia. Consistent with this, several of the Hh pathway components are found in cilia. Hh signaling leads to the accumulation of Smo and GLI2/GLI3 proteins in the cilia (10, 44–47). The transport of Hh signaling components in cilia is presumably mediated by IFT machinery. However, the biochemical evidence that links the Hh signaling components with IFT machinery remains missing. The finding that DZIP1 colocalizes with IFT88 in the basal body and interacts with both IFT88 and GLI3 is the first biochemical evidence that GLI3 is transported by IFT machinery in cilia (Fig. 7B).
Thus far, all known ciliary proteins to regulate Hh signaling do so because they are essential for ciliogenesis or protein and vesicle transport in the cilia (6). Compared with these proteins, DZIP1 is unusual in that it regulates Hh signaling through two different mechanisms. One is by sequestering GLI3 in the cytoplasm, similar to the function of SUFU, and the other is by its regulation of ciliogenesis through the recruitment of cilia appendage and IFT components to the cilia basal body. It is worth noting that the localization of GLI3 to the nucleus in Dzip1 mutant cells is not the result of the loss of primary cilia, because GLI3 in Ift88 mutant cells, which also lack primary cilia, is predominantly in the cytoplasm (Fig. 4). Therefore, the dual functions (the sequestration of GLI3 in the cytoplasm and the regulation of ciliogenesis through the recruitment of cilia appendage and IFT components to the basal body of cilia) appear to be independent of each other and unique to DZIP1. As discussed above, the retention of the GLI3 weak activator in the cytoplasm by DZIP1 ultimately results in a net increase in the output of overall Gli activating function. The formation and maintenance of functional cilia by ciliary proteins, such as DZIP1, are essential for the activation of GLI2FL/GLI3FL proteins. Therefore, it is through this dual mechanism that DZIP1 positively regulates Hh signaling. To our knowledge, DZIP1 is the first ciliary protein that has been identified so far to regulate Hh signaling in this fashion.
Acknowledgments
We thank Drs. Jerome Rattner, Gregory Pazour, and Brian Dynlacht for Ninein, IFT57 and IFT140, and CEP97 antibodies, respectively. Isl1, Hb9, Pax6, Pax7, Nkx2.2, Nkx6.1, and Foxa2 monoclonal antibodies were obtained from the Developmental Studies Hybridoma Bank maintained by the University of Iowa Department of Biological Sciences under contract NO1-HD-7-3263 from NICHD, National Institutes of Health. Procedures involving mice were performed under an approved protocol.
This work was supported, in whole or in part, by National Institutes of Health Grants R01CA111673 and R01GM070820 (to B. W.).
- IFT
- intraflagellar transport
- ES
- embryonic stem
- En
- embryonic day n
- MEF
- mouse embryo fibroblast
- LMB
- leptomycin B
- aa
- amino acids.
REFERENCES
- 1. Jiang J., Hui C. C. (2008) Hedgehog signaling in development and cancer. Dev. Cell 15, 801–812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Stone D. M., Hynes M., Armanini M., Swanson T. A., Gu Q., Johnson R. L., Scott M. P., Pennica D., Goddard A., Phillips H., Noll M., Hooper J. E., de Sauvage F., Rosenthal A. (1996) The tumour-suppressor gene patched encodes a candidate receptor for Sonic hedgehog. Nature 384, 129–134 [DOI] [PubMed] [Google Scholar]
- 3. Fuse N., Maiti T., Wang B., Porter J. A., Hall T. M., Leahy D. J., Beachy P. A. (1999) Sonic hedgehog protein signals not as a hydrolytic enzyme but as an apparent ligand for patched. Proc. Natl. Acad. Sci. U.S.A. 96, 10992–10999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pan Y., Bai C. B., Joyner A. L., Wang B. (2006) Sonic hedgehog signaling regulates Gli2 transcriptional activity by suppressing its processing and degradation. Mol. Cell. Biol. 26, 3365–3377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wang B., Fallon J. F., Beachy P. A. (2000) Hedgehog-regulated processing of Gli3 produces an anterior/posterior repressor gradient in the developing vertebrate limb. Cell 100, 423–434 [DOI] [PubMed] [Google Scholar]
- 6. Goetz S. C., Anderson K. V. (2010) The primary cilium. A signalling centre during vertebrate development. Nat. Rev. 11, 331–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Endoh-Yamagami S., Evangelista M., Wilson D., Wen X., Theunissen J. W., Phamluong K., Davis M., Scales S. J., Solloway M. J., de Sauvage F. J., Peterson A. S. (2009) The mammalian Cos2 homolog Kif7 plays an essential role in modulating Hh signal transduction during development. Curr. Biol. 19, 1320–1326 [DOI] [PubMed] [Google Scholar]
- 8. Liem K. F., Jr., He M., Ocbina P. J., Anderson K. V. (2009) Mouse Kif7/Costal2 is a cilia-associated protein that regulates Sonic hedgehog signaling. Proc. Natl. Acad. Sci. U.S.A. 106, 13377–13382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Corbit K. C., Aanstad P., Singla V., Norman A. R., Stainier D. Y., Reiter J. F. (2005) Vertebrate Smoothened functions at the primary cilium. Nature 437, 1018–1021 [DOI] [PubMed] [Google Scholar]
- 10. Rohatgi R., Milenkovic L., Scott M. P. (2007) Patched1 regulates hedgehog signaling at the primary cilium. Science 317, 372–376 [DOI] [PubMed] [Google Scholar]
- 11. Haycraft C. J., Banizs B., Aydin-Son Y., Zhang Q., Michaud E. J., Yoder B. K. (2005) Gli2 and Gli3 localize to cilia and require the intraflagellar transport protein polaris for processing and function. PLoS Genet. 1, e53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Quinlan R. J., Tobin J. L., Beales P. L. (2008) Modeling ciliopathies. Primary cilia in development and disease. Curr. Top. Dev. Biol. 84, 249–310 [DOI] [PubMed] [Google Scholar]
- 13. Mogensen M. M., Malik A., Piel M., Bouckson-Castaing V., Bornens M. (2000) Microtubule minus-end anchorage at centrosomal and non-centrosomal sites. The role of ninein. J. Cell Sci. 113, 3013–3023 [DOI] [PubMed] [Google Scholar]
- 14. Nakagawa Y., Yamane Y., Okanoue T., Tsukita S., Tsukita S. (2001) Outer dense fiber 2 is a widespread centrosome scaffold component preferentially associated with mother centrioles. Its identification from isolated centrosomes. Mol. Biol. Cell 12, 1687–1697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lange B. M., Gull K. (1995) A molecular marker for centriole maturation in the mammalian cell cycle. J. Cell Biol. 130, 919–927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Graser S., Stierhof Y. D., Lavoie S. B., Gassner O. S., Lamla S., Le Clech M., Nigg E. A. (2007) Cep164, a novel centriole appendage protein required for primary cilium formation. J. Cell Biol. 179, 321–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pedersen L. B., Rosenbaum J. L. (2008) Intraflagellar transport (IFT) role in ciliary assembly, resorption and signalling. Curr. Top. Dev. Biol. 85, 23–61 [DOI] [PubMed] [Google Scholar]
- 18. Silverman M. A., Leroux M. R. (2009) Intraflagellar transport and the generation of dynamic, structurally and functionally diverse cilia. Trends Cell Biol. 19, 306–316 [DOI] [PubMed] [Google Scholar]
- 19. Cheung H. O., Zhang X., Ribeiro A., Mo R., Makino S., Puviindran V., Law K. K., Briscoe J., Hui C. C. (2009) The kinesin protein Kif7 is a critical regulator of Gli transcription factors in mammalian hedgehog signaling. Sci. Signal. 2, ra29. [DOI] [PubMed] [Google Scholar]
- 20. Bershteyn M., Atwood S. X., Woo W. M., Li M., Oro A. E. (2010) MIM and cortactin antagonism regulates ciliogenesis and hedgehog signaling. Dev. Cell 19, 270–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Callahan C. A., Ofstad T., Horng L., Wang J. K., Zhen H. H., Coulombe P. A., Oro A. E. (2004) MIM/BEG4, a Sonic hedgehog-responsive gene that potentiates Gli-dependent transcription. Genes Dev. 18, 2724–2729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Saarikangas J., Mattila P. K., Varjosalo M., Bovellan M., Hakanen J., Calzada-Wack J., Tost M., Jennen L., Rathkolb B., Hans W., Horsch M., Hyvonen M. E., Perala N., Fuchs H., Gailus-Durner V., Esposito I., Wolf E., de Angelis M. H., Frilander M. J., Savilahti H., Sariola H., Sainio K., Lehtonen S., Taipale J., Salminen M., Lappalainen P. (2011) Missing-in-metastasis MIM/MTSS1 promotes actin assembly at intercellular junctions and is required for integrity of kidney epithelia. J. Cell Sci. 124, 1245–1255 [DOI] [PubMed] [Google Scholar]
- 23. Wolff C., Roy S., Lewis K. E., Schauerte H., Joerg-Rauch G., Kirn A., Weiler C., Geisler R., Haffter P., Ingham P. W. (2004) iguana encodes a novel zinc-finger protein with coiled-coil domains essential for Hedgehog signal transduction in the zebrafish embryo. Genes Dev. 18, 1565–1576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sekimizu K., Nishioka N., Sasaki H., Takeda H., Karlstrom R. O., Kawakami A. (2004) The zebrafish iguana locus encodes Dzip1, a novel zinc-finger protein required for proper regulation of Hedgehog signaling. Development 131, 2521–2532 [DOI] [PubMed] [Google Scholar]
- 25. Jia J., Kolterud A., Zeng H., Hoover A., Teglund S., Toftgård R., Liu A. (2009) Suppressor of Fused inhibits mammalian Hedgehog signaling in the absence of cilia. Dev. Biol. 330, 452–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pan Y., Wang C., Wang B. (2009) Phosphorylation of Gli2 by protein kinase A is required for Gli2 processing and degradation and the Sonic Hedgehog-regulated mouse development. Dev. Biol. 326, 177–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ou Y., Rattner J. B. (2000) A subset of centrosomal proteins are arranged in a tubular conformation that is reproduced during centrosome duplication. Cell Motil. Cytoskeleton 47, 13–24 [DOI] [PubMed] [Google Scholar]
- 28. Spektor A., Tsang W. Y., Khoo D., Dynlacht B. D. (2007) Cep97 and CP110 suppress a cilia assembly program. Cell 130, 678–690 [DOI] [PubMed] [Google Scholar]
- 29. Pazour G. J., Baker S. A., Deane J. A., Cole D. G., Dickert B. L., Rosenbaum J. L., Witman G. B., Besharse J. C. (2002) The intraflagellar transport protein, IFT88, is essential for vertebrate photoreceptor assembly and maintenance. J. Cell Biol. 157, 103–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Briscoe J., Pierani A., Jessell T. M., Ericson J. (2000) A homeodomain protein code specifies progenitor cell identity and neuronal fate in the ventral neural tube. Cell 101, 435–445 [DOI] [PubMed] [Google Scholar]
- 31. Pan Y., Wang B. (2007) A novel protein-processing domain in Gli2 and Gli3 differentially blocks complete protein degradation by the proteasome. J. Biol. Chem. 282, 10846–10852 [DOI] [PubMed] [Google Scholar]
- 32. Robert A., Margall-Ducos G., Guidotti J. E., Brégerie O., Celati C., Bréchot C., Desdouets C. (2007) The intraflagellar transport component IFT88/polaris is a centrosomal protein regulating G1-S transition in non-ciliated cells. J. Cell Sci. 120, 628–637 [DOI] [PubMed] [Google Scholar]
- 33. Chen Z., Indjeian V. B., McManus M., Wang L., Dynlacht B. D. (2002) CP110, a cell cycle-dependent CDK substrate, regulates centrosome duplication in human cells. Dev Cell 3, 339–350 [DOI] [PubMed] [Google Scholar]
- 34. Ishikawa H., Kubo A., Tsukita S., Tsukita S. (2005) Odf2-deficient mother centrioles lack distal/subdistal appendages and the ability to generate primary cilia. Nat. Cell Biol. 7, 517–524 [DOI] [PubMed] [Google Scholar]
- 35. Moore F. L., Jaruzelska J., Dorfman D. M., Reijo-Pera R. A. (2004) Identification of a novel gene, DZIP (DAZ-interacting protein), that encodes a protein that interacts with DAZ (deleted in azoospermia) and is expressed in embryonic stem cells and germ cells. Genomics 83, 834–843 [DOI] [PubMed] [Google Scholar]
- 36. Varjosalo M., Li S. P., Taipale J. (2006) Divergence of hedgehog signal transduction mechanism between Drosophila and mammals. Dev. Cell 10, 177–186 [DOI] [PubMed] [Google Scholar]
- 37. Shin S. H., Kogerman P., Lindström E., Toftgárd R., Biesecker L. G. (1999) GLI3 mutations in human disorders mimic Drosophila cubitus interruptus protein functions and localization. Proc. Natl. Acad. Sci. U.S.A. 96, 2880–2884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ding Q., Fukami S., Meng X., Nishizaki Y., Zhang X., Sasaki H., Dlugosz A., Nakafuku M., Hui C. (1999) Mouse suppressor of fused is a negative regulator of sonic hedgehog signaling and alters the subcellular distribution of Gli1. Curr. Biol. 9, 1119–1122 [DOI] [PubMed] [Google Scholar]
- 39. Kogerman P., Grimm T., Kogerman L., Krause D., Undén A. B., Sandstedt B., Toftgård R., Zaphiropoulos P. G. (1999) Mammalian suppressor-of-fused modulates nuclear-cytoplasmic shuttling of Gli-1. Nat. Cell Biol. 1, 312–319 [DOI] [PubMed] [Google Scholar]
- 40. Murone M., Luoh S. M., Stone D., Li W., Gurney A., Armanini M., Grey C., Rosenthal A., de Sauvage F. J. (2000) Gli regulation by the opposing activities of fused and suppressor of fused. Nat. Cell Biol. 2, 310–312 [DOI] [PubMed] [Google Scholar]
- 41. Tay S. Y., Yu X., Wong K. N., Panse P., Ng C. P., Roy S. (2010) The iguana/DZIP1 protein is a novel component of the ciliogenic pathway essential for axonemal biogenesis. Dev. Dyn. 239, 527–534 [DOI] [PubMed] [Google Scholar]
- 42. Glazer A. M., Wilkinson A. W., Backer C. B., Lapan S. W., Gutzman J. H., Cheeseman I. M., Reddien P. W. (2010) The Zn finger protein Iguana impacts Hedgehog signaling by promoting ciliogenesis. Dev. Biol. 337, 148–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kim H. R., Richardson J., van Eeden F., Ingham P. W. (2010) Gli2a protein localization reveals a role for Iguana/DZIP1 in primary ciliogenesis and a dependence of Hedgehog signal transduction on primary cilia in the zebrafish. BMC Biol. 8, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wen X., Lai C. K., Evangelista M., Hongo J. A., de Sauvage F. J., Scales S. J. (2010) Kinetics of hedgehog-dependent full-length Gli3 accumulation in primary cilia and subsequent degradation. Mol. Cell. Biol. 30, 1910–1922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chen M. H., Wilson C. W., Li Y. J., Law K. K., Lu C. S., Gacayan R., Zhang X., Hui C. C., Chuang P. T. (2009) Cilium-independent regulation of Gli protein function by Sufu in Hedgehog signaling is evolutionarily conserved. Genes Dev. 23, 1910–1928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zeng H., Jia J., Liu A. (2010) Coordinated translocation of mammalian Gli proteins and suppressor of fused to the primary cilium. PLoS One 5, e15900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kim J., Kato M., Beachy P. A. (2009) Gli2 trafficking links Hedgehog-dependent activation of Smoothened in the primary cilium to transcriptional activation in the nucleus. Proc. Natl. Acad. Sci. U.S.A. 106, 21666–21671 [DOI] [PMC free article] [PubMed] [Google Scholar]