

Background: The atypical chemokine receptor CXCR7 is highly expressed in various types of cancer.
Results: CXCR7 Nanobodies were generated and show inhibition of β-arrestin2 signaling and secretion of angiogenic CXCL1 in vitro. Anti-CXCR7 Nanobodies reduce tumor growth by inhibiting angiogenesis.
Conclusion: CXCR7 inhibition by Nanobodies inhibit head and neck tumor formation.
Significance: Anti-CXCR7 therapies are potential novel treatments against head and neck cancer.
Keywords: Antibody Engineering, Chemokines, G Protein-coupled Receptors (GPCR), Head and Neck Cancer, Signaling, CXCR7, Nanobodies
Abstract
The chemokine receptor CXCR7, belonging to the membrane-bound G protein-coupled receptor superfamily, is expressed in several tumor types. Inhibition of CXCR7 with either small molecules or small interference (si)RNA has shown promising therapeutic benefits in several tumor models. With the increased interest and effectiveness of biologicals inhibiting membrane-bound receptors we made use of the “Nanobody platform” to target CXCR7. Previously we showed that Nanobodies, i.e. immunoglobulin single variable domains derived from naturally occurring heavy chain-only camelids antibodies, represent new biological tools to efficiently tackle difficult drug targets such as G protein-coupled receptors. In this study we developed and characterized highly selective and potent Nanobodies against CXCR7. Interestingly, the CXCR7-targeting Nanobodies displayed antagonistic properties in contrast with previously reported CXCR7-targeting agents. Several high affinity CXCR7-specific Nanobodies potently inhibited CXCL12-induced β-arrestin2 recruitment in vitro. A wide variety of tumor biopsies was profiled, showing for the first time high expression of CXCR7 in head and neck cancer. Using a patient-derived CXCR7-expressing head and neck cancer xenograft model in nude mice, tumor growth was inhibited by CXCR7-targeting Nanobody therapy. Mechanistically, CXCR7-targeting Nanobodies did not inhibit cell cycle progression but instead reduced secretion of the angiogenic chemokine CXCL1 from head and neck cancer cells in vitro, thus acting here as inverse agonists, and subsequent angiogenesis in vivo. Hence, with this novel class of CXCR7 inhibitors, we further substantiate the therapeutic relevance of targeting CXCR7 in head and neck cancer.
Introduction
Chemokine receptors and their associated ligands form a complex molecular system that regulates leukocyte trafficking and migration during development as well as inflammatory responses (1). CXCR7 is one of the newest additions to the chemokine receptor family, belonging to the superfamily of G protein-coupled receptors. CXCR7 binds with high affinity the chemokines CXCL11 and CXCL12, which were previously thought to exclusively bind to CXCR3 and CXCR4, respectively (2, 3). Unlike other prototypical G protein-coupled receptors, CXCR7 does not signal via Gαi-proteins but rather signals through β-arrestin in a G protein-independent manner. CXCR7 activates downstream signaling pathways, e.g. mitogen-activated protein kinases (MAPK) through recruitment of this scaffold protein (4). Furthermore, CXCR7 constitutively internalizes and recycles back to the surface in a β-arrestin2-dependent manner (5–9).
Importantly, the role of the chemokine system in cancer is gaining attention. The frequent overexpression of chemokines and chemokine receptors in various tumor types and their involvement in proliferation, metastasis, and angiogenesis have brought about new avenues targeting the chemokine receptors (10). Tumors from various origins including breast, lung, prostate, brain, and kidney showed in particular overexpression of CXCR7 (11–14). In some cases, CXCR7 was also shown to induce proliferation (15) and angiogenesis at the primary tumor site due to its expression in tumor cells and associated blood vessels (11). Recent studies demonstrated how CXCR7 directs trans-endothelial migration of cancer cells (16) and highlights the clinical importance of the CXCR4/CXCR7/CXCL12 axis in glioblastoma (17).
CXCR7 can be therapeutically targeted by non-peptidergic small molecules, siRNA as well as conventional antibodies (3, 11, 18). In the present study we identified a novel class of potential therapeutics targeting CXCR7, llama-derived immunoglobulin single variable domains (Nanobodies) specifically directed against CXCR7. We were the first to show that one can therapeutically target the related chemokine receptor CXCR4 both in vitro and in vivo with CXCR4-targeting Nanobodies (19). Thereafter, Nanobodies targeting the intracellular side of the β2 adrenoreceptor were shown to be valuable tools in crystallization of the active state of the β2 adrenoreceptor (20). Nanobodies are novel antibody-based therapeutics derived from the single variable domain (VHH) of heavy chain antibodies found in the Camelidae family, e.g. llamas and camels. Their relatively small size (12–15 kDa) and high solubility allows them to cross tissue barriers more easily than traditional immunoglobulin (e.g. 150-kDa IgG human antibodies). Furthermore, Nanobodies present low immunogenicity, physical stability (21), and are easily produced in prokaryotic or eukaryotic host organisms (22). Using DNA and whole cell immunization, we developed several CXCR7-specific Nanobodies targeting the extracellular site of the receptor that functionally antagonized CXCR7. As for CXCR4 Nanobodies, we also demonstrated that multivalent formatting of Nanobodies improved their potency (19). Using a patient-derived CXCR7 expressing head and neck cancer xenograft model in nude mice, we showed that the CXCR7 Nanobodies inhibit tumor growth by inhibiting angiogenesis. Hence, by introducing this novel class of potential CXCR7 therapeutics, we substantiate the clinical relevance of targeting CXCR7 in head and neck cancer.
EXPERIMENTAL PROCEDURES
Generation of Nanobodies
Llamas were immunized four times with 2-week intervals with either CXCR7-expressing HEK293 cells or with pVAX1-CXCR7DNA (2 mg/injection) via jet injection (Akra DermoJet). Three weeks after the final DNA immunizations, llamas received a boost with whole CXCR7-expressing human kidney cells. After the last genetic immunization as well as after the final cell boost, peripheral blood lymphocytes were collected. Total RNA extracted from peripheral blood B cells was used to amplify Nanobody-encoding fragments. cDNA products were subsequently subcloned into phagemid vectors pAX50 to generate phage display libraries, where the phage particles express individual Nanobodies as a fusion protein with a C-terminal His6-Myc tag and with the Gene-III protein. Selection of the resulting immune libraries was performed by two rounds of panning on CXCR7 virus-like lipoparticles (Integral Molecular) or on CXCR7-expressing Caki and NIH-3T3 cells, alternating cell background. Individual phage clones of selected outputs were generated to verify specific CXCR7 binding in a phage ELISA on CXCR7 virus-like lipoparticles. Binding to cell-expressed CXCR7 was further verified by binding of Nanobodies in crude periplasmatic extracts of HEK293-CXCR7 cells by detecting the associated Myc tag. Selected CXCR7 Nanobodies were recloned in an Escherichia coli expression vector pAX100 and expressed as C-terminal-linked myc-His6-tagged proteins for further characterization. Expression in E. coli was induced by isopropyl 1-thio-β-d-galactopyranoside and allowed to continue for 4 h at 37 °C. After spinning the cell cultures, periplasmic extracts were prepared by freeze-thawing of the cell pellets. Nanobodies were purified from these extracts using immobilized metal affinity chromatography (IMAC) and a buffer exchange to Dulbecco's PBS.
Multivalent Nanobodies were constructed with one or two N-terminal CXCR7-specific building blocks and a C-terminal human serum albumin-specific building block (Alb8), providing the Nanobodies with an extended half-life in vivo. The building blocks are connected with a flexible (GGGGS)7 linker. Different combinations of CXCR7 Nanobodies were explored, combining either two identical CXCR7 Nanobodies or two distinct Nanobodies. Bivalent and trivalent Nanobodies were expressed with His6 tag extension in Pichia pastoris strain XL-33 in small scale productions. Nanobodies were purified from the medium fraction using immobilized metal affinity chromatography and a buffer exchange to Dulbecco's PBS.
Specific binding of purified Nanobody proteins to human CXCR7 was verified by FACS analysis. Hereto serial dilutions of purified proteins (concentration range: 400 nm–180 pm) were incubated with stable HEK-CXCR7 cells for 30 min at 4 °C, and binding was detected using anti-mouse anti-myc (Serotec) and anti-mouse IgG-PE (Jackson ImmunoResearch). Similarly, cross-reactive binding to the mouse orthologues was assessed by transient transfected HEK293 cells with pCDNA3.1-mCXCR7 in FACS analysis.
Cells
NIH-3T3 and HEK293(T) cells were grown in DMEM (PAA) supplemented with 1% penicillin/streptomycin (PAA) in the presence of 10% calf serum (Invitrogen) and fetal bovine serum (Integro), respectively. CXCR7 stable HEK293 and NIH-3T3 cell lines were grown in the presence of 400 μg/ml selection antibiotic G418 (PAA). Transient transfection of mouse CXCR7 was performed in HEK293T cells using the polyethyleneimine method as previously described (23). Head and neck cancer cell lines (22A, 22B) were obtained from T. Carey (University of Michigan), FaDu cells were derived from ATCC, and VU-SCC-OE were generated at the VU University Medical Center. All head and neck cancer cell lines were grown in DMEM supplemented with 25 mm HEPES (Lonza) in the presence of 10% fetal bovine serum and 1% penicillin/streptomycin.
Radioligand Binding
CXCR7-expressing NIH-3T3 cells were seeded in regular growth medium on poly-l-lysine-coated 96-well plates. The following day binding buffer (50 mm Hepes, pH 7.4, 1 mm CaCl2, 5 mm MgCl2, 0.1 m NaCl) supplemented with 0.5% BSA was added to the cells in the absence or presence of either chemokine (10−7 m) or CXCR7-specific nanobodies (10−6 m). Subsequently, radiolabeled 125I-CXCL12 (PerkinElmer Life Sciences) was added to reach a final concentration of 75 pm. Cells were incubated for 3 h at 4 °C then washed twice with binding buffer containing 0.5 m NaCl. After harvesting the samples with lysis buffer, the remaining cell-bound radioactivity was counted.
β-Arrestin2 Recruitment Assay
HEK293T cells were transfected with 1 μg of CXCR7 and 4 μg of β-arrestin2 plasmids genetically fused to Renilla Luciferase (RLuc) and enhanced yellow fluorescent protein, respectively. 24 h post transfection, cells were harvested and seeded on white 96-well plates coated with Poly-l-lysine (Sigma). The following day cells were washed with Hanks' balanced salt solution and further incubated for 30 min in fresh Hanks' balanced salt solution buffer before the measurement of YFP fluorescence. For the antagonism study, the Nanobodies were added to the cells in desired concentrations and incubated for 30 min. The RLuc substrate coelenterazine-h (Promega) was diluted in Hanks' balanced salt solution supplemented with 0.05% BSA and added at a final concentration of 5 μm. After 5 min if incubation, CXCL12 was then added at a final concentration of 31.6 nm and incubated for 15 min before the measurement of fluorescence and luminescence. For the agonistic assay, the Nanobodies were added instead of CXCL12 as described above. Bioluminescence resonance energy transfer (BRET)3 ratio was calculated as the ratio between light emission at 535 and 460 nm and normalized against the maximal BRET ratio signal increase.
Quantitative RT-PCR Analysis
Total RNA was extracted from head and neck cancer cell lines and primary keratinocytes with the RNeasy kit from Qiagen according to the manufacturer's protocol. Messenger (m)RNA was converted into cDNA using the Bio-Rad iScript cDNA synthesis kit. Subsequently, mRNA expression levels were detected with SYBR Green (Bio-Rad) using CXCR7 and β-actin-specific primers from Origene. CXCR7 expression levels were normalized against those of β-actin to allow comparison of the different cell lines.
Immunofluorescence
Fadu or 22A cell-derived xenograft sections (8 μm) were fixed in −20 °C acetone. After blocking with 10% BSA TBS solution, primary antibodies (C1C2 polyclonal anti-CXCR7 antibody 1:25, Genetex; anti-ki67, 1:250, Abcam; anti-CD31, 1:100, BD Pharmingen #550274) were incubated for 1 h at room temperature in blocking buffer. Sections were washed with PBS and incubated for 30 min at room temperature with goat anti-rabbit Alexa 488 or anti-rat Alexa 546 (1:500, Invitrogen). Sections were mounted with DAPI-containing Vectashield medium (Vector Laboratories). Fluorescence was visualized using a Nikon Eclipse TE200 microscope and processed with XM10 camera and Cell̂B imaging software (Olympus). CD31 staining intensities were quantified using Image-Pro Premier (Media Cybernetics), and unpaired t test analysis (p < 0.05) was used to determine the significance with the GraphPad Prism software (San Diego, CA).
Competition FACS
Transiently transfected HEK293T-hCXCR7 cells were incubated simultaneously with 20 nm allophycocyanin-labeled monoclonal antibody 11G8 (R&D Systems) and with a dilution range of Nanobodies or monoclonal antibodies for 2 h at 4 °C using a standard FACS protocol.
Cell Cycle Assay
22A cells were grown on a 6-well plate (75,000 cells/well). The next day cells were synchronized in serum-free medium for 24 h and stimulated for another 24 h on full growth medium with or without stimuli (1 nm CXCL12, 1 μm NB4, 10 μg/ml epidermal growth factor (EGF), or 10 μg/ml Erbitux). Cells were stained with propidium iodine, and cell cycle populations were determined by using the Guava EasyCyte system according to the manufacturer's recommendations (Millipore). The Guava Cell Cycle software was used to determine the cell populations in the different cell cycle phases, and the propidium iodine was quantified from the (%S + %G2M)/%G0G1 ratios as previously described (24).
Human Angiogenesis Antibody Array
22A cells were grown on a 12-well plate (200,000 cells/well). The next day the medium was removed, and cells were incubated on serum-free medium with or without stimuli (1 μm NB4) for 48 h. The supernatant was subsequently collected, centrifuged to remove cellular debris, and used undiluted in a human angiogenesis array (RayBiotech, AAH-ANG-1–4) according to the manufacturer's instructions. Relative amounts of cytokines and chemokines involved in angiogenesis were normalized against positive controls present on the array.
Chemokine ELISA
22A cells were seeded in a 12-well plate (200,000 cells/well). The next day the medium was removed, and cells were incubated with serum-free medium with or without stimuli (1 μm NB4) for 72 h. The supernatant was subsequently collected, centrifuged to remove cellular debris, and used 2-times diluted for the detection by ELISA of human CCL5, CXCL1 (RayBiotech), and CXCL2 (AssaybioTech) chemokines according to the manufacturer's instructions.
Animal Experiment
All animal experiments were conducted according to the NIH principles of laboratory animal care and Dutch national law (“Wet op de Dierproeven” (Stb 1985, 336)), approved by the Dierexperimentencommissie from the VU University Medical Center and performed in compliance with the protocol FaCh 10-01. Head and neck cancer cells (22A) or Fadu cells were injected subcutaneously in the flanks of 8–10-week old female donor nude mice (Hsd, athymic nu/nu, Harlan Laboratories). Xenograft tumors derived from 22A cells were grown to a size of 200–500 mm3 and were subsequently excised, cut in smaller pieces of equal size, and transplanted subcutaneously in the flanks of recipient nude mice. When transplanted tumors properly engrafted, mice were injected intraperitoneally biweekly with either PBS or 1.5 mg of trivalent Nanobody. Tumor growth was measured biweekly with a caliper.
Tumor Screening for CXCR7 Expression
Profiling of CXCR7 protein expression in human tumor biopsies was performed at Oncotest (Freiburg, Germany). Briefly, human primary tumors of variable cancer types were passaged one time in nude mice. Paraffin-embedded tumors were cut into 5-μm sections (with a Leica RM 2135 microtome) and stained with hematoxylin and eosin. Subsequently, one representative region of 1-mm diameter was extracted, and a tissue microarray was prepared as previously described (25). Paraffin-embedded sections were then analyzed for immunohistochemical staining of CXCR7. After removal of paraffin, inactivation of endogenous peroxidase with 3% H2O2 and blocking of unspecific binding with 10% BSA in PBS, the anti-human/mouse CXCR7 monoclonal antibody (Biolegend, clone Mab 8F11) or an isotype control antibody (Biolegend, IgG2b) was incubated at a concentration of 25 μg/ml. The secondary antibody goat anti-mouse biotinylated IgG (Jackson ImmunoResearch) was incubated at a final concentration of 2.8 μg/ml, and detection was performed with the ABC solution and peroxidase substrate of the Vectastain ABC kit (Vector). Tissues were finally counterstained with hematoxylin. The tissue microarray was evaluated semi-quantitatively using a Zeiss Axiovert 35 microscope. Photographs were taken with a Zeiss AxioCam MRc camera. All tumor samples were evaluated in duplicate. Staining was interpreted based on the proportion of positively stained cells as well as on the signal intensity. Samples were grouped in the following categories: 0, no staining (antigen absent); 1, weak staining; 2, moderate staining; 3, strong staining.
RESULTS
Generation of CXCR7 Nanobodies
To ensure native human CXCR7 protein folding and proper antigen presentation, llamas were immunized using two strategies. Similarly to our previous work (19), llamas were immunized with HEK293 cells stably expressing human CXCR7. In parallel, genetic immunizations were done with a plasmid encoding human CXCR7 DNA subsequently followed by a single boost with CXCR7-expressing camel kidney cells. Phage display libraries were generated from the peripheral blood mononuclear cells collected after the final immunizations and used for selection of CXCR7 binding clones using CXCR7-expressing virus-like lipoparticles. CXCR7 binding of individual Nanobodies from selected outputs after two rounds of selection was verified in phage ELISA on virus-like lipoparticles expressing CXCR7 or control vector, and specific interaction was confirmed in FACS on CXCR7-expressing cells. This led to the identification of 78 unique CXCR7-specific Nanobody sequences belonging to 45 distinct Nanobody B-cell lineages (data not shown). Selected CXCR7-specific binders were subcloned into bacterial expression vectors and further analyzed as purified Nanobodies in 125I-CXCL12 displacement assays on CXCR7 expressing NIH-3T3 cells. Purified Nanobodies (10−6 m) displaced 125I-CXCL12 from CXCR7-expressing NIH-3T3 cells to different extents varying from 10–20% up to full displacement (Fig. 1A and Table 1). As a negative control, an unrelated Nanobody showed no displacement of 125I-CXCL12 binding to CXCR7-expressing cells. Next, the CXCR7 Nanobodies showing more than 50% displacement of 125I-CXCL12 from CXCR7-expressing cells were further characterized. We focused on two strong displacers, namely NB2 and NB3, and a weaker displacer NB1 for full binding characterization (Fig. 1B). Increasing concentrations of CXCR7 Nanobodies displaced 125I-CXCL12 in a dose-dependent manner from CXCR7-expressing cells with potencies varying from a nanomolar (pKi NB3 = 8.6 ± 0.2) to submicromolar (pKi NB1 = 7.8 ± 0.3) range (Table 1). Importantly, CXCR7 Nanobodies were able to displace 125I-CXCL12 with a potency comparable to the CXCR7-specific monoclonal IgG antibody 8F11 (pKi = 8.4 ± 0.2) or low molecular compound VUF11403 (pKi = 8.3 ± 0.1) (26). NB1 only partially displaced 125I-CXCL12 from CXCR7-expressing cells. To confirm a specific interaction of CXCR7 Nanobodies to CXCR7, we tested their ability to displace 125I-CXCL12 from cells expressing the related chemokine receptor CXCR4. Although CXCR4-specific inhibitor AMD3100 fully displaces 125I-CXCL12 from CXCR4, we observed no displacement of 125I-CXCL12 from CXCR4-expressing cells with these novel CXCR7 Nanobodies, confirming their specificity toward CXCR7 and not CXCR4 or CXCL12 (Fig. 1C).
FIGURE 1.

Selection and characterization of CXCR7-specific Nanobodies. A, 125I-CXCL12 displacement screening of purified monoclonal Nanobodies (10−6 m) on hCXCR7-expressing NIH-3T3 cells. Unlabeled human CXCL12 and a CXCR7-unrelated Nanobody were used as controls to determine the specificity of the radioligand and control for non-specific effects of Nanobodies. B, 125I-CXCL12 displacement assay on stable hCXCR7-expressing NIH-3T3 cells with cold CXCL12, CXCR7-specific Nanobodies (NB), monoclonal antibody (mAb) 8F11, and small non-peptidergic compound VUF11403. C, 125I-CXCL12 radioligand binding assay on membrane extracts from HEK293T cells transiently transfected with hCXCR4.
TABLE 1.
Affinities (pKi) and potencies (pIC50) of CXCR7-specific reagents in 125I-CXCL12 displacement and CXCL12-induced β-arrestin2 recruitment assays (at least n = 3) on human CXCR7
NA., not applicable.
| CXCR7 reagents |
pKi |
pIC50 |
|||||
|---|---|---|---|---|---|---|---|
| Nanobody | Nanobody structure | Average | S.E. | Displacement | Average | S.E. | Inhibition |
| % | % | ||||||
| NB1 | 7.8 | 0.3 | 60 | NA | NA | 8 | |
| NB2 | 8.0 | 0.1 | 92 | 7.7 | 0.4 | 90 | |
| NB3 | 8.6 | 0.2 | 90 | 8.7 | 0.2 | 96 | |
| NB4 | NB3–35GS-NB1–35GS-Alb8 | 9.3 | 0.1 | 93 | 9.7 | 0.0 | 100 |
| NB5 | NB2–35GS-NB2–35GS-Alb8 | 8.5 | 0.1 | 85 | 8.3 | 0.1 | 100 |
| VUF11403 | 8.3 | 0.1 | 84 | NA | NA. | NA | |
| mAb-8F11 | 8.4 | 0.2 | 79 | 8.7 | 0.1 | 95 | |
CXCR7 Nanobodies Inhibit CXCL12-induced β-Arrestin2 Recruitment
Having confirmed the specific binding of Nanobodies on human CXCR7, we next investigated their inhibitory capacities in the CXCR7/β-arrestin2 signaling axis (4). Besides the two endogenous ligands of CXCR7, CXCL12 and CXCL11, several reported CXCR7-specific low molecular weight molecules are also able to induce β-arrestin2 recruitment toward CXCR7 (4, 9, 26). Thus, we evaluated whether the CXCR7-targeted Nanobodies would behave as CXCR7 agonists or act as antagonists in a β-arrestin2 recruitment assay. For this BRET-based CXCR7/β-arrestin2 assay, cells were transiently transfected with CXCR7-RLuc and β-arrestin2-enhanced YFP and stimulated with either CXCL12, the CXCR7 ligand VUF11403 (26), or CXCR7-targeting Nanobodies. As expected, both CXCL12 and VUF11403 induced the recruitment of β-arrestin2 to CXCR7 (Fig. 2A). However, Nanobodies were unable to induce an agonistic BRET signal at a receptor-saturating concentration of 10−5 m (Fig. 1B), indicating that monovalent Nanobodies are devoid of agonistic properties. Subsequently, the Nanobodies were evaluated for antagonist properties by pretreating cells with the Nanobodies before CXCL12 stimulation (10−7.5 m). As shown in Fig. 2B, Nanobodies NB2 and NB3 effectively inhibited CXCL12-induced β-arrestin2 recruitment to CXCR7 with a high potency (pIC50 = 7.7 ± 0.4 and 8.7 ± 0.2, respectively). However, NB1 was not able to inhibit recruitment of β-arrestin2 to CXCR7, demonstrating that NB2 and NB3, but not NB1, behave as potent antagonists on the CXCR7/β-arrestin 2 axis.
FIGURE 2.
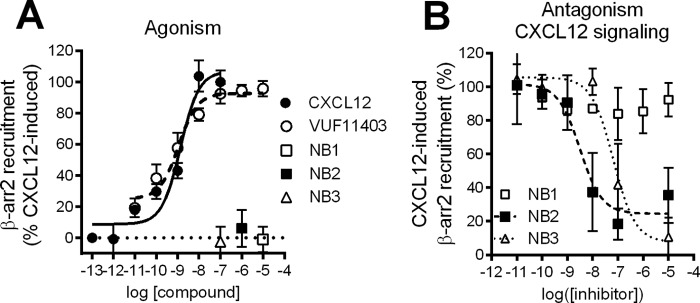
Antagonistic properties of Nanobodies in signaling assays. A, BRET-based assay in CXCR7-RLuc and β-arrestin2-YFP-expressing HEK293T cells stimulated with CXCL12, low molecular weight compound VUF11403, or Nanobody NB1, NB2, or NB3. B, hCXCR7/β-arrestin2 BRET assay in HEK293T cells preincubated with dose-dependent concentrations of NB1, NB2, or NB3 before stimulation with CXCL12 (10−7.5 m).
Formatted Nanobodies Display Enhanced Affinity and Potency
To define whether the identified Nanobodies targeting CXCR7 bind to similar epitopes, we performed FACS-based competition assays with the monoclonal antibody 11G8, known to bind the N terminus of CXCR7 (27). As shown in Fig. 3A, NB1 fully displaced binding of 11G8 to CXCR7, whereas NB2 and NB3 only partially displaced 11G8. This suggests that NB2 and NB3 bind to domains distinct from the epitope recognized by NB1, which is in line with the 125I-CXCL12 displacement data (Fig. 1B). This was further confirmed by the inability of NB1 to displace Alexa647-labeled NB3 from CXCR7-expressing cells (data not shown). Next, we engineered multivalent Nanobody constructs with repetitive GS sequences since this approach had previously led to a gain of function for the CXCR4 Nanobodies (19). In addition, CXCR7 Nanobodies were formatted by genetically linking them to a Nanobody binding serum albumin (referred to as Alb8) to increase their half-life for in vivo studies (28). Using the epitope data, we linked NB1 and NB3 as they bind to different epitopes and coupled them to Alb8, generating the formatted NB4. The formatted NB5 was composed of a bivalent NB2 coupled to Alb8 (Table 1). Binding studies on hCXCR7-expressing NIH-3T3 cells revealed that the trivalent Nanobodies NB4 and NB5 are able to displace radiolabeled 125I-CXCL12 from human CXCR7 (Fig. 3B). Notably, NB4 showed increased affinity (pKi = 9.3 ± 0.1) toward CXCR7 compared with the monovalent Nanobodies (Table 1). To examine whether CXCR7 Nanobodies also cross-react with the mouse counterpart, of potential importance for in vivo studies, we performed a displacement binding experiment on cells expressing mouse CXCR7. NB4 was able to fully displace 125I-CXCL12 from mouse CXCR7 with only a slightly lower potency (pKi = 8.4 ± 0.1) compared with human CXCR7. In contrast, NB5 only partially (70%) displaced 125I-CXCL12 from mouse CXCR7 while being a full displacer on the human receptor (Fig. 3C). Moreover, the affinity of NB5 for the mouse CXCR7 was lower when compared with the human CXCR7 (pKi = 8.5 ± 0.1 for human, pKi = 7.6 ± 0.2 for mouse). Next, the formatted Nanobodies were assayed for their capacity to inhibit CXCL12-induced β-arrestin2 recruitment (Fig. 3D). Both NB4 and NB5 were able to inhibit CXCL12-induced β-arrestin2 recruitment, with NB4 showing a 10-fold higher potency compared with NB5 (pIC50 = 9.7 ± 0.0 and 8.3 ± 0.1, respectively). Overall, NB4 and NB5 constructs display improved affinities and potencies toward CXCR7 compared with their monovalent counterparts.
FIGURE 3.
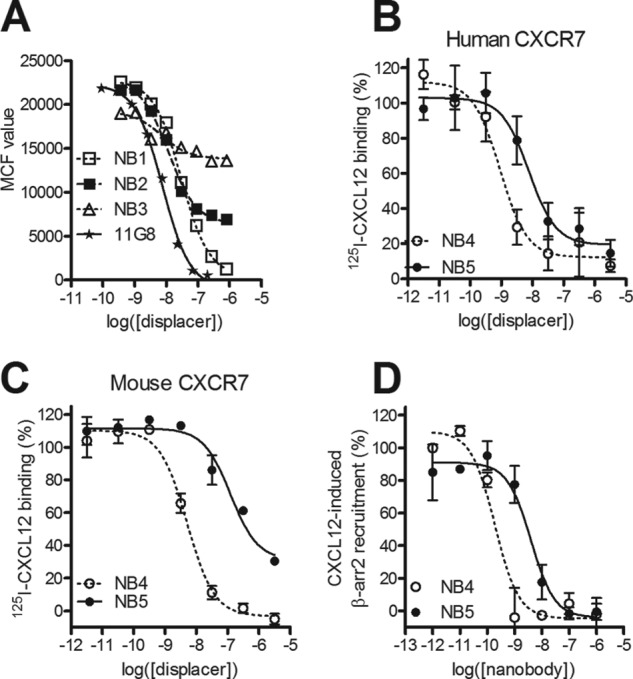
Formatting of Nanobodies for in vivo study. A, FACS-based competition assay with allophycocyanin-labeled monoclonal antibody 11G8 in transiently transfected HEK293T cells with hCXCR7 in the presence of increasing concentrations of Nanobodies. MCF, mean channel fluorescence. B–C, 125I-CXCL12 displacement assay with NB4 (NB3–35GS-NB1-35GS-Alb8) and NB5 (NB2–35GS-NB2–35GS-Alb8) Nanobodies on NIH-3T3 cells stably expressing human CXCR7 (B) or on HEK293T cells transiently transfected with mouse CXCR7 (C). Increasing concentrations of formatted trivalent Nanobodies were used to displace 125I-CXCL12. D, inhibition of CXCL12-induced (10−9 m) β-arrestin2 recruitment by formatted Nanobodies NB4 and NB5.
CXCR7 Is Highly Expressed in Head and Neck Cancer Biopsies
To obtain insight in the expression pattern and role of CXCR7 in human tumor biology and test the novel CXCR7 Nanobodies, we screened several tumor biopsies for CXCR7 protein expression. Immunohistochemistry with the monoclonal IgG antibody 8F11 was performed, and staining intensity was scored to distinguish between CXCR7 non (grade 1)-, low (grade 2)-, or high-expressing (grade 3) tumors (Fig. 4A). As previously documented, CXCR7 staining was not solely membranous but also intracellular (29), which may be due to its intrinsic ability to constitutively internalize and recycle back to the cell surface (6). CXCR7 displayed a non-uniform expression in various human tumors. Biopsies from colon or gastric cancer showed poor expression of CXCR7, namely 17 and 33% of tumors with a staining score of at least 2, respectively (Fig. 4, A and B). In contrast, biopsies from head and neck (Fig. 4, C and D), melanoma, and non-small cell lung cancer displayed a strong CXCR7 protein expression (100, 83, and 93% of tumors stained with a score of at least 2, respectively). These data highlight a potential novel tumorigenic role of CXCR7 in head and neck cancer.
FIGURE 4.

CXCR7 expression in human tumor biopsies. A, expression of CXCR7 protein was determined by immunohistochemistry with the monoclonal antibody 8F11 in human cancer biopsies (non-small cell lung cancer (NSCLC)). Staining intensity was ranked from weak (1) to average (2) and high (3). B, representative marginal CXCR7 immunohistochemical staining in colon cancer biopsy with 8F11 monoclonal anti-CXCR7 antibody. C–D, representative CXCR7 immunohistochemical staining in head and neck cancer biopsy in the absence (C) or presence (D) of anti-CXCR7 antibody.
Head and Neck Cancer Cell Line 22A as CXCR7-expressing in Vitro and in Vivo Model System
To confirm CXCR7 expression in head and neck cancer cells lines, we screened four previously described head and neck squamous cell carcinomas cell lines (30). Quantitative RT-PCR analysis was performed on RNA extracted from 11B, 22A, FaDu, and VU-SCC-OE cell lines. Two of the four tested cell lines displayed CXCR7 mRNA expression, namely the 22A and VU-SCC-OE cell lines (Fig. 5A). Because CXCL12, highly expressed at tumor sites (31), is also known to bind CXCR4, we also determined CXCR4 mRNA expression levels in these cell lines. As can be seen in Fig. 5A, CXCR4 mRNA levels in all four examined cell lines are low compared with CXCR7 mRNA levels. In contrast, primary keratinocytes isolated from healthy tissue do not show any expression of CXCR7 or CXCR4 mRNA (Fig. 5A). To confirm CXCR7 expression at the protein level, we conducted 125I-CXCL12 displacement studies on these cell lines. In accordance to our quantitative RT-PCR data, CXCL12 and CXCL11 were both able to displace 125I-CXCL12 binding to 22A and VU-SCC-OE cell lines. The displacement observed with both CXCL11 and CXCL12 also demonstrates that only CXCR7 and not CXCR4 protein is expressed in these cell lines. As expected, NB4 was also able to inhibit 125I-CXCL12 binding, confirming CXCR7 protein expression on the cell surface (Fig. 5B). To establish an in vivo tumor model, we focused our efforts on the 22A cell line, which had been reported to grow as xenograft in nude mice (32). 22A cells were injected in the flanks of nude mice, and growing viable tumors were measured from 20 days post-injection. At 61 days post-injection, 22A tumors reached an average size of 278 ± 64 mm3 (Fig. 5C). At that stage, CXCR7 protein expression was investigated by immunofluorescence. A rabbit polyclonal anti-CXCR7 antibody (C1C2, GeneTex) was used instead of mouse monoclonal antibodies to avoid the use of a secondary anti-mouse antibody that might react with endogenous murine immunoglobulins. To ensure the specificity of this antibody, Fadu xenograft sections that do not express CXCR7 protein were also stained with the antibody and showed no CXCR7 expression (Fig. 5D). 22A xenograft sections displayed a strong and non-homogenous expression of CXCR7, reflecting the differentiated nature of the tumor with a mixture of epithelial cells (CXCR7-positive) and stromal cells (Fig. 5D). This pattern was similar to what was observed in human biopsies (Fig. 4C). We conclude that 22A cells grown as xenograft express CXCR7 protein.
FIGURE 5.

CXCR7 expression in head and neck cancer cells. A, human head and neck cancer cell lines and primary keratinocytes were tested for CXCR7 and CXCR4 mRNA expression by quantitative RT-PCR. Expression was related to the β-actin content. A.U., arbitrary units; OE, VU-SCC-OE. B, 125I-CXCL12 radioligand binding with head and neck cancer cells. Specific CXCR7 expression was confirmed using the cold chemokines CXCL12 and CXCL11 and CXCR7-specific NB3. TB, total binding. C, growth rate of 22A cells injected subcutaneously in the flank of nude mice. D and E, immunofluorescence staining of CXCR7 protein with C1C2 polyclonal antibody on Fadu-derived (D) and 22A-derived (E) xenograft tumors developed in nude mice.
Formatted CXCR7 Nanobodies Inhibit Secretion of Angiogenic Factor CXCL1 but Not Cell Cycle Progression
Next, we tested whether Nanobodies inhibit cell cycle progression of 22A cells, focusing solely on NB4 due to its enhanced affinity and potency compared with NB5. Stimulation of 22A cells with CXCL12 (10−9 m) did not affect cell cycle progression in a significant manner, nor did NB4 (10−6 m) (Fig. 6A). As a positive control, 22A cells were stimulated with EGF (10 μg/ml) as it was reported earlier that these cells are EGF-sensitive (33). As expected, EGF stimulation led to a significant increase in cell cycle progression. Equally, blocking endogenous EGFR signaling with the EGFR-specific monoclonal antibody Erbitux (10 μg/ml) significantly decreased cell cycle progression. Thus, in contrast to EGFR, CXCR7 did not influence 22A cell cycle progression. Finally, an angiogenesis antibody array was used to determine whether NB4 treatment would affect the secretion of angiogenic factors from 22A cells. 22A cells secrete several angiogenic factors under basal (unstimulated) conditions, which was unaffected by stimulation with 10−9 m CXCL12 (data not shown). Only NB4 treatment led to a significant decrease of basal CXCL1–3 and CCL5 secretion (Fig. 6B). To further confirm these findings, we determined the secretion of CXCL1, CXCL2, and CCL5 by ELISA. We validated the decrease of CXCL1 secretion by treatment with NB4, whereas CXCL2 secretion remained unaffected, and CCL5 secretion was significantly increased (Fig. 6C). Overall, our data demonstrate that CXCR7 displays basal receptor activity associated with the secretion of chemokines, which can be modulated by the CXCR7 Nanobodies in vitro.
FIGURE 6.
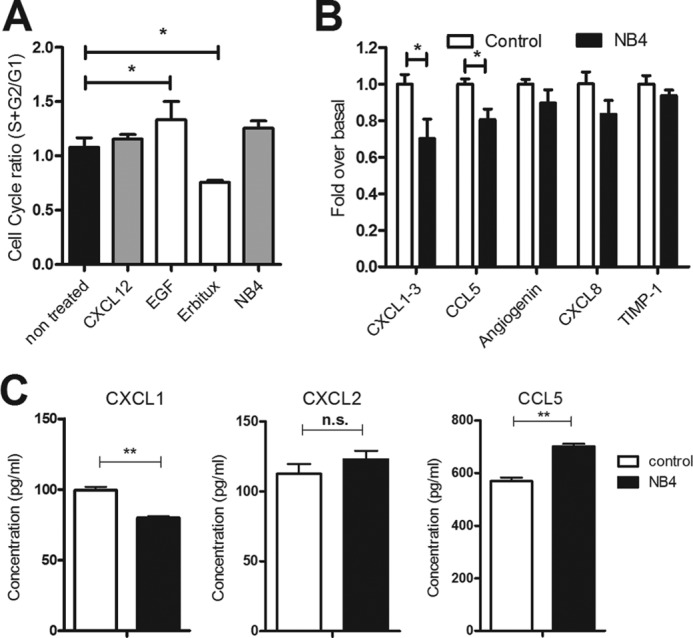
Effect of Nanobodies on cell cycle progression of cancer cells and release of angiogenic factors. A, cell cycle analysis of 22A cells in the absence or presence of CXCR7 (CXCL12 or NB4) or EGFR (EGF ligand or EGFR-specific monoclonal antibody Erbitux) reagents. B, secretion of angiogenic factors from 22A cells in the absence or presence of NB4 (10−6 m). Only CXCL1–3, CCL5, angiogenin CXCL8, and TIMP1 were detected in this array and were, therefore, quantified (p < 0.05 in an unpaired t test comparison). C, validation of angiogenesis array by the use of ELISA. Secretion of CCL5, CXCL1, and CXCL2 was measured in 22A cells in the absence or presence of NB4 (10−6 m) for 72 h. n.s., not significant. *, p < 0.05, **, p < 0.01 in unpaired t test treated versus control cells.
CXCR7 Nanobodies Inhibit Tumor Growth and Angiogenesis in Vivo
After determining the effects of Nanobodies in vitro, we evaluated the therapeutic potential of CXCR7 Nanobodies in vivo. To ensure that mice from different groups (treated versus non-treated) presented similar initial tumor sizes for the therapy experiment, we performed tumor transplantations. First, donor nude mice were initially subcutaneously injected with 2 × 106 22A cells in their flanks. Tumors were grown to a size of 200–500 mm3 and subsequently extracted, cut in smaller pieces of equal size, and transplanted subcutaneously in recipient nude mice. When tumors engrafted and started growing, mice were randomly distributed into five groups that were injected biweekly with 400 μl of PBS without or with 1.5 mg of CXCR7-specific Nanobodies. Over a period of 50 days of therapy, the control (PBS) and NB5-treated groups grew tumors to a similar extent (Fig. 7A). However, mice treated with NB4 displayed a slower tumor growth and significantly smaller size compared with PBS-injected mice at the end of the therapy experiment (PBS tumors size = 274 ± 47 mm3, NB4 tumors size = 119 ± 30 mm3). Because during the therapy period, no weight loss was observed for any Nanobody-injected animals, the tumor growth inhibitory effect of NB4 could not be attributed to toxicity issues. To identify the in vivo mode of action of Nanobodies, tumors were stained for the angiogenesis marker CD31 (Fig. 7B). CD31 staining was present in the periphery of the tumor, and CD31 staining pattern was strongly reduced in NB4-treated mice compared with PBS-treated mice. Quantification of the staining intensities in 10 independent tumors from both PBS- and NB4-treated groups confirmed that CD31 is significantly decreased in mice receiving NB4 therapy (Fig. 7C).
FIGURE 7.
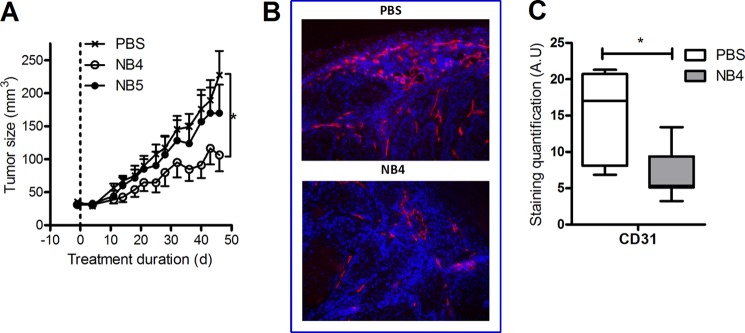
Nanobodies reduce in vivo xenograft growth by reducing angiogenesis. A, nude mice transplanted with 22A tumors were treated biweekly for 7 weeks with PBS (cross), NB4 (open circle), or NB5 (closed circle). Tumor growth was followed by measuring xenograft size with a caliper and was significantly slower in mice treated with NB4 compared with PBS or NB5. B–C, tumor sections from PBS- or NB4-treated mice were stained for the endothelial cell marker CD31. The staining was quantified, and significantly less CD31 staining was observed in NB4-treated tumors compared with PBS-treated tumors (p < 0.05 in an unpaired t test comparison) (C). A.U., arbitrary units.
DISCUSSION
Nanobodies represent a novel class of potential antibody-based therapeutics and have been successfully developed against several drug targets. Llama-derived immunoglobulin single variable domain antibodies have proven to be an excellent platform to use in cancer drug research, either as therapy or as a diagnostic tool (34). Therapeutic Nanobodies have been generated against cancer-specific drug targets such as the receptor tyrosine kinases EGFR/Erbb1 (35–38), HER2 (39), c-Met (28), and VEGFR2 (40) and more recently against the chemokine receptor CXCR4 (19). Based on our experience with CXCR4 (19), we identified therapeutic and high affinity Nanobodies against the other CXCL12 receptor, i.e. CXCR7 (Fig. 1). After in vivo whole cell and DNA immunization, 125I-CXCL12 radioligand displacement screening on whole CXCR7-expressing cells allowed us to select potent Nanobodies with affinities in the nanomolar range. To functionally characterize these Nanobodies, we used a β-arrestin2 BRET recruitment assay, as CXCR7 is devoid of the classical G protein-mediated signaling (4). Interestingly, unlike our CXCR7-specific small molecule VUF11403 (26), the majority of Nanobodies act as antagonists on CXCR7 and do not induce recruitment of β-arrestin (Fig. 2). As such, the low molecular weight agonist and antibody-derived therapeutics present opposite effects on CXCR7-mediated recruitment of β-arrestin2.
Previously, we observed that different CXCR4 Nanobodies generated from whole cell immunization bind to distinct epitopes, and coupling of Nanobodies increases their potency (19). Differential displacement of the allophycocyanin-labeled monoclonal antibody 11G8, known to bind the N terminus of CXCR7 (27), by the Nanobodies (Fig. 3) and the inability of NB1 to displace NB3 from CXCR7-expressing cells indicate that these Nanobodies recognize distinct epitopes. Additionally, the partial displacement of 125I-CXCL12 from CXCR7 by NB1 and the inability of NB1 to effectively inhibit CXCL12-induced β-arrestin2 recruitment to the receptor point toward allosteric properties for this specific Nanobody. By coupling of Nanobodies with distinct binding properties (NB1 and NB3), biparatopic Nanobody (NB4) was created with increased affinity and potency.
The chemokine receptor CXCR7 is currently considered as a drug target in oncology due to its overexpression in a wide range of tumors, e.g. glioblastoma (13), hepatocellular carcinoma (41), bladder (42), and cervical cancer (43). In our search for an appropriate CXCR7-expressing in vivo tumor model, we evaluated human tumor biopsies from various origins for CXCR7 expression by immunostaining with the 8F11 monoclonal antibody. Our results indicate that CXCR7 is highly expressed in melanoma and mammary and non-small cell lung cancer tissue (Fig. 4A) as previously described (11, 44). Moreover, we show that colon and gastric tumor tissue express relatively low levels of CXCR7 compared with the other tumor tissues analyzed. Importantly, we show a high incidence of CXCR7 protein expression in head and neck tumor biopsies, in line with previous studies where CXCR7 expression was detected in oral carcinoma patient material (45). Several human head and neck squamous cell carcinomas cells analyzed in our studies showed high expression of CXCR7, as detected by radioligand binding and anti-CXCR7 antibody staining (Fig. 5). The selected patient-derived head and neck squamous cell carcinomas 22A cell line appears a suitable model system for CXCR7, inducing tumor formation in a xenograft model, showing elevated levels of CXCR7 expression and not CXCR4 in these cells (Fig. 5). Most importantly, we show that the CXCR7 Nanobody NB4 reduces tumor growth in vivo (Fig. 7). The fact that NB5 is unable to reduce tumor growth can likely be attributed to its 10-fold lower affinity and potency to inhibit CXCL12 binding and signaling on the human CXCR7 receptor when compared with NB4. We cannot exclude a potential effect of NB4 on mouse stromal cells due to its cross-reactivity with mouse CXCR7. However, staining of 22A-derived mouse xenograft and human biopsies seem to exclude the presence of CXCR7 protein in tumor stromal cells. Overall, the inhibitory effect of NB4 strengthens a potential role for CXCR7 in oncogenesis and in particular head and neck cancer.
Cell cycle progression of 22A cells in vitro was not affected by treatment with NB4 or the chemokine CXCL12, ruling out a direct involvement of CXCR7 in head and neck cancer cell proliferation. This lack of involvement of CXCR7 in cellular proliferation was previously observed in other cancer types, in particular in glioma cells (13). In vitro, we demonstrated that inhibition of CXCR7 by NB4 in 22A cells reduces secretion of the angiogenic factor CXCL1 (Fig. 5). CXCL1 was shown to be important in tumor-associated angiogenesis in melanoma, non-small cell lung cancer, Kaposi's sarcoma, and colorectal cancer (46–49). Most importantly, a correlation is found between CXCL1 expression and angiogenesis in oral cancer patients as well as increased expression of CXCL1 in patients suffering from oral tongue squamous cell carcinoma (20, 50). Our findings confirm that CXCR7 is able to promote angiogenesis and signaling in a ligand-independent manner, which is an interesting characteristic of this atypical chemokine receptor (15). The CXCR7 Nanobodies are able to inhibit this basal activity, thus acting here as inverse agonists. CXCR7 expression in PC3 prostate cancer cells was also shown to display constitutive active properties (15). The significant increase in CCL5 secretion, not observed in the antibody array, is an unexpected observation that needs further investigation. In vivo, we confirmed that NB4 reduces tumor angiogenesis significantly (Fig. 7). Further research is required to dissect the exact molecular mechanisms by which NB4 decreases angiogenesis in head and neck tumors.
In summary, using the Nanobody platform we generated Nanobodies against the oncogenic chemokine receptor CXCR7. These llama-derived single domain antibodies were functionally able to inhibit CXCL12 binding, block β-arrestin2 recruitment to CXCR7, and decrease secretion of angiogenic factors in head and neck cancer cell lines. Notably, we demonstrate for the first time the enhanced CXCR7 expression in head and neck cancer biopsies and derived cell lines. Treatment of head and neck cancer xenografts with CXCR7 Nanobodies reduced tumor growth by inhibiting blood vessel formation. We demonstrate that CXCR7 is a novel drug target against this devastating disease.
Footnotes
- BRET
- bioluminescence resonance energy transfer
- EGFR
- EGF receptor.
REFERENCES
- 1. Scholten D. J., Canals M., Maussang D., Roumen L., Smit M. J., Wijtmans M., de Graaf C., Vischer H. F., Leurs R. (2012) Pharmacological modulation of chemokine receptor function. Br. J. Pharmacol. 165, 1617–1643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Balabanian K., Lagane B., Infantino S., Chow K. Y., Harriague J., Moepps B., Arenzana-Seisdedos F., Thelen M., Bachelerie F. (2005) The chemokine SDF-1/CXCL12 binds to and signals through the orphan receptor RDC1 in T lymphocytes. J. Biol. Chem. 280, 35760–35766 [DOI] [PubMed] [Google Scholar]
- 3. Burns J. M., Summers B. C., Wang Y., Melikian A., Berahovich R., Miao Z., Penfold M. E., Sunshine M. J., Littman D. R., Kuo C. J., Wei K., McMaster B. E., Wright K., Howard M. C., Schall T. J. (2006) A novel chemokine receptor for SDF-1 and I-TAC involved in cell survival, cell adhesion, and tumor development. J. Exp. Med. 203, 2201–2213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rajagopal S., Kim J., Ahn S., Craig S., Lam C. M., Gerard N. P., Gerard C., Lefkowitz R. J. (2010) β-Arrestin- but not G protein-mediated signaling by the “decoy” receptor CXCR7. Proc. Natl. Acad. Sci. U.S.A. 107, 628–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Naumann U., Cameroni E., Pruenster M., Mahabaleshwar H., Raz E., Zerwes H. G., Rot A., Thelen M. (2010) CXCR7 functions as a scavenger for CXCL12 and CXCL11. PLoS ONE 5, e9175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Luker K. E., Steele J. M., Mihalko L. A., Ray P., Luker G. D. (2010) Constitutive and chemokine-dependent internalization and recycling of CXCR7 in breast cancer cells to degrade chemokine ligands. Oncogene 29, 4599–4610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Luker K. E., Gupta M., Steele J. M., Foerster B. R., Luker G. D. (2009) Imaging ligand-dependent activation of CXCR7. Neoplasia 11, 1022–1035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zabel B. A., Wang Y., Lewén S., Berahovich R. D., Penfold M. E., Zhang P., Powers J., Summers B. C., Miao Z., Zhao B., Jalili A., Janowska-Wieczorek A., Jaen J. C., Schall T. J. (2009) Elucidation of CXCR7-mediated signaling events and inhibition of CXCR4-mediated tumor cell transendothelial migration by CXCR7 ligands. J. Immunol. 183, 3204–3211 [DOI] [PubMed] [Google Scholar]
- 9. Canals M., Scholten D. J., de Munnik S., Han M. K., Smit M. J., Leurs R. (2012) Ubiquitination of CXCR7 controls receptor trafficking. PLoS ONE 7, e34192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Balkwill F. R. (2012) The chemokine system and cancer. J. Pathol. 226, 148–157 [DOI] [PubMed] [Google Scholar]
- 11. Miao Z., Luker K. E., Summers B. C., Berahovich R., Bhojani M. S., Rehemtulla A., Kleer C. G., Essner J. J., Nasevicius A., Luker G. D., Howard M. C., Schall T. J. (2007) CXCR7 (RDC1) promotes breast and lung tumor growth in vivo and is expressed on tumor-associated vasculature. Proc. Natl. Acad. Sci. U. S. A. 104, 15735–15740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang J., Shiozawa Y., Wang J., Wang Y., Jung Y., Pienta K. J., Mehra R., Loberg R., Taichman R. S. (2008) The role of CXCR7/RDC1 as a chemokine receptor for CXCL12/SDF-1 in prostate cancer. J. Biol. Chem. 283, 4283–4294 [DOI] [PubMed] [Google Scholar]
- 13. Hattermann K., Held-Feindt J., Lucius R., Müerköster S. S., Penfold M. E., Schall T. J., Mentlein R. (2010) The chemokine receptor CXCR7 is highly expressed in human glioma cells and mediates antiapoptotic effects. Cancer Res. 70, 3299–3308 [DOI] [PubMed] [Google Scholar]
- 14. Gahan J. C., Gosalbez M., Yates T., Young E. E., Escudero D. O., Chi A., Garcia-Roig M., Satyanarayana R., Soloway M. S., Bird V. G., Lokeshwar V. B. (2012) Chemokine and chemokine receptor expression in kidney tumors. Molecular profiling of histological subtypes and association with metastasis. J. Urol. 187, 827–833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Singh R. K., Lokeshwar B. L. (2011) The IL-8-regulated chemokine receptor CXCR7 stimulates EGFR signaling to promote prostate cancer growth. Cancer Res. 71, 3268–3277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zabel B. A., Lewén S., Berahovich R. D., Jaén J. C., Schall T. J. (2011) The novel chemokine receptor CXCR7 regulates trans-endothelial migration of cancer cells. Mol. Cancer 10, 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Liu C., Pham K., Luo D., Reynolds B. A., Hothi P., Foltz G., Harrison J. K. (2013) Expression and functional heterogeneity of chemokine receptors CXCR4 and CXCR7 in primary patient-derived glioblastoma cells. PLoS ONE 8, e59750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kollmar O., Rupertus K., Scheuer C., Nickels R. M., Haberl G. C., Tilton B., Menger M. D., Schilling M. K. (2010) CXCR4 and CXCR7 regulate angiogenesis and CT26.WT tumor growth independent from SDF-1. Int. J. Cancer 126, 1302–1315 [DOI] [PubMed] [Google Scholar]
- 19. Jähnichen S., Blanchetot C., Maussang D., Gonzalez-Pajuelo M., Chow K. Y., Bosch L., De Vrieze S., Serruys B., Ulrichts H., Vandevelde W., Saunders M., De Haard H. J., Schols D., Leurs R., Vanlandschoot P., Verrips T., Smit M. J. (2010) CXCR4 Nanobodies (VHH-based single variable domains) potently inhibit chemotaxis and HIV-1 replication and mobilize stem cells. Proc. Natl. Acad. Sci. U.S.A. 107, 20565–20570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ye H., Yu T., Temam S., Ziober B. L., Wang J., Schwartz J. L., Mao L., Wong D. T., Zhou X. (2008) Transcriptomic dissection of tongue squamous cell carcinoma. BMC Genomics 9, 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Harmsen M. M., De Haard H. J. (2007) Properties, production, and applications of camelid single-domain antibody fragments. Appl. Microbiol. Biotechnol. 77, 13–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Klarenbeek A., Maussang D., Blanchetot C., Saunders M., van der Woning S., Smit M., de Haard H., Hofman E. (2012) Targeting chemokines and chemokine receptors with antibodies. Drug Discov. Today 9, e237-e244 [DOI] [PubMed] [Google Scholar]
- 23. Maussang D., Vischer H. F., Schreiber A., Michel D., Smit M. J. (2009) Pharmacological and biochemical characterization of human cytomegalovirus-encoded G protein-coupled receptors. Methods Enzymol. 460, 151–171 [DOI] [PubMed] [Google Scholar]
- 24. Maussang D., Verzijl D., van Walsum M., Leurs R., Holl J., Pleskoff O., Michel D., van Dongen G. A., Smit M. J. (2006) Human cytomegalovirus-encoded chemokine receptor US28 promotes tumorigenesis. Proc. Natl. Acad. Sci. U.S.A. 103, 13068–13073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Smith V., Wirth G. J., Fiebig H. H., Burger A. M. (2008) Tissue microarrays of human tumor xenoggrafts: characterization of proteins involved in migration and angiogenesis for applications in the development of targeted anti-cancer agents. Cancer Genomics Proteomics 5, 263–273 [PMC free article] [PubMed] [Google Scholar]
- 26. Wijtmans M., Maussang D., Sirci F., Scholten D. J., Canals M., Mujić-Delić A., Chong M., Chatalic K. L., Custers H., Janssen E., de Graaf C., Smit M. J., de Esch I. J., Leurs R. (2012) Synthesis, modeling and functional activity of substituted styrene-amides as small-molecule CXCR7 agonists. Eur. J. Med. Chem. 51, 184–192 [DOI] [PubMed] [Google Scholar]
- 27. Shimizu S., Brown M., Sengupta R., Penfold M. E., Meucci O. (2011) CXCR7 protein expression in human adult brain and differentiated neurons. PLoS ONE 6, e20680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Vosjan M. J., Vercammen J., Kolkman J. A., Stigter-van Walsum M., Revets H., van Dongen G. A. (2012) Nanobodies targeting the hepatocyte growth factor. Potential new drugs for molecular cancer therapy. Mol. Cancer Ther. 11, 1017–1025 [DOI] [PubMed] [Google Scholar]
- 29. Xu H., Wu Q., Dang S., Jin M., Xu J., Cheng Y., Pan M., Wu Y., Zhang C., Zhang Y. (2011) Alteration of CXCR7 expression mediated by TLR4 promotes tumor cell proliferation and migration in human colorectal carcinoma. PLoS ONE 6, e27399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lansford C. D., Grenman R., Bier H., Somers K. D., Kim S. Y., Whiteside T. L., Clayman G. L., Welkoborsky H.-J., Carey T. E. (2002) Head and Neck Cancers Human Cell Culture (Masters J. R. W., Palsson B., eds) pp. 185–255, Springer, The Netherlands [Google Scholar]
- 31. Kryczek I., Wei S., Keller E., Liu R., Zou W. (2007) Stroma-derived factor (SDF-1/CXCL12) and human tumor pathogenesis. Am. J. Physiol. Cell Physiol. 292, C987–C995 [DOI] [PubMed] [Google Scholar]
- 32. Senkal C. E., Ponnusamy S., Rossi M. J., Sundararaj K., Szulc Z., Bielawski J., Bielawska A., Meyer M., Cobanoglu B., Koybasi S., Sinha D., Day T. A., Obeid L. M., Hannun Y. A., Ogretmen B. (2006) Potent antitumor activity of a novel cationic pyridinium-ceramide alone or in combination with gemcitabine against human head and neck squamous cell carcinomas in vitro and in vivo. J. Pharmacol. Exp. Ther. 317, 1188–1199 [DOI] [PubMed] [Google Scholar]
- 33. Helman E. E., Newman J. R., Dean N. R., Zhang W., Zinn K. R., Rosenthal E. L. (2010) Optical imaging predicts tumor response to anti-EGFR therapy. Cancer Biol. Ther. 10, 166–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Vaneycken I., D'huyvetter M., Hernot S., De Vos J., Xavier C., Devoogdt N., Caveliers V., Lahoutte T. (2011) Immuno-imaging using Nanobodies. Curr. Opin. Biotechnol. 22, 877–881 [DOI] [PubMed] [Google Scholar]
- 35. Omidfar K., Rasaee M. J., Modjtahedi H., Forouzandeh M., Taghikhani M., Golmakani N. (2004) Production of a novel camel single-domain antibody specific for the type III mutant EGFR. Tumour. Biol. 25, 296–305 [DOI] [PubMed] [Google Scholar]
- 36. Roovers R. C., Laeremans T., Huang L., De Taeye S., Verkleij A. J., Revets H., de Haard H. J., van Bergen en Henegouwen P. M. (2007) Efficient inhibition of EGFR signaling and of tumour growth by antagonistic anti-EFGR Nanobodies. Cancer Immunol. Immunother. 56, 303–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Huang L., Gainkam L. O., Caveliers V., Vanhove C., Keyaerts M., De Baetselier P., Bossuyt A., Revets H., Lahoutte T. (2008) SPECT imaging with 99mTc-labeled EGFR-specific nanobody for in vivo monitoring of EGFR expression. Mol. Imaging Biol. 10, 167–175 [DOI] [PubMed] [Google Scholar]
- 38. Tijink B. M., Laeremans T., Budde M., Stigter-van Walsum M., Dreier T., de Haard H. J., Leemans C. R., van Dongen G. A. (2008) Improved tumor targeting of anti-epidermal growth factor receptor Nanobodies through albumin binding. Taking advantage of modular nanobody technology. Mol. Cancer Ther. 7, 2288–2297 [DOI] [PubMed] [Google Scholar]
- 39. Vaneycken I., Devoogdt N., Van Gassen N., Vincke C., Xavier C., Wernery U., Muyldermans S., Lahoutte T., Caveliers V. (2011) Preclinical screening of anti-HER2 Nanobodies for molecular imaging of breast cancer. FASEB J. 25, 2433–2446 [DOI] [PubMed] [Google Scholar]
- 40. Behdani M., Zeinali S., Khanahmad H., Karimipour M., Asadzadeh N., Azadmanesh K., Khabiri A., Schoonooghe S., Habibi Anbouhi M., Hassanzadeh-Ghassabeh G., Muyldermans S. (2012) Generation and characterization of a functional Nanobody against the vascular endothelial growth factor receptor-2. Angiogenesis cell receptor. Mol. Immunol. 50, 35–41 [DOI] [PubMed] [Google Scholar]
- 41. Zheng K., Li H. Y., Su X. L., Wang X. Y., Tian T., Li F., Ren G. S. (2010) Chemokine receptor CXCR7 regulates the invasion, angiogenesis, and tumor growth of human hepatocellular carcinoma cells. J. Exp. Clin. Cancer Res. 29, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hao M., Zheng J., Hou K., Wang J., Chen X., Lu X., Bo J., Xu C., Shen K. (2012) Role of chemokine receptor CXCR7 in bladder cancer progression. Biochem. Pharmacol. 84, 204–214 [DOI] [PubMed] [Google Scholar]
- 43. Schrevel M., Karim R., ter Haar N. T., van der Burg S. H., Trimbos J. B., Fleuren G. J., Gorter A., Jordanova E. S. (2012) CXCR7 expression is associated with disease-free and disease-specific survival in cervical cancer patients. Br. J. Cancer 106, 1520–1525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sánchez-Martín L., Estecha A., Samaniego R., Sánchez-Ramón S., Vega M. Á., Sánchez-Mateos P. (2011) The chemokine CXCL12 regulates monocyte-macrophage differentiation and RUNX3 expression. Blood 117, 88–97 [DOI] [PubMed] [Google Scholar]
- 45. Xia J., Wang J., Chen N., Dai Y., Hong Y., Chen X., Cheng B. (2011) Expressions of CXCR7/ligands may be involved in oral carcinogenesis. J. Mol. Histol. 42, 175–180 [DOI] [PubMed] [Google Scholar]
- 46. Dhawan P., Richmond A. (2002) Role of CXCL1 in tumorigenesis of melanoma. J. Leukoc. Biol. 72, 9–18 [PMC free article] [PubMed] [Google Scholar]
- 47. Arenberg D. A., Polverini P. J., Kunkel S. L., Shanafelt A., Hesselgesser J., Horuk R., Strieter R. M. (1997) The role of CXC chemokines in the regulation of angiogenesis in non-small cell lung cancer. J. Leukoc. Biol. 62, 554–562 [DOI] [PubMed] [Google Scholar]
- 48. Lane B. R., Liu J., Bock P. J., Schols D., Coffey M. J., Strieter R. M., Polverini P. J., Markovitz D. M. (2002) Interleukin-8 and growth-regulated oncogene α mediate angiogenesis in Kaposi's sarcoma. J. Virol. 76, 11570–11583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wang D., Wang H., Brown J., Daikoku T., Ning W., Shi Q., Richmond A., Strieter R., Dey S. K., DuBois R. N. (2006) CXCL1 induced by prostaglandin E2 promotes angiogenesis in colorectal cancer. J. Exp. Med. 203, 941–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Shintani S., Ishikawa T., Nonaka T., Li C., Nakashiro K., Wong D. T., Hamakawa H. (2004) Growth-regulated oncogene-1 expression is associated with angiogenesis and lymph node metastasis in human oral cancer. Oncology 66, 316–322 [DOI] [PubMed] [Google Scholar]