

Background: A nonsense mutation in cereblon, which results in the loss of the last 24 amino acids in the protein, causes mental retardation.
Results: This mutant form of cereblon undergoes autoubiquitination by a CRL4 E3 ligase complex, leading to enhanced proteasomal degradation.
Conclusion: The protein level of the mutant cereblon can be rescued by proteasome inhibition.
Significance: These findings provide an approach for the treatment of mental retardation associated with this cereblon mutation.
Keywords: E3 Ubiquitin Ligase, Genetic Diseases, Mutant, Protein Degradation, Ubiquitin, Cereblon, Mental Retardation, Nonsense Mutation, Proteasome Inhibition, Ubiquitination
Abstract
A nonsense mutation in cereblon (CRBN) causes autosomal recessive nonsyndromic mental retardation. Cereblon is a substrate receptor for the Cullin-RING E3 ligase complex and couples the ubiquitin ligase to specific ubiquitination targets. The CRBN nonsense mutation (R419X) results in a protein lacking 24 amino acids at its C terminus. Although this mutation has been linked to mild mental retardation, the mechanism by which the mutation affects CRBN function is unknown. Here, we used biochemical and mass spectrometric approaches to explore the function of this mutant. We show that the protein retains its ability to assemble into a Cullin-RING E3 ligase complex and catalyzes the ubiquitination of CRBN-target proteins. However, we find that this mutant exhibits markedly increased levels of autoubiquitination and is more readily degraded by the proteasome than the wild type protein. We also show that the level of the mutant protein can be restored by a treatment of cells with a clinically utilized proteasome inhibitor, suggesting that this agent may be useful for the treatment of mental retardation associated with the CRBN R419X mutation. These data demonstrate that enhanced autoubiquitination and degradation account for the defect in CRBN activity that leads to mental retardation.
Introduction
Mental retardation occurs in 1–3% of the population in the United States (1) and is currently untreatable. In most cases, the genetic causes of mental retardation are highly heterogeneous (2); single gene mutations have been identified only in a few instances. Understanding how genetic lesions affect the function of the encoded proteins can suggest the potential therapeutic approaches.
Recently, a new genetic cause for a mild form of mental retardation was discovered in five nuclear families (3). This form of mental retardation is otherwise nonsyndromic, exhibits an autosomal recessive pattern of inheritance, and is associated with an IQ between 50 and 70 (4). Single strand polymorphism analysis and DNA sequencing showed that this form of mental retardation is caused by a single nucleotide mutation (C→T mutation) in the cereblon (CRBN) gene (3). This mutation introduces a premature stop codon, which terminates protein translation at arginine 419. The corresponding protein, CRBN R419X, lacks the last 24 amino acids compared with the full-length protein that contains 442 amino acids. Because the mutation exhibits an autosomal recessive pattern of inheritance, it is likely to inactivate the protein. However, it is unknown how this mutation affects the function of CRBN.
CRBN is thought to function as a substrate receptor for the Cullin 4-RING ubiquitin ligase (CRL4) complex. CRL4 is a complex comprising CUL4A or CUL4B, DDB1 (damage-specific DNA-binding protein 1), and the RING finger protein ROC1 (5). DDB1 can interact with a variety of substrate receptors, including damage-specific DNA-binding protein 2 (DDB2) and VS5 V (6, 7). Each of these substrate receptors is thought to couple the CRL4 complex to different target proteins. Ubiquitin is then transferred from an E2 ubiquitin-conjugating enzyme to target proteins. The crystal structures and biochemical experiments of two of these E3 ligases, CRL4-DDB2 (8) and CRL4-VS5 V (6), demonstrated that DDB2 and VS5 V are the substrate receptors, which recognize a set of target proteins for ubiquitination. A structural analysis showed CRBN is one of the substrate receptors (6), which contains a DDB1 binding domain similar to those in DDB2 and VS5 V. Ito et al. (9) further confirmed that CRBN directly interacts with DDB1 and competes with DDB2 for binding with DDB1. Their data and our data in this work also showed that CRBN indeed forms a complex with ROC1, CUL4A, and DDB1. These results support the idea that CRBN is a substrate receptor of the CRL4 E3 ligase.
It has been reported that CRBN can directly interact with several proteins, such as AMP-activated protein kinase (10), the large conductance calcium-activated potassium channel (11), voltage-gated chloride channels (12), and the 20 S core proteasome subunit β type 4 (13). None of these proteins has yet been shown to be targeted by CRL4 as a result of their binding to CRBN.
Here, we explore the mechanism by which the premature stop codon in CRBN affects its function. The stop codon is not predicted to destabilize the CRBN mRNA by making it a target for nonsense-mediated decay. Instead, we show that CRBN R419X is expressed in cells and binds the CRL4 complex to mediate the ubiquitination of target proteins. However, we find that CRBN R419X exhibits markedly increased autoubiquitination and degrades faster than the wild type (WT) protein. Furthermore, we show that widely used proteasome inhibitors can restore levels of CRBN R419X in cells, suggesting a potential therapeutic strategy for this form of mental retardation.
EXPERIMENTAL PROCEDURES
cDNA Cloning
Total RNA was isolated from human embryonic kidney (HEK) 293T cells using TRIzol reagent, and the first strand cDNA was synthesized using the first strand cDNA synthesis kit (Invitrogen). The cDNAs for WT, R419X, and 1–338 CRBN were amplified by PCR and cloned into a pRRL lentiviral vector with or without FLAG and StrepII tag at the N terminus. Human AMPKα1 and BKCa plasmids were purchased from Addgene, amplified by PCR, and cloned to pcDNA3.1 with a 3×FLAG, 2×HA, and His6 tags at their C termini. The CUL4A, CUL4A(Δ), and His6-Ub plasmids were generously provided by Dr. Pengbo Zhou (Weill Medical College of Cornell University). The ubiquitin mutants were cloned to the pRRL vector. The positive clones were confirmed by sequencing.
Calcium Phosphate Transfection
HEK293T cells were grown in high glucose DMEM supplied with 10% fetal bovine serum (FBS) and 100 units/ml penicillin and 100 μg/ml streptomycin. Cells were split in a 1:6 ratio on the day before the transfection and transfected using the standard calcium phosphate transfection protocol with the indicated amount of plasmids. For the control samples, an EGFP3 plasmid or an empty plasmid was used to balance the total amount of plasmids to ensure the equal expression of heterologous proteins in cells. Cells were grown for an additional 48 h before drug treatments and cellular lysate preparation.
Thalidomide Inhibition of CRBN Ubiquitination
Thalidomide and a proteasome inhibitor, MG132, were dissolved in DMSO. HEK293T cells expressing WT and R419X CRBN were treated with thalidomide at the indicated concentration, or DMSO, for 1 h, and MG132 or DMSO was added to the cells for an additional 3 h before lysis.
Affinity Purification of FLAG-StrepII (FS)-CRBN WT and R419X
Cells expressing FS-CRBN WT and FS-CRBN R419X were treated as indicated and then washed with ice-cold PBS and lysed in ice-cold lysis buffer containing 150 mm NaCl, 50 mm Tris-HCl, pH 7.4, 1% Triton X-100, 0.1% SDS with freshly added protease inhibitor mixture (Roche Applied Science) and 5 mm chloroacetamide. The lysates were centrifuged at 17,900 × g for 10 min and incubated with 50 μl of 50% Strep-Tactin beads overnight at 4 °C. The beads were washed with lysis buffer four times, each with a 7-min incubation at 4 °C. The tagged CRBN (WT and R419X) was eluted twice with 40 μl of 5 mm desthiobiotin in lysis buffer with a 20-min constant shaking at room temperature. The purification of the CRBN complex was performed using the same procedure but with a lysis and washing buffer containing 150 mm NaCl, 50 mm Tris-HCl, pH 8.0, and 1 mm EDTA.
Purification of BKCa and AMPKα1
C-terminal 3×FLAG, 2×HA, and His6-tagged BKCa and AMPKα1 were coexpressed with a control plasmid, StrepII-tagged CRBN WT, or R419X mutant in the presence or absence of the dominant negative CUL4A, CUL4A(Δ) in HEK293T cells. Cells are treated with MG132 for 16 h and lysed in a denaturing buffer containing 8 m urea, 50 mm phosphate, 0.5 m NaCl, 20 mm imidazole, pH 8, and 5 mm fresh chloroacetamide. The cell lysate was incubated with 50 μl of TALON resin for 4 h and washed with denaturing buffer six times. The purified proteins were eluted with 200 mm imidazole in the denaturing buffer and diluted with double distilled water for SDS-PAGE and immunoblotting.
SDS-PAGE and Immunoblotting
Affinity-purified samples or cell lysates were mixed with LDS sample buffer, heated at 100 °C for 10 min, centrifuged, separated by SDS-PAGE, and transferred to polyvinylidene fluoride membrane (Millipore). Blots were blocked with 5% nonfat milk and incubated with appropriate primary antibodies for 1 h at room temperature and washed with PBST (0.1% Tween 20 in PBS) three times. The blots were further incubated with secondary antibodies for 1 h at room temperature and washed with PBST four times. The blots were incubated with ECL prime reagents (GE Healthcare), and chemiluminescence was recorded by a quantitative gel imaging station (Bio-Rad ChemiDoc system).
Making Viral Particles Expressing WT and R419X CRBN
HEK293T cells were split in a 1:4 dilution in 10-cm plates the day before the transfection. The media were replaced with fresh media 1–3 h before the transfection. For each plate, 20 μg of pRRL IRES EGFP plasmid, which expresses either CRBN WT or R419X mutant, 10 μg of pLP1, 5 μg of pLP2, 6 μg of pVSV-G, and 86.8 μl of 2 m CaCl2 solution (Clontech), were combined, and the volume was adjusted to 700 μl with double distilled water. The plasmid mixture was added dropwise to 700 μl of 2×HBS (Clontech) with low speed vortexing and incubated at room temperature for 20 min. The mixture was added to cells dropwise and mixed by rotating back and forth for 10 s, and the plates were placed in a humidified incubator with 5% CO2 at 37 °C. After 3–4 h of incubation, the media were replaced with 6 ml of fresh media. After 24 h, the conditioned media were collected (stored at 4 °C), and 6 ml of fresh media were added to the plates. After an additional 24 h, the media were collected and combined with previously collected media, centrifuged for 5 min at 1000 rpm at room temperature, and filtered through a 0.45-μm syringe filter. The viral particles were precipitated by centrifugation at 22,000 rpm and 22 °C for 2 h in an ultracentrifuge. The pellet was resuspended in 150 μl of 1% BSA in PBS and shaken at room temperature for 30 min. The viral particles were frozen at −80 °C until further use.
Generation of HEK293T Cell Lines Expressing WT and R419X CRBN
HEK293T cell lines expressing WT and R419X CRBN were established by transducing cells with the corresponding lentiviral particles in 24-well plates. The infection efficiency was calculated based on the flow cytometry using the EGFP signal, which was encoded after the IRES in the pRRL plasmid. More than 95% of cells expressed WT CRBN or the R419X mutant.
Immunofluorescence
HEK293T cells expressing either WT or R419X CRBN were plated in 24-well plates and treated with DMSO or MG132 (10 μm) for 4 h. The immunostaining was carried out according to the protocol previously described (14). Briefly, cells were washed with PBS, pH 7.4, and fixed with 4% (w/v) paraformaldehyde in PBS for 15 min at room temperature. Cells were washed again with PBS three times, permeabilized with 0.1% Triton X-100 in PBS for 10 min, and blocked with 2% BSA in PBS containing 0.1% Triton X-100 for 1 h at room temperature. CRBN was stained with a rabbit anti-FLAG antibody (Hangzhou Hua'an Biotechnology) in 2% BSA in PBS containing 0.1% overnight at 4 °C and washed with PBST three times. Cells were further incubated with FITC-conjugated donkey anti-rabbit immunoglobulin G antibody (Jackson ImmunoResearch) for 1 h at room temperature in the dark and washed with PBST three times. The samples were incubated with DAPI in PBS. The immunofluorescence was measured with an inverted microscope (Olympus IX71).
Measurement of Degradation Rate for WT and R419X CRBN
The HEK293T cells expressing WT or R419X CRBN were plated in 10-cm plates. Cycloheximide (100 μm) was added to the cells when the cells were ∼80% confluent, and cells were lysed at different time points. The cell lysate was separated on SDS-PAGE, and CRBN was quantified using the signal in the anti-FLAG Western blot using the Bio-Rad ChemiDoc system and normalized with β-actin. The relative amount CRBN was averaged from three independent experiments.
Identification of CRBN Ubiquitination Sites
Twenty 10-cm plates of HEK293T cells were transiently transfected with 5 μg of pRRL FS-CRBN IRES EGFP and 5 μg of pcDNA His6-Ub. Cells were treated with 10 μm MG132 for 4 h, washed with ice-cold PBS, and lysed under denaturing conditions with 8 m urea. Ubiquitination sites were identified as was described previously (15). In brief, the ubiquitinated proteins were purified via the His6 tag at the N terminus of Ub using TALON resin under denaturing conditions. The proteins were reduced with dithiothreitol, blocked with chloroacetamide, and digested with sequencing grade modified trypsin (Promega). The peptide digest was desalted by C18 column and lyophilized. The diglycine-lysine-containing peptides were enriched using immobilized diglycyl-lysine antibody and analyzed by mass spectrometry according to the method described previously (15).
RESULTS
Premature Stop Codon Is Not Predicted to Make CRBN a Nonsense-mediated Decay (NMD) Target
Gene mutations that introduce a premature stop codon usually result in a reduction in the encoded transcript due to NMD (16). NMD occurs when the stop codon lies more than 50–55 nucleotides upstream of a splicing-generated exon-exon junction (17, 18). Most protein-coding genes contain stop codons in the terminal exon, which allows these transcripts evade NMD (19–26). However, the R419X premature stop codon in CRBN simply repositions that stop codon within exon 11, the last exon in CRBN (Fig. 1). This suggests that the encoded mRNA would not be targeted by NMD and that the transcript would be translated into a truncated protein.
FIGURE 1.
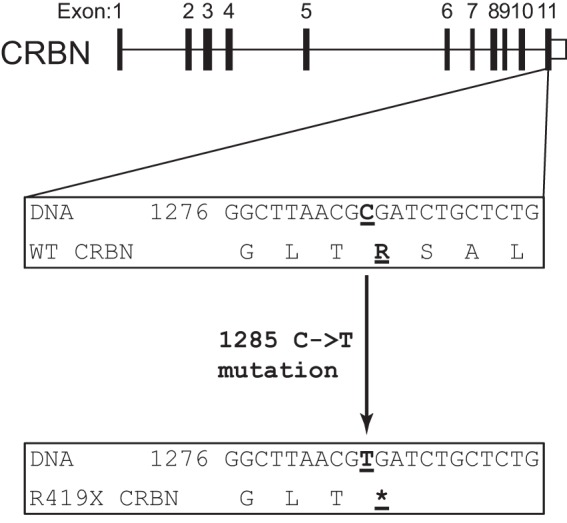
cereblon (CRBN) R419X transcript is not predicted to undergo nonsense-mediated mRNA decay. The exon-intron structure of the cereblon gene is indicated. The R419X nonsense mutation is located in the final exon. In the wild type (WT) sequence (middle), the normal C nucleotide is underlined. In the mutant cereblon gene, a mutation of C to T results in the introduction of a stop codon (bottom). Because the mutation introduces a nonsense mutation in the final exon, the encoded transcript is not predicted to undergo nonsense-mediated decay. The encoded protein is predicted to be missing the last 24 amino acids. Asterisk represents the stop codon.
Nonsense Mutation in CRBN Does Not Disrupt the Formation of the CRL4-CRBN E3 Ligase Complex
Because the autosomal recessive pattern of inheritance suggests that the mutant CRBN should be functionally inactive, we asked whether it could bind CRL4 and reconstitute a CRL4-CRBN E3 ligase complex (Fig. 2A). In the case of CRL4 bound to a different substrate receptor, VS5 V, the binding of VS5 V to CRL4 requires the C-terminal domain of VS5 V (7). We therefore asked if the CRBN R419X mutation, which loses C-terminal residues, prevents the association of CRBN with CRL4. To test this, we expressed FLAG-StrepII-tagged WT and R419X CRBN in HEK293T cells and measured the coprecipitation of CRBN-binding proteins through Strep-Tactin affinity purification and immunoblotting. In these experiments, pulldown of FS-CRBN coprecipitated endogenous DDB1, CUL4A, and ROC1 (Fig. 2B). However, in HEK293T cells expressing FS-CRBN R419X, Strep-Tactin pulldown also resulted in coprecipitation of DDB1, CUL4A, and ROC1 (Fig. 2B). In mock-transfected cells, Strep-Tactin pulldown did not coprecipitate any of these proteins. These data suggest that the R419X mutation does not prevent the association of the mutant protein with its CRL4-binding partners that constitute the E3 ligase complex.
FIGURE 2.
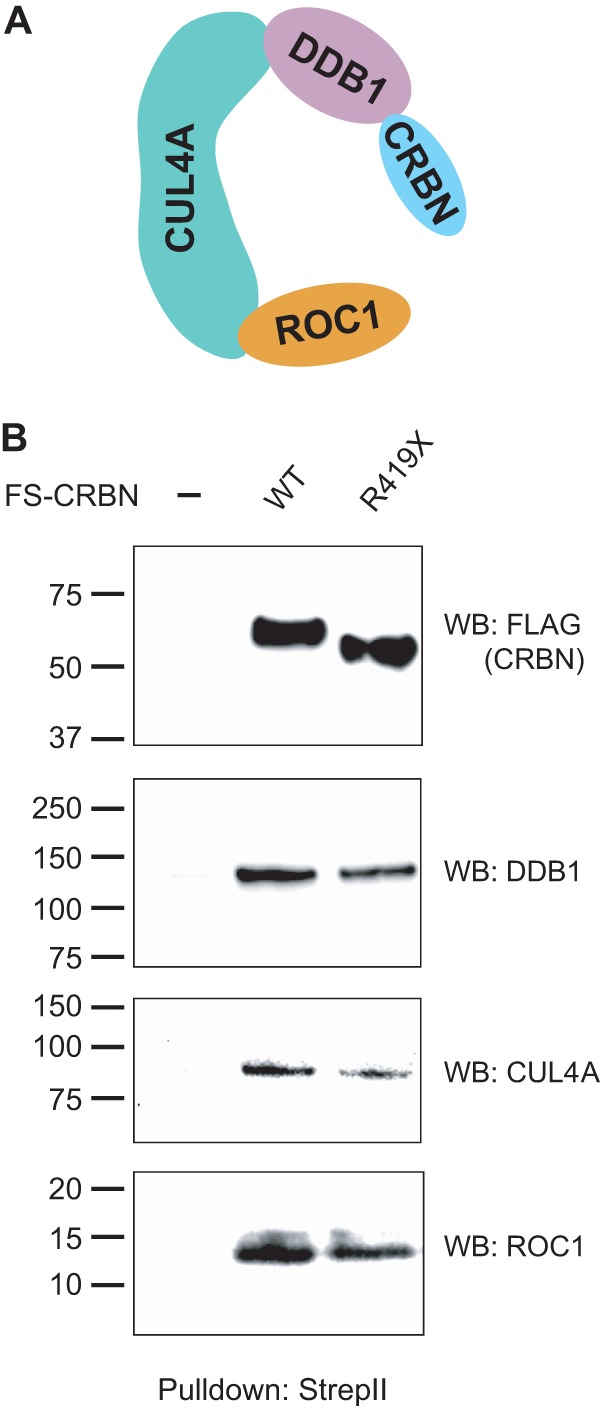
CRBN R419X can assemble into a CRL4 E3 ligase complex. A, schematic illustration of the ubiquitin E3 ligase complex, CRL4-CRBN, composed of damage-specific DNA-binding protein 1 (DDB1), CUL4A, X-box protein (ROC1), and CRBN. CRBN is the substrate receptor that targets proteins for ubiquitination. The CRL4 complex (DDB1, CUL4A, and ROC1) can interact with diverse substrate receptors. B, CRBN R419X binds the CRL4 E3 ligase. To test whether the CRBN mutant forms an E3 ligase as is seen with WT CRBN, HEK293T cells were transfected with a plasmid expressing an N-terminal FLAG-StrepII-tagged full-length human CRBN (FS-CRBN) or the nonsense mutant, FS-CRBN R419X. The tagged CRBN was affinity-purified with Strep-Tactin beads that specifically interact with the StrepII tag under native conditions. The purified protein complex was separated on an SDS-PAGE, and the binding of endogenous CRL4 was measured by immunoblotting with anti-DDB1, anti-CUL4A, and anti-ROC1 antibodies. Both WT CRBN and CRBN R419X assemble into CRL4 complexes. WB, Western blot.
CRBN Mutation Does Not Affect the E3 Ligase Activity of CRL4-CRBN
We next asked if the premature stop codon in CRBN affects the ability of the CRL4-CRBN E3 ligase to ubiquitinate its target proteins. In CUL4A ubiquitin ligases, substrate receptors such as DDB2 and CRBN couple the E3 ligase to specific targets (6). Therefore, the truncation in CRBN might affect the ability of CRBN to bind to its targets as demonstrated in other proteins (27–30). Although specific targets of CRL4-CRBN-mediated ubiquitination have not been identified, binding partners of CRBN have been described (10–13, 31). These proteins are therefore possible targets of the CRL4-CRBN E3 ligase complex. In these experiments, we expressed two CRBN-binding proteins, BKCa and AMPKα1, as FLAG- and His6-tagged fusion proteins in HEK293T cells, and we measured the level of ubiquitination in these proteins following coexpression with StrepII-tagged CRBN. Coexpression of BKCa with CRBN markedly enhanced the levels of BKCa ubiquitination as measured by anti-ubiquitin Western blotting of the eluate from TALON resin purification (Fig. 3, A and B). Similarly, in the case of AMPKα1, coexpression with CRBN resulted in increased levels of ubiquitinated AMPKα1. These data indicate that both BKCa and AMPKα1 are ubiquitinated in a CRBN-dependent manner.
FIGURE 3.

CRBN R419X forms an active CRL4-CRBN E3 ligase. A, CRL4-CRBN ubiquitinates CRBN-interacting proteins, BKCa and AMPKα1. To test whether the CRBN R419X mutant can ubiquitinate CRL4-CRBN substrates, we sought to identify targets of the CRL4-CRBN E3 ligase complex. To do so, we asked whether potential targets exhibit increased ubiquitination upon expression of CRBN. HEK293T cells were transfected with plasmids expressing BKCa-3×FLAG-2×HA-His6 (BKCa-tag) and AMPKα1–3×FLAG-2×HA-His6 (AMPKα1-tag) in the absence or in the presence of StrepII-CRBN WT and R419X. The tagged BKCa and AMPKα1 were purified on TALON resin under denaturing conditions, and the ubiquitination level was determined by anti-ubiquitin (P4D1) immunoblotting. Upon cotransfection with WT and R419X CRBN, both BKCa and AMPKα1 exhibited a significant increase in their ubiquitination. This suggests that BKCa and AMPKα1 are substrates of the CRL4-CRBN E3 ligase and the premature termination mutation does not significantly affect the E3 ligase activity. B, quantification of the relative ubiquitination level of BKCa and AMPKα1 in the presence of WT or R419X CRBN. As with WT CRBN, CRBN R419X induces the ubiquitination of both BKCa and AMPKα1, indicating that it can form a functional CRL4-CRBN R419X E3 ligase complex. WB, Western blot.
We next asked if the ubiquitination of BKCa and AMPKα1 is impaired by the premature stop codon in CRBN. To test this idea, we coexpressed CRBN R419X rather than WT CRBN. However, coexpression of CRBN R419X also resulted in ubiquitination of BKCa and AMPKα1 (Fig. 3, A and B). Taken together, these data indicate that the truncated CRBN assembles into a CRL4 E3 ligase complex and mediates ubiquitination of target proteins.
To further ask whether BKCa and AMPKα1 are indeed ubiquitinated by CRL4 E3 ligases, we coexpressed tagged BKCa and AMPKα1 with WT CRBN alone or with WT CRBN and the dominant negative CUL4A, CUL4A(Δ). This dominant negative mutant comprises CUL4A with a C-terminal truncation. This mutant binds DDB1 but not ROC1 (32). Therefore, CUL4A(Δ) interferes with the assembly of a CRL4 complex. Upon expression of WT CRBN, both BKCa and AMPKα1 have increased ubiquitination levels (Fig. 4A). However, after the coexpression of CUL4A(Δ), their ubiquitination level was reduced to the level seen in cells without CRBN expression. These results suggest that BKCa and AMPKα1 are indeed ubiquitinated by CRL4 E3 ligases, although we cannot completely rule out possible contributions by other E3 ligases.
FIGURE 4.

BKCa and AMPKα1 are ubiquitinated by the CRL4 E3 ligase. A, BKCa and AMPKα1 are ubiquitinated by CRL4 E3 ligases. BKCa-3×FLAG-2×HA-His6 and AMPKα1–3×FLAG-2×HA-His6 were coexpressed with WT CRBN in the absence or presence of a dominant negative CUL4A, CUL4A(Δ), in HEK293T, cells and the cells were treated with MG132 (10 μm, 16 h). The ubiquitinated BKCa and AMPKα1 were purified with TALON resin under denaturing conditions and blotted with an anti-ubiquitin antibody. FLAG Western blotting is used as a loading control. The results showed that their ubiquitination level is increased upon the expression of CRBN, and this increase is diminished when CUL4A(Δ) is expressed. B, quantification of the relative ubiquitination level of BKCa and AMPKα1 in the presence of WT CRBN or WT CRBN and CUL4A(Δ). These data indicate that the ubiquitination of BKCa and AMPKα1 by CRBN is due to its binding to the CRL4 E3 ligase. WB, Western blot.
CRBN R419X Is More Susceptible to Ubiquitination and Degradation
We next asked if the R419X nonsense mutation affects the ubiquitination of CRBN itself. CRBN undergoes ubiquitination (9), a phenomenon that has been seen in many E3 ligases (33–37). To test this, we expressed WT or mutant FS-CRBN and monitored the level of ubiquitination following affinity purification of tagged CRBN under stringent washing conditions. Because the CRBN R419X expresses less efficiently than CRBN (discussed in more detail below), we adjusted the level of the expression plasmids to ensure that equal expression levels were obtained in cells. In the case of WT CRBN, the ubiquitination level increases following treatment with MG132 (Fig. 5A). This treatment allows ubiquitinated proteins to accumulate by inhibiting their proteasomal degradation. This result is consistent with previous observations (9). However, pulldown of CRBN R419X revealed noticeably higher levels of ubiquitination (Fig. 5A).
FIGURE 5.
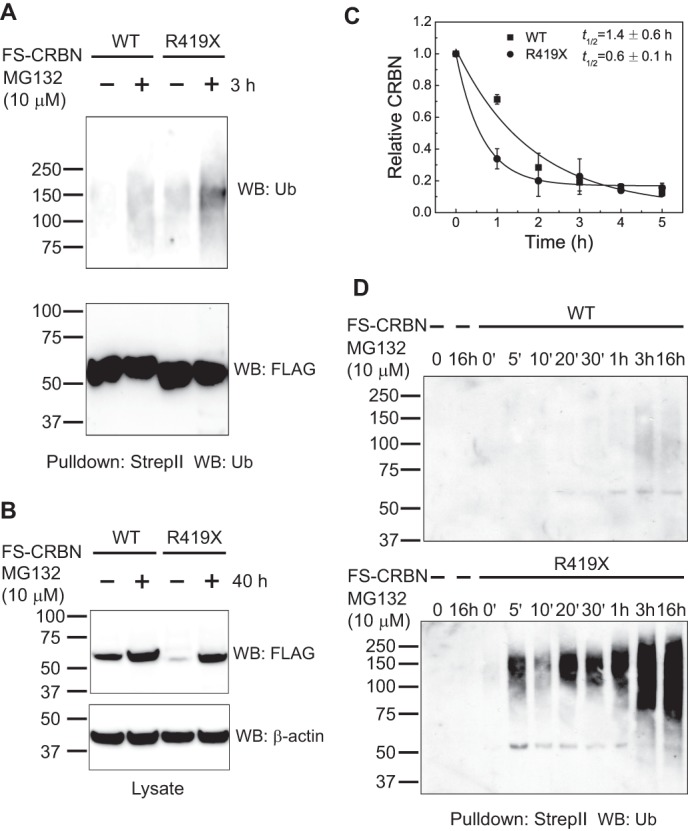
CRBN R419X is more susceptible to ubiquitination and degradation. A, CRBN R419X is more susceptible to ubiquitination. To test whether CRBN R419X exhibits altered levels of ubiquitination, HEK293T cells were transfected with FS-CRBN WT or FS-CRBN R419X and treated with a proteasome inhibitor, MG132 (10 μm, 3 h). The amount of ubiquitinated CRBN was detected by Strep-Tactin pulldown followed by anti-ubiquitin immunoblotting. The Western blot signal indicates that CRBN R419X exhibits higher levels of ubiquitination than WT CRBN. Anti-FLAG immunoblotting is shown as a loading control for the CRBN level. B, CRBN R419X protein levels significantly increase upon proteasome inhibition. To further test the idea that CRBN R419X is targeted for ubiquitination to a larger degree than WT CRBN, we asked if CRBN R419X protein levels exhibit increases upon proteasome inhibition. HEK293T cells were transfected with FS-CRBN WT or FS-CRBN R419X and then treated with a proteasome inhibitor, MG132 (10 μm, 40 h). Cells lysate was blotted with an anti-FLAG antibody. WT CRBN levels are slightly increased after proteasome inhibition, whereas CRBN R419X levels exhibit marked increases upon proteasome inhibition and can rise to a similar level as the WT counterpart. C, CRBN R419X is more labile in cells. The increased ubiquitination of CRBN R419X suggests that it might be more efficiently targeted for proteasomal degradation. To test this, HEK293T cells expressing WT and R419X CRBN were grown for 2 days and then treated with 100 μm cycloheximide to block protein synthesis. Cell lysates were harvested at the indicated time points, and the remaining amount of CRBN was determined by quantitative anti-FLAG Western blotting (WB). The results shown are an average from experiments repeated three times. R419X CRBN exhibits a significantly shorter half-life than the WT protein. D, CRBN R419X mutant is ubiquitinated much faster than the WT protein. Our data suggest that CRBN R419X is more rapidly or efficiently ubiquitinated by CRL4. We examined this by measuring ubiquitin levels in WT and R419X CRBN. The FS-tagged WT and R419X CRBN were expressed in HEK293T cells by calcium phosphate transfection. Cells were treated with MG132 (10 μm) for the indicated time and lysed in denaturing conditions. The tagged proteins were purified by Strep-Tactin beads and immunoblotted with an anti-ubiquitin antibody. As can be seen, R419X exhibits higher levels of ubiquitination at earlier time points than WT.
We next asked if CRBN R419X is degraded more readily than WT CRBN. To test this possibility, we compared the expression level of WT CRBN and CRBN R419X under basal conditions and following treatment with MG132. Upon transfection of 10 μg of plasmid expressing either WT CRBN or CRBN R419X into HEK293T cells, lower levels of CRBN R419X were detected in lysates (Fig. 5B, 1st and 3rd lanes). To determine whether the lower level of CRBN R419X reflects a higher level of protein degradation, we monitored the protein levels following treatment with MG132. In these experiments, we observed a considerable enhancement in the levels of CRBN R419X following MG132 treatment. However, MG132 treatment resulted in a much weaker increase in WT CRBN levels. Treatment of CRBN R419X-expressing cells with MG132 resulted in CRBN R419X achieving the same expression level as WT CRBN (Fig. 5B). These experiments indicate that the steady-state levels of CRBN R419X are reduced in cells due to a higher degree of proteasomal degradation.
To further explore the idea that CRBN R419X is more readily degraded than WT CRBN, we monitored their half-lives in cells. In these experiments, we used HEK293T cells stably expressing FS-CRBN WT or R419X mutant. These cell lines were prepared by transduction with lentivirus expressing the corresponding proteins. Then, we treated cells with cycloheximide to inhibit new protein synthesis and monitored CRBN levels over time. The levels of the epitope-tagged proteins were measured by quantitative Western blot using an anti-FLAG antibody. In these experiments, WT CRBN exhibited a half-life of 1.4 h, although CRBN R419X exhibited a half-life of 0.6 h (Fig. 5C). We also examined the rate of ubiquitination of WT and R419X CRBN and found that R419X is ubiquitinated much faster than WT CRBN (Fig. 5D). Taken together, these data show that CRBN R419X is more readily ubiquitinated and degraded than WT CRBN, which results in lower CRBN R419X levels in cells.
CRBN R419X Is Autoubiquitinated through CRL4 E3 Ligase
In many cases, the ubiquitin transferase activity of a ubiquitin ligase can result in ubiquitin transfer to the ubiquitin ligase itself or its constituent components (33–37). We therefore asked if the ubiquitination of CRBN R419X reflects autoubiquitination or ubiquitination via some other E3 ligases. To test this, we measured CRBN R419X ubiquitination when the CRL4 complex was inhibited by expression of a dominant negative CUL4A, CUL4A(Δ). We first asked if the dominant negative protein affects CRBN R419X protein expression levels, which is a reflection of protein ubiquitination and degradation. Expression of the full-length CUL4A resulted in nearly complete loss of CRBN R419X, suggesting that increased CRL4 activity leads to enhanced CRBN R419X degradation. However, overexpression of CUL4A(Δ) increases CRBN R419X level significantly (Fig. 6A). These data suggest that the amount of CRL4 activity controls the stability of CRBN R419X.
FIGURE 6.

Degradation and ubiquitination of CRBN R419X is mediated by CRL4. A, CRBN R419X expression level is regulated by cullin-RING E3 ligases. To test whether the CRL4 complex regulates CRBN itself, we monitored CRBN R419X levels upon activation or inhibition of CRL4. In these experiments, CRBN R419X was coexpressed with the full-length (FL) and dominant negative CUL4A, CUL4A(Δ), and CRBN R419X levels were determined by anti-FLAG immunoblotting. Expression of full-length CUL4A results in a near total loss of CRBN R419X. However, inhibition of the CRL4 complex with CUL4A(Δ) results in an increase in CRBN R419X levels. β-Actin levels are shown as a loading control, and anti-CUL4A immunoblotting is used to show the expression of the CUL4A constructs. Note: the weak signal at ∼75 kDa in the 1st and 3rd lanes of the middle image is from the endogenous CUL4A. B, ubiquitination of CRBN R419X is mediated by CRL4. To further test whether the ubiquitination of CRBNR419X is mediated by CRL4s, we monitored CRBN R419X ubiquitination following activation and inhibition of CRL4 using full-length and dominant negative CUL4A. In these experiments, cells were treated for 3 h with 10 μm MG132 to allow accumulation of ubiquitinated CRBN R419X. CRBN R419X was purified by Strep-Tactin and blotted with anti-ubiquitin and anti-FLAG antibodies. The ubiquitination level of CRBN R419X is increased upon expression of full-length CUL4A, although it is reduced when coexpressed with the dominant negative CUL4A. These results suggest that CRBN R419X ubiquitination and degradation is mediated by CRL4s, presumably when CRBN R419X is bound in a CRL4-CRBN R419X complex. C, CRBN R419X is ubiquitinated with Lys-48-linkage specific polyubiquitin chains. The FS-CRBN R419X was expressed in HEK293T cells. Cells were treated with either DMSO or MG132 and lysed in denaturing conditions. The tagged proteins were purified by Strep-Tactin beads and immunoblotted with a ubiquitin antibody recognizing Lys-48 polyubiquitin chain (clone, Apu2). Upon MG132 treatment, the Lys-48-linked polyubiquitin chain on CRBN is significantly increased. D, CRBN R419X ubiquitination is reduced upon coexpressing a K48R-ubiquitin mutant. FLAG-StrepII-tagged CRBN R419X was coexpressed with a control plasmid, WT-Ub, K48R-Ub, or K63R-Ub, and purified with Strep-Tactin beads. The ubiquitinated proteins were blotted with an anti-ubiquitin antibody. FLAG blotting was used as a loading control. Upon coexpression of K48R-Ub, CRBN R419X exhibited slightly reduced ubiquitination, although its ubiquitination level is increased when the WT and K63R-Ub are expressed. Note: the ubiquitination of the 1st lane, with a control plasmid, and the residual ubiquitination in the 3rd lane are from the endogenous ubiquitin. These data support the idea that the ubiquitination in CRBN R419X is indeed with the Lys-48-linked polyubiquitin chains and leads to its degradation. E, quantification of CRBN R419X ubiquitination in D. F, localization of R419X CRBN is not significantly altered upon proteasome inhibition. To test whether the ubiquitination causes the change of subcellular localization of CRBN R419X, we used HEK293 cells expressing WT and R419X CRBN for immunostaining with an anti-FLAG antibody after DMSO or MG132 treatment (10 μm, 4 h). The subcellular localization of the WT and R419X CRBN is not changed upon the MG132 treatment in HEK293T cells. Red, FLAG (CRBN WT or R419X mutant); blue, DAPI for nuclei; scale bar, 10 μm. WB, Western blot.
We next directly measured the effect of CRL4 activation and inhibition on CRBN R419X ubiquitination levels. Immunoprecipitation of CRBN R419X followed by anti-ubiquitin Western blotting showed that the ubiquitination of CRBN R419X is significantly increased after the expression of full-length CUL4A and is reduced after overexpression of CUL4A(Δ) (Fig. 6B). These results suggest that CRBN R419X is ubiquitinated in a CRL4 E3 ligase-dependent manner, consistent with its autoubiquitination. However, it should be noted that we cannot completely rule out the possibility that other E3 ligases contribute to CRBN R419X ubiquitination.
We next asked if the ubiquitination observed on CRBN reflects ubiquitin modifications that target CRBN for proteasomal degradation. Ubiquitin can be conjugated onto lysine residues in proteins in the context of polyubiquitin chains with different chain linkages, determined by the Lys residues in ubiquitin that connects the ubiquitins. The different chain linkages can affect the stability, activity, localization, or other functions of the modified proteins (38). Proteins that are conjugated to ubiquitin chains with Lys-48 linkages are typically targeted for proteasomal degradation (15). To determine whether the ubiquitin conjugates on CRBN contain this specific chain linkage, we pulled down exogenously expressed FS-CRBN R419X from HEK293T cells and Western blotted using an antibody specific for ubiquitin conjugates with a Lys-48 linkage. Our experiments indicate that CRBN R419X is conjugated to Lys-48-linked polyubiquitin chains (Fig. 6C).
To further test if CRBN R419X is modified by a Lys-48-containing polyubiquitin chain, we monitored the incorporation of wild type ubiquitin (WT-Ub) or ubiquitin constructs incapable of forming Lys-48 (K48R-Ub) or Lys-63 (K63R-Ub) linkages. These mutants contain lysine to arginine mutations that prevent chain-specific linkages. After expression of these ubiquitin mutants, CRBN R419X was affinity-purified and blotted with an anti-ubiquitin antibody (Fig. 6, D and E). The detected ubiquitination level for CRBN R419X is significantly increased when coexpressed with the WT- and K63R-Ub. However, the ubiquitination level of R419X CRBN is slightly reduced when it is expressed with the K48R-Ub. This indicates that the ubiquitin chains on CRBN R419X contain Lys-48-linked ubiquitin. These data further support the idea that CRBN R419X exhibits the Lys-48-linked form of ubiquitination, which typically leads to degradation.
We next asked whether the ubiquitination of CRBN R419X alters its subcellular localization. For this, we used HEK293T cells expressing WT and R419X mutant and treated them with DMSO or MG132 for 4 h. The cells were fixed, permeabilized, and stained with an anti-FLAG antibody for CRBN. The immunostaining showed, at least in HEK293T cells, that both the WT and R419X mutant are localized in cytosol and nucleus. The proteasome inhibitor, which causes an accumulation of ubiquitinated proteins, does not alter their localization (Fig. 6F), although it causes the significant increase of ubiquitination level for R419X mutant (Fig. 5A).
Lysines in the Thalidomide-binding Domain of CRBN Are Ubiquitinated in the Truncated Protein
We next sought to understand the basis for the increased ubiquitination of CRBN R419X. To do so, we mapped the ubiquitination sites in CRBN using the ubiquitin remnant profiling technique developed by our group (15). In this approach, WT CRBN and His6-tagged ubiquitin were overexpressed in HEK293T cells, which were treated with MG132 to enable the accumulation of the ubiquitinated proteins. The ubiquitinated proteins were pulled down with immobilized metal affinity chromatography via the tagged ubiquitin. Following pulldown, ubiquitinated proteins were digested with trypsin, and peptides containing lysine residues with the diglycine ubiquitin remnant were affinity-purified using an anti-diglycyl lysine antibody and identified by tandem mass spectrometry. Using this approach, we identified two ubiquitination sites in CRBN, Lys-300 and Lys-413 (see Fig. 7A for MS/MS spectra of the peptides containing ubiquitination sites). In addition to these residues, ubiquitination sites in CRBN have also been reported in proteome-wide analyses of protein ubiquitination in different cell lines (39). These previous analyses identified lysines 43, 166, 269, and 300 ubiquitinated. All the identified ubiquitination sites are depicted in Fig. 7B.
FIGURE 7.

CRBN R419X exhibits increased ubiquitination in its C-terminal domain. A, MS/MS spectra of the ubiquitin remnant-containing peptides derived from CRBN. Two ubiquitination sites, Lys-300 and Lys-413, were identified from ubiquitin-remnant profiling experiments. The b- and y-ions are labeled in the MS/MS spectra and in the peptide sequences. The ubiquitinated lysine residues are indicated. The Spectrum Mill search score, the percentage of the scored peak intensity, measured mass, charge state, and Δmass for the peptide in the top panel are 11.39, 68.9%, 1453.8857 Da, +3, 0.6 ppm; those for the peptide in the bottom panel are 11.28, 66.0%, 1579.7831 Da, +3, 9.2 ppm. B, schematic illustration of the ubiquitination sites in CRBN that were identified by tandem MS from our experiments and previous proteomic experiments. Besides Lys-300 and Lys-413, other ubiquitination sites, such as Lys-43, Lys-166, and Lys-269, were identified. The C-terminal truncation introduced by the R419X mutation in CRBN is indicated with a jagged line, and the thalidomide (Thal)-binding domain (339–442) is shown. C, 339–418 domain is the major ubiquitination domain in CRBN. To understand which portion of CRBN exhibits increased ubiquitination as a result of the R419X mutation, we monitored ubiquitination in CRBN mutants truncated at different positions in the protein. HEK293T cells were transfected with plasmids expressing FS-CRBN WT, FS-CRBN R419X, or FS-CRBN(1–338) and then treated with 10 μm MG132 for 3 h. Cells were lysed, and ubiquitin levels in the CRBN constructs were assessed in the Strep-Tactin pulldown by anti-ubiquitin Western blotting. The R419X protein is readily ubiquitinated, but this is lost in the 1–338 construct that lacks the C-terminal ubiquitination site. These data indicate these lysine residues within residues 339–418 exhibit increased ubiquitination in the R419X mutant. D, CRBN(1–338) mutant can interact with DDB1 to form an E3 ligase complex. C shows that the CRBN(1–338) mutant is not efficiently ubiquitinated. We thought this could be due to its inability to bind CRL4. To rule this out, we tested whether this mutant can still bind its interacting partner DDB1. FS-tagged WT (WT) and CRBN(1–338) were expressed in HEK293T cells. Cells were lysed using native conditions. The tagged proteins were purified by Strep-Tactin beads under native conditions and immunoblotted with DDB1 and FLAG antibodies. The experiment shows that CRBN(1–338) can interact with DDB1 at a similar level as that of the WT CRBN, which suggests that the reduction of ubiquitination of CRBN(1–338) is indeed through the reduction of ubiquitination on the lysine residues but not the inability to form a CRL4 E3 ligase complex. Note: the experiments were done in parallel and imaged simultaneously.
Because Lys-300 and Lys-413 are both near the C terminus of the protein, they therefore may be more highly influenced by the premature stop codon at residue 419. To determine whether ubiquitination of these residues accounts for the high level of CRBN R419X ubiquitination, we expressed WT CRBN, as well as CRBN R419X and CRBN(1–338) in HEK293T cells, and monitored the ubiquitination levels following immunoprecipitation. As expected, CRBN R419X exhibited higher ubiquitination levels than the WT protein. However, the CRBN(1–338) mutant exhibited base-line ubiquitination levels (Fig. 7C). The reduced ubiquitination of CRBN(1–338) is not due to the inability of the protein to bind to CRL4 because immunoprecipitation of CRBN(1–338) under native conditions confirmed the presence of DDB1 in the precipitated complex (Fig. 7D), which interacts with CUL4A and ROC1 to form an E3 ligase with CRBN(1–338). These data suggest that ubiquitination sites at the C-terminal lysines in the CRBN R419X are the ones that are preferentially ubiquitinated.
It should be noted that the ubiquitination level of the R419X mutant with the Lys-413 mutated to Arg (K413R/R419X CRBN) is not substantially reduced compared with that of the R419X mutant (data not shown). This may be due to ubiquitination of adjacent lysine residues that occurred after mutation of the preferred lysine residue. This is a relative common phenomenon after mutation of lysines that are ubiquitination sites in proteins (40, 41). Further experiments are needed to identify these sites to study the biological function of ubiquitination in the R419X mutant.
Thalidomide Inhibits the Ubiquitination of WT CRBN but Not CRBN R419X
We next considered the possibility that the high level of ubiquitination seen in the CRBN R419X mutant could be reduced by treatment with thalidomide, a therapeutic drug currently used for the treatment of several diseases, including multiple myeloma (9). Thalidomide has previously been shown to reduce the autoubiquitination of CRBN in various cell types (39). Therefore, thalidomide may be useful for reducing the increased levels of autoubiquitination in CRBN R419X. To test this idea, we expressed FS-tagged WT CRBN and CRBN R419X and monitored the autoubiquitination level seen following treatment with thalidomide. In these experiments, cells were treated with MG132 for 3 h after 1 h of pretreatment with thalidomide to allow accumulation of the ubiquitinated form of CRBN, and then CRBN was affinity-purified to measure ubiquitin conjugates on CRBN by anti-ubiquitin Western blotting. Following treatment with thalidomide, the ubiquitination level of WT CRBN was markedly reduced in a dose-dependent manner. However, thalidomide treatment did not reduce the levels of ubiquitination in CRBN R419X (Fig. 8, A and B). These data indicate that CRBN R419X is unaffected by thalidomide treatment.
FIGURE 8.

Restoration of CRBN R419X levels by bortezomib. A, thalidomide (Thal) inhibits the ubiquitination of WT CRBN but not CRBN R419X. To test whether thalidomide could block the enhanced autoubiquitination of CRBN R419X, HEK293T cells expressing either WT or R419X CRBN were treated with DMSO, 10 or 100 μm thalidomide for 1 h. Ubiquitinated protein was then allowed to accumulate by treating cells with 10 μm MG132 for 3 h. CRBN was purified with Strep-Tactin beads under denaturing conditions and immunoblotted with anti-ubiquitin or anti-FLAG antibody (loading control). This experiment showed that the ubiquitination level of WT CRBN is reduced by thalidomide treatment, but the ubiquitination level of CRBN R419X was unaffected by thalidomide. B, quantification of the relative ubiquitination level after thalidomide treatment for WT CRBN and R419X mutant. C, clinically utilized proteasome inhibitor, bortezomib, significantly increases the cellular level of CRBN R419X. To test whether bortezomib inhibits CRBN degradation, HEK293T cells were transfected with WT and R419X CRBN and treated with DMSO or bortezomib (Btz, 1 μm) for 24 h. Lysates were separated by SDS-PAGE, and CRBN levels were determined by immunoblotting with an anti-FLAG antibody. The Western blot (WB) showed that bortezomib slightly increases the protein level of WT CRBN, although it significantly increases the level of CRBN R419X. The right panels show the relative quantification of the WT and R419X CRBN after bortezomib treatment. These data suggest that bortezomib may be useful for restoring CRBN R419X levels in patients with this mutation.
Bortezomib Restores the Level of CRBN R419X
We next considered the possibility that clinically useful proteasome inhibitors might be able to restore the expression levels of CRBN R419X. We focused on bortezomib, which is currently used in the treatment of multiple myeloma (42) and mantle cell lymphoma (33, 36). Treatment of HEK293T cells expressing FS-CRBN with bortezomib resulted in a slight increased level of CRBN (Fig. 8, B and C). However, bortezomib resulted in a more substantial increase in the levels of CRBN R419X (Fig. 8, B and C). This increase is more robust than that of the WT protein. This result suggests that bortezomib treatment can lead to an increase and potentially renormalization of CRBN activity levels in cells expressing the CRBN R419X mutant. Hence, bortezomib or other proteasome inhibitors under clinical development may be potential therapeutic treatments for restoring the levels of CRBN in cells expressing CRBN R419X.
DISCUSSION
Although the premature termination codon in CRBN has been linked to mild mental retardation, the mechanism by which this mutation affects CRBN has, until now, not been explored. In many cases, premature stop codons lead to reduced mRNA stability due to NMD. However, the R419X mutation is present in the final exon of CRBN and is therefore not expected to trigger NMD of the mutant transcript. Thus, the truncated protein is expected to be translated in cells. We show that this mutant protein is capable of forming a CRL4-CRBN E3 ligase and promoting the ubiquitination of different CRL4-CRBN targets. Our data indicate that the mutation affects CRBN function by promoting its autoubiquitination and degradation. As a result, the R419X mutation leads to CRBN deficiency in cells.
Our data suggest that the R419X mutation interferes with the normal regulation of CRBN autoubiquitination. Autoubiquitination of E3 ligase components has important roles for the activity of these complexes (43). Autoubiquitination of a substrate receptor and its subsequent degradation may be important to enable the replacement of one adaptor for another, enabling CRL4 to form other active E3 ligases (44–46). The C terminus of CRBN seems to have an important role as an autoinhibitory domain that normally limits the ubiquitination rate of CRBN. The loss of the C-terminal 24 amino acids in CRBN R419X results in unrestrained ubiquitination. Because this ubiquitination constitutes Lys-48-linked polyubiquitin chains, this results in CRBN degradation. Thus, the C terminus comprises a ubiquitin autoinhibitory domain that is removed as a result of the R419X mutation. Further truncation of the mutant significantly reduces the ubiquitination level of the protein, which suggests the presence of a ubiquitination domain between amino acids 339 and 418. These results suggest that there are two functional domains, an autoinhibitory domain and a ubiquitination domain, at the C terminus of CRBN.
It is important to note that it is still currently unclear how CRBN deficiency leads to mental retardation. Because of the subtle nature of the human phenotype, it is likely that it will be difficult to model this disease in cultured cells. Nevertheless, the disease is likely due to CRBN deficiency that is caused by enhanced CRL4-mediated autoubiquitination. The reduction in CRBN levels could affect neuronal function by either reducing the levels of CRL4-CRBN activity or by reducing the levels of CRL4-independent CRBN functions. Indeed, there is evidence that CRBN can influence the proteasome activity by binding one of its subunits (13), which may not be involved in CRL4. It will be important to determine the specific functional roles of CRBN to gain insight into how this protein influences neuronal function.
Although our data suggest a potential route to increase CRBN levels, at present it is not known if restoration of CRBN levels in patients will result in improved cognitive performance. It is possible that CRBN deficiency during embryonic development results in neurodevelopmental abnormalities that cannot be restored by augmenting CRBN levels later in development. However, recent studies focusing on Fragile X mental retardation and tuberous sclerosis have suggested that postnatal restoration of protein function can restore some cognitive phenotypes (47). Thus, it will be important to determine whether augmentation of CRBN levels can also improve cognitive performance in patients harboring CRBN mutations.
Our results indicate that therapeutic strategies that prevent the autoubiquitination or degradation of CRBN can restore the levels of CRBN R419X. Our experiments focused on proteasome inhibitors, one of which is currently in use for treatment of multiple myeloma. However, more selective inhibitors are likely to have fewer side effects. Thalidomide has previously been shown to prevent the ubiquitination of CRBN. Because this drug binds to the C terminus of CRBN, which is affected in CRBN R419X, this drug likely is incapable of binding CRBN R419X and as a result cannot prevent its ubiquitination. Another approach could be to use selective inhibitors that target CRL4. However, currently there are no drugs that selectively target this complex. Nevertheless, a family of inhibitors that target other cullin-RING E3 ligases have been described (15), suggesting that CRL4 inhibitors may also be possible for increasing CRBN R419X levels.
Acknowledgments
The lentiviral system was a generous gift from Livio Pellizzoni (Columbia University). The full-length and dominant negative CUL4As and His6-ubiquitin plasmids were from Pengbo Zhou (Weill Medical College of Cornell University). We are grateful to Michael S. Cohen and Alessia Deglincerti for assistance in molecular biology and plasmid transfection and Steven S. Gross (Weill Medical College) and Yuliang Ma (Weill Medical College) for helpful discussions in MS/MS analysis. The mass spectrometry work was performed at the Weill Medical College of Cornell University mass spectrometry core facility using instrumentation supported by National Institutes of Health Grants RR19355 and RR22615.
This work was supported, in whole or in part, by National Institutes of Health Grants MH086128 from NIMH (to S. R. J.) and T32CA062948 from NCI (to G. X.). This work was also supported by the National High-tech Research and Development Program of China 973-projects (2012CB947602), National Natural Science Foundation of China Grant 31270874, a project funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (to G. X.).
- EGFP
- enhanced GFP
- NMD
- nonsense-mediated decay
- Ub
- ubiquitin
- IRES
- internal ribosome entry site.
REFERENCES
- 1. Centers for Disease Control and Prevention (CDC) (1996) State-specific rates of mental retardation–United States, 1993. MMWR Morb. Mortal. Wkly Rep. 45, 61–65 [PubMed] [Google Scholar]
- 2. Chelly J., Khelfaoui M., Francis F., Chérif B., Bienvenu T. (2006) Genetics and pathophysiology of mental retardation. Eur. J. Hum. Genet. 14, 701–713 [DOI] [PubMed] [Google Scholar]
- 3. Higgins J. J., Pucilowska J., Lombardi R. Q., Rooney J. P. (2004) A mutation in a novel ATP-dependent Lon protease gene in a kindred with mild mental retardation. Neurology 63, 1927–1931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Higgins J. J., Rosen D. R., Loveless J. M., Clyman J. C., Grau M. J. (2000) A gene for nonsyndromic mental retardation maps to chromosome 3p25-pter. Neurology 55, 335–340 [DOI] [PubMed] [Google Scholar]
- 5. Jackson S., Xiong Y. (2009) CRL4s: the CUL4-RING E3 ubiquitin ligases. Trends Biochem. Sci. 34, 562–570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Angers S., Li T., Yi X., MacCoss M. J., Moon R. T., Zheng N. (2006) Molecular architecture and assembly of the DDB1-CUL4A ubiquitin ligase machinery. Nature 443, 590–593 [DOI] [PubMed] [Google Scholar]
- 7. Lin G. Y., Paterson R. G., Richardson C. D., Lamb R. A. (1998) The V protein of the paramyxovirus SV5 interacts with damage-specific DNA-binding protein. Virology 249, 189–200 [DOI] [PubMed] [Google Scholar]
- 8. Fischer E. S., Scrima A., Böhm K., Matsumoto S., Lingaraju G. M., Faty M., Yasuda T., Cavadini S., Wakasugi M., Hanaoka F., Iwai S., Gut H., Sugasawa K., Thomä N. H. (2011) The molecular basis of CRL4DDB2/CSA ubiquitin ligase architecture, targeting, and activation. Cell 147, 1024–1039 [DOI] [PubMed] [Google Scholar]
- 9. Ito T., Ando H., Suzuki T., Ogura T., Hotta K., Imamura Y., Yamaguchi Y., Handa H. (2010) Identification of a primary target of thalidomide teratogenicity. Science 327, 1345–1350 [DOI] [PubMed] [Google Scholar]
- 10. Lee K. M., Jo S., Kim H., Lee J., Park C. S. (2011) Functional modulation of AMP-activated protein kinase by cereblon. Biochim. Biophys. Acta 1813, 448–455 [DOI] [PubMed] [Google Scholar]
- 11. Jo S., Lee K. H., Song S., Jung Y. K., Park C. S. (2005) Identification and functional characterization of cereblon as a binding protein for large-conductance calcium-activated potassium channel in rat brain. J. Neurochem. 94, 1212–1224 [DOI] [PubMed] [Google Scholar]
- 12. Hohberger B., Enz R. (2009) Cereblon is expressed in the retina and binds to voltage-gated chloride channels. FEBS Lett. 583, 633–637 [DOI] [PubMed] [Google Scholar]
- 13. Lee K. M., Lee J., Park C. S. (2012) Cereblon inhibits proteasome activity by binding to the 20 S core proteasome subunit β type 4. Biochem. Biophys. Res. Commun. 427, 618–622 [DOI] [PubMed] [Google Scholar]
- 14. Ren H., Fu K., Mu C., Zhen X., Wang G. (2012) L166P mutant DJ-1 promotes cell death by dissociating Bax from mitochondrial Bcl-XL. Mol. Neurodegener. 7, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Xu G., Paige J. S., Jaffrey S. R. (2010) Global analysis of lysine ubiquitination by ubiquitin remnant immunoaffinity profiling. Nat. Biotechnol. 28, 868–873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chang Y. F., Imam J. S., Wilkinson M. F. (2007) The nonsense-mediated decay RNA surveillance pathway. Annu. Rev. Biochem. 76, 51–74 [DOI] [PubMed] [Google Scholar]
- 17. Maquat L. E. (2004) Nonsense-mediated mRNA decay: splicing, translation and mRNP dynamics. Nat. Rev. Mol. Cell Biol. 5, 89–99 [DOI] [PubMed] [Google Scholar]
- 18. Nagy E., Maquat L. E. (1998) A rule for termination-codon position within intron-containing genes: when nonsense affects RNA abundance. Trends Biochem. Sci. 23, 198–199 [DOI] [PubMed] [Google Scholar]
- 19. Baumann B., Potash M. J., Köhler G. (1985) Consequences of frameshift mutations at the immunoglobulin heavy chain locus of the mouse. EMBO J. 4, 351–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Baserga S. J., Benz E. J., Jr. (1988) Nonsense mutations in the human β-globin gene affect mRNA metabolism. Proc. Natl. Acad. Sci. U.S.A. 85, 2056–2060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Urlaub G., Mitchell P. J., Ciudad C. J., Chasin L. A. (1989) Nonsense mutations in the dihydrofolate reductase gene affect RNA processing. Mol. Cell. Biol. 9, 2868–2880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mashima Y., Murakami A., Weleber R. G., Kennaway N. G., Clarke L., Shiono T., Inana G. (1992) Nonsense-codon mutations of the ornithine aminotransferase gene with decreased levels of mutant mRNA in gyrate atrophy. Am. J. Hum. Genet. 51, 81–91 [PMC free article] [PubMed] [Google Scholar]
- 23. Enssle J., Kugler W., Hentze M. W., Kulozik A. E. (1993) Determination of mRNA fate by different RNA polymerase II promoters. Proc. Natl. Acad. Sci. U.S.A. 90, 10091–10095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Belgrader P., Maquat L. E. (1994) Nonsense but not missense mutations can decrease the abundance of nuclear mRNA for the mouse major urinary protein, while both types of mutations can facilitate exon skipping. Mol. Cell. Biol. 14, 6326–6336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cheng J., Belgrader P., Zhou X., Maquat L. E. (1994) Introns are cis effectors of the nonsense-codon-mediated reduction in nuclear mRNA abundance. Mol. Cell. Biol. 14, 6317–6325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Carter M. S., Li S., Wilkinson M. F. (1996) A splicing-dependent regulatory mechanism that detects translation signals. EMBO J. 15, 5965–5975 [PMC free article] [PubMed] [Google Scholar]
- 27. Tiso M., Tejero J., Panda K., Aulak K. S., Stuehr D. J. (2007) Versatile regulation of neuronal nitric oxide synthase by specific regions of its C-terminal tail. Biochemistry 46, 14418–14428 [DOI] [PubMed] [Google Scholar]
- 28. Zheng J., Yang X., Harrell J. M., Ryzhikov S., Shim E. H., Lykke-Andersen K., Wei N., Sun H., Kobayashi R., Zhang H. (2002) CAND1 binds to unneddylated CUL1 and regulates the formation of SCF ubiquitin E3 ligase complex. Mol. Cell 10, 1519–1526 [DOI] [PubMed] [Google Scholar]
- 29. Liu L., Lee S., Zhang J., Peters S. B., Hannah J., Zhang Y., Yin Y., Koff A., Ma L., Zhou P. (2009) CUL4A abrogation augments DNA damage response and protection against skin carcinogenesis. Mol. Cell 34, 451–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Haberland J., Becker J., Gerke V. (1997) The acidic C-terminal domain of rna1p is required for the binding of Ran·GTP and for RanGAP activity. J. Biol. Chem. 272, 24717–24726 [DOI] [PubMed] [Google Scholar]
- 31. Wu F., Xing J., Wang S., Li M., Zheng C. (2011) Screening and identification of host factors interacting with UL14 of herpes simplex virus 1. Med. Microbiol. Immunol. 200, 203–208 [DOI] [PubMed] [Google Scholar]
- 32. Chen X., Zhang Y., Douglas L., Zhou P. (2001) UV-damaged DNA-binding proteins are targets of CUL-4A-mediated ubiquitination and degradation. J. Biol. Chem. 276, 48175–48182 [DOI] [PubMed] [Google Scholar]
- 33. Amemiya Y., Azmi P., Seth A. (2008) Autoubiquitination of BCA2 RING E3 ligase regulates its own stability and affects cell migration. Mol. Cancer Res. 6, 1385–1396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chen A., Kleiman F. E., Manley J. L., Ouchi T., Pan Z. Q. (2002) Autoubiquitination of the BRCA1·BARD1 RING ubiquitin ligase. J. Biol. Chem. 277, 22085–22092 [DOI] [PubMed] [Google Scholar]
- 35. Lai Z., Yang T., Kim Y. B., Sielecki T. M., Diamond M. A., Strack P., Rolfe M., Caligiuri M., Benfield P. A., Auger K. R., Copeland R. A. (2002) Differentiation of Hdm2-mediated p53 ubiquitination and Hdm2 autoubiquitination activity by small molecular weight inhibitors. Proc. Natl. Acad. Sci. U.S.A. 99, 14734–14739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yang Y., Fang S., Jensen J. P., Weissman A. M., Ashwell J. D. (2000) Ubiquitin protein ligase activity of IAPs and their degradation in proteasomes in response to apoptotic stimuli. Science 288, 874–877 [DOI] [PubMed] [Google Scholar]
- 37. Zhou P., Howley P. M. (1998) Ubiquitination and degradation of the substrate recognition subunits of SCF ubiquitin-protein ligases. Mol. Cell 2, 571–580 [DOI] [PubMed] [Google Scholar]
- 38. Wilkinson K. D., Tashayev V. L., O'Connor L. B., Larsen C. N., Kasperek E., Pickart C. M. (1995) Metabolism of the polyubiquitin degradation signal: structure, mechanism, and role of isopeptidase T. Biochemistry 34, 14535–14546 [DOI] [PubMed] [Google Scholar]
- 39. Singhal S., Mehta J., Desikan R., Ayers D., Roberson P., Eddlemon P., Munshi N., Anaissie E., Wilson C., Dhodapkar M., Zeddis J., Barlogie B. (1999) Antitumor activity of thalidomide in refractory multiple myeloma. N. Engl. J. Med. 341, 1565–1571 [DOI] [PubMed] [Google Scholar]
- 40. Bhat K. P., Truax A. D., Greer S. F. (2010) Phosphorylation and ubiquitination of degron proximal residues are essential for class II transactivator (CIITA) transactivation and major histocompatibility class II expression. J. Biol. Chem. 285, 25893–25903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Treier M., Staszewski L. M., Bohmann D. (1994) Ubiquitin-dependent c-Jun degradation in vivo is mediated by the δ domain. Cell 78, 787–798 [DOI] [PubMed] [Google Scholar]
- 42. Damaj G., Lefrère F., Delarue R., Varet B., Furman R., Hermine O. (2003) Thalidomide therapy induces response in relapsed mantle cell lymphoma. Leukemia 17, 1914–1915 [DOI] [PubMed] [Google Scholar]
- 43. Bosu D. R., Kipreos E. T. (2008) Cullin-RING ubiquitin ligases: global regulation and activation cycles. Cell Div. 3, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Silva A. J., Ehninger D. (2009) Adult reversal of cognitive phenotypes in neurodevelopmental disorders. J. Neurodev. Disord. 1, 150–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zeier Z., Kumar A., Bodhinathan K., Feller J.A., Foster T. C., Bloom D. C. (2009) Fragile X mental retardation protein replacement restores hippocampal synaptic function in a mouse model of fragile X syndrome. Gene Ther. 16, 1122–1129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ehninger D., Matynia A., Silva A. J. (2005) Trafficking in emotions. Nat. Neurosci. 8, 548–550 [DOI] [PubMed] [Google Scholar]
- 47. Soucy T. A., Smith P. G., Milhollen M. A., Berger A. J., Gavin J. M., Adhikari S., Brownell J. E., Burke K. E., Cardin D. P., Critchley S., Cullis C. A., Doucette A., Garnsey J. J., Gaulin J. L., Gershman R. E., Lublinsky A. R., McDonald A., Mizutani H., Narayanan U., Olhava E. J., Peluso S., Rezaei M., Sintchak M. D., Talreja T., Thomas M. P., Traore T., Vyskocil S., Weatherhead G. S., Yu J., Zhang J., Dick L. R., Claiborne C. F., Rolfe M., Bolen J. B., Langston S. P. (2009) An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature 458, 732–736 [DOI] [PubMed] [Google Scholar]