

Background: OxLDL and the high level of hyaluronan are major triggering factors of atherosclerosis.
Results: The oxLDL load of the aortic human smooth muscle cells (SMC) via the scavenger receptor LOX-1 causes ER stress, overexpression of HAS2, and hyaluronan deposition.
Conclusion: the oxidized sterols driven into the SMC by oxLDL have a role in hyaluronan metabolism.
Significance: oxLDL influences extracellular matrix hyaluronan.
Keywords: Atherosclerosis, Cholesterol, Glycosaminoglycan, Scavenger Receptor, Vascular Smooth Muscle Cells
Abstract
Thickening of the vessel in response to high low density lipoprotein(s) (LDL) levels is a hallmark of atherosclerosis, characterized by increased hyaluronan (HA) deposition in the neointima. Human native LDL trapped within the arterial wall undergoes modifications such as oxidation (oxLDL). The aim of our study is to elucidate the link between internalization of oxLDL and HA production in vitro, using human aortic smooth muscle cells. LDL were used at an effective protein concentration of 20–50 μg/ml, which allowed 80% cell viability. HA content in the medium of untreated cells was 28.9 ± 3.7 nmol HA-disaccharide/cell and increased after oxLDL treatment to 53.9 ± 5.6. OxLDL treatments doubled the transcripts of HA synthase HAS2 and HAS3. Accumulated HA stimulated migration of aortic smooth muscle cells and monocyte adhesiveness to extracellular matrix. The effects induced by oxLDL were inhibited by blocking LOX-1 scavenger receptor with a specific antibody (10 μg/ml). The cholesterol moiety of LDL has an important role in HA accumulation because cholesterol-free oxLDL failed to induce HA synthesis. Nevertheless, cholesterol-free oxLDL and unmodified cholesterol (20 μg/ml) induce only HAS3 transcription, whereas 22,oxysterol affects both HAS2 and HAS3. Moreover, HA deposition was associated with higher expression of endoplasmic reticulum stress markers (CHOP and GRP78). Our data suggest that HA synthesis can be induced in response to specific oxidized sterol-related species delivered through oxLDL.
Introduction
Atherosclerosis, usually classified as a chronic inflammatory disease of the blood vessel wall, is the most common cause of cardiovascular diseases. The localized inflammation causes the thickening of the arterial wall due to the increased deposition of extracellular matrix (ECM).3 The newly formed ECM traps lipoprotein and inflammatory/growth factors from the circulation within the vessel wall (1). The atheromatous plaque is therefore formed by a central core of cholesterol crystals and foam cells encircled by altered ECM.
Despite the many cardiovascular risk factors reported in published guidelines and reduction strategies, recent studies underscored again the relevance of plasma lipoproteins (LDL) in the pathogenesis of atherosclerosis (2). In particular, the ratio of the protein moieties of the lipoproteins ApoB/ApoA1 (reflecting LDL/HDL amounts) and the quantification of the circulating LDL-cholesterol are at the moment the routine risk factors evaluated in the daily clinical practice. The chronic and repeated exposure to pathogenic risk factors triggers a complex interplay between circulating species, such as monocytes and lymphocytes, and various cell types, resident within the arterial wall, including endothelial cells and smooth muscle cells (SMC).
In the healthy arterial vessel wall, endothelial cells regulate the vascular tone, the thrombosis/fibrinolysis balance, and the recruitment of inflammatory cells; most of these abilities are due to ECM that is enriched with hyaluronan (HA), which forms the glycocalyx. Although the HA within the arterial wall is present in physiological conditions predominantly in the tunica adventitia produced by vascular SMC, during the atherosclerotic process HA, is highly synthesized and also deposited in the neointima (3–5), thus contributing to the thickening of the arterial wall.
HA is a nonsulfated glycosaminoglycan that is present in ECMs as a polyanionic chain of up to 107 Da. Several cell types, including aortic SMC (AoSMC), produce HA either in physiological or in pathological conditions. HA is the only glycosaminoglycan synthesized at the plasma membrane (6) from cytoplasmic UDP-sugar precursors. The HA synthases (HASs) are a family of three members (HAS1, HAS2, and HAS3), each one characterized by expression timelines and regulation at transcriptional and/or post-transcriptional levels (7, 8). HA, both secreted and pericellular, is known to have a pivotal role in cell migration, proliferation, and leukocyte adhesion in the atherosclerotic plaques (9, 10). Inflammation processes are strongly influenced by HA (11). Merrilees et al. (12) demonstrated in an in vivo model that monocytes localize in the atherosclerotic plaque in an HA-dependent manner and that the HA-rich ECM is promoted by the presence of oxidized LDL (oxLDL). Given the numerous oxidative events associated with the development of an inflammatory atherosclerotic plaque (13), LDL, which accumulate early in the intima primarily from enhanced retention by the glycosaminoglycans (1, 12, 14), are converted into aggregated LDL (aggLDL) (15) and/or oxLDL (16). OxLDL, in addition to the proper oxidized lipid products, also contains lysophospholipids, hydrolytic derivatives of oxidized phospholipids, prostanoids, isoprostanoids, leukotrienes, which are derived from the oxidation of arachidonic acid (17), and oxysterols (18), which contribute to reactive oxygen species generation and amplification of the steps described thus far (19).
Although the majority of SMC in the arterial wall are contained within the medial layer, a significant number exist within the intima as well. Those SMC retain a certain degree of plasticity, altering their expression of cell-specific genes in response to many stimuli, i.e. switching from the “contractile” to the “synthetic” phenotypic state. This phenotypic switch can be triggered by several atherogenic stimuli, such as ECM composition, cytokines, shear stress, reactive oxygen species, and lipids, and is associated with different behaviors of SMC (20). Moreover, those SMC can also give rise to a significant number of lipid-laden cells, although the larger portion of the foam cells derives from macrophages (20, 21). The lipid load can induce endoplasmic reticulum (ER) stress (22, 23) that is associated with oxidative stress (24) and can enhance atherosclerosis progression.
In this study, we used a cellular model to investigate the correlation between the exposure of AoSMC to oxLDL and the accumulation of HA. In particular, we analyzed at the molecular level the mechanisms that induced AoSMC to produce an HA-enriched ECM, the role of the oxLDL in delivering molecules that can trigger the phenotypic switch, and the eventual contribution of cholesterol to this early atherogenic event.
EXPERIMENTAL PROCEDURES
Cell Culture
Human AoSMC were purchased from Lonza and grown in the complete SmGm2 culture medium (Lonza, Basel, Switzerland) supplemented with 5% FBS as described previously (25). 1.5 × 105 cells were seeded in a 6-well plate at 70–80% confluence. They were incubated for 48 h in DMEM with 0.2% FBS medium and afterward supplemented with SmGm2, 5% FBS and incubated for a further 48 h with different concentrations of native, oxidized, and aggregated LDL (nLDL, oxLDL, and aggLDL, respectively). Cell viability was evaluated with trypan blue staining. Monocyte U937 cells were grown in suspension culture in RPMI 1640 medium containing 10% FBS and routinely cultured at 1–10 × 105 cells/ml.
LDL Isolation
Native LDL (nLDL, d = 1.019–1.063 g/ml) were purified from plasma of normocholesterolemic volunteers kindly provided by the “Ospedale di Circolo” following the method described in Ref. 26. OxLDL were obtained by incubation of 100 μg/ml nLDL with 5 μm CuSO4 at 37 °C for 16–18 h, and the reaction was then stopped with EDTA 1 mm. The oxLDL were desalted by filtration in PD-10 columns (GE Healthcare) and concentrated with Centriprep concentrators (10 kDa NMWL, nominal molecular weight limit) (Amicon). AggLDLs were obtained by vortexing nLDL for 5 min at room temperature (15). The nLDLs were treated overnight with 5 mm methyl-β-cyclodextrin (Sigma) (27) at 37 °C with gentle mixing, and excess of methyl-β-cyclodextrin was eliminated by filtration on PD-10 columns to obtain the cholesterol-free form of LDL (CF-nLDL). CF-nLDLs were then subjected to oxidation as described above, and oxidized cholesterol-free LDLs (CF-oxLDLs) were obtained. All LDL preparations were sterilized by filtration through 0.2-μm filters. Protein contents were determined by the Bradford assay, and total cholesterol was quantified by a colorimetric enzymatic assay (Olympus Cholesterol OSR6516). The purity of LDL preparations was assessed by agarose gel electrophoresis. Briefly, 50 μg of total proteins were added with the same volume of Sudan black (Sigma) 2 mg/ml in ethanol, 2% glycerol, incubated at room temperature in the dark for 1 h, loaded in a 0.5% agarose gel in TAE buffer (40 mm Tris, 20 mm acetic acid, 1 mm EDTA), and run with 0.05 m thiobarbituric acid buffer pH 8.5, 100 V for 30 min. Anti-scavenger receptor LOX-1 antibody was added at a final concentration of 10 μg/ml.
Cholesterol Loading
Cholesterol was delivered to AoSMC in a complex with (2-hydroxypropyl)-β-cyclodextrin solution (CD) (Sigma), a cyclic oligosaccharide that solubilizes cholesterol and can be used as a “carrier” molecule. The mixture was prepared as described previously (27) with a final concentration of 10 μg/ml cholesterol. When CD-cholesterol complex was added to cell culture, the final CD concentration was 2.5 mm, which was therefore added to AoSMC as a further control.
LXR Assays
(22)(R)-Hydrocholesterol (Sigma) was added to the culture medium at a final concentration of 7.5 μm for 48 h.
DiI Labeling
nLDL and oxLDL were labeled with DiI (1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate, Molecular Probes) following the procedure described previously (28). Briefly, LDL preparations at 1 mg/ml were incubated with DiI 3 mg/ml in DMSO overnight at 37 °C, recovered by ultracentrifugation, and concentrated. Images of AoSMC after a 24-h treatment with 20 μg/ml nLDL-DiI or oxLDL-DiI, in the absence or presence of anti-LOX-1 antibody (10 μg/ml), were captured using fluorescence microscopy (Olympus), and the cell fluorescence was quantified using the ImageJ software (58) and expressed as corrected total cell fluorescence.
Gene Expression
Total RNA was extracted from AoSMC cultures with TRIzol (Invitrogen, Milan, Italy), digested with Turbo DNase (Ambion, Monza, Italy), retrotranscribed by the High Capacity cDNA synthesis kit (Applied Biosystems, Monza, Italy), and amplified on an ABI Prism 7000 instrument (Applied Biosystems) using the TaqMan universal PCR master mix (Applied Biosystems) following the manufacturer's instructions. The following human TaqMan gene expression assays were used: HAS1 (Hs00155410), HAS2 (Hs00193435_m1), HAS3 (Hs00193436_m1), CHOP (Hs01090850_m1), GRP78 (Hs00946084_g1), and the housekeeping gene β-actin (Hs99999903_m1). The relative quantification of gene expression was determined by comparing 2−ΔΔCt (29). For semiquantitative RT-PCR analyses of LDL scavenger receptors, cDNAs were amplified using the following primers: LOX-1 (forward, 5′-GCCATTCCGAAATCAAGAAA-3′ and reverse, 5′-AGGAGTCATCAGGAGGAGCA-3′, SR-PSOX (forward, 5′-TACACGAGGTTCCAGCTCCT-3′ and reverse, 5′-TACCATGTTGTCAGGGGTCA-3′), SR-AI (forward, 5′-GGAACACATGAGCAACATGG-3′ and reverse, 5′-CCAAGCTCCTACAGACGACC-3′), CD36 (forward, 5′-GGCTGAGCAAGGTTGACTTC-3′ and reverse, 5′-CCTCCTTATCCTTTGAGCCC-3′), LRP1 (forward, 5′-ACCACCAGCTACCTCATTGG-3′ and reverse, 5′-TTGTTCTCGCACTTGAATCG-3′), and housekeeping β-actin (forward, 5′-CTCTTCCAGCCTTCCTTCCT-3′ and reverse, 5′-ATGCTATCACCTCCCCTGTG-3′). Amplifications of cDNA were done using the first strand cDNA synthesis mixture, 0.1 μm forward and reverse primers, 200 μm dNTPs, and RedTaq polymerase (Sigma) in its own buffer. Reaction mixtures were subjected to 35 cycles with the following parameters: denaturation at 94 °C for 30 s, annealing at 52 °C for 40 s, and elongation at 72 °C for 1 min.
HA Determination
HA released into the medium was quantified by HPLC analysis as described previously (30). HA in the pericellular coating was determined as in Ref. 25, quantified after particle exclusion assay using ImageJ software, and expressed as the percentage of total area (pericellular coating area/(cell area + pericellular coating area)×100).
Cell Mobility Assay
Migration assay was done as described previously in Ref. 25 with quantification by Image J software as follows. Wound area was measured at time 0 (t0) and after 24 h (t24), and the wound closure was determined by the equation: ((wound area t0 − wound area t24)/wound area t0)×100. AoSMC cultures were treated with 4-methylumbelliferone in DMSO at a final concentration of 1 mm (25).
Monocyte Adhesion Assay
Adhesion assay was performed using the monocyte U937 cell line as described previously in Ref. 31. Briefly, U937 were labeled with fluorescent green CytoTracker (Invitrogen, Milan, Italy) vital stain and plated on AoSMC incubated with oxLDL. As control, AoSMC were pretreated with 2 units/μl Streptomyces hyaluronidase (Seikagaku Corp., Tokyo, Japan). After PBS washing, the adherent cells were quantified under a fluorescent microscope (Olympus, Segrate, Italy).
Statistical Analysis
GraphPad Prism version 5.01 for Windows and GraphPad Software were used for statistical analysis. The number of independent replicate experiments is indicated in each figure legend. Data are presented as means of independent experiments ± S.E. Statistical significance was tested with analysis of variance followed by Bonferroni's post hoc test, or in Fig. 5B, by unpaired Student's t test. Statistically significant values of p are reported in the figure legends.
FIGURE 5.

OxLDL treatment induces ER stress in AoSMC. A and B, quantification of transcripts coding for CHOP (A) and GRP78 (B), ER stress markers, with respect to control (CNT) in AoSMC treated for 24 h with 20 μg of nLDL or oxLDL in the absence (−) or presence (+) of 10 μg/ml LOX-1 antibody. Values are mean ± S.E. of triplicates of two independent experiments, ***, p = 0.001. C, quantitative RT-PCR analysis of HAS2 and HAS3 expression of CD-cholesterol and (22)(R)-hydrocholesterol treatment in AoSMC, each reported with respect to its control AoSMC (CNT+CD and CNT, respectively). Values are mean ± S.E. of two independent experiments, *, p < 0.05, **, p < 0.01, ***, p < 0.001.
RESULTS
Oxidized LDL Increase HA Secretion in AoSMC by Enhancing HAS2 and HAS3 mRNA Levels
To investigate how LDL particles may affect glycosaminoglycans secreted by AoSMC and contribute to the onset of atherosclerosis, we subjected AoSMC to 48 h of treatment with different LDLs. Increasing amounts of nLDL, oxLDL, and aggLDL were added to cultured cells, and the cell viability was evaluated after 48 h (Fig. 1A). Nontoxic concentrations spanned the range of 10–50 μg of LDL proteins/ml for all the preparations, with an average of more than 80% of total vital cells after the exposure. At higher concentrations, oxLDL and aggLDL were toxic to AoSMC, with the highest effect obtained with oxLDL. Treatment with 20–50 μg/ml of either nLDL or aggLDL did not modify HA secretion into the culture medium beyond control level (Fig. 1B). In contrast, oxLDLs at these concentrations significantly increased HA secretion, from 28.9 ± 3.7 nmol Δ-HA/cell of the control to 53.9 ± 5.6 and 61.0 ± 2.8 nmol Δ-HA/cell in 20 and 50 μg/ml oxLDL treatments, respectively (Fig. 1B). Interestingly, the 20–50 μg/ml concentrations of oxLDL were able to produce a similar effect on HA secretion. Because aggLDL did not influence the levels of either HA or CS (data not shown), they were not tested further. AoSMC pericellular coat was also increased after 48 h of treatment with oxLDL as compared with control and nLDL-treated cultures (Fig. 1C). The increase was mainly constituted by HA because hyaluronidase digestion restored the sizes to control levels as shown by quantification after particle exclusion assays (Fig. 1C). Again, the coating sizes at the two concentrations of oxLDL were nearly the same. Therefore, in the following experiments, only the lowest active amount (20 μg/ml) was used. Because AoSMC can express three hyaluronan synthases (32), we measured the levels of their mRNAs. HAS1 mRNA was not significantly detected in AoSMC cultures (data not shown). The expression of HAS2 in untreated AoSMC was about 30-fold higher as compared with HAS3 (Fig. 1D, lowest panel). HAS2 and HAS3 mRNA levels were both doubled after 48 h of oxLDL exposure (Fig. 1D, upper panels) as compared with control and nLDL treatments. The enhanced HA synthesis could therefore be due to the increases of the hyaluronan synthases 2 and 3 transcripts.
FIGURE 1.

Effect of different LDL particles on AoSMC viability, HA, secretion, and HAS expression. A, viability of AoSMC after 48 h of treatment with increasing amounts of nLDL (empty circles), oxLDL (empty squares), and aggLDL (filled triangles) was evaluated by trypan blue staining and expressed as the percentage of the total cell number. B, AoSMC were treated with different amounts (20 and 50 μg of LDL proteins/ml) of nLDL (striped bars), oxLDL (filled bars), or aggLDL (dotted bars) for 48 h in complete medium. Media were collected, and glycosaminoglycans were precipitated and quantified by HPLC. Amounts of Δ-HA disaccharides from hyaluronidase digests of HA secreted into the media after the treatments are shown with respect to control (white bar). Values are mean ± S.E. of triplicates, **, p < 0.01, ***, p < 0.001. C, quantification of HA pericellular coating after 24 h of treatment with nLDL and oxLDL. Values are mean ± S.E. of triplicates, ***, p < 0.01, ***, p < 0.001. D, quantitative RT-PCR analysis of HAS expression in AoSMC treated with or without (CNT) 20 μg of nLDL (striped bars) or oxLDL (filled bars). RNA from AoSMC was analyzed for HAS2 (empty bars) and HAS3 (filled bars) expression using β-actin as housekeeping gene. The result is expressed as relative expression of the HAS3 and HAS2 genes in the treatments with respect to the untreated AoSMC (CNT). The lower panel represents the relative expression of HAS3 with respect to HAS2 and for each gene, with respect to CNT. Data are mean ± S.E. of three independent experiments **, p < 0.01, ***, p < 0.001.
Increased HA Affects in Vitro Motility and AoSMC ECM Adhesiveness
Cell migration, a critical event in atherosclerosis development, can be regulated by HA (25). We therefore tested whether oxLDL can affect cell motility by scratch assays. Exposure to oxLDL increased the migration ability ∼41 and 33% as compared with control and nLDL treatments, respectively. The increased migration depends on HA because treatment with 4-methylumbelliferone, an inhibitor of HA synthesis (25), restored initial migration levels (Fig. 2A). In addition, HA produced by oxLDL treatment enhanced adhesion of U937 monocytic cells (Fig. 2B). Digestion with hyaluronidase significantly decreased the monocyte adhesion, but did not completely revert the adhesion to control level, probably due to the unmasking of other adhesion molecules (i.e. integrins).
FIGURE 2.
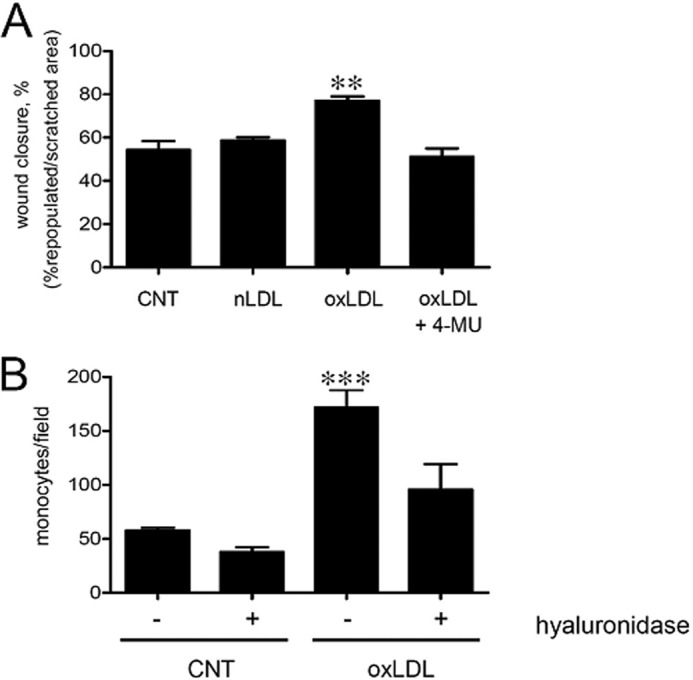
OxLDL modulates migration and ECM adhesive properties of AoSMC. A, evaluation of AoSMC migration ability by scratch assay. AoSMC were serum-deprived (0.2% FBS) for 48 h and scratched with a pipette tip, and new complete medium was added with or without (CNT) 20 μg of nLDL, oxLDL, or oxLDL + 1 mm 4-methylumbelliferone (4-MU). The migration was followed up to 24 h, and the mobility rate was calculated with respect to time 0 for each treatment as the percentage of the repopulated area with respect to the starting scratched area, using the ImageJ software. Results are expressed as mean ± S.E. of three independent experiments, ** p < 0.01. B, U937 monocyte adhesion assays were done on AoSMC alone or treated with 20 μg of oxLDL. After 48 h, fluorescent U937 cells were added for 20 min at 37 °C, washed, and counted under a fluorescence microscope. As control, cells were treated with hyaluronidase. The experiments were repeated three times. Results are expressed as number of adherent U937 cells per field and are shown as mean ± S.E., ***, p < 0.001.
LOX-1 Scavenger Receptor Mediates oxLDL Internalization and HA Secretion
A number of scavenger receptors involved in cholesterol metabolism are known to bind modified LDL and to be expressed by human AoSMC (20, 33). SR-PSOX and LOX-1 specifically recognize oxLDL, and SR-AI and CD36 bind oxLDL and also acetylated LDL. In addition, LDL receptor-related protein 1 (LRP1) is known to have a specific role in uptake of aggLDL (15). We therefore screened by PCR the expression of various scavenger receptors in our cell model (Fig. 3A) and found that CD36 and LRP1 were already present on untreated cells. LOX-1 and SR-PSOX, the specific oxLDL receptors, together with SR-AI, were very poorly expressed by resting AoSMC (the primers were tested using U937 cells RNA, data not shown). Nevertheless, expression of scavenger receptors may be induced by exposure to modified LDLs. Indeed, after oxLDL exposure, LOX-1 was the only receptor to be up-regulated, in good agreement with data from literature (34), whereas no up-regulation was detected following nLDL exposure. The expression of SR-PSOX, SR-AI, CD36, and LRP1 was not increased upon oxLDL exposure.
FIGURE 3.
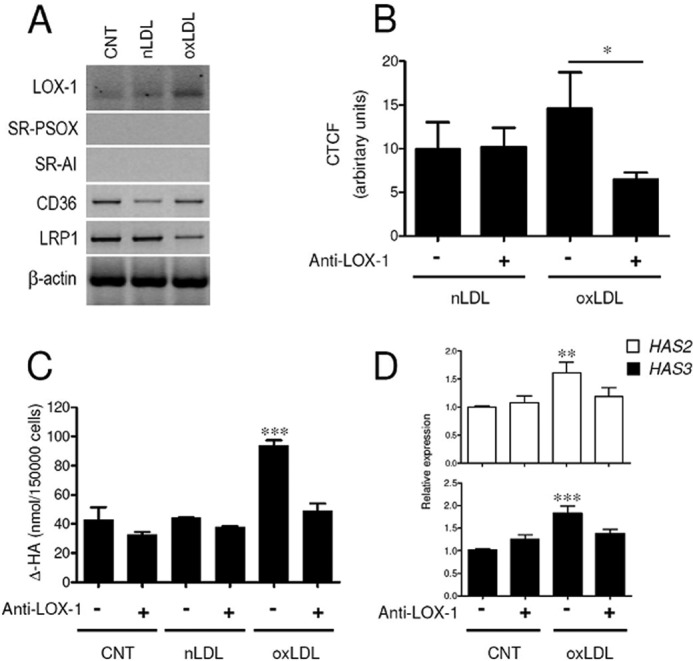
LOX-1 scavenger receptor is responsible for the internalization of oxLDL and mediates their effect. A, RT-PCR analysis of scavenger receptors LOX-1, SR-PSOX, SR-AI, CD36, and LRP1 and of housekeeping gene β-actin in untreated AoSMC and after exposure to 20 μg of nLDL or oxLDL. B, quantification of corrected total cell fluorescence (CTCF) in AoSMC treated with 20 μg of nLDL-DiI and oxLDL-DiI in the absence (−) or presence (+) of anti-LOX-1 antibody. Results are expressed as corrected total cell fluorescence and represented as mean ± S.E., *, p < 0.05. C, quantification by HPLC analysis of ΔHA disaccharides of hyaluronidase digests of HA secreted in culture medium of AoSMC untreated (CNT) or treated with 20 μg of nLDL or oxLDL. Cells were subjected (+) or not (−) to the simultaneous treatment with anti-LOX-1 antibody. D, quantitative RT-PCR analysis of HAS2 and HAS3 expression of CNT- and oxLDL-treated AoSMC from C. Results are expressed as relative expression of the HAS3 and HAS2 genes in the treatments with respect to the untreated AoSMC (CNT) (− anti-LOX-1). Values are mean ± S.E. of triplicates of two independent experiments, **, p < 0.01, ***, p < 0.001.
LOX-1 receptor is recognized as the major player in internalization of oxLDL in a variety of cells (35, 36), and it is known to be up-regulated in response to oxLDL exposure. Thus, we investigated the involvement of LOX-1 in oxLDL uptake and HA secretion in AoSMC by blocking with a specific antibody that was added to cell cultures at time 0 and re-added at the same concentration after 24 h to overcome any degradation of the antibody. To evaluate uptake, AoSMC were treated with DiI-labeled nLDL or oxLDL, and the DiI fluorescence was quantified after 24 h (Fig. 3B). Both nLDL-DiI and oxLDL-DiI were taken up as assessed by fluorescence intensity measured within the cells. Labeled oxLDL-DiI was internalized with a higher rate as compared with nLDL-DiI although the increment in total fluorescence was not statistically different. It is noteworthy that the simultaneous presence of anti-LOX-1 antibody significantly affected the total fluorescence of oxLDL-DiI-treated AoSMC but had no effect on nLDL-DiI-treated cells.
In addition, when cells were exposed to oxLDL in the presence of anti-LOX-1 antibody, the amount of HA within the culture medium was strongly reduced to concentrations found in untreated cells (Fig. 3C). HA release by untreated and nLDL-treated cells was not affected by the presence of LOX-1 antibody. Scavenger receptor LOX-1 appears to be the preferential way by which oxLDL enter the cells and influence HA secretion. Moreover, in the context of oxLDL exposure, treatment with anti-LOX-1 antibody reduced the expression of HAS2 and, with a lesser extent, of HAS3 (Fig. 3D). Treatment with a control antibody (anti-α-tubulin) had no significant effect on either HAS2 or HAS3 mRNA levels (data not shown).
HAS Expression Depends on Oxidized Sterol-related Species in oxLDL
To assess whether the excess of cholesterol delivered through oxLDL was responsible for the increased HA synthesis, we prepared cholesterol-depleted oxLDL by treatment with methyl-β-cyclodextrin, which specifically removes cholesterol from membranes and lipoproteins (27). The particles obtained, CF-nLDL, were then oxidized and named CF-oxLDL. They were then stained with Sudan black and run on an agarose gel to check their mobility (Fig. 4A, inset). Indeed, CF-nLDL and CF-oxLDL showed higher electrophoretic mobilities as compared with nLDL and oxLDL. Total cholesterol content diminished as was confirmed by quantification. The ratio of cholesterol/proteins, which spanned the range 0.9–1.6 in all nLDL and oxLDL preparations used, was decreased to 0.1–0.47 in CF-nLDL and CF-oxLDL. Neither CF-oxLDL nor CF-nLDL stimulated HA release in the medium (Fig. 4A) as compared with levels of untreated AoSMC (CNT).
FIGURE 4.
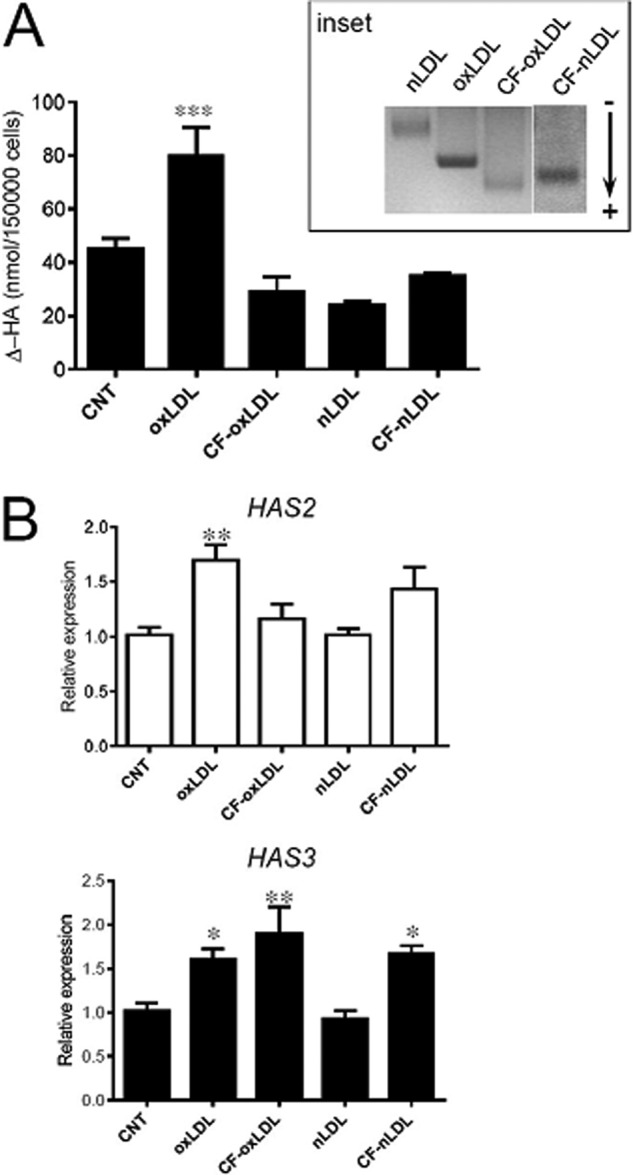
HAS expression depends on oxidized sterol-related species in oxLDL. A, quantification by HPLC analysis of Δ-HA disaccharides of hyaluronidase digests of HA secreted in culture medium of controls (CNT) and treated AoSMC as indicated (20 μg of proteins each). Values are mean ± S.E. of triplicates of three independent experiments, ***, p = 0.001. Electrophoretic mobility of nLDL, oxLDL, and cholesterol-free LDL forms on agarose gel are shown in the inset, and the direction of migration is indicated by an arrow. B, quantitative RT-PCR analysis of HAS2 and HAS3 expression of samples depicted in A. Results are expressed as relative expression of HAS3 and HAS2 for each gene with respect to its control (CNT). Values are mean ± S.E. of three independent experiments, *, p < 0.05, **, p < 0.01.
Quantitative RT-PCR analysis of HAS expression showed that HAS2 was not increased by exposure to CF-oxLDL (Fig. 4B) with respect to its control. On the other hand, HAS3 expression appeared not to be modulated in the same fashion because a higher level of mRNA was measured in CF-oxLDL- and CF-nLDL-treated AoSMC. The different response of HAS3 transcription with respect to HAS2 not related to the HA production suggests a possible alternative role of the enzyme in cells behavior, e.g. adhesion or motility mechanisms.
OxLDL Treatment Induces Endoplasmic Reticulum Stress in AoSMC
Because lipid load and excess of cholesterol might lead to ER stress (22), we determined whether oxLDL exposure may modulate levels of CHOP and GRP78, marker genes for the ER membrane stress in AoSMC. Because ER stress is a fast event, the transcript levels were checked after 24 h of exposure. OxLDL induced an up-regulation of both CHOP and GRP78 24 h after the treatment, whereas nLDL showed no effect. Interestingly, the induction of ER stress was abolished when cells were simultaneously exposed to LOX-1 antibody (Fig. 5, A and B).
The HAS2 and HAS3 induction seems therefore related to an oxidized sterol-related specie driven inside the cells without regulation by the oxLDL. To assess this hypothesis, we used both unmodified cholesterol and the sterol 22(R)-hydrocholesterol, targeting the LXR/RXR sterol sensor system, to investigate their involvement in the expression of the enzymes. In fact, liver X receptors (LXR) of the nuclear receptor superfamily are factors that regulate the transcription of several genes involved in cholesterol metabolism.
AoSMC were exposed to CD-cholesterol, a complex used as a sterol donor to induce cholesterol loading of cell membranes (37, 38), using as control CD-treated AoSMC (Fig. 5C) (CNT + CD), with the evidence that cholesterol alone increased only the HAS3 expression ∼3.2-fold. A similar situation, with an even more significant effect on both HAS2 and HAS3 gene expression, was evidenced using the (22)R-hydrocholesterol, hereinafter named 22,oxysterol (39, 40) (Fig. 5C) in which we had an increase of 8.8- and 9.3-fold, respectively.
DISCUSSION
The results of our work demonstrate that internalization of oxLDL by AoSMC via scavenger receptor LOX-1 increases the synthesis and secretion of HA through regulation of the HAS2 gene, and possibly the HAS3 gene, at the transcriptional level and that this effect is due to oxidized sterol overload of the cell. LDL retention at the subendothelial level is one of the first events triggering atherosclerosis, with the concurrent involvement of SMC for the disease onset and progression (20, 41). It has been shown that, together with macrophages, AoSMC overloaded with cholesterol contribute to foam cell formation (21), highlighting the emerging role of this cell type in early atherosclerosis. AoSMC are known to highly synthesize HA, which is one of the major components of ECM that regulates a variety of processes (e.g. proliferation, adhesion, and migration) in physiological conditions. It is also known to be accumulated in ECMs in vivo both in human (9, 42) and in animal models of atherosclerosis (5). Thus, AoSMC are a good model to explore the link between HA deposition in atherosclerotic plaque and the LDL/cholesterol altered metabolism as well as macrophages (43). LDL chemical modifications (aggregation and oxidation) are processes occurring in vivo as a consequence of LDL trapping within the arterial wall. Oxidation is the most representative modification, and several enzymatic and nonenzymatic oxidant candidates have been described to be involved (16). Oxidation may affect both lipid and protein moieties of the particles, and as a result, a series of biologically active species including peroxides, aldehydes, and oxysterols may be produced (44, 45). Intracellular and cell membrane cholesterol contents are finely regulated by a feedback system (46) involving: regulated nLDL uptake by LDL receptor; a fine balance between biosynthesis and efflux (47); cholesterol acylation/deacylation; and cholesterol intracellular trafficking between organelle membranes (mainly ER, lysosomes, and plasma membrane) (48, 49). Nevertheless, modified LDL can escape the regulated uptake and enter the cell via a series of unregulated scavenger receptors (20, 33). In particular, scavenger receptor LOX-1 expression was reported to be altered in atherosclerotic lesions (34, 35). Here we show that LOX-1 is a preferential receptor through which oxLDL is internalized by AoSMC. Incorporated oxLDL significantly decreased when the receptor was blocked by a specific antibody. Other scavenger receptors may contribute to the uptake because fluorescence associated with oxLDL is not completely erased by blocking of LOX-1. OxLDL internalization in the absence of antibody was not significantly increased with respect to nLDL uptake most likely because a longer time is necessary for oxLDL to induce overexpression of the LOX-1 receptor. Entrance of nLDL instead was independent of LOX-1.
HA accumulation in both culture medium and pericellular coating followed the stimulation with oxLDL. The HA-enriched ECM of the AoSMC is responsible for their altered migratory ability and the increased monocyte adhesion. Although both HAS2 and HAS3 transcripts were found increased concomitantly with oxLDL treatment, HAS2 and HAS3 expression is not followed by HA production due to the same mechanism.
In fact, HA was reduced to CNT level when oxLDL capture was blocked by anti-LOX-1 antibody concomitantly with HAS2 and HAS3 transcript reduction, but after exposure to CF-oxLDL, HA decreased to CNT level due to HAS2 but not HAS3 down-regulation. Moreover, HAS3 transcripts were significantly higher with respect to CNT following both oxLDL (1.6-fold) (blocked by the anti-LOX-1 antibody, Fig. 3D) and CF-oxLDL (1.9-fold) exposure, although the latter was not reflected in an increment in HA levels. The possible explanation of this finding is that HAS2 is the main hyaluronan synthase expressed by AoSMC. In the same cell model, our group previously demonstrated by siRNA that HAS2 accounted for 70% of total HA secretion versus 30% by HAS3 (25). HAS3 contribution to total HA content is therefore secondary. Moreover, several studies on AoSMC pointed out the pivotal role in HA production of the post-transduction regulation of the enzymes (7, 50, 51).
HAS2 in vitro induction is the first evidence of a transcriptional control of HA levels in response to oxLDL stimuli. Other studies already suggested the tight link between regulation of HAS2 and the production of an abnormal atherosclerotic-related ECM as found in monocyte-to-macrophage differentiation (43) and by atherosclerosis development in an in vivo model (52). Although this evidence, together with the high level of HA found in plaques in vivo, suggests that the synthetic enzymes (HASs) could be targets to inhibit atherosclerosis and neointimal expansion, Nagy et al. (3) demonstrated that inhibition of HA synthesis with 4-methylumbelliferone in proatherosclerotic (apoE knock-out) mice accelerated atherosclerosis because of the glycocalyx damage. A potential pharmacological treatment based on HA synthesis regulation should therefore be pinpointed on the HAS expression pathway rather than on enzymes/substrates control.
Previous studies highlighted the link between HA synthesis and cholesterol metabolism in AoSMC and in cancer cells (37, 53). In both studies, cholesterol depletion from plasma membrane by methyl-β-cyclodextrin was associated with decreased HA accumulation, suggesting a diminished stability of HASs in membrane microdomains. Our results suggest that membrane cholesterol content itself, and therefore stabilization of the enzymes in raft domains, may not be the only reason for HA modulation. In fact, in our cell model, the membrane loading with CD-cholesterol complex actually resulted in overexpression of the enzyme HAS3, which does not seem to be responsible for the HA augmented release in the medium. Nevertheless, we cannot exclude that HAS3 is stabilized on the membrane and might play a different role, for example synthesizing the pericellular HA used for cellular adhesion and/or migration. It remains to be elucidated whether HAS2 and HAS3 activation by oxLDL may be direct (i.e. induction of specific regulator elements on the proximal promoter of HAS genes) or indirect, with the activation of other pathways that, in turn, lead to gene expression. The main cholesterol sensor/regulator in cells are the ER-bound SREBP complex and the LXR/RXR nuclear receptor. The SREBP activation as a transcription factor is usually due to a low level of free cellular cholesterol (46).
It is known that oxLDL can induce ER stress in vascular cells (54) and that excessive cholesterol uptake may induce ER stress in macrophages (55). Moreover, oxLDL promote in AoSMC an ECM enriched in versican and most likely HA (which can account for increased monocyte binding in vitro (12)). Nevertheless, the existence of clear evidence that links oxLDL effects on ER stress and on ECM modification is still lacking. LDL-cholesterol (both nLDL and oxLDL) was reported to be released as unesterified cholesterol in the endosomes that can be driven either to the plasma membrane or to a lesser extent to the ER (48, 49). Cholesterol loading in the ER membrane can establish a stress condition that may induce the activation of SREBPs (22). Our results are consistent with this fascinating hypothesis. In fact, oxLDL treatment of AoSMC was associated with ER stress induction as demonstrated by CHOP and GRP78 increased mRNA levels and with enhanced HA secretion (50). In addition, the CF-oxLDL had no effect on HA accumulation.
Concerning ER stress activation of SREBPs, the HAS2 promoter has been proposed to have potential SREBP-2 binding sites, which can activate up-regulation of HA synthases (37). However, no clear evidence in support of such a hypothesis has been presented so far, and recent analysis of SREBP ChIP-seq in the human HAS2 locus did not highlight specific peaks within the gene promoter (56). Other pathways involved in regulation of cholesterol homeostasis may be responsible for HAS2 activation, one possibility being the oxysterol receptor LXR (57). In fact, oxLDL are reported to include several oxidized species, in particular oxysterols (18). Such species may either directly activate an LXR pathway due to specific LXR response elements present on HAS2 promoter (data not shown) or cause an oxidative stress, which may result in increased deposition of HA because secretion of HA has been described as a mechanism of cell rescue in response to stress (50). Upon the addition of 22,oxysterols, the strongest of the endogenous LXR agonists, both HAS2 an HAS3 were highly expressed, reproducing the effect of the oxLDL. In our model, the cell metabolic condition (e.g. the abundance of UDP-sugars), the post-translational processing of the HASs (e.g. phosphorylation or O-GlcNAcylation), and RNA transduction control (microRNA) or enzyme maturation through cell membrane (6) are possible regulatory points in which the nonsterol moiety of the LDL has a critical role in HA final production.
In conclusion, oxLDL induce secretion of an HA-enriched ECM by AoSMC with severe pro-inflammatory properties (e.g. enhanced migration capacity and adhesiveness). The molecular events at the basis of this behavior are triggered by a higher entrance of LDL particles that have been modified by oxidation. Further events, such as ER stress and activation of the LXR/RXR nuclear receptor, contribute to expression of HA synthases 2 and 3.
Acknowledgments
We acknowledge “Centro Grandi Attrezzature per la Ricerca Biomedica,” Università degli Studi dell'Insubria, for the availability of instruments and “Ospedale di Circolo - Fondazione Macchi” in Varese, Italy and Prof. Francesco Pallotti for providing plasma of healthy donors and for technical assistance in cholesterol quantification. We thank Ileana Badi, Ph.D., from the Laboratory of Vascular Biology and Regenerative Medicine, Centro Cardiologico Monzino, Milan, Italy for database analyses.
This work was supported by a grant from PRIN 2009 (to E. K.).
- ECM
- extracellular matrix
- HA
- hyaluronan
- HAS
- hyaluronan synthase
- LOX-1
- lectin-like oxidized LDL receptor-1
- SR-PSOX
- scavenger receptor for phosphatidylserine and oxidized lipoprotein
- SR-AI
- scavenger receptor AI
- CD36
- class B scavenger receptor
- CD
- (2-hydroxypropyl)-β-cyclodextrin
- CF
- cholesterol-free
- CHOP
- C/EBP homologous protein
- C/EBP
- CCAAT-enhancer-binding protein
- GRP78
- glucose-regulated protein 78
- SREBP
- sterol regulatory element binding protein
- aggLDL
- aggregated LDL
- nLDL
- native LDL
- oxLDL
- oxidized LDL
- ER
- endoplasmic reticulum
- SMC
- smooth muscle cells
- AoSMC
- aortic smooth muscle cells
- LXR
- liver X receptors
- RXR
- retinoid X receptors
- DiI
- 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate
- DMSO
- dimethyl sulfoxide
- CNT
- control.
REFERENCES
- 1. Vijayagopal P., Figueroa J. E., Fontenot J. D., Glancy D. L. (1996) Isolation and characterization of a proteoglycan variant from human aorta exhibiting a marked affinity for low density lipoprotein and demonstration of its enhanced expression in atherosclerotic plaques. Atherosclerosis 127, 195–203 [DOI] [PubMed] [Google Scholar]
- 2. Badimon L., Vilahur G. (2012) LDL-cholesterol versus HDL-cholesterol in the atherosclerotic plaque: inflammatory resolution versus thrombotic chaos. Ann. N.Y. Acad. Sci. 1254, 18–32 [DOI] [PubMed] [Google Scholar]
- 3. Nagy N., Freudenberger T., Melchior-Becker A., Röck K., Ter Braak M., Jastrow H., Kinzig M., Lucke S., Suvorava T., Kojda G., Weber A. A., Sörgel F., Levkau B., Ergün S., Fischer J. W. (2010) Inhibition of hyaluronan synthesis accelerates murine atherosclerosis: novel insights into the role of hyaluronan synthesis. Circulation 122, 2313–2322 [DOI] [PubMed] [Google Scholar]
- 4. Nakashima Y., Fujii H., Sumiyoshi S., Wight T. N., Sueishi K. (2007) Early human atherosclerosis: accumulation of lipid and proteoglycans in intimal thickenings followed by macrophage infiltration. Arterioscler. Thromb. Vasc. Biol. 27, 1159–1165 [DOI] [PubMed] [Google Scholar]
- 5. Riessen R., Wight T. N., Pastore C., Henley C., Isner J. M. (1996) Distribution of hyaluronan during extracellular matrix remodeling in human restenotic arteries and balloon-injured rat carotid arteries. Circulation 93, 1141–1147 [DOI] [PubMed] [Google Scholar]
- 6. Vigetti D., Genasetti A., Karousou E., Viola M., Clerici M., Bartolini B., Moretto P., De Luca G., Hascall V. C., Passi A. (2009) Modulation of hyaluronan synthase activity in cellular membrane fractions. J. Biol. Chem. 284, 30684–30694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Vigetti D., Deleonibus S., Moretto P., Karousou E., Viola M., Bartolini B., Hascall V. C., Tammi M., De Luca G., Passi A. (2012) Role of UDP-N-acetylglucosamine (GlcNAc) and O-GlcNAcylation of hyaluronan synthase 2 in the control of chondroitin sulfate and hyaluronan synthesis. J. Biol. Chem. 287, 35544–35555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Karousou E., Kamiryo M., Skandalis S. S., Ruusala A., Asteriou T., Passi A., Yamashita H., Hellman U., Heldin C. H., Heldin P. (2010) The activity of hyaluronan synthase 2 is regulated by dimerization and ubiquitination. J. Biol. Chem. 285, 23647–23654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Toole B. P., Wight T. N., Tammi M. I. (2002) Hyaluronan-cell interactions in cancer and vascular disease. J. Biol. Chem. 277, 4593–4596 [DOI] [PubMed] [Google Scholar]
- 10. Evanko S. P., Angello J. C., Wight T. N. (1999) Formation of hyaluronan- and versican-rich pericellular matrix is required for proliferation and migration of vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 19, 1004–1013 [DOI] [PubMed] [Google Scholar]
- 11. Jiang D., Liang J., Noble P. W. (2011) Hyaluronan as an immune regulator in human diseases. Physiol. Rev. 91, 221–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Merrilees M. J., Beaumont B. W., Braun K. R., Thomas A. C., Kang I., Hinek A., Passi A., Wight T. N. (2011) Neointima formed by arterial smooth muscle cells expressing versican variant V3 is resistant to lipid and macrophage accumulation. Arterioscler. Thromb. Vasc. Biol. 31, 1309–1316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cave A. C., Brewer A. C., Narayanapanicker A., Ray R., Grieve D. J., Walker S., Shah A. M. (2006) NADPH oxidases in cardiovascular health and disease. Antioxid. Redox Signal. 8, 691–728 [DOI] [PubMed] [Google Scholar]
- 14. Khalil M. F., Wagner W. D., Goldberg I. J. (2004) Molecular interactions leading to lipoprotein retention and the initiation of atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 24, 2211–2218 [DOI] [PubMed] [Google Scholar]
- 15. Llorente-Cortés V., Martínez-González J., Badimon L. (2000) LDL receptor-related protein mediates uptake of aggregated LDL in human vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 20, 1572–1579 [DOI] [PubMed] [Google Scholar]
- 16. Stocker R., Keaney J. F., Jr. (2004) Role of oxidative modifications in atherosclerosis. Physiol. Rev. 84, 1381–1478 [DOI] [PubMed] [Google Scholar]
- 17. Leonarduzzi G., Gamba P., Gargiulo S., Biasi F., Poli G. (2012) Inflammation-related gene expression by lipid oxidation-derived products in the progression of atherosclerosis. Free Radic. Biol. Med. 52, 19–34 [DOI] [PubMed] [Google Scholar]
- 18. Schroepfer G. J., Jr. (2000) Oxysterols: modulators of cholesterol metabolism and other processes. Physiol. Rev. 80, 361–554 [DOI] [PubMed] [Google Scholar]
- 19. Chang M. Y., Han C. Y., Wight T. N., Chait A. (2006) Antioxidants inhibit the ability of lysophosphatidylcholine to regulate proteoglycan synthesis. Arterioscler. Thromb. Vasc. Biol. 26, 494–500 [DOI] [PubMed] [Google Scholar]
- 20. Doran A. C., Meller N., McNamara C. A. (2008) Role of smooth muscle cells in the initiation and early progression of atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 28, 812–819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rong J. X., Shapiro M., Trogan E., Fisher E. A. (2003) Transdifferentiation of mouse aortic smooth muscle cells to a macrophage-like state after cholesterol loading. Proc. Natl. Acad. Sci. U.S.A. 100, 13531–13536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Colgan S. M., Hashimi A. A., Austin R. C. (2011) Endoplasmic reticulum stress and lipid dysregulation. Expert Rev. Mol. Med. 13, e4. [DOI] [PubMed] [Google Scholar]
- 23. Dickhout J. G., Lhoták S̆., Hilditch B. A., Basseri S., Colgan S. M., Lynn E. G., Carlisle R. E., Zhou J., Sood S. K., Ingram A. J., Austin R. C. (2011) Induction of the unfolded protein response after monocyte to macrophage differentiation augments cell survival in early atherosclerotic lesions. FASEB J. 25, 576–589 [DOI] [PubMed] [Google Scholar]
- 24. Lu J., Mitra S., Wang X., Khaidakov M., Mehta J. L. (2011) Oxidative stress and lectin-like ox-LDL-receptor LOX-1 in atherogenesis and tumorigenesis. Antioxid. Redox Signal. 15, 2301–2333 [DOI] [PubMed] [Google Scholar]
- 25. Vigetti D., Rizzi M., Viola M., Karousou E., Genasetti A., Clerici M., Bartolini B., Hascall V. C., De Luca G., Passi A. (2009) The effects of 4-methylumbelliferone on hyaluronan synthesis, MMP2 activity, proliferation, and motility of human aortic smooth muscle cells. Glycobiology 19, 537–546 [DOI] [PubMed] [Google Scholar]
- 26. Albertini R., Ramos P., Giessauf A., Passi A., De Luca G., Esterbauer H. (1997) Chondroitin 4-sulphate exhibits inhibitory effect during Cu2+-mediated LDL oxidation. FEBS Lett. 403, 154–158 [DOI] [PubMed] [Google Scholar]
- 27. Christian A. E., Haynes M. P., Phillips M. C., Rothblat G. H. (1997) Use of cyclodextrins for manipulating cellular cholesterol content. J. Lipid Res. 38, 2264–2272 [PubMed] [Google Scholar]
- 28. Reynolds G. D., St Clair R. W. (1985) A comparative microscopic and biochemical study of the uptake of fluorescent and 125I-labeled lipoproteins by skin fibroblasts, smooth muscle cells, and peritoneal macrophages in culture. Am. J. Pathol. 121, 200–211 [PMC free article] [PubMed] [Google Scholar]
- 29. Vigetti D., Moretto P., Viola M., Genasetti A., Rizzi M., Karousou E., Pallotti F., De Luca G., Passi A. (2006) Matrix metalloproteinase 2 and tissue inhibitors of metalloproteinases regulate human aortic smooth muscle cell migration during in vitro aging. FASEB J. 20, 1118–1130 [DOI] [PubMed] [Google Scholar]
- 30. Karousou E. G., Militsopoulou M., Porta G., De Luca G., Hascall V. C., Passi A. (2004) Polyacrylamide gel electrophoresis of fluorophore-labeled hyaluronan and chondroitin sulfate disaccharides: application to the analysis in cells and tissues. Electrophoresis 25, 2919–2925 [DOI] [PubMed] [Google Scholar]
- 31. Vigetti D., Genasetti A., Karousou E., Viola M., Moretto P., Clerici M., Deleonibus S., De Luca G., Hascall V. C., Passi A. (2010) Proinflammatory cytokines induce hyaluronan synthesis and monocyte adhesion in human endothelial cells through hyaluronan synthase 2 (HAS2) and the nuclear factor-κB (NF-κB) pathway. J. Biol. Chem. 285, 24639–24645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Vigetti D., Viola M., Karousou E., Rizzi M., Moretto P., Genasetti A., Clerici M., Hascall V. C., De Luca G., Passi A. (2008) Hyaluronan-CD44-ERK1/2 regulate human aortic smooth muscle cell motility during aging. J. Biol. Chem. 283, 4448–4458 [DOI] [PubMed] [Google Scholar]
- 33. Goyal T., Mitra S., Khaidakov M., Wang X., Singla S., Ding Z., Liu S., Mehta J. L. (2012) Current concepts of the role of oxidized LDL receptors in atherosclerosis. Curr Atheroscler. Rep., in press [DOI] [PubMed] [Google Scholar]
- 34. Mitra S., Goyal T., Mehta J. L. (2011) Oxidized LDL, LOX-1 and atherosclerosis. Cardiovasc. Drugs Ther. 25, 419–429 [DOI] [PubMed] [Google Scholar]
- 35. Kataoka H., Kume N., Miyamoto S., Minami M., Moriwaki H., Murase T., Sawamura T., Masaki T., Hashimoto N., Kita T. (1999) Expression of lectinlike oxidized low-density lipoprotein receptor-1 in human atherosclerotic lesions. Circulation 99, 3110–3117 [DOI] [PubMed] [Google Scholar]
- 36. Moriwaki H., Kume N., Sawamura T., Aoyama T., Hoshikawa H., Ochi H., Nishi E., Masaki T., Kita T. (1998) Ligand specificity of LOX-1, a novel endothelial receptor for oxidized low density lipoprotein. Arterioscler. Thromb. Vasc. Biol. 18, 1541–1547 [DOI] [PubMed] [Google Scholar]
- 37. Sakr S. W., Potter-Perigo S., Kinsella M. G., Johnson P. Y., Braun K. R., Goueffic Y., Rosenfeld M. E., Wight T. N. (2008) Hyaluronan accumulation is elevated in cultures of low density lipoprotein receptor-deficient cells and is altered by manipulation of cell cholesterol content. J. Biol. Chem. 283, 36195–36204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Levitan I., Christian A. E., Tulenko T. N., Rothblat G. H. (2000) Membrane cholesterol content modulates activation of volume-regulated anion current in bovine endothelial cells. J. Gen. Physiol. 115, 405–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Morello F., Saglio E., Noghero A., Schiavone D., Williams T. A., Verhovez A., Bussolino F., Veglio F., Mulatero P. (2009) LXR-activating oxysterols induce the expression of inflammatory markers in endothelial cells through LXR-independent mechanisms. Atherosclerosis 207, 38–44 [DOI] [PubMed] [Google Scholar]
- 40. Lee S., Wang P. Y., Jeong Y., Mangelsdorf D. J., Anderson R. G., Michaely P. (2012) Sterol-dependent nuclear import of ORP1S promotes LXR regulated trans-activation of apoE. Exp. Cell Res. 318, 2128–2142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Skålén K., Gustafsson M., Rydberg E. K., Hultén L. M., Wiklund O., Innerarity T. L., Borén J. (2002) Subendothelial retention of atherogenic lipoproteins in early atherosclerosis. Nature 417, 750–754 [DOI] [PubMed] [Google Scholar]
- 42. Kolodgie F. D., Burke A. P., Farb A., Weber D. K., Kutys R., Wight T. N., Virmani R. (2002) Differential accumulation of proteoglycans and hyaluronan in culprit lesions: insights into plaque erosion. Arterioscler. Thromb. Vasc. Biol. 22, 1642–1648 [DOI] [PubMed] [Google Scholar]
- 43. Chang M. Y., Chan C. K., Braun K. R., Green P. S., O'Brien K. D., Chait A., Day A. J., Wight T. N. (2012) Monocyte-to-macrophage differentiation: synthesis and secretion of a complex extracellular matrix. J. Biol. Chem. 287, 14122–14135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hevonoja T., Pentikäinen M. O., Hyvönen M. T., Kovanen P. T., Ala-Korpela M. (2000) Structure of low density lipoprotein (LDL) particles: basis for understanding molecular changes in modified LDL. Biochim. Biophys. Acta 1488, 189–210 [DOI] [PubMed] [Google Scholar]
- 45. Yoshida H., Kisugi R. (2010) Mechanisms of LDL oxidation. Clin. Chim. Acta 411, 1875–1882 [DOI] [PubMed] [Google Scholar]
- 46. Goldstein J. L., DeBose-Boyd R. A., Brown M. S. (2006) Protein sensors for membrane sterols. Cell 124, 35–46 [DOI] [PubMed] [Google Scholar]
- 47. Yancey P. G., Bortnick A. E., Kellner-Weibel G., de la Llera-Moya M., Phillips M. C., Rothblat G. H. (2003) Importance of different pathways of cellular cholesterol efflux. Arterioscler. Thromb. Vasc. Biol. 23, 712–719 [DOI] [PubMed] [Google Scholar]
- 48. Ikonen E. (2008) Cellular cholesterol trafficking and compartmentalization. Nat. Rev. Mol. Cell Biol. 9, 125–138 [DOI] [PubMed] [Google Scholar]
- 49. Steck T. L., Lange Y. (2010) Cell cholesterol homeostasis: mediation by active cholesterol. Trends Cell Biol. 20, 680–687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Vigetti D., Rizzi M., Moretto P., Deleonibus S., Dreyfuss J. M., Karousou E., Viola M., Clerici M., Hascall V. C., Ramoni M. F., De Luca G., Passi A. (2011) Glycosaminoglycans and glucose prevent apoptosis in 4-methylumbelliferone-treated human aortic smooth muscle cells. J. Biol. Chem. 286, 34497–34503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Vigetti D., Clerici M., Deleonibus S., Karousou E., Viola M., Moretto P., Heldin P., Hascall V. C., De Luca G., Passi A. (2011) Hyaluronan synthesis is inhibited by adenosine monophosphate-activated protein kinase through the regulation of HAS2 activity in human aortic smooth muscle cells. J. Biol. Chem. 286, 7917–7924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chai S., Chai Q., Danielsen C. C., Hjorth P., Nyengaard J. R., Ledet T., Yamaguchi Y., Rasmussen L. M., Wogensen L. (2005) Overexpression of hyaluronan in the tunica media promotes the development of atherosclerosis. Circ. Res. 96, 583–591 [DOI] [PubMed] [Google Scholar]
- 53. Kultti A., Kärnä R., Rilla K., Nurminen P., Koli E., Makkonen K. M., Si J., Tammi M. I., Tammi R. H. (2010) Methyl-β-cyclodextrin suppresses hyaluronan synthesis by down-regulation of hyaluronan synthase 2 through inhibition of Akt. J. Biol. Chem. 285, 22901–22910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Majors A. K., Austin R. C., de la Motte C. A., Pyeritz R. E., Hascall V. C., Kessler S. P., Sen G., Strong S. A. (2003) Endoplasmic reticulum stress induces hyaluronan deposition and leukocyte adhesion. J. Biol. Chem. 278, 47223–47231 [DOI] [PubMed] [Google Scholar]
- 55. Feng B., Yao P. M., Li Y., Devlin C. M., Zhang D., Harding H. P., Sweeney M., Rong J. X., Kuriakose G., Fisher E. A., Marks A. R., Ron D., Tabas I. (2003) The endoplasmic reticulum is the site of cholesterol-induced cytotoxicity in macrophages. Nat. Cell Biol. 5, 781–792 [DOI] [PubMed] [Google Scholar]
- 56. Seo Y. K., Jeon T. I., Chong H. K., Biesinger J., Xie X., Osborne T. F. (2011) Genome-wide localization of SREBP-2 in hepatic chromatin predicts a role in autophagy. Cell Metab. 13, 367–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Calkin A. C., Tontonoz P. (2012) Transcriptional integration of metabolism by the nuclear sterol-activated receptors LXR and FXR. Nat. Rev. Mol. Cell Biol. 13, 213–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Burgess A., Vigneron S., Brioudes E., Labbé J.-C., Lorca T., Castro A. (2010) Loss of human Greatwall results in G2 arrest and multiple mitotic defects due to deregulation of the cyclin B-Cdc2/PP2A balance. Proc. Natl. Acad. Sci. U.S.A. 107, 12564–12569 [DOI] [PMC free article] [PubMed] [Google Scholar]