

Background: Phosphatidylserine-expressing platelets do not have active integrin αIIbβ3 but somehow retain fibrinogen.
Results: The adhesive α-granule proteins fibrinogen and thrombospondin are concentrated in a fibrin polymerization-dependent “cap” on phosphatidylserine-expressing platelets that promotes their incorporation into thrombi.
Conclusion: This suggests a revised model for the adhesive properties of phosphatidylserine-expressing platelets.
Significance: The role of phosphatidylserine-expressing platelets in thrombus formation and its mechanism are re-evaluated.
Keywords: Fibrinogen, Phosphatidylserine, Platelets, Thrombospondin, Transglutaminases, Integrin αIIbβ3, Platelet Aggregation
Abstract
Strongly activated “coated” platelets are characterized by increased phosphatidylserine (PS) surface expression, α-granule protein retention, and lack of active integrin αIIbβ3. To study how they are incorporated into thrombi despite a lack of free activated integrin, we investigated the structure, function, and formation of the α-granule protein “coat.” Confocal microscopy revealed that fibrin(ogen) and thrombospondin colocalized as “cap,” a single patch on the PS-positive platelet surface. In aggregates, the cap was located at the point of attachment of the PS-positive platelets. Without fibrin(ogen) retention, their ability to be incorporated in aggregates was drastically reduced. The surface fibrin(ogen) was strongly decreased in the presence of a fibrin polymerization inhibitor GPRP and also in platelets from a patient with dysfibrinogenemia and a fibrinogen polymerization defect. In contrast, a fibrinogen-clotting protease ancistron increased the amount of fibrin(ogen) and thrombospondin on the surface of the PS-positive platelets stimulated with collagen-related peptide. Transglutaminases are also involved in fibrin(ogen) retention. However, platelets from patients with factor XIII deficiency had normal retention, and a pan-transglutaminase inhibitor T101 had only a modest inhibitory effect. Fibrin(ogen) retention was normal in Bernard-Soulier syndrome and kindlin-3 deficiency, but not in Glanzmann thrombasthenia lacking the platelet pool of fibrinogen and αIIbβ3. These data show that the fibrin(ogen)-covered cap, predominantly formed as a result of fibrin polymerization, is a critical mechanism that allows coated (or rather “capped”) platelets to become incorporated into thrombi despite their lack of active integrins.
Introduction
Physiological platelet activation leads to formation of a procoagulant phosphatidylserine (PS)-expressing5 platelet subpopulation (1–3) that binds coagulation factors to promote coagulation and retains surface α-granule proteins but paradoxically has inactivated integrin αIIbβ3 (4–7) and cannot bind fibrinogen (8). Despite this defect, this subpopulation was detected in thrombi (9–11). These procoagulant and poorly aggregating platelets have been called coated platelets (1) (because of the α-granule protein coat), SCIP (sustained calcium-induced platelet morphology) platelets (5), necrotic platelets (3), or simply procoagulant platelets (2).
The function and the mechanism of formation of the α-granule protein coat are poorly understood, which complicates understanding of the role of the coated platelets themselves. Although our previous study hypothesized that it can serve to attach coated to non-coated platelets and allow their incorporation into aggregates (8), this hypothesis has not been tested. The vast majority of published results for coated platelets were obtained using flow cytometry (4, 6, 12–16), so the structure and localization of the coat were not studied. One hypothesis for coat formation is that proteins are cross-linked by platelet-derived thrombin-activated factor XIII (fXIII) and/or tissue transglutaminase (tTG) (4). However, fXIII knock-out mice exhibit normal coated platelet formation (13). Another hypothesis is the conversion of platelet α-granule fibrinogen into fibrin associated with platelets (10), with the possible entanglement of other proteins in the fibrin mesh. In favor of this, strong platelet stimulation without thrombin (e.g. by a glycoprotein VI agonist convulxin or a PAR1 agonist SFLLRN) leads to the formation of a phosphatidylserine-expressing subpopulation without the coat (17). Finally, binding to αIIbβ3 or another receptor is an obvious possibility at least for fibrinogen (and thrombospondin, another major α-granule protein). Other proteins such as factor Va can also use specific binding mechanisms (18).
Our study focuses on the imaging of the fibrin(ogen) coat on the PS-positive platelet surface, on the functional role of this coat and on the mechanisms of its formation. The three main findings are as follows: (i) α-granule proteins form a single localized cap rather than a uniformly spread coat; (ii) it allows the coated platelets to bind to non-coated ones and become incorporated into a growing thrombus; (iii) fibrin(ogen) and thrombospondin localize to this cap mainly due to fibrin polymerization.
EXPERIMENTAL PROCEDURES
Reagents
The following materials were obtained from the sources shown in parentheses: convulxin (Pentapharm, Basel, Switzerland); human thrombin and fXIIIa (Hematologic Technologies, Essex Junction, VT); prostaglandin E1 (MP Biochemicals, Irvine, CA); Fura-Red, R-phycoerythrin (PE)-, and FITC-conjugated annexin V (Molecular Probes, Eugene, OR); Alexa Fluor 647-conjugated annexin V (Biolegend, San Diego, CA); PPACK (Calbiochem, San Diego, CA); FITC-conjugated anti-human fibrinogen antibody (Labvision, Fremont, CA); PerCP (peridinin-chlorophyll-protein complex)-conjugated CD61 antibody, PE-conjugated CD62P antibody (BD Biosciences); anti-FXIIIA Ig (clone AC-1A1) (Thermo Scientific, Rockford, IL); anti-Fg Ig (clone 15H12) (Abcam, Cambridge, UK); Gly-Pro-Arg-Pro (GPRP), Gly-Pro-Pro-Pro (GPPP) (Bachem, Bubendorf, Switzerland). T101 (ZEDIRA GmbH, Darmstadt, Germany); ancistron (Technologia-Standart, Barnaul, Russia). Integrin αIIbβ3 antagonist Monafram® (F(ab′)2 fragment of a monoclonal antibody that blocks αIIbβ3 activity) was prepared as described (19). Collagen-related peptide (CRP) was kindly provided by Prof. R. W. Farndale (University of Cambridge, Cambridge, UK). All other reagents were from Sigma-Aldrich. FXIIIa was additionally dialyzed prior to experiments to remove glycerol.
Patients
The Glanzmann thrombasthenia patient (15) whose platelets were used for the majority of the experiments lacked integrin αIIbβ3 as determined by flow cytometry (type 1). She possessed a homozygous p.Asp314Tyr mutation in the ITGB3 gene. Her platelets failed to aggregate or to support clot retraction. The patient had a severe bleeding syndrome with frequent hematomas and purpuras and had a history of blood transfusion with platelet concentrates. Another patient (whose platelets were used for experiments in Fig. 5, B and C) had little residual integrin by flow cytometry; the platelets also did not aggregate, and the patient had a history of bleeding during surgery and platelet concentrate transfusion. Kindlin-3 deficient patient was described previously as having leukocyte adhesion deficiency disease type III characterized by both immunodeficiency (20) and severe Glanzmann-type bleeding. It was caused by a homozygous mutation (c.310-2A.C) in the FERMT3 gene that led to an open reading frameshift (p.Asn54ArgfsX142) that prevented kindlin-3 expression. Kindlin-3-deficient platelets expressed integrin αIIbβ3, but it failed to become activated, and the platelets did not aggregate in response to either ADP or collagen. The Bernard-Soulier syndrome patient had a mutation in the CD42a (glycoprotein IX) gene (p.Asn61Ser) leading to zero GPIb expression, giant platelets and thrombocytopenia (2.1 × 107 ml−1), and reduced ristocetin-induced agglutination. Atypically, this Bernard-Soulier syndrome patient had a normal bleeding time (∼5 min) and no clinical manifestations. The patient with dysfibrinogenemia had a homozygous fibrinogen Metz mutation (p.Arg16Cys) in the Aα chain preventing fibrinopeptide A release, so that no fibrin clot formation was observed, although fibrinopeptide B was released normally. Platelet aggregation was normal. The patient suffered from severe bleeding and had a history of fibrinogen transfusions. (The last transfusion was 2 years before this study.) The patient with severe fXIII deficiency (fXIII < 1%) had a homozygous T866 insertion in exon 6 (p.Pro255SerfsX16).
FIGURE 5.

Testing the formation of fibrin(ogen) cap using platelets from patients with inherited bleeding disorders. A and B, platelets from healthy donors and patients were activated at 20,000 μl−1 with 100 nm thrombin for 15 min, labeled with fluorochrome-labeled annexin V and anti-fibrinogen antibody, and analyzed by flow cytometry. Abbreviations are as follows: healthy, healthy donors; DF, dysfibrinogenemia; FXIIIdef, factor XIII deficiency. A, dot plots represent binding of anti-fibrinogen antibody versus annexin V for platelets from healthy donor and patients stimulated at 20,000 μl−1 with 100 nm thrombin. B, the quantity of fibrin(ogen) on the surface of the PS-positive platelets from dysfibrinogenemia patient is significantly decreased, whereas the PS-positive platelets from patient with factor XIII deficiency had normal fibrin(ogen) retention. C, same as in A, except for activation with 100 nm thrombin plus 20 μg/ml CRP. D, the same as in B, except for activation with 100 nm thrombin plus 20 μg/ml CRP. All results were normalized on the value of fluorescence of anti-fibrinogen antibody bound to the fibrin(ogen) on the surface of PS-positive platelets from healthy donors obtained by thrombin alone stimulation. The data for healthy donors are means ± S.D. for n = 3 experiments with platelets from different donors. E, total levels of fibrinogen and fXIII in patient plasma and platelet lysates determined by Western blotting. Additional samples are as follows: low fibrinogen, a patient with low plasma fibrinogen (<1 mg/ml); high fibrinogen, a patient with high plasma fibrinogen (>10 mg/ml); hypofibrinogenemia; purified fibrinogen; healthy controls.
Platelet Isolation
Platelets were isolated from freshly drawn blood of healthy adult volunteers or patients. Investigations were performed in accordance with the Declaration of Helsinki, and written informed consent was obtained from all patients and control donors, or from their parents. Briefly, blood was collected into 3.8% sodium citrate, pH 5.5, at a 9:1 blood/anticoagulant volume ratio and supplemented with prostaglandin E1 (1 μm) and apyrase (0.1 unit/ml) to prevent platelet activation. It was centrifuged to obtain platelet-rich plasma that was supplemented with 3.8% sodium citrate, pH 5.5, at a 1:3 citrate/plasma ratio to lower pH. Platelets were concentrated by centrifugation at 400 × g for 5 min (or 1000 × g for 5 min), resuspended in buffer A (150 mm NaCl, 2.7 mm KCl, 1 mm MgCl2, 0.4 mm NaH2PO4, 20 mm HEPES, 5 mm glucose, 0.5% bovine serum albumin) and subjected to gel filtration on a chromatography column packed with Sepharose CL-2B and equilibrated with buffer A.
Flow Cytometry
Platelets at indicated concentrations were activated by incubation with agonists in buffer A with 2.5 mm CaCl2 for 15 min in the presence of labeling antibodies, diluted 10-fold, and immediately analyzed in a FACSCalibur (BD Biosciences) or Accuri C6 (Accuri Cytometers, Ann Arbor, MI) flow cytometer. The acquired data were processed using a WinMDI (version 2.8, Joseph Trotter, Scripps Research Institute, La Jolla, CA) or CFlow software (Accuri Cytometers). Compensation was done for all types of two-color and three-color analysis. To quantify thrombospondin or fibrin(ogen) binding, activated platelets were additionally incubated with anti-thrombospondin antibody or FITC-anti-fibrinogen antibody for 5 min and then (for the anti-thrombospondin sample) with secondary antibody for an additional 5 min, fixed in 1.5% formalin in PBS for 20 min, diluted 15-fold, and analyzed by flow cytometry.
Flow Cytometry Aggregation Assay
The ability of different subpopulations to aggregate was evaluated essentially as described (8). Platelets at 50,000/μl were activated by incubation with agonists in buffer A with 2.5 mm CaCl2 in the presence of FITC-annexin V and PE-CD62P or CD61-PerCP for 15 min in the presence or absence of 200 μg/ml Monafram with and without shaking (600 rpm in an MS1 Minishaker (IKA, Staufen, Germany)). Then, samples were diluted 10-fold and analyzed in a FACSCalibur flow cytometer. Alternatively, platelets at 50,000/μl were activated with agonists in buffer A with 2.5 mm CaCl2 in the presence of PE-annexin V and CD42b-FITC. This was followed by addition of 1 μm PPACK and 1 mg/ml fibrinogen. Next, platelets were shaken (600 rpm) for 5 min, diluted 10-fold, and analyzed in a flow cytometer.
Confocal Microscopy
Glass coverslips (24 × 24 mm, Heinz Herenz, Hamburg, Germany) were cleaned with potassium dichromate, rinsed with distilled water, and dried, and the clean coverslips were coated with 20 mg/ml fibrinogen in buffer A for 40 min at room temperature, rinsed with distilled water, and then assembled as part of the flow chamber. In the experiments with non-aggregated platelets, samples were prepared as described above. In the experiments with aggregates, platelets at 50,000/μl were activated by incubation with 100 nm thrombin and 10 μg/ml CRP in buffer A with 2.5 mm CaCl2 and fluorescently labeled annexin V and CD61 for 15 min. Then, 1 μm PPACK and 1 mg/ml fibrinogen were added; the platelets were vortexed for 3 min and allowed to spread on the fibrinogen surface in the chamber for 5 min. Confocal images were acquired with an Axio Observer Z1 microscope (Carl Zeiss, Jena, Germany) with a 100× objective. For Fig. 5B, a Leica DMI 6000 SP5 confocal microscope (Leica Microsystems SAS, Nanterre, France) with a resonant scanner and 100× objective was used.
SDS-PAGE
To visualize the products of tTG activity, we used 7.5% SDS-PAGE in reducing conditions as described (21). One mg/ml fibrinogen was cross-linked by 50 nm tTG in buffer 10 mm Tris, 150 mm NaCl, 5 mm CaCl2, 1 mm DTT; pH 8.0. Some samples contained 200 μm T101, 10 mm GPRP, 10 mm GPPP, or 25 mm EDTA as a positive control for the inhibitor of transglutaminase activity. After incubation for 30 min at 37 °C, the reaction was stopped with 25 mm EDTA. Samples were mixed with 3× loading buffer, and 7.5 μl of each sample were run in Tris-glycine-SDS running buffer. Gels were stained with Coomassie R-250.
Western Blot
Platelets were resuspended in T3EN buffer (10 mm Tris-HCl, pH 7.0, 150 mm NaCl, 3 mm EDTA) at 1 μl/106 platelets and lysed by adding 10 mm Tris-HCl, pH 7.0, 30 mm N-ethylmaleimide, 12% SDS at a 1:5 ratio. Samples were boiled for 5 min and then spun at 14000 rpm for 5 min at 4 °C. Ten μg of total protein were loaded on a 10% gel and analyzed by SDS-PAGE. Immunoblotting was performed using anti-FXIIIA Ig or anti-Fg Ig and revealed by chemiluminescence.
Whole Blood Thrombus Formation on Collagen in Flow Chambers
Glass coverslips (24 × 24 mm, Heinz Herenz, Hamburg, Germany) were cleaned with potassium dichromate, rinsed with distilled water, and dried. Then, the coverslips were coated with 200 μg/ml native fibrillar type I collagen (Chrono-Log, Havertown, PA) in buffer A for 60 min at room temperature in a humid chamber, rinsed with distilled water, dried at 37 °C, and then assembled as a part of the flow chamber (slit depth of 80 μm). Flow chambers with tubings were blocked with buffer A for at least 30 min before the perfusion of blood. Blood from healthy donors was collected into 3.8% sodium citrate, pH 5.5, at a 9:1 blood/anticoagulant volume ratio and recalcified with 7.5 mm CaCl2 (providing physiological free ionized calcium concentration) upon entry into the flow chamber. Recalcified whole blood was perfused for 2 min at shear rate of 500 s−1 over the collagen. Thereafter, the flow chamber was post-perfused at the same shear rate for a further 2 min with buffer A containing 2.5 mm CaCl2, 1 μm PPACK, and annexin V-Alexa Fluor 647 to detect phosphatidylserine exposure. Confocal images of thrombi were acquired with an Axio Observer Z1 microscope.
Statistics
All experiments were performed at least in triplicate with platelets from different donors except for studies with patients. Comparisons were carried out with the paired Student's t test. Statistical significance was set as p < 0.05. Values are reported as mean ± S.D. unless specified otherwise.
RESULTS
Morphological Characterization of the α-Granule Protein Coat
A typical experiment on the formation of the fibrinogen coat in the PS-expressing subpopulation in presented in Fig. 1A. Washed platelets were activated with thrombin, labeled with annexin V and anti-human fibrinogen antibody, and analyzed at indicated time points by flow cytometry. In agreement with previous reports, this leads to the appearance of a distinct PS-positive subpopulation that have retained secreted fibrin(ogen). Both the mean fluorescence and quantity increased steadily with time over the course of 30 min before reaching a plateau.
FIGURE 1.

Localization of fibrin(ogen) in a single cap on the surface of the PS-positive platelets. A, platelets from healthy donors were activated at 50,000 μl−1 with 100 nm thrombin, dual-labeled with annexin V, and anti-fibrin(ogen) antibody, and analyzed by flow cytometry at indicated timepoints. Dot plots of anti-fibrin(ogen) antibody versus annexin V fluorescence show the kinetics of the two subpopulations formation and fibrinogen coat development on their surface. The box indicates the PS-positive subpopulation. B, platelets from healthy donors were activated at 50,000 μl−1 with 100 nm thrombin and labeled with anti-fibrin(ogen) antibody and annexin V. Representative confocal images of DIC, Alexa Fluor 647 (red) fluorescence of annexin V, FITC (green) fluorescence of anti-fibrinogen antibody, and an overlay are shown. All panels represent the same field of view. Three optical cross-sections of a platelet separated by 0.6 μm (layer 1 and layer 2) and 1.2 μm (layer 2 and layer 3) are shown. C, three-dimensional rendering of the same PS-positive platelet stained with Alexa Fluor 647-conjugated annexin V (pink) and FITC-conjugated anti-fibrinogen antibody (green). See also supplemental Movie 1. D, platelets were activated at 50,000 μl−1 with 100 nm thrombin for 15 min and labeled with anti-fibrinogen antibody (green) and annexin V (red). Confocal images were collected and then averaged to see all layers at once. There are 11 PS-positive platelets, and a single fibrin(ogen) cap is clearly seen in nine of the platelets. In A–D, typical experiments of n = 3 with platelets from different donors are shown. For microscopy experiments in B–D, a total of 52 PS-positive platelets were scanned, and 85% had a single cap; 15% had none.
To study the structure of this fibrin(ogen) coat on the platelet surface, these activated platelets labeled with annexin V and anti-human fibrinogen antibody were imaged by confocal microscopy. Intriguingly, the coat of the PS-positive platelets was not uniformly spread over the platelet surface but rather was located in a single cap (Fig. 1B). A three-dimensional reconstruction of it is shown in Fig. 1C (see also supplemental Movie 1). The annexin V concentration in the cap was also high and increased compared with the rest of the platelet surface (Fig. 1B), indicating that the cap is phospholipid-based. It was clear that the cap contained fibrin(ogen) at a density vastly exceeding that on the rest of the platelet surface. Each procoagulant platelet had only one cap, and 85% of platelets possessed a cap (Fig. 1D). The cap was formed early in the platelet activation coinciding with the overall shape change (data not shown). In the PS-negative platelets, fibrinogen was distributed differently, with several points of fluorescence over the surface in agreement with previous reports.
Significantly, both the procoagulant platelets in general and their caps lacked unoccupied active integrins (supplemental Fig. S1). Residual αIIbβ3 itself was detected more or less uniformly over the whole platelet with little concentration in the cap (Fig. 2A). Interestingly, the α-granule membrane glycoprotein, P-selectin, was also distributed over the whole platelet surface in a manner different from that of fibrin(ogen) (Fig. 2B). In contrast, glycoprotein Ib was concentrated in the cap (Fig. 2C).
FIGURE 2.
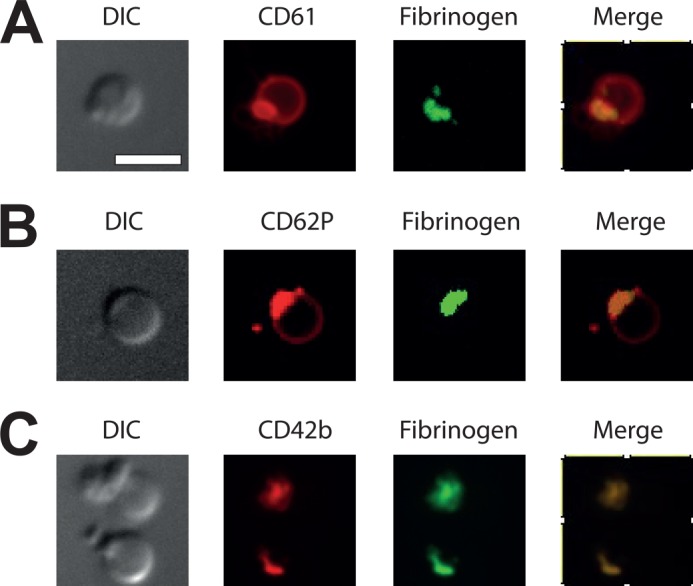
Distribution of membrane proteins on the procoagulant platelets. Normal platelets were activated at 50,000 μl−1 with thrombin at 100 nm, labeled with indicated antibodies, and imaged with confocal microscopy. A, integrin αIIbβ3 (CD61); B, P-selectin (CD62P); C, glycoprotein Ib (CD42b). Scale bar, 5 μm.
Functional Significance of the Fibrin(ogen) Cap
We have previously hypothesized that coated platelets can be incorporated into aggregates despite their lack of activated integrins because of their fibrin(ogen) coat that could interact with activated integrin αIIbβ3 molecules on non-coated platelets. To study this, platelets were activated either with thrombin (producing ∼5% PS+ platelets) or thrombin plus CRP (∼70% PS+ platelets) in the presence or absence of an αIIbβ3 antagonist Monafram for 15 min and shaken during activation to induce aggregation. Samples with and without shaking were analyzed by flow cytometry, and the percentage of platelets lost from the suspension after shaking (calculated from the single platelet histogram) was assessed. In agreement with previous reports, thrombin-stimulated platelets readily formed aggregates, although potent activation with thrombin plus CRP resulted in less aggregation in the sample (Fig. 3A). Addition of Monafram abolished both the thrombin-induced and thrombin/CRP-induced aggregation (Fig. 3A). These results demonstrate that the fibrin(ogen) cap on the PS+/Ca+ platelet surface cannot itself provide the basis for the aggregate formation and that αIIbβ3 is always involved.
FIGURE 3.

Elucidation of the possible role of fibrin(ogen) coat in platelet aggregation. A, platelets were stimulated at 50,000 μl−1 with 100 nm thrombin or 100 nm thrombin plus 30 μg/ml CRP for 15 min in the presence or absence of Monafram with and without shaking (600 rpm) and analyzed by flow cytometry. A number of platelets leaving the suspension after the shaking was determined using calibration beads. Means ± S.E. for n = 3 experiments with different donors are shown. B, platelets (50,000 μl−1) were stimulated with 100 nm thrombin plus 10 μg/ml CRP or 10 μg/ml CRP alone for 15 min, supplemented with 1 μm PPACK and 1 mg/ml fibrinogen, shaken (600 rpm) for 5 min, and analyzed by flow cytometry. A number of platelets from different subpopulations leaving the suspension after the shaking was determined using calibration beads. Means ± S.E. for n = 4 experiments with different donors are shown. C, representative confocal images of DIC, PerCP (red) fluorescence of CD61, Alexa Fluor 647 (green) fluorescence of annexin V, and their overlay for a typical aggregate of platelets stimulated with thrombin and CRP essentially as described in B are shown. Scale bar, 5 μm.
To test whether the fibrin(ogen) cap is responsible for the coated platelet incorporation into the aggregates, platelets were stimulated with thrombin plus CRP (to produce PS+ platelets with high fibrin(ogen)) or CRP alone (to obtain PS+ platelets with low fibrin(ogen)) for 15 min, supplemented with PPACK and fibrinogen, shaken for 5 min at 600 rpm, and analyzed by flow cytometry. The percentages of platelets from the three different subpopulations (excluding aggregates from the analysis) that left the suspension after shaking were assessed (15). As shown in Fig. 3B, the PS− and the recently reported PS+/Ca− platelets (15) aggregated equally well regardless of the type of activation, whereas the proaggregatory ability of coated PS+/Ca+ platelets decreased 5-fold in the absence of a fibrin(ogen) coat on their surface. This indicates that the fibrin(ogen) coat on the surface of PS+/Ca+ platelets plays an important role in their involvement into aggregates with PS- and PS+/Ca− platelets.
To directly observe aggregate formation, we used confocal microscopy. Fig. 3C demonstrates a typical structure of heterogeneous platelet aggregates. Interestingly, the procoagulant balloon-shaped PS-expressing platelets surround the aggregate of the non-coated PS-negative platelets. In the aggregates, the cap was located at the point of attachment of the procoagulant platelets (supplemental Movie 2).
Testing the “Fibrin Polymerization” and the “Transglutaminase” Hypotheses
To test whether fibrin polymerization during platelet activation is essential for this cap formation, we performed experiments in the presence or absence of 15 mm fibrin polymerization inhibitor GPRP, using GPPP as a negative control (Fig. 4). This significantly decreased the amount of fibrin(ogen) on the surface of PS-positive platelets stimulated with thrombin plus CRP (Fig. 4A) or thrombin alone (Fig. 4E), whereas GPPP had no effect. As another line of evidence, we stimulated platelets with CRP in the presence of the fibrinogen-clotting protease ancistron at a concentration equivalent to 15 NIH units of thrombin. Ancistron dramatically increased the amount of fibrin(ogen) on the surface of PS-positive platelets produced with CRP that normally do not have a coat (Fig. 4B). Ancistron alone did not produce PS-positive platelets in control experiments and did not affect the number of the PS-positive platelets (data not shown). Thus, these data indicate that fibrin polymerization is both necessary (the inhibitor data) and sufficient (the ancistron data) to form the fibrin(ogen) coat on the surface of the PS-positive platelets.
FIGURE 4.

Testing the mechanism of the fibrin(ogen) cap formation pharmacologically. A–E, platelets from healthy donors were activated at 50,000 μl−1 in 20 μl with 100 nm thrombin plus 20 μg/ml CRP in the presence or absence of 15 mm of fibrin polymerization inhibitor GPRP or GPPP as a negative control peptide (A), 20 μg/ml CRP in the presence or absence of ancistron at a concentration equivalent to 15 NIH of thrombin (B), 100 nm thrombin in the presence or absence 200 μm of transglutaminase inhibitor T101 (C), 20 μg/ml CRP in the presence or absence of 500 nm tTG (D); 100 nm thrombin in the presence of GPRP or GPPP at 5 mm (E). Samples were labeled with fluorochrome-labeled anti-fibrinogen antibodies and annexin V and analyzed by flow cytometry. Histograms indicate the amount of fibrinogen on the surface of PS-positive platelets, depending on the presence or absence of pharmacological agent during platelet activation. For each type of activation, all results were normalized on the value of fluorescence of anti-fibrinogen antibody bound to the fibrin(ogen) on the surface of PS-positive platelets obtained by agonist stimulation alone. The data show means ± S.D. for n = 3 experiments with platelets from different donors. Asterisks indicate statistical significance.
To test the alternative hypothesis that platelet-secreted transglutaminases are able to cross-link fibrin(ogen) on the surface of the PS-positive platelets, a pan-transglutaminase inhibitor T101 was added prior to platelet activation with thrombin (Fig. 4C). This caused a small, not statistically significant (p = 0.14), decrease in the amount of fibrin(ogen) on the surface of the PS-positive platelets (Fig. 4C) and did not influence the percentage of these platelets (data not shown). In addition, we activated platelets with CRP in the absence or presence of exogenous tTG (Fig. 4D). Addition of tTG significantly increased the amount of fibrin(ogen) on the PS+ platelet surface (Fig. 4D) without affecting their percentage. Similar results were obtained with exogenous fXIIIa (data not shown). Control experiments (supplemental Fig. S2) confirmed that T101 indeed effectively reduces the tTG activity, whereas GPRP and GPPP do not affect it. Interestingly, thrombin activity was not absolutely required for the cap; CRP-stimulated platelets did produce distinct caps, although the concentration of fibrin(ogen) in them was much lower than in the presence of thrombin (data not shown).
We next conducted experiments using platelets from patients with inherited disorders leading to dysfibrinogenemia and fXIII deficiency. Dysfibrinogenemia is a coagulation disorder caused by a polymerization defect that results in abnormal fibrin formation. Metz β-fibrin clots are more fragile than those of a normal αβ-fibrin clot and are comprised almost entirely of a fine network of fibers. Importantly, fibrinogen-dependent platelet aggregation is not affected. First, platelets from healthy donors and patients with dysfibrinogenemia and factor XIII deficiency were stimulated with thrombin (Fig. 5, A and B) or thrombin plus CRP (Fig. 5, C and D). The fibrin(ogen) retention on the PS-positive platelets was significantly decreased in the dysfibrinogenemia patient under all conditions. In contrast, PS-positive platelets from the fXIII-deficient patient had a normal amount of fibrin(ogen) on their surface (Fig. 5). Western blotting confirmed that the total levels of fibrinogen and factor XIII in the lysates of platelets from a healthy donor and the patient with dysfibrinogenemia were indistinguishable; whereas the fXIII-deficient patient lacked fXIII but had a normal fibrinogen content (Fig. 5E). Thus, this patient model confirms the critical importance of fibrin polymerization in the fibrin(ogen) retention, whereas fXIII does not seem to be critical. However, contribution of other transglutaminases such as tTG cannot be excluded.
Formation of the Thrombospondin Cap
To test whether other α-granule proteins can be associated with the fibrin(ogen) mesh during platelet activation, we labeled platelets with antibodies against thrombospondin. Platelets were either stimulated with thrombin in the presence or absence of the fibrin polymerization inhibitor GPRP (15 mm), using GPPP as a negative control (Fig. 6A) or with CRP using the fibrinogen-cleaving protease ancistron at concentrations equivalent to 15 NIH of thrombin (Fig. 6B). GPRP did not affect the amount of thrombospondin on the surface of PS-positive platelets (Fig. 6A). However, in dysfibrinogenemia platelet retention of thrombospondin was decreased significantly (supplemental Fig. S3). In addition, ancistron increased the amount of thrombospondin on the surface of PS-positive platelets produced with CRP (Fig. 6B) by ∼3-fold, similarly to its effect on fibrin(ogen). This suggests that fibrin polymerization might be important for retention of thrombospondin as well as of fibrin.
FIGURE 6.

Formation of the thrombospondin cap and its localization. Platelets from healthy donors were activated at 50,000 μl−1 with 100 nm thrombin plus 20 μg/ml CRP in the presence or absence of 15 mm of fibrin polymerization inhibitor GPRP or GPPP as a negative control peptide (A) and 20 μg/ml CRP in the presence or absence of ancistron standard solution at concentrations equivalent to 15 NIH of thrombin (B). Samples were labeled with fluorochrome-labeled anti-fibrinogen antibodies and annexin V and analyzed by flow cytometry. Histograms indicate the amount of fibrinogen on the surface of PS-positive platelets, depending on the presence or absence of pharmacological agent during platelet activation. For each type of activation, all of the results were divided to the value of fluorescence of anti-fibrinogen antibody bound to the fibrin(ogen) on the surface of PS-positive platelets obtained by agonist stimulation alone. The data show means ± S.D. for n = 5 experiments with platelets from different donors. C, platelets from healthy donors were activated at 50,000 μl−1 with 100 nm thrombin plus 20 μg/ml CRP. Representative confocal images of DIC (C, upper panel), FITC (green) fluorescence of annexin V, PE (red) fluorescence of thrombospondin antibody, and an overlay of the three or DIC (C, lower panel), PE (red) fluorescence of annexin V, FITC (green) fluorescence of anti-fibrinogen antibody, and an overlay of the three are shown. D, platelets from healthy donors were activated at 50,000 μl−1 with 20 μg/ml CRP in the presence of 100 nm thrombin (D, upper panel) or ancistron standard solution at concentrations equivalent to 15 NIH of thrombin (D, lower panel). Representative confocal images of DIC, FITC (green) fluorescence of anti-fibrinogen antibody, PE (red) fluorescence of thrombospondin antibody, and an overlay of the three are shown. Microscopy experiments were reproduced n = 3 times with platelets from different donors. A total of 86 platelets were scanned, and 93% of them had a thrombospondin cap that was co-localized with fibrinogen in all cases.
Confocal microscopy revealed that localization of thrombospondin on the surface of the PS-positive platelets produced by stimulation with thrombin plus CRP also looks like a cap (Fig. 6C). Moreover, it co-localized with the fibrin(ogen) cap on the surface of the PS-positive platelets stimulated with either thrombin plus CRP or CRP in the presence of ancistron (Fig. 6D). These results strongly suggest that thrombospondin associates by direct interactions within the fibrin(ogen) cap.
Identification of the Surface Component Responsible for Fibrin(ogen) Retention
To study the role of the major platelet receptors in the fibrin(ogen) retention, we studied platelets from a series of patients with inherited platelet disorders. These included type I Glanzmann thrombasthenia (severe deficiency of αIIbβ3 and platelet Fg (22)), leukocyte adhesion deficiency disease type III syndrome (deficiency of kindlin-3 due to mutations in FERMT3; kindlin-3 is a critically important molecule involved in activation and signaling of αIIbβ3), and the Bernard-Soulier syndrome (deficiency of GPIb/IX/V). Platelets were activated with thrombin (Fig. 7, A and B) or thrombin plus CRP (Fig. 7, C and D). The amount of fibrin(ogen) on the PS-positive platelets in patients with Bernard-Soulier syndrome or kindlin-3 deficiency was normal, whereas that on platelets from the Glanzmann thrombasthenia patient was extremely low. Flow cytometry of permeabilized platelets confirmed that total level of fibrinogen in the platelets from this Glanzmann thrombasthenia patient was indeed low, whereas fibrinogen levels were abundant in platelets of the kindlin-3-deficient patient, suggesting that αIIbβ3 activation is not required for the surface expression of the secreted fibrinogen.
FIGURE 7.

Searching for a possible substrate of the fibrin(ogen) cap. A and B, platelets from healthy donors or patients were activated at 20,000 μl−1 with 100 nm thrombin for 15 min, labeled with fluorochrome-labeled annexin V and anti-fibrinogen antibody and analyzed by flow cytometry. The abbreviations are as follows: healthy, healthy donors; GT, Glanzmann thrombasthenia; K3def, kindlin-3 deficiency; BSS, Bernard-Soulier syndrome. A, dot plots represent binding of anti-fibrinogen antibody versus annexin V for platelets from healthy donor and patients stimulated at 20,000 μl−1 with 100 nm thrombin. B, amount of fibrin(ogen) on the surface of PS-positive platelets from healthy donors and donors with inherited platelet disorders. C, the same as in A except for the activation with 100 nm thrombin plus 20 μg/ml CRP for 15 min. D, the same as in B except for the activation with 100 nm thrombin plus 20 μg/ml CRP for 15 min. All results were normalized on the value of fluorescence of anti-fibrinogen antibody bound to the fibrin(ogen) on the surface of PS-positive platelets from healthy donors obtained by thrombin alone stimulation. The data for healthy donors are means ± S.D. for n = 3 experiments with platelets from different donors.
To evaluate whether the cap is a phenomenon specific for the suspension-activated platelets, experiments were also performed on fibrinogen-attached platelets (supplemental Fig. S4A); the cap was formed on substrate-attached procoagulant platelets as well as on the suspension-activated ones. We also analyzed the distribution of thrombospondin on Glanzmann thrombasthenia platelets and found that the cap was present judging by DIC, annexin V, and thrombospondin staining (supplemental Fig. S4B); only trace fibrinogen was present in these caps that could be observed only with a maximal laser intensity (supplemental Fig. S4C). This suggests that adhesive protein clustering is the key event independent of the nature of the membrane receptor involved. Furthermore, we studied formation of caps on annexin V-positive platelets detected in thrombi formed under flow of whole blood over the collagen surface. These platelets also possessed a cap-like structure enriched with annexin V (supplemental Fig. S5).
DISCUSSION
This study aimed to investigate the mechanisms of fibrin(ogen) coat formation on the surface of PS-positive platelets, its structure, and its physiological role. Our main findings are as follows: (i) the fibrin(ogen) coat of the PS-positive platelets is not uniformly spread over the platelet surface but rather located in a single PS-rich cap, where it is co-localized with thrombospondin; (ii) this fibrin(ogen) cap promotes the incorporation of the coated platelets into aggregates; (iii) both transglutaminase-dependent fibrinogen cross-linking and thrombin-induced fibrin polymerization are able to retain fibrin(ogen) on the surface of the activated procoagulant platelets; (iv) the cap, at least for thrombospondin, is formed independently of αIIbβ3; and (v) the cap is observed not only in suspension-activated but also in surface- and thrombus-attached PS-positive platelets.
The coating of the PS-positive platelet with α-granule proteins is an intriguing enigma of procoagulant platelets. Although the procoagulant ‘balloon-shaped’ platelets have been observed by microscopy for some time (5, 8, 23), and there have been reports that annexin V is sometimes distributed non-uniformly on their surface (7, 24), we are not aware of any attempts to use confocal microscopy to focus on their ‘coating’ aspect, whose study has been dominated by flow cytometry (4, 12–15, 17). There was a report on patched fibrinogen distribution on the platelet surface (25); although that phenomenon could be related to the cap described here, the previous study has marked differences: it used weak stimulation with ADP so that there were no coated platelets, there usually were numerous patches instead of single cap, and there was a continuous slow redistribution of fibrinogen in an integrin αIIbβ3-dependent manner, ultimately resulting in its internalization. Our finding that the secreted α-granule protein coat organizes into a cap, a small ball-like structure polarized to a distinct zone, is the most unexpected result of the present study. Labeling of this structure with annexin V suggests that it is definitely phospholipid membrane-based and could be related to the phosphatidylserine non-uniformity phenomenon described previously (24). Nevertheless, unlike the distribution of fibrin(ogen), annexin V binding also remained over the whole of the procoagulant platelet surface; there is also no evidence that PS enrichment in the cap is linked by a causal relationship with fibrin(ogen) retention.
Despite the fact that the activated platelet coat was first reported a decade ago (4, 12), its function has also remained obscure. We have recently reported that coated platelets can be incorporated into aggregates in an unusual manner: they can bind to non-coated platelets but not with each other (8). The data of the present study directly show that procoagulant platelets without the cap lose the ability to be incorporated into aggregates and that the cap is located exactly at the point of platelet attachment. This indicates that the fibrin(ogen) coat/cap is responsible for endowing coated platelets with their proaggregatory ability. It could be a protective mechanism preventing procoagulant platelets from forming aggregates by themselves; it could also be noted that the concentrating of fibrin(ogen) into a single cap makes coated platelet favor a location “on the border” of an aggregate, which could possibly explain their suggested role in limiting thrombus growth (26), as well as in its consolidation via fibrin generation. It is also known that fibrin aggregates formed in the circulating blood during low grade intravascular coagulation are largely removed by the reticulo-endothelial system (27). It is also possible that the fibrin(ogen) coat serves another function: elimination of the procoagulant platelets from the circulation.
In contrast to the structure and function of the coat, the mechanism of the α-granule protein attachment to the surface of coated platelets has been studied more extensively but has remained a subject of debate. Our results support the hypothesis of Munnix and co-workers (10) who suggested that fibrin polymerization and entrapment (or direct binding) of other proteins in the fibrin net could explain this attachment. Several lines of evidence, including inhibition of fibrin polymerization with GPRP, stimulation of fibrin formation by ancistron (when thrombin is not present), and experiments with platelets from a patient with a polymerization defect (dysfibrinogenemia), all suggest that this simple mechanism plays a significant role in the fibrin(ogen) retention. Interestingly, thrombospondin retention on the procoagulant platelets was also decreased in dysfibrinogenemia (although GPRP did not inhibit it). Based on previous reports on thrombospondin incorporation into fibrin clots (28), fibrin formation may well facilitate thrombospondin retention on procoagulant platelets. In agreement with the experiments of Jobe et al. (13) on fXIII-knock-out mice, we did not see any effects of human fXIII deficiency on coated platelet formation. Their study also suggested a role of tTg, based on its increased expression in the fXIII-deficient platelets; in our experiments, inhibition of tTG did not exert a strong effect indicating that native tTG is probably not strongly involved.
Notwithstanding, the predominant role of fibrin polymerization does not rule out other mechanisms completely. We have shown that tTG was able to directly induce a high level of fibrinogen retention on the PS-positive platelets activated with CRP in the absence of thrombin, supporting suggestions of Dale et al. (4) and Jobe et al. (13). It is also possible that the role of platelet-derived transglutaminase(s) may increase in vivo, where platelet secretion is more important due to the increased platelet local concentration within the thrombus.
Although polymerization or cross-linking could explain high levels of the proteins on the coated platelet surface, surface receptors on the membrane probably mediate their initial attachment. In line with this, an important question is the mechanism responsible for αIIbβ3 inactivation in this subpopulation where both calpain-dependent and independent mechanisms have been proposed (4, 9). Another appealing hypothesis is the “blocking” of active integrin αIIbβ3 by fibrinogen (and thrombospondin), which could explain both increased quantities of fibrinogen (and thrombospondin) and the inability of PAC-1 to bind to these platelets. Fibrin(ogen) retention was normal on the platelets from a Bernard-Soulier syndrome patient (suggesting that glycoprotein Ib is not critical). There was an absolute requirement for αIIbβ3 as shown for platelets of the type I Glanzmann thrombasthenia patient. Platelets of the kindlin-3-deficient patient had not only normal coating but also normal amount of α-granule fibrinogen, suggesting that αIIbβ3 activation is not required for fibrinogen capture during circulation or its retention after activation. This would further suggest a direct role for fibrin polymerization in cap formation; significantly, previous studies have shown that the interaction of fibrin with αIIbβ3 can occur in the absence of Fg binding (29). The thrombospondin cap was observed even in Glanzmann thrombasthenia patients. In fact, αIIbβ3 continued to be observed over the whole platelet, with little concentration in the cap, and the same was observed for P-selectin. In contrast, GPIb was more significantly concentrated in the cap, perhaps because of its known ability to bind to fibrin (30) or of the presence of VWF. Thrombospondin has been shown to bind to CD36 and to CD47 (31) and is known to form clusters on normal and Glanzmann thrombasthenia platelets, although these are smaller in the absence of fibrinogen (32). Thrombospondin incorporation is also known to improve fibrin clot formation (33).
All of this allows us to propose a new model for the protein retention in the cap. In this model, membrane receptors bind proteins such as thrombospondin or fibrinogen (and it is likely that some of these proteins come out of the granules already associated with the receptors); the proteins subsequently cluster or cross-link on the platelet surface to form the cap region. The newly formed fibrin clot allows a further interaction with thrombospondin and serves as a kind of positive feedback, improving retention of the attached proteins and possibly trapping others as well.
In a recent study of the distribution of procoagulant proteins in thrombi formed under venous shear rate (34), it is possible to identify non-uniform patches of proteins, although the conditions are vastly different and that study analyzed exogenous rather than secreted proteins. Preliminary experiments with surface-attached platelets and whole blood thrombi of the present study also indicate, however, that the cap is not a phenomenon restricted to the suspension-activated platelets but rather an always present component of the PS-positive platelets. The next step of our work is to analyze the role of cap formation in ex vivo models of thrombosis.
Acknowledgments
We thank Prof. R. Farndale for supplying CRP. We are grateful to Dr. N. V. Zakharova for helpful discussions and careful reading of the manuscript.
This work was supported by Russian Foundation for Basic Research Grants 11-04-00303, 12-04-00438, 12-04-31401, 12-04-31788, 12-04-32246, 12-04-31873, 12-04-33055, and 13-04-00401; by the Russian Academy of Sciences Presidium Basic Research Programs “Molecular and Cellular Biology,” “Basic Science for Medicine,” and “Molecular Mechanisms of Physiologic Functions”; and by Russian Federation President Fellowship for Young Scientists SP-2274.2012.4.

This article contains supplemental Figs. S1–S5 and Movies 1 and 2.
- PS
- phosphatidylserine
- CRP
- collagen-related peptide
- fXIII
- factor XIII
- tTG
- tissue transglutaminase
- GPIb
- glycoprotein Ib
- PE
- R-phycoerythrin
- PPACK
- Phe-Pro-Arg-chloromethylketone
- DIC
- differential interference contrast.
REFERENCES
- 1. Dale G. L. (2005) Coated-platelets: an emerging component of the procoagulant response. J. Thromb. Haemost. 3, 2185–2192 [DOI] [PubMed] [Google Scholar]
- 2. Heemskerk J. W., Mattheij N. J., Cosemans J. M. (2013) Platelet-based coagulation: different populations, different functions. J. Thromb. Haemost. 11, 2–16 [DOI] [PubMed] [Google Scholar]
- 3. Jackson S. P., Schoenwaelder S. M. (2010) Procoagulant platelets: are they necrotic? Blood 116, 2011–2018 [DOI] [PubMed] [Google Scholar]
- 4. Dale G. L., Friese P., Batar P., Hamilton S. F., Reed G. L., Jackson K. W., Clemetson K. J., Alberio L. (2002) Stimulated platelets use serotonin to enhance their retention of procoagulant proteins on the cell surface. Nature 415, 175–179 [DOI] [PubMed] [Google Scholar]
- 5. Kulkarni S., Jackson S. P. (2004) Platelet factor XIII and calpain negatively regulate integrin αIIbβ3 adhesive function and thrombus growth. J. Biol. Chem. 279, 30697–30706 [DOI] [PubMed] [Google Scholar]
- 6. Panteleev M. A., Ananyeva N. M., Greco N. J., Ataullakhanov F. I., Saenko E. L. (2005) Two subpopulations of thrombin-activated platelets differ in their binding of the components of the intrinsic factor X-activating complex. J. Thromb. Haemost. 3, 2545–2553 [DOI] [PubMed] [Google Scholar]
- 7. Mattheij N. J., Gilio K., van Kruchten R., Jobe S. M., Wieschhaus A. J., Chishti A. H., Collins P., Heemskerk J. W., Cosemans J. M. (2013) Dual mechanism of integrin αIIbβ3 closure in procoagulant platelets. J. Biol. Chem. 288, 13325–13336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yakimenko A. O., Verholomova F. Y., Kotova Y. N., Ataullakhanov F. I., Panteleev M. A. (2012) Identification of different proaggregatory abilities of activated platelet subpopulations. Biophys. J. 102, 2261–2269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Munnix I. C., Kuijpers M. J., Auger J., Thomassen C. M., Panizzi P., van Zandvoort M. A., Rosing J., Bock P. E., Watson S. P., Heemskerk J. W. (2007) Segregation of platelet aggregatory and procoagulant microdomains in thrombus formation: regulation by transient integrin activation. Arterioscler. Thromb. Vasc. Biol. 27, 2484–2490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Munnix I. C., Cosemans J. M., Auger J. M., Heemskerk J. W. (2009) Platelet response heterogeneity in thrombus formation. Thromb. Haemost. 102, 1149–1156 [DOI] [PubMed] [Google Scholar]
- 11. Hayashi T., Mogami H., Murakami Y., Nakamura T., Kanayama N., Konno H., Urano T. (2008) Real-time analysis of platelet aggregation and procoagulant activity during thrombus formation in vivo. Pflugers Arch. 456, 1239–1251 [DOI] [PubMed] [Google Scholar]
- 12. Alberio L., Safa O., Clemetson K. J., Esmon C. T., Dale G. L. (2000) Surface expression and functional characterization of α-granule factor V in human platelets: effects of ionophore A23187, thrombin, collagen, and convulxin. Blood 95, 1694–1702 [PubMed] [Google Scholar]
- 13. Jobe S. M., Leo L., Eastvold J. S., Dickneite G., Ratliff T. L., Lentz S. R., Di Paola J. (2005) Role of FcRγ and factor XIIIA in coated platelet formation. Blood 106, 4146–4151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kotova Y. N., Ataullakhanov F. I., Panteleev M. A. (2008) Formation of coated platelets is regulated by the dense granule secretion of adenosine 5′diphosphate acting via the P2Y12 receptor. J. Thromb. Haemost. 6, 1603–1605 [DOI] [PubMed] [Google Scholar]
- 15. Topalov N. N., Yakimenko A. O., Canault M., Artemenko E. O., Zakharova N. V., Abaeva A. A., Loosveld M., Ataullakhanov F. I., Nurden A. T., Alessi M. C., Panteleev M. A. (2012) Two types of procoagulant platelets are formed upon physiological activation and are controlled by integrin α(IIb)β(3). Arterioscler. Thromb. Vasc. Biol. 32, 2475–2483 [DOI] [PubMed] [Google Scholar]
- 16. Kempton C. L., Hoffman M., Roberts H. R., Monroe D. M. (2005) Platelet heterogeneity: variation in coagulation complexes on platelet subpopulations. Arterioscler. Thromb. Vasc. Biol. 25, 861–866 [DOI] [PubMed] [Google Scholar]
- 17. Topalov N. N., Kotova Y. N., Vasil'ev S. A., Panteleev M. A. (2012) Identification of signal transduction pathways involved in the formation of platelet subpopulations upon activation. Br. J. Haematol. 157, 105–115 [DOI] [PubMed] [Google Scholar]
- 18. Fager A. M., Wood J. P., Bouchard B. A., Feng P., Tracy P. B. (2010) Properties of procoagulant platelets: defining and characterizing the subpopulation binding a functional prothrombinase. Arterioscler. Thromb. Vasc. Biol. 30, 2400–2407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mazurov A. V., Pevzner D. V., Antonova O. A., Byzova T. V., Khaspekova S. G., Semenov A. V., Vlasik T. N., Samko A. N., Staroverov I. I., Ruda M. Y. (2002) Safety, inhibition of platelet aggregation and pharmacokinetics of Fab′2 fragments of the anti-glycoprotein IIb-IIIa monoclonal antibody FRaMon in high-risk coronary angioplasty. Platelets 13, 465–477 [DOI] [PubMed] [Google Scholar]
- 20. Robert P., Canault M., Farnarier C., Nurden A., Grosdidier C., Barlogis V., Bongrand P., Pierres A., Chambost H., Alessi M. C. (2011) A novel leukocyte adhesion deficiency III variant: kindlin-3 deficiency results in integrin- and nonintegrin-related defects in different steps of leukocyte adhesion. J. Immunol. 186, 5273–5283 [DOI] [PubMed] [Google Scholar]
- 21. Sugimura Y., Hosono M., Wada F., Yoshimura T., Maki M., Hitomi K. (2006) Screening for the preferred substrate sequence of transglutaminase using a phage-displayed peptide library: identification of peptide substrates for TGASE 2 and Factor XIIIA. J. Biol. Chem. 281, 17699–17706 [DOI] [PubMed] [Google Scholar]
- 22. Disdier M., Legrand C., Bouillot C., Dubernard V., Pidard D., Nurden A. T. (1989) Quantitation of platelet fibrinogen and thrombospondin in Glanzmann's thrombasthenia by electroimmunoassay. Thromb. Res. 53, 521–533 [DOI] [PubMed] [Google Scholar]
- 23. Heemskerk J. W., Vuist W. M., Feijge M. A., Reutelingsperger C. P., Lindhout T. (1997) Collagen but not fibrinogen surfaces induce bleb formation, exposure of phosphatidylserine, and procoagulant activity of adherent platelets: evidence for regulation by protein tyrosine kinase-dependent Ca2+ responses. Blood 90, 2615–2625 [PubMed] [Google Scholar]
- 24. Briedé J. J., Heemskerk J. W., Hemker H. C., Lindhout T. (1999) Heterogeneity in microparticle formation and exposure of anionic phospholipids at the plasma membrane of single adherent platelets. Biochim. Biophys. Acta 1451, 163–172 [DOI] [PubMed] [Google Scholar]
- 25. Peerschke E. I. (1995) Bound fibrinogen distribution on stimulated platelets. Examination by confocal scanning laser microscopy. Am. J. Pathol. 147, 678–687 [PMC free article] [PubMed] [Google Scholar]
- 26. Jobe S. M., Wilson K. M., Leo L., Raimondi A., Molkentin J. D., Lentz S. R., Di Paola J. (2008) Critical role for the mitochondrial permeability transition pore and cyclophilin D in platelet activation and thrombosis. Blood 111, 1257–1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Handagama P., Scarborough R. M., Shuman M. A., Bainton D. F. (1993) Endocytosis of fibrinogen into megakaryocyte and platelet α-granules is mediated by α IIb β 3 (glycoprotein IIb-IIIa). Blood 82, 135–138 [PubMed] [Google Scholar]
- 28. Bale M. D., Westrick L. G., Mosher D. F. (1985) Incorporation of thrombospondin into fibrin clots. J. Biol. Chem. 260, 7502–7508 [PubMed] [Google Scholar]
- 29. Nurden A. T., Fiore M., Nurden P., Pillois X. (2011) Glanzmann thrombasthenia: a review of ITGA2B and ITGB3 defects with emphasis on variants, phenotypic variability, and mouse models. Blood 118, 5996–6005 [DOI] [PubMed] [Google Scholar]
- 30. Hantgan R. R., Hindriks G., Taylor R. G., Sixma J. J., de Groot P. G. (1990) Glycoprotein Ib, von Willebrand factor, and glycoprotein IIb:IIIa are all involved in platelet adhesion to fibrin in flowing whole blood. Blood 76, 345–353 [PubMed] [Google Scholar]
- 31. Yamauchi Y., Kuroki M., Imakiire T., Uno K., Abe H., Beppu R., Yamashita Y., Kuroki M., Shirakusa T. (2002) Opposite effects of thrombospondin-1 via CD36 and CD47 on homotypic aggregation of monocytic cells. Matrix Biol. 21, 441–448 [DOI] [PubMed] [Google Scholar]
- 32. Hourdillé P., Hasitz M., Belloc F., Nurden A. T. (1985) Immunocytochemical study of the binding of fibrinogen and thrombospondin to ADP- and thrombin-stimulated human platelets. Blood 65, 912–920 [PubMed] [Google Scholar]
- 33. Bale M. D., Mosher D. F. (1986) Effects of thrombospondin on fibrin polymerization and structure. J. Biol. Chem. 261, 862–868 [PubMed] [Google Scholar]
- 34. Berny M. A., Munnix I. C., Auger J. M., Schols S. E., Cosemans J. M., Panizzi P., Bock P. E., Watson S. P., McCarty O. J., Heemskerk J. W. (2010) Spatial distribution of factor Xa, thrombin, and fibrin(ogen) on thrombi at venous shear. PLoS. One 5, e10415. [DOI] [PMC free article] [PubMed] [Google Scholar]