

Background: Antibody KM33 blocks factor VIII (FVIII) endocytosis and phospholipid binding.
Results: Hydrogen-deuterium exchange mass spectrometry reveals that KM33 binds C1 domain spikes 2092–2093 and 2158–2159. Glycosylated FVIII-R2159N shows reduced endocytosis and decreased binding to phospholipid membranes with low phosphatidylserine content.
Conclusion: Spikes 2092–2093 and 2158–2159 modulate FVIII endocytosis and phospholipid binding.
Significance: Novel insight is obtained about the role of the C1 domain for FVIII biology.
Keywords: Endocytosis, Enzyme Kinetics, Epitope Mapping, Factor VIII, Mass Spectrometry (MS), Phospholipid, Surface Plasmon Resonance (SPR), von Willebrand Factor
Abstract
The C1 domain of factor VIII (FVIII) has been implicated in binding to multiple constituents, including phospholipids, von Willebrand factor, and low-density lipoprotein receptor-related protein (LRP). We have previously described a human monoclonal antibody called KM33 that blocks these interactions as well as cellular uptake by LRP-expressing cells. To unambiguously identify the apparent “hot spot” on FVIII to which this antibody binds, we have employed hydrogen-deuterium exchange mass spectrometry. The results showed that KM33 protects FVIII regions 2091–2104 and 2157–2162 from hydrogen-deuterium exchange. These comprise the two C1 domain spikes 2092–2093 and 2158–2159. Spike 2092–2093 has been demonstrated recently to contribute to assembly with lipid membranes with low phosphatidylserine (PS) content. Therefore, spike 2158–2159 might serve a similar role. This was assessed by replacement of Arg-2159 for Asn, which introduces a motif for N-linked glycosylation. Binding studies revealed that the purified, glycosylated R2159N variant had lost its interaction with antibody KM33 but retained substantial binding to von Willebrand factor and LRP. Cellular uptake of the R2159N variant was reduced both by LRP-expressing U87-MG cells and by human monocyte-derived dendritic cells. FVIII activity was virtually normal on membranes containing 15% PS but reduced at low PS content. These findings suggest that the C1 domain spikes 2092–2093 and 2158–2159 together modulate FVIII membrane assembly by a subtle, PS-dependent mechanism. These findings contribute evidence in favor of an increasingly important role of the C1 domain in FVIII biology.
Introduction
Activated coagulation factor VIII (FVIIIa) is a cofactor that assembles with activated factor IX (FIXa)3 on lipid membranes that expose phosphatidylserine (PS) in the outer leaflet (1). This complex effectively generates activated factor X, ultimately leading to blood clot formation at sites of vascular injury. Current treatment of hemophilia A patients, who lack functional factor VIII (FVIII), involves intravenous infusion with either recombinant or plasma-derived FVIII (2). This treatment is, however, limited by the particularly effective clearance of FVIII from the circulation. Moreover, about 20% of patients develop antibodies against FVIII (3). The molecular determinants that drive the assembly of FVIII with membranes, including those of cells involved in clearance and immune response, remain incompletely understood.
FVIII is a multidomain protein that comprises a heavy chain (domains A1-A2-B) that is non-covalently linked to a light chain (domains A3-C1-C2) (1). The recently solved crystal structures of B domain-deleted FVIII have revealed that the three A domains are organized as a compact trimer in which the A1 and A3 domains form the base that carries the A2 domain on top. The trimer is supported by the two C domains, which are aligned in parallel (4, 5). As such, the structure of FVIII is highly homologous to that of coagulation factor V (FV), the cofactor of activated factor X in the prothrombinase complex (6–8). Both in FVIII and FV, the two C domains each display two characteristic β-hairpin loops, generally referred to as fatty feet or spikes (9, 10). Initially, FVIII interaction with membranes has been mainly attributed to the C2 domain (10–13). However, the tandem orientation of the two C domains in the crystal structures suggests that the C1 domain could also mediate membrane binding. Indeed, mutagenesis studies provided direct evidence that the C1 domain contributes to phospholipid binding as well and that this particularly involves the spike containing residues Lys-2092 to Phe-2093 (14, 15).
Besides lipid binding, the C1 domain residues 2092–2093 also contribute to the uptake of FVIII by cells that express low-density lipoprotein receptor-related protein (LRP) and by monocyte-derived dendritic cells (16, 17). Moreover, the same spike contributes to FVIII immunogenicity in a murine model of hemophilia A (17). The same membrane-binding spike also contributes to the interaction of FVIII with the antibody KM33 (14, 17). This anti-C1 domain antibody has been derived from the immune repertoire of a patient with severe hemophilia A (18) and is of particular interest because it also inhibits, besides FVIII activity, the interaction with von Willebrand factor (VWF) and LRP, cellular uptake, and immunogenicity (16, 17, 19). Apparently, the epitope of this antibody provides a “hot spot” for multiple macromolecular interactions and biological processes involving FVIII. Although the affinity of the KM33 antibody for FVIII is greatly dependent on C1 domain spike 2092–2093 (14), it seems unlikely that this putative hot spot would be limited to merely these two amino acids.
In the present study, we have employed hydrogen-deuterium exchange mass spectrometry (HDX-MS) to characterize the binding epitope of antibody KM33. This technology exploits the characteristic that HD exchange in a protein can be followed by mass spectrometry upon replacement of H2O by D2O in the aqueous solvent (20). Sites on FVIII that contribute to antibody binding should display reduced HD exchange in the FVIII-antibody complex. This approach revealed that the epitope of antibody KM33 comprises, besides spike 2092–2093, the second C1 domain spike of residues 2158–2159. We therefore explored the role of this spike by introducing a glycan in position 2159. The purified R2159N substitution variant was analyzed for a variety of FVIII interactions, with a particular focus on those interactions that are known to be inhibited by the KM33 antibody. The presence of the glycan in position 2159 proved predominantly apparent in assembly in the presence of membranes with low PS content.
EXPERIMENTAL PROCEDURES
Materials
DMEM-F12 was from Lonza (Walkersville, MD). FCS was from HyClone (Thermo Fisher Scientific, Rockford, IL). Chicken egg L-α-phosphatidylcholine, L-α-phosphatidylethanolamine (transphosphatidylated), and porcine brain L-α-phosphatidylserine (PS) were from Avanti Polar Lipids Inc. (Alabaster, AL). Tris (2-carboxyethyl)-phosphine hydrochloride was from Calbiochem, EMD Millipore Chemicals. The activated factor X substrate S-2765 containing the thrombin inhibitor I-2581 was from Chromogenix (Milan, Italy). All other chemicals were from Merck (Darmstadt, Germany).
Proteins
Monoclonal antibodies CLB-EL14 (EL14), CLB-KM33 (KM33), CLB-CAg12, CLB-CAg9, and CLB-CAg117 have been described before (14, 21, 22). ESH4 was obtained from American Diagnostica (Greenwich, CT). The construct of FVIII lacking the B domain residues 746–1639 (referred to as wild-type or FVIII-WT throughout this work) in the pcDNA3.1(+) vector has been described previously (22). The substitution R2159N was introduced by QuikChange mutagenesisTM using forward primer 5′-CCAACTCATTATAGCATTAATAGCACTCTTCGCATGGAG-3′ and reverse primer 5′-CTCCATGCGAAGAGTGCTATTAATGCTATAATGAGTTGG-3′. The coding regions of the constructs were verified by sequence analysis. Sequence reactions were performed with a BigDye terminator sequencing kit (Applied Biosystems, Foster City, CA). HEK293 cell lines stably expressing B domain-deleted FVIII and the R2159N variant were produced as described (23) and grown in DMEM-F12 medium supplemented with 10% FCS. Recombinant FVIII-WT and the R2159N variant were purified and analyzed as described using CLB-VK34 IgG1 coupled to CNBr-Sepharose 4B as an affinity matrix (16, 24). Recombinant VWF was prepared as described previously (25). LRP cluster II was expressed in baby hamster kidney cells and purified as described (26). Human FX, FIXa, and thrombin were purified and active site-titrated as described (27–29). BSA was from Merck (Darmstadt, Germany).
HDX Mass Spectrometry
HDX-MS was performed as described before with minor adjustments (30). For these studies, recombinant FVIII Turoctocog alfa (previously named N8) was produced in CHO cells as described previously (31). The molecule consists of a heavy chain of 88 kDa including a 21-amino acid residue-truncated B domain and a light chain of 79 kDa. It contains four N-glycosylation sites of which two are complex biantennary glycans and two are high-mannose structures (31). FVIII and KM33 were dialyzed to a buffer comprising 20 mm imidazole (pH 7.3), 10 mm CaCl2, and 150 mm NaCl, and the proteins were kept at 2 °C until the start of the experiment. HDX was initiated by incubating 3 μm FVIII in the absence or presence of 4.5 μm KM33 in the same buffer in 98% D2O instead of H2O. At four time intervals, i.e. 10, 100, 1000, and 10,000 s, HDX was quenched by the addition of an equal volume of ice-cold quenching buffer containing 1.35 m Tris (2-carboxyethyl)-phosphine hydrochloride, resulting in a final pH of 2.6. Samples were analyzed using the Waters SYNAPT G2 HDMS in combination with the nanoACQUITY UPLC with HDX technology. The analytical column was an ACQUITY UPLC BEH C18 1.7 μm (1.0 × 100 mm) with a 9-min gradient of 8–40% CH3CN. The trap column was an ACQUITY VanGuard Precolumn, BEH C18 1.7 μm (2.1 × 5 mm). Online pepsin digestion was performed using a 2.1 × 30 mm immobilized pepsin column (Applied Biosystems Inc.). MSE data were collected for all analyses. Undeuterated analyses were processed using ProteinLynx Global Server 2.5 with IdentityE informatics generating a peptic peptide map of FVIII on the basis of retention time, mobility drift time, intensity, fragment ions, and mass accuracy. DynamX was used to measure the deuterium uptake of each peptide as a function of deuterium exposure time. The deuterium uptake curves were plotted in a function of time for comparative analysis of FVIII in the absence and presence of KM33 antibody.
Tandem Mass Tag Labeling
Tandem mass tag (TMT) labeling and mass spectrometry analysis were performed as described by Bloem et al. (24), with the exception that proteins were labeled for 15 min and that the reaction was terminated by the addition of a 500-fold molar excess of hydroxylamine. FVIII-WT was modified with TMT-126, whereas FVIII-R2159N was modified with TMT-127. The identity of the peptides, including TMT-labeled lysine residues and the TMT-127/TMT-126 ratio thereof, were assessed employing Peaks Studio 6.0 build software. The protein database uniprot-organism_9606_AND_keyword_kw-0181.fasta was used for peptide identification. Search criteria allowed a peptide parent mass error tolerance of 30 ppm; a fragment mass error tolerance of 0.8 Da; and modifications including cysteine carbamidomethylation (57.02 Da), methionine oxidation (15.99 Da), and lysine-TMT labeling (225.16 Da).
Surface Plasmon Resonance Analysis
Surface plasmon resonance (SPR) analysis was performed using a BIAcore 3000 biosensor (Biacore AB, Uppsala, Sweden) essentially as described (32). For assessment of FVIII-VWF interaction, recombinant VWF (9 fmol/mm2) was immobilized onto a CM5 sensor chip using the amine coupling method according to the instructions of the manufacturer. FVIII-WT or FVIII-R2159N were passed over the chip at varying concentrations (0.4–240 nm) for 240 s in a buffer containing 150 mm NaCl, 5 mm CaCl2, 0.005% (v/v) Tween 20, and 20 mm Hepes (pH 7.4) at 25 °C with a flow rate of 20 μl/min. The sensor chip surface was regenerated three times after each experiment using the same buffer containing 1 m NaCl. Binding to VWF was corrected for binding in absence of VWF. Binding data during the association phase were fitted in a one-phase exponential association model, and response at equilibrium (Req) was plotted as a function of the FVIII concentration. For FVIII-LRP or antibody binding studies, FVIII-WT and FVIII-R2159N were bound at a density of 9 fmol/mm2 to the immobilized anti-C2-domain antibody EL14 IgG4 (39 fmol/mm2). Antibodies KM33, CLB-CAg 9, CLB-CAg12, and ESH4 (100 nm) were passed over immobilized FVIII-WT or FVIII-R2159N at a flow rate of 20 μl/min in a buffer containing 20 mm Hepes (pH 7.4), 150 mm NaCl, 5 mm CaCl2, and 0.005% (v/v) Tween 20 at 25 °C. Receptor binding was assessed by passing LRP cluster II at varying concentrations (0.2–200 nm) over the immobilized FVIII. Association was fitted in a one-phase exponential model, and Req was plotted against the LRP cluster II concentration as described (32).
FVIII Cellular Uptake by U87-MG Cells and Dendritic Cells
The cellular uptake of FVIII and the R2159N variant was studied in U87-MG cells (16) and human monocyte-derived dendritic cells (17) essentially as described. U87-MG cells (HTB-14, ATCC) were cultured on collagen type I-coated surface in DMEM-F12 containing 10% FCS. Cells were washed with 150 mm NaCl, 10 mm KCl, 5 mm CaCl2, 2 mm MgSO4, and 10 mm Hepes (pH 7.4) and subsequently incubated with FVIII-WT or FVIII-R2159N (0–200 nm) in the same buffer for 30 min at 37 °C. After washing with ice-cold TBS containing 0.5% (w/v) BSA, cells were trypsinized and collected in TBS/10% FCS and subsequently fixed using 0.37% paraformaldehyde in PBS for 15 min at room temperature. FVIII was detected employing the anti-C2 domain antibody EL14 that had been labeled with DyLight 549 (Thermo Fisher Scientific). Cells were permeabilized employing 0.25% saponin in TBS containing 1% (w/v) BSA and incubated with labeled EL14 antibody. Cells were washed twice with TBS/0.5% (w/v) BSA and analyzed by flow cytometry using a BD Biosciences LSR II flow cytometer as described (16, 17).
Factor X Activation Studies
FX activation was assessed in the presence of phospholipid vesicles of varying PS content. These contained PS/L-α-phosphatidylethanolamine (transphosphatidylated)/L-α-phosphatidylcholine in a molar ratio of 15/20/65 (15% PS) or 5/20/85 (5% PS) and were prepared by sonication in 40 mm Tris-HCl (pH 7.8), 150 mm NaCl. FX was incubated with FIXa and FVIII-WT or FVIII-R2159N (0.3 nm) in the presence of thrombin (1 nm) and phospholipids in a buffer containing 40 mm Tris-HCl (pH 7.8), 150 mm NaCl, 1.5 mm CaCl2, and 0.2% (w/v) BSA at 25 °C in a final volume of 40 μl. After varying time points (1–3 min), the reaction was terminated by adding 50 μl of the same buffer containing 16 mm EDTA, and finally 10 μl of 1.8 mm S-2765 was added to quantify activated factor X (14, 24).
RESULTS
FVIII Regions That Contribute to Antibody KM33 Binding
HDX-MS was employed to identify the epitope of KM33 on FVIII. After digestion of FVIII by pepsin, a total of 482 peptides was identified. These covered 95% of the FVIII primary sequence with 88% sequence coverage of the C1 domain. HDX of FVIII in the presence and absence of KM33 was first allowed for 10 s. Of these peptides, 98% displayed an exchange pattern that was unaffected by the binding of KM33, suggesting that amino acid residues within these regions do not contribute to KM33 binding. In contrast, a limited number of peptides did show protection from HDX in the presence of KM33. As an example, Fig. 1A shows the isotope distribution of peptides 2076–2090, 2076–2094, and 2149–2162. These displayed the typical shift in m/z value after 10 s of incubation of FVIII with D2O in the absence of KM33. For the latter two, however, this was markedly less pronounced in the presence of the antibody. HDX was subsequently followed at prolonged time intervals, i.e. 100, 1000, and 10,000 s. Fig. 1B shows the time-dependent deuterium incorporation for peptides 2076–2090, 2076–2094, and 2149–2162. In contrast to the first peptide, the other two did display time-dependent protection from HDX. In total, the FVIII region that displayed prolonged protection from deuterium incorporation in the presence of antibody KM33 included 10 peptic peptides that clustered on two distinct regions in the C1 domain. One cluster comprised the partially overlapping peptides 2075–2095, 2076–2094, 2077–2095, 2078–2095, 2081–2096, and 2090–2105, whereas the other comprised peptides 2148–2161, 2148–2162, 2149–2162, and 2152–2162. The cluster comprising residues 2075–2105 could be further confined because two partially overlapping peptides did not show any change in deuterium incorporation level in the presence of KM33. These were peptide 2076–2090 (see Fig. 1, A and B) and 2102–2113 (not shown). Taking into account that deuterium atoms attached in the first two residues may be lost during protein processing and peptide separation (33), the latter peptide allows exclusion of residues 2105–2113. Together, this delineates the residues that are protected by KM33 to amino acid residues 2091–2104. Similarly, the cluster spanning residues 2148–2162 could be confined to residues 2157–2162 because the overlapping peptide 2149–2156 did not display any change in deuterium incorporation (not shown). In conclusion, the residues protected by HD exchange comprise amino acids 2091–2104 and 2157–2162, which are located in two loops at the bottom of the FVIII C1 domain (see Fig. 1C).
FIGURE 1.

FVIII peptides 2076–2094 and 2149–2162, but not 2076–2090, exhibit reduced HD exchange in the presence of antibody KM33. A, the mass over charge ratio of the FVIII peptic peptides covering residues 2076–2094 and 2149–2162 that were dissolved in H2O in the absence of KM33 (black spectra, top panel), in D2O (cyan spectra, center panel), or in D2O in the presence of KM33 (red spectra, bottom panel). B, the relative level of deuterium incorporation (Da) of FVIII peptides 2076–2090, 2076–2094, and 2149–2162 as a function of the incubation time in D2O in the presence (red lines and circles) or absence (cyan lines and squares) of KM33. C, the obtained peptic peptides mapped onto the crystal structure of FVIII (PDB code 2R7E). Gray areas are uncovered by the peptide map. The deuterium incorporation level upon KM33 binding was identified to be unchanged for cyan areas and reduced for red areas. The inset zooms in on the FVIII C1 domain.
Glycosylation at FVIII Position 2159 Completely Abolishes the Interaction with KM33
Regions 2091–2104 and 2157–2162 comprise residues that are located on opposite sides of the cylindrically shaped C1 domain (Fig. 2A). Therefore, it seems unlikely that all individual residues in these regions contribute to KM33 binding. Within regions 2091–2104, C1 domain spike 2092–2093 provides a major contribution to KM33 binding (14). Within region 2157–2162, residues 2158–2159 constitute a second spike of the C1 domain. Because both spikes are at the same side of the C1 domain, this opens the possibility that KM33 requires both spikes for effective FVIII binding (Fig. 2A). This was addressed by replacing Arg-2159 for Asn. This substitution introduces an N-linked glycosylation motif. FVIII-R2159N was expressed in human 293 cells, and SDS-PAGE analysis of the purified variant revealed a reduced mobility of the FVIII light chain corresponding with an increased apparent molecular mass of ∼3 kDa (Fig. 2B). SPR analysis revealed that glycosylation at position 2159 had fully eliminated binding to antibody KM33, whereas the association with control antibodies CLB-CAg 9 (anti-A2 domain), CLB-CAg12 (anti-A3 domain), and ESH4 (anti-C2 domain) remained unaffected (Fig. 1C). This suggests that KM33 has a discontinuous epitope that comprises not only C1 domain spike 2092–2093 but also spike 2158–2159.
FIGURE 2.

Glycosylation in C1 domain spike 2158–2159 abolishes the interaction with KM33. A, peptides 2076–2094 (turquoise) and 2149–2162 (blue) are located at opposite sites of the C1 domain of FVIII (from PDB code 2R7E). C1 domain spikes 2092–2093 and 2158–2159 are shown in yellow and red, respectively. B, FVIII-WT and the R2159N variant separated by 7.5% SDS-PAGE under reducing conditions. Protein was visualized by silver staining. The light chain of FVIII appears on the gel as a doublet of approximately 80 kDa. C, SPR analysis of antibody binding to FVIII-R2159N and FVIII-WT. Antibodies included CLB-CAg9 (anti-A2 domain), CLB-CAg12 (anti-A3 domain), ESH4 (anti-C2 domain), and KM33. An SPR analysis was performed as described under “Experimental Procedures.”
Glycosylation at FVIII Position 2159 Retains VWF Interaction
We observed previously that FVIII binding to VWF is effectively inhibited by antibody KM33 (19). Within its epitope, Arg-2159 has been implicated previously in VWF interaction (15). We therefore analyzed the interaction of FVIII-R2159N with VWF by SPR. As shown in Fig. 3, the R2159N variant did bind to VWF, although the extent of association was approximately 50% reduced. Normal FVIII and FVIII-R2159N both failed to display satisfactory fits into a single exponential association model. Consequently, its remains difficult to conclude whether the apparent equilibrium dissociation constant (KD) is affected by glycosylation at position 2159 (see Fig. 3B, inset). It seems evident, however, that the R2159N variant has retained substantial VWF binding.
FIGURE 3.

Effect of glycosylation at FVIII position 2159 on VWF binding. FVIII-WT (A) or FVIII-R2159N (B) were passed over immobilized VWF on a CM5 sensor chip as described under “Experimental Procedures.” Inset, binding response at equilibrium (Req) was plotted as a function of the FVIII-WT (●) and FVIII-R2159N (○) concentration.
Glycosylation at FVIII Position 2159 Reduces LRP Binding
We have established previously that C1 domain residues 2092–2093 contribute to the LRP-dependent endocytosis of FVIII (16). We now employed an SPR analysis to assess the interaction between R2159N and LRP ligand binding cluster II, which mediates the binding of LRP to FVIII (34, 35). To this end, LRP cluster II was passed over immobilized FVIII-WT and R2159N (Fig. 4, A and B). The results reveal that cluster II binding of the R2159N variant is reduced. The apparent KD was estimated from a plot of Req against the LRP cluster II concentration (Fig. 3B, inset) and was 41 ± 3 nm (mean ± S.D.) for FVIII-WT and 6-fold higher (263 ± 21 nm) for FVIII-R2159N. Apparently, the introduction of a glycan in one of the KM33-binding loops in the C1 domain reduces the affinity for LRP.
FIGURE 4.

Effect of glycosylation at FVIII position 2159 on LRP binding. FVIII-WT (A) or FVIII-R2159N (B) were immobilized via the anti-C2 domain antibody EL14 on a CM5 sensor chip, and LRP cluster II was passed over as described under “Experimental Procedures.” Inset, binding response at equilibrium (Req) was plotted as a function of the FVIII-WT (●) and FVIII-R2159N (○) concentration.
Glycosylation at FVIII Position 2159 Reduces Cellular Uptake
We observed previously that antibody KM33 effectively inhibits FVIII uptake by LRP-expressing U87-MG cells (16) and by dendritic cells (36). We therefore investigated whether the elimination of KM33 binding by glycosylation at position 2159 would affect cellular uptake. FVIII-WT and the R2159N variant were incubated with U87-MG cells for 30 min, and uptake was estimated by flow cytometry. In comparison with FVIII-WT, uptake of FVIII-R2159N was reduced by ∼35% (Fig. 5A). A similar effect was observed for dendritic cells, which displayed a more pronounced reduction of FVIII-R2159N uptake (Fig. 5B). These data suggest that C1 domain spike 2158–2159 contributes to cellular uptake of FVIII.
FIGURE 5.

Effect of glycosylation at FVIII position 2159 on cellular uptake. FVIII-WT and FVIII-R2159N were incubated with U87-MG cells (A) and dendritic cells (B) for 30 min at 37 °C, and cellular uptake was analyzed as described under “Experimental Procedures.” ●, FVIII-WT; ○, FVIII-R2159N. Data are shown as mean ± S.D. from three experiments.
FVIII-R2159N Retains FVIII Cofactor Activity
Antibody KM33 has been established as being a strong inhibitor of FVIII biological activity (18). Inserting a glycan in part of its epitope, therefore, might interfere with FVIII function. This was tested using purified proteins and phospholipid vesicles of varying PS content. On membranes containing 15% PS, FVIII-R2159N proved virtually normal, with only a slight reduction of Vmax and similar Km (Fig. 6A). In agreement with these data, FIXa titration curves reflected that FVIII-R2159N displays a normal affinity for FIXa in assembling the FX activating complex (Fig. 6C). On membranes containing 5% PS, however, Km proved to be increased 3-fold and Vmax to be reduced 5-fold (Fig. 6B). Moreover, the apparent affinity for FIXa on these membranes was reduced 4-fold (Fig. 6D). These data suggest that the presence of the glycan in C1 domain spike 2158–2159 does not affect FVIII activity per se but makes FVIII more dependent on the PS content of the membrane whereon it assembles with FIXa to activate FX.
FIGURE 6.

Effect of glycosylation at FVIII position 2159 on FX activation. A and B, FX (25–450 nm) was incubated with FVIII (0.3 nm), thrombin (1 nm), FIXa (16 nm), and phospholipid vesicles (25 μm) containing 15% PS (A) or 5% PS (B) as described under Experimental Procedures.” ●, FVIII-WT; ○, FVIII-R2159N. Calculated values for Km and Vmax were derived assuming standard Michaelis-Menten kinetics. C and D, 200 nm FX was activated by 1–32 nm FIXa in the presence of 0.3 nm FVIII, 1 nm thrombin, and 25 μm phospholipid vesicles comprising 15% PS (C) or 5% PS (D). ●, FVIII-WT; ○, FVIII-R2159N. Apparent KD and Bmax were obtained by fitting the data to a hyperbola. Data represent mean ± S.D. from at least three experiments.
Glycosylation at Position 2159 Increases PS Dependence of FVIII Cofactor Activity
The lipid-dependence of FVIII-R2159N was further investigated at limiting phospholipid concentrations. A lipid titration experiment using vesicles containing 15% PS demonstrated that half-maximal FX activation occurred at ∼1 μm phospholipids, whereas 3 μm was required for FVIII-R2159N (Fig. 7A). These apparent KD values cannot be taken to represent “true” KD values for FVIII-lipid interaction because of the low FVIII concentration (0.3 nm) in these experiments and the presence of FIXa and FX, which compete for binding to the same surface. Nevertheless, these data reflect that assembly of the FX activating complex at limiting lipid concentrations is slightly hindered by the presence of the glycan at position 2159. This became much more prominent on membranes containing 5% PS (Fig. 7B). Apparently, the glycan in C1 domain spike 2158–2159 interferes with assembly of the FX activating complex on membranes with low PS content while leaving complex assembly at higher PS content virtually unaffected.
FIGURE 7.

Lipid dependence of FX activation in the presence of normal FVIII and FVIII-R2159N. 0.3 nm FVIII-WT (●) or 0.3 nm FVIII-R2159N (○) was preincubated with 1 nm thrombin, 16 nm FIXa, and 0–20 μm phospholipid vesicles comprising 15% PS (A) or 5% PS (B). After 1 min, the reaction was started by the addition of FX (200 nm final concentration), and activated factor X formation was monitored as described under “Experimental Procedures.” Data represent mean ± S.D. from at least three experiments.
Glycosylation at Position 2159 Does Not Hinder C1 Domain Spike 2092–2093
The observed PS dependence of FVIII-R2159N (Fig. 7B) is similar to that of FVIII with substitutions in membrane-binding spike 2092–2093 (14). The close proximity of the glycan to this spike (see Fig. 2) opens the possibility that the glycan effectively shields C1 domain residues Lys-2092 to Phe-2093 and, thereby, causes the typical PS dependence associated with this spike. This was addressed by TMT labeling of surface-exposed lysine residues and mass spectrometry analysis of peptides containing Lys-2092. FVIII-WT was labeled with TMT-126 and FVIII-R2159N with TMT-127. A ratio of 127/126 lower than 1 would be indicative for a lower labeling efficiency of Lys-2092 in FVIII-R2159N and, thus, for reduced exposure of spike 2092–2093. After chymotrypsin digestion, the FVIII peptide 2080-IIHGIKTQGARQKTMTF-2093 was identified with a Δppm of −4.5 (Fig. 8, A and B). Fragmentation of these ions by higher energy collision dissociation generates a reporter ion from the TMT label that allows quantitative comparison. By this approach, the 127/126 ratio of Lys-2092 in peptide 2080–2093 was found to be 0.95 (Fig. 8C). Thus, glycosylation at position 2159 does not reduce the labeling efficiency of Lys-2092.
FIGURE 8.
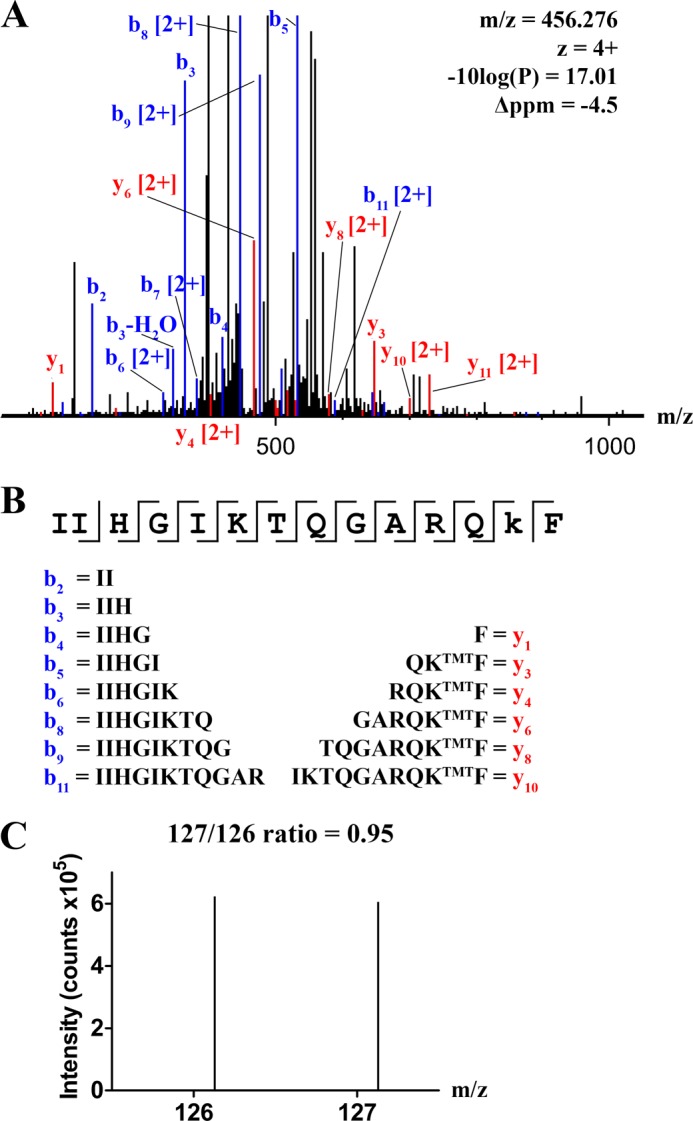
Surface exposure of Lys-2092 in normal FVIII and FVIII-R2159N. FVIII-WT and FVIII-R2159N were labeled with TMT tags and analyzed by mass spectrometry as described under “Experimental Procedures.” A, the collision-induced dissociation fragmentation spectrum of peptide 2080-IIHGIKTQGARQKTMTF-2093. Y ions are displayed in red and recovered b ions in blue. Values of m/z, z, −10log(P), and Δppm are indicated. B, the peptide sequence, including the assigned y and b ions. C, higher energy collision-induced dissociation fragmentation spectrum of peptide 2080-IIHGIKTQGARQKTMTF-2093. The peaks at m/z 126.13 and 127.13 represent the labeling efficiency of Lys-2092 in FVIII-WT and FVIII-R2159N, respectively.
DISCUSSION
In this study, we used HDX-MS to map the epitope of the anti-C1 domain antibody KM33. This was of particular interest because this antibody is known to interfere with multiple FVIII functional interactions, including VWF binding, LRP interaction, cellular uptake, and FVIII cofactor activity in the activation of FX by FIXa (14, 16, 17, 19). The epitope was found to be discontinuous, comprising two loops in the C1 domain (Fig. 1). We obtained evidence that the core of this epitope is composed of the two spikes at the bottom of these loops, which contain residues 2092–2093 and 2158–2159, respectively (Fig. 2). Previously, we found spike Lys-2092 to Phe-2093 to be involved in binding to LRP, in cellular uptake, and in FVIII activity on membranes of low PS content (14, 16, 17). We now observed that spike Ile-2158 to Arg-2159 serves a similar role. These findings are on the basis of the functional properties of the FVIII-R2159N variant, which carries a glycan in position 2159 (Fig. 2). Interestingly, the presence of this glycan interferes in the same interactions as antibody KM33 does. This particularly applies to interaction with VWF (Fig. 3), LRP cluster II (Fig. 4), and cellular uptake by U87-MG cells and dendritic cells (Fig. 5). However, the effect of the glycan is less pronounced than that of the antibody bound to the same part of the C1 domain. This is not surprising in view of the size difference between the antibody and a glycan of ∼3 kDa (Fig. 2). Nevertheless, our findings suggest that the bottom of the C1 domain contributes to a number of FVIII functional interactions.
It is remarkable that although the KM33 antibody is a strong inhibitor of FVIII activity, the introduction of the glycan in position 2159 did not inhibit FVIII activity per se (Figs. 6, A and C, and 7A). The glycan, however, did increase the PS dependence of this variant, as has been found previously for FVIII variants with amino acid substitutions in the other spike in the C1 domain in positions 2092–2093 (14, 15). Apparently, both spikes serve similar functions in the assembly of FVIII with PS-containing lipid membranes. At low PS content, membrane assembly was impaired significantly (Figs. 6, B and D, and 7B), suggesting that membrane binding cannot be fully mediated by the C2 domain alone under these conditions of limiting PS exposure. This supports the view that both C domains support membrane interaction in a cooperative manner (15) and is also compatible with the finding that a FVIII deletion mutant lacking the C2 domain still displays substantial FVIII cofactor activity (37).
One question is whether the phenotype of FVIII-R2159N is due to steric effects because of the glycan or specific to the lack of Arg in position 2159. In this regard, it is interesting that Gilbert et al. (15) described that an R2159A substitution variant displays nearly normal FX activation kinetics but without the typical sensitivity for low PS . One explanation for this apparent discrepancy might be that the glycan shields the 2092–2093 spike and, thereby, causes the typical PS dependence associated with this spike. However, our TMT labeling studies (Fig. 8) indicate that, despite the glycosylation at position 2159, Lys-2092 is fully accessible for labeling. Apparently, the glycan increases PS sensitivity by a mechanism that is similar to but distinct from that of spike 2092–2093. The most likely interpretation of these findings is that binding to PS-containing membranes involves both spikes 2092–2092 and 2158–2159. In this regard, it is of interest that human FV has been found to be partially glycosylated at Asn-2181, which is in one of the two membrane-binding spikes of the C2 domain (38, 39). The glycosylated FV variant displays lower affinity for PS-containing membranes and reduced FV activity. This provides additional evidence that both in FV and FVIII, the two C domains together support membrane interaction and that each C domain contributes therein by two membrane-binding spikes. In the presence of sufficient PS, however, individual spikes may be dispensable for full cofactor function.
As for VWF interaction, our SPR studies revealed that FVIII-R2159N had retained substantial VWF binding (Fig. 3). The hemophilia A database reports various Arg-2159 substitutions that are associated with reduced levels of FVIII and with mild hemophilia A, thus being compatible with reduced VWF interaction (40). In this regard, it is interesting that the R2159A substitution variant described by Gilbert et al. (15) displayed an apparent lack of VWF binding . This may seem at variance with the residual binding of FVIII-R2159N in our SPR studies. The reason for this discrepancy remains unclear but might be related to methodological differences between SPR and competition studies with normal FVIII as reported (15).
With regard to FVIII cellular uptake, it remains unclear whether the glycosylated spike 2158–2159 reduces assembly with the endocytic receptor LRP on the cell membrane. Although the R2159N variant displays reduced binding to LRP cluster II in our SPR studies, it remains questionable whether this is really due to a difference in affinity (Fig. 3). The interaction of FVIII with LRP is thought to involve a pattern of specific Lys residues distributed over the A3-C1 region of FVIII (41). It therefore seems possible that our binding data reflect some sterical hindrance by the glycan at position 2159 in LRP assembly (Fig. 4) and cellular uptake (Fig. 5). If so, this should involve an interactive site other than that located in spike 2092–2093 (16, 17) because that would be incompatible with our observation that Lys-2092 is normally accessible in the glycosylated variant (Fig. 8). The precise mechanism of FVIII uptake by LRP-expressing cells and dendritic cells remains to be identified. Possibly, assembly on the cell membrane involves a mechanism similar to that on membranes of low PS content (Fig. 7), for instance as an initial step preceding the assembly with LRP or other endocytic receptors.
It would be interesting to have more information on the structure and, more importantly, the orientation of the glycan at position 2159. A typical 3-kDa biantennary glycan could protrude toward the membrane surface but may also fold aside toward the C2 domain. As such, it may disturb the parallel orientation of the two C domains, as seen in the apparently rigid crystal structure (4, 5), and, thereby, affect the cooperative membrane binding by the spikes of the two C domains. Structural studies using protein dynamics will be needed to unravel this subtle PS-dependent process in more detail.
Footnotes
- FIXa
- activated factor IX
- PS
- phosphatidylserine
- FVIII
- factor VIII
- FV
- factor V
- LRP
- low-density lipoprotein receptor-related protein
- VWF
- von Willebrand factor
- HDX
- hydrogen-deuterium exchange
- TMT
- tandem mass tag
- SPR
- surface plasmon resonance.
REFERENCES
- 1. Fay P. J. (2004) Activation of factor VIII and mechanisms of cofactor action. Blood Rev. 18, 1–15 [DOI] [PubMed] [Google Scholar]
- 2. Hoyer L. W. (1994) Hemophilia A. N. Engl. J. Med. 330, 38–47 [DOI] [PubMed] [Google Scholar]
- 3. Scharrer I., Bray G. L., Neutzling O. (1999) Incidence of inhibitors in haemophilia A patients. A review of recent studies of recombinant and plasma-derived factor VIII concentrates. Haemophilia 5, 145–154 [DOI] [PubMed] [Google Scholar]
- 4. Shen B. W., Spiegel P. C., Chang C.-H., Huh J.-W., Lee J.-S., Kim J., Kim Y.-H., Stoddard B. L. (2008) The tertiary structure and domain organization of coagulation factor VIII. Blood 111, 1240–1247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ngo J. C., Huang M., Roth D. A., Furie B. C., Furie B. (2008) Crystal structure of human factor VIII. Implications for the formation of the factor IXa-factor VIIIa complex. Structure 16, 597–606 [DOI] [PubMed] [Google Scholar]
- 6. Kane W. H., Davie E. W. (1986) Cloning of a cDNA coding for human factor V, a blood coagulation factor homologous to factor VIII and ceruloplasmin. Proc. Natl. Acad. Sci. U.S.A. 83, 6800–6804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nicolaes G. A., Dahlbäck B. (2002) Factor V and thrombotic disease. Arterioscler. Thromb. Vasc. Biol. 22, 530–538 [DOI] [PubMed] [Google Scholar]
- 8. Adams T. E., Hockin M. F., Mann K. G., Everse S. J. (2004) The crystal structure of activated protein C-inactivated bovine factor Va. Implications for cofactor function. Proc. Natl. Acad. Sci. U.S.A. 101, 8918–8923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Macedo-Ribeiro S., Bode W., Huber R., Quinn-Allen M. A., Kim S. W., Ortel T. L., Bourenkov G. P., Bartunik H. D., Stubbs M. T., Kane W. H., Fuentes-Prior P. (1999) Crystal structures of the membrane-binding C2 domain of human coagulation factor V. Nature 402, 434–439 [DOI] [PubMed] [Google Scholar]
- 10. Pratt K. P., Shen B. W., Takeshima K., Davie E. W., Fujikawa K., Stoddard B. L. (1999) Structure of the C2 domain of human factor VIII at 1.5 A resolution. Nature 402, 439–442 [DOI] [PubMed] [Google Scholar]
- 11. Mertens K., Celie P. H., Kolkman J. A., Lenting P. J. (1999) Factor VIII-factor IX interactions. Molecular sites involved in enzyme-cofactor complex assembly. Thromb. Haemost. 82, 209–217 [PubMed] [Google Scholar]
- 12. Stoilova-McPhie S., Villoutreix B. O., Mertens K., Kemball-Cook G., Holzenburg A. (2002) 3-Dimensional structure of membrane-bound coagulation factor VIII. Modeling of the factor VIII heterodimer within a 3-dimensional density map derived by electron crystallography. Blood 99, 1215–1223 [DOI] [PubMed] [Google Scholar]
- 13. Gilbert G. E., Kaufman R. J., Arena A. A., Miao H., Pipe S. W. (2002) Four hydrophobic amino acids of the factor VIII C2 domain are constituents of both the membrane-binding and von Willebrand factor-binding motifs. J. Biol. Chem. 277, 6374–6381 [DOI] [PubMed] [Google Scholar]
- 14. Meems H., Meijer A. B., Cullinan D. B., Mertens K., Gilbert G. E. (2009) Factor VIII C1 domain residues Lys-2092 and Phe-2093 contribute to membrane binding and cofactor activity. Blood 114, 3938–3946 [DOI] [PubMed] [Google Scholar]
- 15. Lü J., Pipe S. W., Miao H., Jacquemin M., Gilbert G. E. (2011) A membrane-interactive surface on the factor VIII C1 domain cooperates with the C2 domain for cofactor function. Blood 117, 3181–3189 [DOI] [PubMed] [Google Scholar]
- 16. Meems H., van den Biggelaar M., Rondaij M., van der Zwaan C., Mertens K., Meijer A. B. (2011) C1 domain residues Lys-2092 and Phe-2093 are of major importance for the endocytic uptake of coagulation factor VIII. Int. J. Biochem. Cell Biol. 43, 1114–1121 [DOI] [PubMed] [Google Scholar]
- 17. Wroblewska A., van Haren S. D., Herczenik E., Kaijen P., Ruminska A., Jin S. Y., Zheng X. L., van den Biggelaar M., ten Brinke A., Meijer A. B., Voorberg J. (2012) Modification of an exposed loop in the C1 domain reduces immune responses to factor VIII in hemophilia A mice. Blood 119, 5294–5300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. van den Brink E. N., Turenhout E. A., Bovenschen N., Heijnen B. G., Mertens K., Peters M., Voorberg J. (2001) Multiple VH genes are used to assemble human antibodies directed toward the A3-C1 domains of factor VIII. Blood 97, 966–972 [DOI] [PubMed] [Google Scholar]
- 19. Meems H. (2011) New Insight into the C1 Domain of Coagulation Factor VIII. Ph.D. thesis, Utrecht University [Google Scholar]
- 20. Wales T. E., Engen J. R. (2006) Hydrogen exchange mass spectrometry for the analysis of protein dynamics. Mass Spectrom. Rev. 25, 158–170 [DOI] [PubMed] [Google Scholar]
- 21. Stel H. V. (1984) Monoclonal Antibodies against Factor VIII-von Willebrand Factor. Ph.D. thesis, University of Amsterdam [Google Scholar]
- 22. van den Biggelaar M., Meijer A. B., Voorberg J., Mertens K. (2009) Intracellular cotrafficking of factor VIII and von Willebrand factor type 2N variants to storage organelles. Blood 113, 3102–3109 [DOI] [PubMed] [Google Scholar]
- 23. Fribourg C., Meijer A. B., Mertens K. (2006) The interface between the EGF2 domain and the protease domain in blood coagulation factor IX contributes to factor VIII binding and factor X activation. Biochemistry 45, 10777–10785 [DOI] [PubMed] [Google Scholar]
- 24. Bloem E., Meems H., van den Biggelaar M., van der Zwaan C., Mertens K., Meijer A. B. (2012) Mass spectrometry-assisted study reveals that lysine residues 1967 and 1968 have opposite contribution to stability of activated factor VIII. J. Biol. Chem. 287, 5775–5783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. van den Biggelaar M., Bierings R., Storm G., Voorberg J., Mertens K. (2007) Requirements for cellular co-trafficking of factor VIII and von Willebrand factor to Weibel-Palade bodies. J. Thromb. Haemost. 5, 2235–2242 [DOI] [PubMed] [Google Scholar]
- 26. Bovenschen N., Mertens K., Hu L., Havekes L. M., van Vlijmen B. J. (2005) LDL receptor cooperates with LDL receptor-related protein in regulating plasma levels of coagulation factor VIII in vivo. Blood 106, 906–912 [DOI] [PubMed] [Google Scholar]
- 27. Mertens K., Bertina R. M. (1980) Pathways in the activation of human coagulation factor X. Biochem. J. 185, 647–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lenting P. J., ter Maat H., Clijsters P. P., Donath M.-J., van Mourik J. A., Mertens K. (1995) Cleavage at arginine 145 in human blood coagulation factor IX converts the zymogen into a factor VIII binding enzyme. J. Biol. Chem. 270, 14884–14890 [DOI] [PubMed] [Google Scholar]
- 29. Mertens K., van Wijngaarden A., Bertina R. M. (1985) The role of factor VIII in the activation of human blood coagulation factor X by activated factor IX. Thromb. Haemost. 54, 654–660 [PubMed] [Google Scholar]
- 30. Andersen M. D., Faber J. H. (2011) Structural characterization of both the non-proteolytic and proteolytic activation pathways of coagulation factor XIII studied by hydrogen-deuterium exchange mass spectrometry. Int. J. Mass Spectrom. 302, 139–148 [Google Scholar]
- 31. Thim L., Vandahl B., Karlsson J., Klausen N. K., Pedersen J., Krogh T. N., Kjalke M., Petersen J. M., Johnsen L. B., Bolt G., Nørby P. L., Steenstrup T. D. (2010) Purification and characterization of a new recombinant factor VIII (N8). Haemophilia 16, 349–359 [DOI] [PubMed] [Google Scholar]
- 32. van den Biggelaar M., Sellink E., Klein Gebbinck J. W., Mertens K., Meijer A. B. (2011) A single lysine of the two-lysine recognition motif of the D3 domain of receptor-associated protein is sufficient to mediate endocytosis by low-density lipoprotein receptor-related protein. Int. J. Biochem. Cell Biol. 43, 431–440 [DOI] [PubMed] [Google Scholar]
- 33. Bai Y., Milne J. S., Mayne L., Englander S. W. (1993) Primary structure effects on peptide group hydrogen exchange. Proteins 17, 75–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lenting P. J., Neels J. G., van den Berg B. M., Clijsters P. P., Meijerman D. W., Pannekoek H., van Mourik J. A., Mertens K., van Zonneveld A.-J. (1999) The light chain of factor VIII comprises a binding site for low density lipoprotein receptor-related protein. J. Biol. Chem. 274, 23734–23739 [DOI] [PubMed] [Google Scholar]
- 35. Meijer A. B., Rohlena J., van der Zwaan C., van Zonneveld A. J., Boertjes R. C., Lenting P. J., Mertens K. (2007) Functional duplication of ligand-binding domains within low-density lipoprotein receptor-related protein for interaction with receptor associated protein, α2-macroglobulin, factor IXa and factor VIII. Biochim. Biophys. Acta 1774, 714–722 [DOI] [PubMed] [Google Scholar]
- 36. Herczenik E., van Haren S. D., Wroblewska A., Kaijen P., van den Biggelaar M., Meijer A. B., Martinez-Pomares L., ten Brinke A., Voorberg J. (2012) Uptake of blood coagulation factor VIII by dendritic cells is mediated via its C1 domain. J. Allergy Clin. Immunol. 129, 501–509 [DOI] [PubMed] [Google Scholar]
- 37. Wakabayashi H., Griffiths A. E., Fay P. J. (2010) Factor VIII lacking the C2 domain retains cofactor activity in vitro. J. Biol. Chem. 285, 25176–25184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Nicolaes G. A., Villoutreix B. O., Dahlbäck B. (1999) Partial glycosylation of Asn2181 in human factor V as a cause of molecular and functional heterogeneity. Modulation of glycosylation efficiency by mutagenesis of the consensus sequence for N-linked glycosylation. Biochemistry 38, 13584–13591 [DOI] [PubMed] [Google Scholar]
- 39. Kim S. W., Ortel T. L., Quinn-Allen M. A., Yoo L., Worfolk L., Zhai X., Lentz B. R., Kane W. H. (1999) Partial glycosylation at asparagine-2181 of the second C-type domain of human factor V modulates assembly of the prothrombinase complex. Biochemistry 38, 11448–11454 [DOI] [PubMed] [Google Scholar]
- 40. Liu M. L., Shen B. W., Nakaya S., Pratt K. P., Fujikawa K., Davie E. W., Stoddard B. L., Thompson A. R. (2000) Hemophilic factor VIII C1- and C2-domain missense mutations and their modeling to the 1.5-angstrom human C2-domain crystal structure. Blood 96, 979–987 [PubMed] [Google Scholar]
- 41. van den Biggelaar M., van der Zwaan C., Zuurveld M. G., Mertens K., Meijer A. B. (2011) Low density lipoprotein receptor-related protein (LRP) cluster II interacts with the factor (F) VIII light chain via an extended surface comprising multiple lysine residues. J. Thromb. Haemost. 9, Suppl. S2, 821 (abstr.) [Google Scholar]