

Background: Human 2-Cys peroxiredoxins (Prxs) are differentially susceptible to inactivation by H2O2.
Results: Engineered Prx2 and Prx3 variants demonstrate that C-terminal residues modulate the extent of hyperoxidation.
Conclusion: Rapid disulfide bond formation protects Prx3 from inactivation.
Significance: The reactivity of Prx3 with H2O2 is important for understanding its protective role in the mitochondria.
Keywords: Mass Spectrometry (MS), Mitochondria, Peroxiredoxin, Redox, Thiol
Abstract
Peroxiredoxins (Prxs) detoxify peroxides and modulate H2O2-mediated cell signaling in normal and numerous pathophysiological contexts. The typical 2-Cys subclass of Prxs (human Prx1–4) utilizes a Cys sulfenic acid (Cys-SOH) intermediate and disulfide bond formation across two subunits during catalysis. During oxidative stress, however, the Cys-SOH moiety can react with H2O2 to form Cys sulfinic acid (Cys-SO2H), resulting in inactivation. The propensity to hyperoxidize varies greatly among human Prxs. Mitochondrial Prx3 is the most resistant to inactivation, but the molecular basis for this property is unknown. A panel of chimeras and Cys variants of Prx2 and Prx3 were treated with H2O2 and analyzed by rapid chemical quench and time-resolved electrospray ionization-TOF mass spectrometry. The latter utilized an on-line rapid-mixing setup to collect data on the low seconds time scale. These approaches enabled the first direct observation of the Cys-SOH intermediate and a putative Cys sulfenamide (Cys-SN) for Prx2 and Prx3 during catalysis. The substitution of C-terminal residues in Prx3, residues adjacent to the resolving Cys residue, resulted in a Prx2-like protein with increased sensitivity to hyperoxidation and decreased ability to form the intermolecular disulfide bond between subunits. The corresponding Prx2 chimera became more resistant to hyperoxidation. Taken together, the results of this study support that the kinetics of the Cys-SOH intermediate is key to determine the probability of hyperoxidation or disulfide formation. Given the oxidizing environment of the mitochondrion, it makes sense that Prx3 would favor disulfide bond formation as a protection mechanism against hyperoxidation and inactivation.
Introduction
Peroxiredoxins (Prxs)5 are ubiquitous, highly expressed antioxidant enzymes that can convert hydrogen peroxide (H2O2), peroxynitrite (ONOO−), and lipid peroxides (ROOH) to water. Although this function was originally thought to be primarily protective in nature, Prxs also play a key role in modulating H2O2-mediated cell signaling in normal and pathophysiological contexts, including cell growth, differentiation, adrenal steroidogenesis, neurodegeneration, and cancer (1–5). Human cells contain six Prx isoforms with differences in subcellular localization and the content of Cys residues (6). The typical 2-Cys or Prx1 subclass (human Prx1–4) contains two catalytic Cys residues on each monomer of an obligate homodimer (Fig. 1A). Under normal conditions, the peroxidatic Cys residue (Cys-SPH) attacks a H2O2 molecule to form a sulfenic acid intermediate (Cys-SPOH). Subsequent structural rearrangements enable the active site to transition from the fully folded (FF) to a locally unfolded conformation. The Cys-SPH residue can then form a disulfide with the resolving Cys residue (Cys-SRH), located near the C terminus of the adjacent subunit. This disulfide (SP-SR) is reduced by the thioredoxin-thioredoxin reductase system. Additionally, during the catalytic cycle, an interchange between dimeric and higher order oligomeric states occurs, with the reduced decamer typically being the most active form (7, 8).
FIGURE 1.
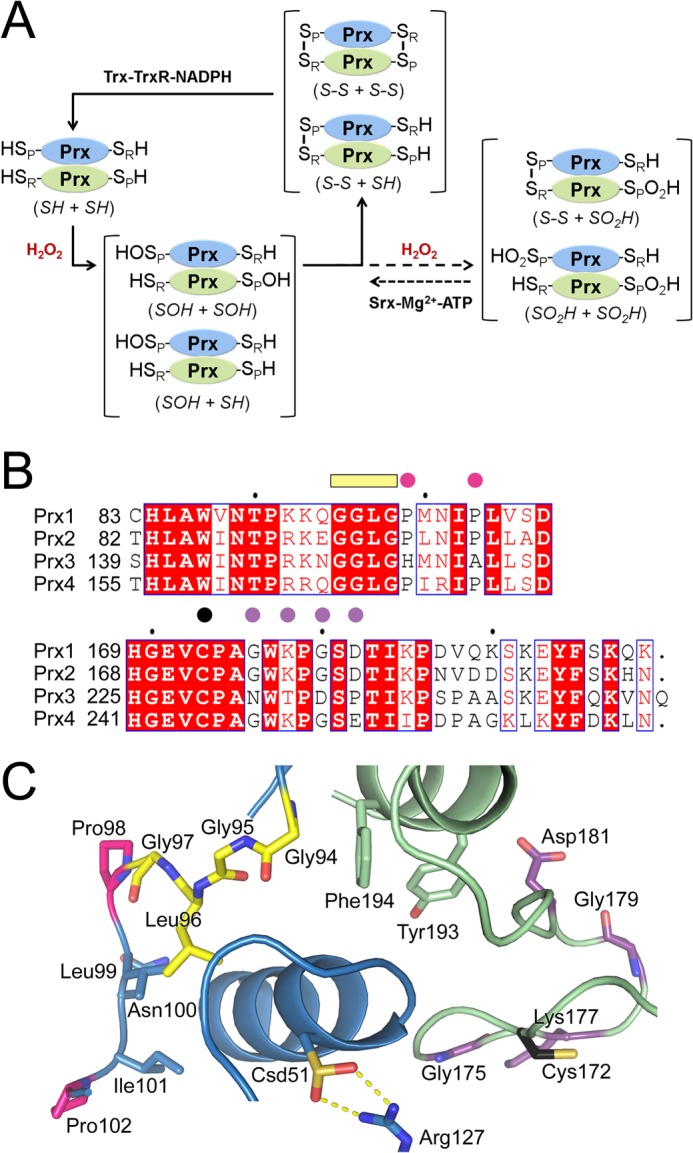
Key residues involved in 2-Cys Prx catalysis and hyperoxidation. A, 2-Cys Prx catalytic cycle showing oxidation, hyperoxidation, and repair by sulfiredoxin. The monomers of the obligate Prx homodimer are shown in blue and green. Depending on the concentration of peroxide present, one or both of the peroxidatic Cys residues (Cys-SPH) may be oxidized to the Cys sulfenic acid (Cys-SPOH) or hyperoxidized to the Cys sulfinic acid (Cys-SPO2H). The resolving Cys residue, Cys-SRH, is located near the C terminus and forms an intermolecular disulfide bond with the Cys-SPH residue during normal catalysis. Reduction of this disulfide and the Cys-SPO2H moiety is performed by the thioredoxin-thioredoxin reductase-NAPDH (Trx-Trx-NADPH) system and sulfiredoxin (Srx), respectively. The abbreviation used within the main text for each species is indicated in italics. B, sequence alignment of key residues within the active site. The following motifs and residues are highlighted: GGLG motif, yellow bar; residue differences between the Prxs, pink and purple circles; Cys-SRH residue, black circle. C, active site of hyperoxidized, human Prx2. The same coloring scheme from B is used. The peroxidatic Cys is hyperoxidized and labeled as Csd51. The Cys-SRH residue for Prx2 is Cys-172. PDB code 1QMV (19).
Under conditions of high oxidative stress, a second H2O2 molecule can react with the Cys-SPOH moiety to form a Cys sulfinic acid (Cys-SPO2H) moiety within some Prx isoforms (9). This hyperoxidation of the Prx molecule results in inactivation and is thought to enable H2O2 to modulate the activity of a variety of other proteins, including phosphatases and the master redox transcription factor Nrf2 (10–12). Repair of the Prx molecules by sulfiredoxin restores the peroxidase activity, lowers peroxide levels, and terminates subsequent downstream signaling events (9, 13–15). However, the susceptibility of human 2-Cys Prxs to hyperoxidation varies greatly, with the cytoplasmic Prx1 and Prx2 being more susceptible than the mitochondrial Prx3 (16). The resistance of Prx3 to hyperoxidation is consistent with its ability to maintain function within a highly oxidative environment, but the molecular basis for this characteristic is not known. Moreover, a detailed analysis of Prx3 is needed to understand its ability to protect the murine heart from the damage caused by myocardial infarction and cancer cells from apoptosis-inducing drugs (2, 17, 18).
An alignment of human Prx1–4 reveals that Prx3 has a unique primary sequence near the GGLG motif within the active site region (Fig. 1B) and near the C terminus. The structure of hyperoxidized Prx2 (Fig. 1C) illustrates the proximity of these regions to the Cys-SPH residue (19). In particular, the GGLG motif interacts with the C-terminal helix of the adjacent Prx subunit, which contains the Tyr and Phe residues of the YF motif. This specific interaction is postulated to slow the rate of formation of the intermolecular disulfide intermediate (SP-SR) during catalysis, enabling hyperoxidation to occur (6, 10). The changes in the Prx3 sequence in the proximity of the Cys-SRH residue and the YF motif have been postulated to alter the interaction with the rest of the Prx molecule, resulting in a decreased susceptibility to hyperoxidation (9, 16). Therefore, changes in both regions in Prx3 may result in its unique biochemical and physiological properties.
In this study, a panel of Prx2 and Prx3 variants and chimeras was analyzed to investigate the contribution of the observed sequence changes near the GGLG motif and the C terminus to hyperoxidation. Previous reports have used long time scales, nonreducing SDS-PAGE, two-dimensional PAGE, and Western blotting to monitor the hyperoxidation of Prx molecules (16, 20, 21). In contrast, the data presented herein was collected using a combination of rapid chemical quench and time-resolved ESI-TOF mass spectrometry methods to facilitate analysis under both denaturing and native conditions (22, 23). These improvements and the strategic use of Cys variants have enabled the direct observation of the Cys-SPOH intermediate during catalysis. Moreover, the stability of this intermediate in Prx2 is supported by the time-dependent formation of a putative Cys-sulfenamide (−SN) intermediate. Changing the C-terminal residues of Prx2 and Prx3 had the largest impact on resistance to hyperoxidation. The residues near the GGLG motif appeared to play a minimal role. Although Prx3 could be converted into a Prx2-like molecule and vice versa, the transformations were incomplete suggesting that additional residues, regions of the protein, and perhaps the equilibrium of the oligomeric states may also be involved in regulating the ease of hyperoxidation. Altogether, the data support that the unique C-terminal residues of Prx3 facilitate the rapid formation of the SP-SR disulfide, thus protecting the enzyme from inactivation.
EXPERIMENTAL PROCEDURES
Protein Expression and Purification
The human PRDX2 and PRDX3 genes were subcloned into the pET17 (Novagen) and pTYB21 (New England Biolabs), respectively, in a manner that ultimately resulted in the mature form of each protein without any additional N- or C-terminal residues. This was necessary as additional residues at either location could negatively impact catalytic activity. All Prx variants were created using the QuikChange site-directed mutagenesis method (Stratagene) with the appropriate primers. All proteins were expressed in BL21-Gold (DE3) Escherichia coli cells (New England Biolabs).
For the Prx2 variants (WT, PP → HA (P98H and P102A), C2S (C70S and C172S), CT (G175N, K177T, G179D, and D181P), PP → HA + CT), the E. coli cells were grown at 37 °C until an A600 of 0.8 and induced with 0.5 mm isopropyl 1-thio-β-d-galactopyranoside at 25 °C for 4–5 h. Given the absence of an affinity tag, the purification required four chromatographic steps. The cells were lysed in 100 ml of 20 mm HEPES, pH 7.9, 100 mm NaCl, 1 mm EDTA containing protease inhibitors (PMSF and benzamidine; both at 0.1 mm) using an Emulsiflex C5 homogenizer (Avestin, Inc.). This mixture was then centrifuged, and the supernatant was treated with 2.5% streptomycin sulfate followed by centrifugation. Ammonium sulfate was added to a final concentration of 20% to the supernatant, and the solution was filtered. This solution was loaded onto a phenyl-Sepharose high performance (Low Sub) column (GE Healthcare) and eluted with a 600-ml linear gradient to buffer without ammonium sulfate. The fractions corresponding to the Prx molecule, as determined by SDS-PAGE, were dialyzed into 20 mm Tris, pH 7.9, subsequently loaded onto a Q-Sepharose FF column (GE Healthcare), and eluted with a 600-ml linear gradient to 500 mm NaCl. The Prx fractions were pooled and dialyzed into 7 mm potassium phosphate, pH 7.0. The dialysate was subsequently loaded onto a CHT ceramic hydroxyapatite column (Bio-Rad) and eluted with a 600-ml linear gradient to 400 mm potassium phosphate, pH 7.0. The Prx2-containing fractions were concentrated to 5 ml and loaded onto a Superdex 200 column equilibrated with 20 mm HEPES, pH 7.5, 100 mm NaCl. The Prx fractions were pooled, concentrated, flash-frozen with liquid nitrogen, and stored at −80 °C until use. All Prx2 proteins were stored in a buffer without dithiothreitol (DTT) with the exception of Prx2C2S, which was stored in 20 mm HEPES, pH 7.5, 100 mm NaCl, and 10 mm DTT.
For the Prx3 variants (WT, HA → PP (H155P and A159P), C2S (C127S and C229S), CT (N232G, T234K, D236G, and P238D), HA → PP + CT), the E. coli cells were grown at 37 °C until an A600 of 0.8 and induced with 0.5 mm isopropyl 1-thio-β-d-galactopyranoside at 18 °C for 16 h. Expression from the pTYB21 vector results in the addition of an N-terminal chitin binding domain contained within an intein sequence, enabling the self-processing and removal of the chitin binding domain-intein tag after incubation with DTT. The cells were lysed in 150 ml of 20 mm Tris, pH 8.5, 500 mm NaCl, and 1 mm EDTA containing protease inhibitors (PMSF and benzamidine; both at 0.1 mm). The supernatant was loaded onto a chitin column (New England Biolabs) and extensively washed. Intein-mediated cleavage was initiated by equilibrating the column with 20 mm Tris, pH 8.5, 500 mm NaCl, 1 mm EDTA, and 50 mm DTT followed by a 40-h incubation at room temperature. The mature form of Prx3, residues 62–255, was eluted from the column, dialyzed against 20 mm Mes, pH 6.5, 1 mm DTT and subsequently loaded onto a Q-Sepharose FF column (GE Healthcare) and eluted with a 600-ml (0–50%) linear gradient to 1 m NaCl. The Prx3-containing fractions were concentrated and purified further using the Superdex 200 column, as described for the Prx2 variants. All Prx3 variants were stored in 20 mm HEPES, pH 7.5, 100 mm NaCl with the exception of Prx3 WT, which had 10 mm DTT.
Preparation of Samples for Mass Spectrometry Analysis
Immediately prior to analysis, the Prx variants were thawed and reduced with 10 mm DTT at room temperature for 30 min. DTT was removed by passing the protein solution through a Bio-Gel P6 spin column (Bio-Rad) pre-equilibrated with either 50 mm Tris buffer, pH 7.5, or 50 mm ammonium acetate, pH 6.9. Protein concentrations were determined, in duplicate at a minimum, using the absorbance at 280 nm and the theoretical extinction coefficients for each protein (Prx2 WT, 20,460 m−1 cm−1; Prx2-C2S, 21,430 m−1 cm−1; Prx2-CT, 21,555 m−1 cm−1; Prx2 PP → HA, 21,555 m−1 cm−1; Prx2 PP → HA + CT, 21,555 m−1 cm−1; Prx3 WT, 20,065 m−1 cm−1; Prx3-C2S, 19,940 m−1 cm−1; Prx3 CT, 20,065 m−1 cm−1; Prx3 HA → PP, 20,065 m−1 cm−1; Prx3 HA → PP + CT, 20,065 m−1 cm−1) calculated by the ExPASy ProtParam tool. The protein samples were immediately diluted and analyzed using the chemical quench and time-resolved methods described below.
Mass Spectrometry Data Collection and Analysis
For the chemical quench experiments, each DTT-free Prx protein was diluted further in 50 mm Tris, pH 7.5, to a final concentration of 50 μm. Oxidation was initiated by the addition of 0.8 eq of standardized H2O2 (ϵ240 = 43.6 m−1 cm−1) to the protein solution. The solution was incubated at 25 °C in a Thermomixer (Eppendorf) with gentle mixing. In control experiments, all conditions were the same as above except the same volume of H2O instead of H2O2 was used. At 30 s incubation time, the sample was applied to a Bio-Gel P6 spin column pre-equilibrated with 0.03% formic acid in H2O to quench the oxidation reaction. The flow-through was then used directly for ESI-TOF MS analysis.
In the comparative time-resolved experiments using the Prx2 and Prx3 variants, protein oxidation was performed using an on-line rapid-mixing setup. The experimental setup contained two Hamilton syringes as follows: one containing 100 μm DTT-free Prx variant, and the other containing 100 μm H2O2, both in 50 mm ammonium acetate, pH 6.9. The syringes were individually connected to separate fused silica capillaries and simultaneously advanced using a syringe pump (KD Scientific). The solutions were combined through a zero dead volume-mixing tee (Upchurch Scientific) into a connecting fused silica capillary (volume, 0.362 μl). The mixture was then continuously flowed into an ESI needle (volume, 1.269 μl) inserted in a stainless steel electrospray probe for ESI-TOF MS analysis. Varying flow rates were applied to achieve reaction time points lower than 30 s.
All ESI-TOF MS data were recorded in a positive ion mode on an Agilent MSD TOF system with the following settings: capillary voltage (VCap) 3500 V, nebulizer gas (N2) 30 pounds/inch2, drying gas (N2) 5.0 liter min−1; fragmentor 140 V; gas temperature 325 °C. The chemical quench samples were injected for analysis by ESI-TOF MS at a flow rate of 25 μl min−1 from a 250-μl syringe via a syringe pump. For the time-resolved experiments, the samples were injected as described above. The averaged MS spectra were deconvoluted using the Agilent MassHunter workstation software Version B.01.03. Data for the Prx2-C2S variant were fitted using SigmaPlot Version 11.0 (Systat Software Inc) and KinTek Explorer (KinTek Corp.) based on a simple kinetic model E + S ↔ EI; EI + S ↔ EP, where E is Prx-C2S; S is H2O2; EI is the Prx-C2S-SPOH, and EP is Prx-C2S-SPO2H.
RESULTS AND DISCUSSION
Hyperoxidation of Wild-type Prx2 and Prx3
Although human Prx2 and Prx3 exhibit second order rate constants of ∼107 m−1 s−1 with H2O2, these enzymes represent divergent 2-Cys Prx molecules with respect to their susceptibility to hyperoxidation of the catalytic Cys-SPH residue to Cys sulfinic acid (Cys-SPO2H) (24). To evaluate this difference, a panel of human Prx2 and Prx3 variants was expressed and purified from E. coli. Importantly, the expression construct for each protein was designed with the requirement that no affinity tags or additional N- and C-terminal residues remain at the final step of purification, as these can greatly influence the oligomeric state and peroxidase activity (8, 25). Although Prx2 was readily expressed and purified without affinity tags, Prx3 was more problematic requiring the screening of a variety of expression tags and an evaluation of their ease of removal by proteases. In the end, only an N-terminal chitin binding domain-intein fusion led to sufficient expression levels for all variants analyzed, resulting in a mature N terminus at residue 62 following DTT treatment.
Previous in vitro and cellular studies have used gel-based and Western blotting methods to monitor the hyperoxidation of Prx2 and Prx3 (16, 20, 21). Although these low resolution techniques do illustrate the differences in reactivity with H2O2, they have missed critical reaction intermediates that may shed light into the molecular mechanism of resistance to hyperoxidation in Prx3. Quantitative ESI-TOF mass spectrometry approaches were used in this study to dissect the appearance and disappearance of reaction intermediates (Fig. 1) associated with oxidation (Cys-SPOH, M + 16) and hyperoxidation (Cys-SPO2H, M + 32). A key feature of this approach has been to pre-reduce the samples with DTT and to desalt immediately prior to analysis. Moreover, all data presented herein were collected without the presence of DTT or other external reductant like thioredoxin in the reaction mixture. This simplification prevents the Prx molecule from cycling and enables partial-turnover analysis of Prx oxidation.
Reduced Prx2 or Prx3 (50 μm) was mixed with 0.8 eq of H2O2 at pH 7.5 and incubated for 30 s. The reaction was chemically quenched by passing the sample through a desalting column equilibrated with 0.03% formic acid in H2O and immediately analyzed by ESI-TOF mass spectrometry (Fig. 2). The addition of H2O2 to Prx2 results in the conversion of the reduced monomer (SH, M) to the hyperoxidized monomer (SO2H, M + 32) and two intermolecular disulfide-linked species, the oxidized (SS + SH, M + M − 2) and hyperoxidized (SS + SO2H, M + M + 30) dimers (Fig. 1; all theoretical and experimental mass values are given in Table 1). In contrast, the addition of H2O2 to Prx3 results in the same species but with more of the reduced (SH) monomer remaining.
FIGURE 2.

Susceptibility of wild-type Prx2 and Prx3 to hyperoxidation. Chemical quench and ESI-TOF MS were used to assess the oxidation state of each protein (50 μm) following treatment with 0.8 eq of H2O2 for 30 s at pH 7.5. The 1st column of panels shows the full spectra for Prx2 and Prx3 with and without H2O2 treatment. The panels to the right show a close-up view of the mass ranges encompassing the monomeric and dimeric species. See Fig. 1A for the abbreviations used for each species. All theoretical and experimental mass values (± S.D.) are given in Table 1; amu, atomic mass units.
TABLE 1.
Theoretical and experimental mass values for the different oxidation states of Prx2 and Prx3 variants
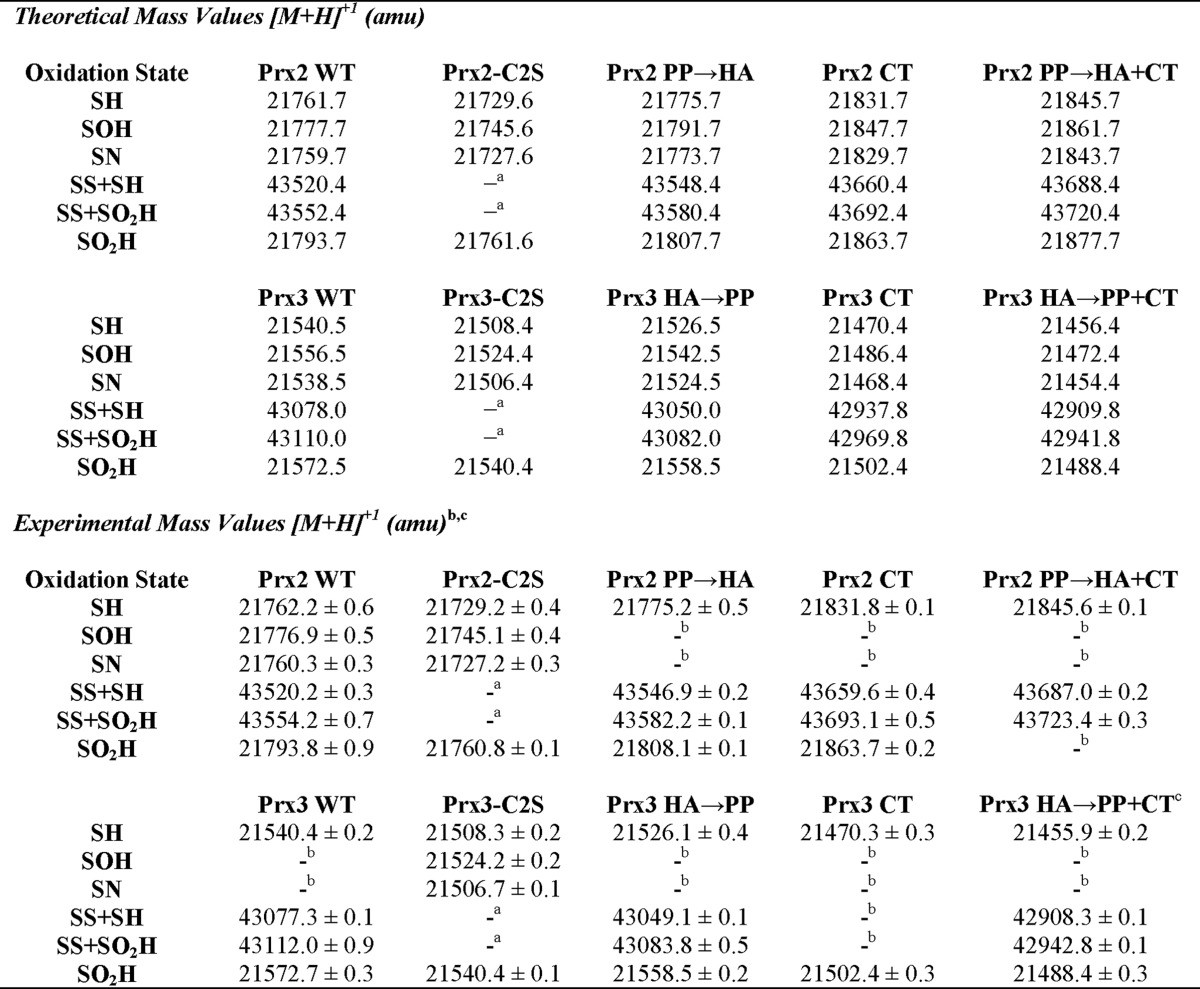
a The removal of the Cys-SRH residue by mutagenesis prevents the possibility of this species forming. See text for details.
b Species was not observed. Please see text for experimental details, as some species can only be captured with the time-resolved approach. Not all Prx2 and Prx3 variants were analyzed with the latter approach.
c All mass values were determined in triplicate except for Prx3 HA → PP + CT, which was performed in duplicate, due to the paucity of material available.
With the substoichiometric addition of H2O2, the presence of reduced monomer and monomeric and dimeric species containing the hyperoxidized Cys-SPO2H moiety for both Prx2 and Prx3 is counterintuitive. The Cys-SPH residue should react quickly with H2O2 to form the Cys-SPOH intermediate and then transition to the SP-SR dimeric species. Based on other biochemical and molecular modeling studies, however, there is evidence for one Cys-SPH residue to be more reactive than the other within the Prx homodimer (26–28). In addition, in order for the Cys-SPOH species to react with a second molecule of H2O2, the active site must be in the FF conformation (6). Thus, it appears that under these nonturnover reaction conditions a small subpopulation of Prx active sites maintains the Cys-SPOH intermediate in the FF conformation, leading to hyperoxidation.
In summary, these analyses of the wild-type (WT) proteins are consistent with cell-based and in vitro studies showing that Prx3 is more resistant to hyperoxidation (16, 20, 21). Moreover, the concentrations of Prx and H2O2 used were directly comparable with those found within cells (29, 30). These experiments demonstrate that hyperoxidation of Prx2 and Prx3 can occur on a physiologically relevant time scale without catalytic cycling when the concentration of H2O2 is similar to the amount of Prx protein. These observations also support the notion that the lifetime or stability of the Cys-SPOH intermediate is crucial to enable subsequent hyperoxidation, but this species has not been directly observed during Prx turnover before (10, 31).
Time-resolved ESI-TOF MS Analysis of Early Reaction Intermediates
The tracking of the formation of the Cys-SPOH species in Prxs was first studied using molecular probes specific for this functional group, including dimedone (32). Advances in the development of chemical probes have revolutionized the isolation and identification of other proteins that form a Cys sulfenic acid within cells exposed to a variety of stress conditions (6, 33–35). The reactivity of these probes is, however, not high enough to capture the transient Cys-SPOH intermediate during the Prx reaction cycle. The reported reaction rates vary from 0.003 to 1.65 min−1 at a saturating concentration of dimedone (36). Even when using the low pH chemical quench method and ESI-TOF MS at a 30-s time point (Fig. 2), the Cys-SPOH species was not captured for WT Prx2 and Prx3. Therefore, a more rapid analysis of the reaction intermediates is necessary.
Time-resolved ESI-TOF MS experiments, employing an on-line, rapid-mixing setup, were used to monitor the formation of the Cys-SPOH species for Prx2 and Prx3 during catalysis, i.e. under native conditions. In this approach, the Prx proteins were pre-reduced with DTT, desalted into a volatile buffer, and loaded into a Hamilton syringe. The samples were then mixed on-line in an equimolar ratio (100 μm each) with H2O2 at varying flow rates (10–80 μl/min) to achieve the acquisition of mass spectra at short reaction time points (1–15 s). With this 30-fold reduction of the reaction time scale, the detection of the Cys-SPOH intermediate at pH 7.5 was still not possible.
Decreasing the reaction pH to 6.9, however, enabled the detection of the Cys-SPOH species for wild-type (WT) Prx2 at 1.5 s (Fig. 3), even when the majority of the protein was present as the oxidized dimer (SS + SH), as expected. Importantly, under these reaction conditions and short time scale, the SS + SO2H species was not consistently observed. By 5.8 s the Cys-SPOH intermediate was consumed, and a new peak emerged with a mass consistent with the formation of a Cys sulfenamide (Cys-SPN, M − 2) (Table 1), a novel finding to our knowledge for human Prx proteins. At this point, we cannot determine whether the putative sulfenamide species exists in solution as a minor species or is a result of the dehydration process when the protein ion transitions to the gas phase during MS analysis. Nonetheless, the ability to capture this putative intermediate, even as a minor product, supports that the Cys-SPOH species is stabilized in Prx2.
FIGURE 3.

Time-resolved ESI-TOF MS analysis of wild-type Prx2 and Prx3 during catalysis. Each variant was treated with an equimolar concentration of H2O2 followed by the continuous analysis of reaction intermediates at pH 6.9. Left panel, representative deconvoluted spectra for Prx2 at different reaction time points. The full spectra and a close-up of the region around the monomer are shown. The series of shoulder peaks are consistent with the presence of Na+ adducts (+22 Da). See Table 1 for mass details for the different species. amu, atomic mass units. Right panel, full spectra for Prx3.
In contrast, only the reduced monomer (SH) and the oxidized dimer (SS + SH) were observed in the mass spectra for WT Prx3 in the 1.0–5.0-s reaction time range. The absence of the Cys-SPOH and sulfenamide intermediates for Prx3 supports that the lifetime of the Cys-SPOH intermediate is considerably shorter than that for Prx2, as a consequence of rapid intermolecular disulfide bond formation (SP-SR). Thus, the presence of the Cys-SRH residue and the ability to form the intermolecular SP-SR species appears to greatly impact the lifetime of the Cys-SPOH intermediate.
Hyperoxidation of Prx2 and Prx3 Cys Variants
To dissect the contribution of the Cys-SRH residue and SP-SR intermediate formation to the hyperoxidation of Prx2 and Prx3, the Cys-SRH residue and one other noncatalytic Cys residue were mutated to Ser (Prx2-C2S, C70S and C172S; Prx3-C2S, C127S and C229S; numbering scheme based on full-length gene sequence). These mutations leave only the Cys-SPH residue for each protein, and therefore the dimeric SP-SR species cannot form, and the potential for unwanted thiol-disulfide exchange reactions is removed.
An analysis of the variants at pH 7.5, with the addition of 2 eq of H2O2 for 30 s (Fig. 4), was performed using the chemical quench method coupled with ESI-TOF mass spectrometry. The Prx3-C2S variant remained in the reduced state while the Prx2-C2S variant was fully hyperoxidized, further highlighting the intrinsic differences between Prx2 and Prx3. This comparison clearly demonstrates that the mutation of the Cys-SRH residue has a significant impact on the reactivity of Prx3 with H2O2. One likely explanation is that the active site for the Prx3-C2S variant is predominantly in the locally unfolded conformation, thus preventing the reaction with H2O2. These observations are consistent with the decrease in hyperoxidation observed when mutating the Cys-SRH residue to Ser or Ala in other eukaryotic Prxs (37–39).
FIGURE 4.

Susceptibility of Prx2-C2S and Prx3-C2S to hyperoxidation. Chemical quench and ESI-TOF MS were used to assess the oxidation state of each protein (50 μm) following treatment with 2 eq of H2O2 for 30 s at pH 7.5. These Prx2 and Prx3 variants contain only the Cys-SPH residue and cannot form the intermolecular disulfide reaction intermediate. Therefore, the deconvoluted spectra focus on the monomeric species, as indicated. amu, atomic mass units.
The Prx2 and Prx3 Cys variants were also analyzed by time-resolved ESI-TOF MS at pH 6.9 to evaluate the formation of reaction intermediates. For Prx2-C2S, the addition of 1 eq of H2O2 resulted in the formation of Cys-SPOH species at 1.2 s (Fig. 5A). By 15 s, three species were present as follows: Cys-SPOH, Cys-SPN, and Cys-SPO2H. At the 600-s time point, the hyperoxidized species predominated. Additional time points were collected, and the relative abundance for each species to the total signal was converted to concentration to generate a plot (Fig. 5B) of the reduced, oxidized, and hyperoxidized species versus time. The intensities for the Cys-SPOH and Cys-SPN intermediates were combined, as the Cys-SPN intermediate can only form from the Cys-SPOH. A global fit of the data using KinTek Explorer was used to determine the following rate constants: kSH → SOH, 2.0 × 104 m−1 s−1; kSOH → SO2H, 1.1 × 103 m−1 s−1. A separate exponential fit to the formation of the Cys-SPO2H species yielded the kSH → SO2H rate constant of 9.2 × 102 m−1 s−1, consistent with the conversion of the Cys-SPOH intermediate to the Cys-SPO2H species being the rate-limiting step in Prx2-C2S hyperoxidation. Importantly, the kSH → SOH rate constant of ∼104 Prx2-C2S is consistent with the rate reported for turnover for the WT Prx2 (∼107 m−1 s−1), especially given the suboptimal pH conditions and the mutation of the Cys-SRH residue (24, 37–40).
FIGURE 5.

Time-resolved ESI-TOF MS analysis of the Prx2-C2S and Prx3-C2S variants during catalysis. A, representative deconvoluted spectra for Prx2-C2S at the indicated reaction time points. The protein was treated with an equimolar concentration of H2O2 (50 μm of each final) at pH 6.9 followed by the analysis of reaction mixture with ESI-TOF mass spectrometry. The spectra are focused on the following species: Cys-SPH, Cys-SPOH, Cys-SPO2H, and a putative Cys-sulfenamide (Cys-SPN) intermediate (Table 1). B, global kinetic modeling of the Prx2-C2S kinetic data. The plot shows the determined kinetic profiles for the –SPH and –SPO2H and the combined –SPOH/–SPN species, as the –SPN intermediate logically originates from the –SPOH species. C, deconvoluted spectra for Prx3-C2S treated with H2O2 for 600 s. amu, atomic mass units.
One caveat to the Prx2-C2S studies was the unanticipated observation of more oxidization than expected, considering the equimolar proportion of H2O2 added. It is unclear why this occurred for only this Prx2 variant and not others in this study, but it could have been formed during MS analysis. Nonetheless, the data for Prx2-C2S are consistent with the increased lifetime of the Cys-SPOH intermediate and the inability to form the normal SP-SR intermolecular disulfide. In marked contrast, the Cys-SPOH, Cys-SPN, and Cys-SPO2H species were observed at similar levels at 600 s for Prx3-C2S (Fig. 5C), in line with the amount of H2O2 added. Given the significantly lowered reactivity of the Prx3-C2S variant, the rates for the formation of the different oxidized and hyperoxidized species were not calculated.
An orthogonal analysis of the hyperoxidation of WT Prx2 and Prx3 was recently reported while our studies were in review (41). This analysis focused on determining the rate of hyperoxidation, kSOH → SO2H (k2 in their scheme), and the rate of disulfide formation, kSOH → SP-SR (k3 in their scheme). Our kSOH → SO2H value of 1.1 × 103 m−1 s−1, for Prx2-C2S at a suboptimal pH, is comparable with 1.2 × 104 m−1 s−1 for the WT enzyme. Moreover, our chemical quench and time-resolved MS data (Figs. 2 and 3) directly illustrate the rapid formation of the SP-SR intermediate for Prx3, consistent with the ∼10-fold higher kSOH → SP-SR rate for Prx3 (20 s−1 versus 2 s−1 for Prx2) and the oxidation of the Cys-SPOH species being the rate-limiting step for hyperoxidation. Taken together, the data support the importance of rate of formation of the SP-SR species in controlling susceptibility to hyperoxidation, as originally proposed by Wood et al. (10). As described below, a panel of Prx2 and Prx3 chimeras of the active site and C-terminal region, near the Cys-SRH residue, was evaluated in an effort to determine which residues unique to Prx3 may impart the unique resistance to hyperoxidation.
Hyperoxidation of C-terminal and GGLG Motif Chimeras of Prx2 and Prx3
As briefly described earlier, the packing of the C-terminal YF-containing helix against the GGLG motif (Fig. 1C) is a prominent feature of eukaryotic Prxs. This interaction and the resultant stabilization of the active site are thought to slow the rate of formation of the intermolecular SS intermediate during catalysis enabling hyperoxidation (6, 10). In fact, the mutation and truncation of the C terminus results in an increased resistance to hyperoxidation in other Prxs (37, 42–44). The appendage of a C terminus from a Prx molecule sensitive to hyperoxidation to one that is normally resistant can also result in an increase in sensitivity to hyperoxidation (43). Similar studies have not been performed with human Prx2 and Prx3 in an effort to address their differences in hyperoxidation.
A sequence alignment (Fig. 1B) of human Prx1–4 reveals that two Pro residues, Pro-98 and Pro-102 of Prx2, are substituted to His and Ala in Prx3, respectively. Their position next to the GGLG motif suggests that these Pro residues may be important for the positioning of this motif to interact with the C terminus of the adjacent Prx subunit. Four additional differences between Prx2 and Prx3 were identified adjacent to the Cys-SRH residue (16). In this region, Gly-175, Lys-177, Gly-179, and Asp-181 of Prx2 are substituted with Asn, Thr, Asp, and Pro in Prx3, respectively. A panel of Prx2 and Prx3 variants was generated where these sequence differences were swapped as a group to generate chimeras. The panel was evaluated using the same experimental conditions as for the WT proteins and using the chemical quench method. Importantly, these variants all contain the Cys-SRH residue and can therefore undergo normal catalytic cycling.
Following the addition of 0.8 eq H2O2 for 30 s, the Prx2 GGLG region chimera (Prx2 PP → HA) (Fig. 6A) had a comparable profile to WT Prx2, with prominent monomeric and dimeric species containing the Cys-SPO2H moiety. In contrast, the Prx2 C-terminal chimera (Prx2 CT) was more resistant to hyperoxidation, as indicated by the lack of formation of the Cys-SPO2H monomeric species and the presence of the SS + SH and SS + SO2H species. The combination of the variants, Prx2 PP → HA + CT, did not result in a further increase in protection from hyperoxidation. These observations support that the sequence changes near the GGLG motif of Prx2 do not influence hyperoxidation. The changing of the C-terminal residues of Prx2 to those of Prx3, however, resulted in a Prx3-like protein with an increased resistance to hyperoxidation.
FIGURE 6.

Susceptibility of Prx2 and Prx3 GGLG and C-terminal chimeras to hyperoxidation. A, Prx2 variants: WT, PP → HA (P98H and P102A); CT (G175N, K177T, G179D, and D181P); PP → HA + CT. B, Prx3 variants: WT, HA → PP (H155P and A159P); CT (N232G, T234K, D236G, and P238D); HA → PP + CT. Chemical quench ESI-TOF analyses were performed with 0.8 eq of H2O2 for 30 s at pH 7.5. A close-up of the mass range for the monomeric (left panel) and dimeric (right panel) species is presented within each panel (Table 1). amu, atomic mass units.
The analysis of the Prx3 HA → PP chimera revealed an increase in hyperoxidation, i.e. the monomeric and dimeric species containing the Cys-SPO2H moiety increased (Fig. 6B) over the WT protein. The Prx3 CT chimera was even more sensitive to hyperoxidation, as only the monomeric Cys-SO2H species was observed in addition to a complete loss of SS + SH and SS + SO2H species. The combination of the GGLG and CT variants, Prx3 HA → PP + CT, yielded a similar increase in the monomeric SO2H species and a small increase in the SS-containing species. It is unclear at this time how the combination of the two sets of mutations could lead to a compensatory effect. The analysis of the Prx2 and Prx3 chimeras supports that the C terminus of Prx3 is the primary determinant to the resistance of the WT enzyme to hyperoxidation and that the residues near the GGLG motif can modulate this resistance to some degree. Moreover, the lack of the dimeric SS + SH and SS + SO2H for the Prx3 CT variant suggests that the active site is stabilized in the FF conformation, causing the Cys-SPOH species to be readily hyperoxidized. It is interesting to note that none of the Prx2 and Prx3 chimeras exhibited a full transformation in their sensitivity or resistance to hyperoxidation. This finding suggests that other regions of the proteins and their dynamic oligomeric states may also influence the ease of hyperoxidation.
Potential Implications of Cys-Sulfenamide Formation
The observation of a putative Cys-SPN intermediate for WT Prx2 and Prx2-C2S (Figs. 3 and 5) supports a Cys-SPOH-Cys-SPN equilibrium that prolongs the lifetime of the Cys-SPOH intermediate and increases its susceptibility to hyperoxidation. However, the inability to observe the Cys-SPOH and Cys-SPN intermediates for WT Prx3 also supports the rapid formation of the SP-SR disulfide and the resistance of this protein to hyperoxidation.
The low abundance of the Prx2 Cys-SPN species, under the current reaction conditions, contrasts sharply with other proteins that utilize the Cys-SN intermediate, chiefly protein phosphatase PTP1B, the distal domain of receptor PTPα, and OhrR (45–48). For example, unequivocal electron density was observed for this intermediate in PTP1B and PTPα crystals treated with H2O2. For OhrR, treatment of the protein with cumene hydroperoxide in solution resulted in complete conversion to the sulfenamide, which was subsequently verified by a 2-Da shift in the intact protein and tryptic fragment masses, although the latter was still technically challenging given the reversibility of the adduct.
Although the limited formation of the Prx2 Cys-SN intermediate does raise some doubts concerning its biological relevance, an analysis of its potential likely structure is warranted and informative. For PTP1B, the five-membered Cys-SN intermediate occurs between Cys-215 and the backbone amide group of Ser-216. This intermediate is thought to protect Cys-215 from hyperoxidation and irreversible inactivation and to facilitate glutathionylation (45, 46). Thus, we were surprised to observe the Cys-SPN species for Prx2 because it is more sensitive to hyperoxidation than Prx3. It is certainly possible that the Cys-SPN intermediate of Prx2 is readily collapsed back to the Cys-SPOH species by the addition of a water molecule. This scenario would extend the lifetime of the Cys-SPOH species enabling hyperoxidation.
Inspection of the Prx2 active site (Fig. 1C) and the residues surrounding the Cys-51-SPH residue reveals that Prx2 would not be able to form a backbone-mediated Cys-SPN intermediate similar to PTP1B. Cys-51 is adjacent to the conserved amino acid Pro-52, which lacks an amide proton and cannot attack the sulfenic acid moiety. Based on studies with synthetic peptides, it is possible that the Cys-SPN formation in Prx2 is mediated through the amine groups of a Lys or Arg side chain (49). Arg-127, a conserved residue, is the only residue adjacent to Cys-51 that could be involved in Cys-SN formation. Additional solution and structural studies are clearly needed to establish the kinetic competence of the Cys-SN species in Prx2.
Conclusions
This study supports that human Prx3 is more resistant to hyperoxidation by H2O2 than Prx2, as a consequence of rapid intermolecular disulfide formation between the Cys-SPH and Cys-SRH residues. Residues adjacent to Cys-229-SRH of Prx3 (Asn-232, Thr-234, Asp-236, and Pro-238) are unique and influence the lifetime of the Cys-SPOH intermediate and SP-SR formation. These observations attest to the dynamic nature of the C terminus and its ability to modulate catalysis, hyperoxidation, and ultimately repair by sulfiredoxin (9, 10, 13). Given the highly oxidizing environment of the mitochondria, it makes sense that the Prx3 molecule would favor rapid SP-SR formation to protect the Cys-SPH residue. Nonetheless, hyperoxidation of Prx3 and its subsequent repair by sulfiredoxin does occur within the mitochondrion and plays a crucial role in adrenal steroidogenesis (5).
Acknowledgments
We thank Jill Clodfelter, Lauren Filipponi, and Lynnette Johnson for their technical expertise; Candice Summitt for work on the Prx2 CT variant; and Leslie Poole for critical reading of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grant R01 GM072866 from NIGMS (to W. T. L.) and Grant R01 CA136810 from NCI (to C. M. F.).
- Prx
- peroxiredoxin
- FF
- fully folded
- ESI
- electrospray ionization.
REFERENCES
- 1. Rhee S. G., Chae H. Z., Kim K. (2005) Peroxiredoxins: a historical overview and speculative preview of novel mechanisms and emerging concepts in cell signaling. Free Radic. Biol. Med. 38, 1543–1552 [DOI] [PubMed] [Google Scholar]
- 2. Song I. S., Kim H. K., Jeong S. H., Lee S. R., Kim N., Rhee B. D., Ko K. S., Han J. (2011) Mitochondrial peroxiredoxin III is a potential target for cancer therapy. Int. J. Mol. Sci. 12, 7163–7185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Järvelä S., Rantala I., Rodriguez A., Kallio H., Parkkila S., Kinnula V. L., Soini Y., Haapasalo H. (2010) Specific expression profile and prognostic significance of peroxiredoxins in grade II-IV astrocytic brain tumors. BMC Cancer 10, 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lee K. W., Lee D. J., Lee J. Y., Kang D. H., Kwon J., Kang S. W. (2011) Peroxiredoxin II restrains DNA damage-induced death in cancer cells by positively regulating JNK-dependent DNA repair. J. Biol. Chem. 286, 8394–8404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kil I. S., Lee S. K., Ryu K. W., Woo H. A., Hu M. C., Bae S. H., Rhee S. G. (2012) Feedback control of adrenal steroidogenesis via H2O2-dependent, reversible inactivation of peroxiredoxin III in mitochondria. Mol. Cell 46, 584–594 [DOI] [PubMed] [Google Scholar]
- 6. Hall A., Nelson K., Poole L. B., Karplus P. A. (2011) Structure-based insights into the catalytic power and conformational dexterity of peroxiredoxins. Antioxid. Redox. Signal. 15, 795–815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wood Z. A., Poole L. B., Hantgan R. R., Karplus P. A. (2002) Dimers to doughnuts: redox-sensitive oligomerization of 2-cysteine peroxiredoxins. Biochemistry 41, 5493–5504 [DOI] [PubMed] [Google Scholar]
- 8. Parsonage D., Youngblood D. S., Sarma G. N., Wood Z. A., Karplus P. A., Poole L. B. (2005) Analysis of the link between enzymatic activity and oligomeric state in AhpC, a bacterial peroxiredoxin. Biochemistry 44, 10583–10592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lowther W. T., Haynes A. C. (2011) Reduction of cysteine sulfinic acid in eukaryotic, typical 2-Cys peroxiredoxins by sulfiredoxin. Antioxid. Redox. Signal. 15, 99–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wood Z. A., Poole L. B., Karplus P. A. (2003) Peroxiredoxin evolution and the regulation of hydrogen peroxide signaling. Science 300, 650–653 [DOI] [PubMed] [Google Scholar]
- 11. Rhee S. G., Woo H. A., Kil I. S., Bae S. H. (2012) Peroxiredoxin functions as a peroxidase and a regulator and sensor of local peroxides. J. Biol. Chem. 287, 4403–4410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ray P. D., Huang B. W., Tsuji Y. (2012) Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal. 24, 981–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jönsson T. J., Johnson L. C., Lowther W. T. (2008) Structure of the sulphiredoxin-peroxiredoxin complex reveals an essential repair embrace. Nature 451, 98–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Woo H. A., Jeong W., Chang T. S., Park K. J., Park S. J., Yang J. S., Rhee S. G. (2005) Reduction of cysteine sulfinic acid by sulfiredoxin is specific to 2-Cys peroxiredoxins. J. Biol. Chem. 280, 3125–3128 [DOI] [PubMed] [Google Scholar]
- 15. Jeong W., Bae S. H., Toledano M. B., Rhee S. G. (2012) Role of sulfiredoxin as a regulator of peroxiredoxin function and regulation of its expression. Free Radic. Biol. Med. 53, 447–456 [DOI] [PubMed] [Google Scholar]
- 16. Cox A. G., Pearson A. G., Pullar J. M., Jönsson T. J., Lowther W. T., Winterbourn C. C., Hampton M. B. (2009) Mitochondrial peroxiredoxin 3 is more resilient to hyperoxidation than cytoplasmic peroxiredoxins. Biochem. J. 421, 51–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Matsushima S., Ide T., Yamato M., Matsusaka H., Hattori F., Ikeuchi M., Kubota T., Sunagawa K., Hasegawa Y., Kurihara T., Oikawa S., Kinugawa S., Tsutsui H. (2006) Overexpression of mitochondrial peroxiredoxin-3 prevents left ventricular remodeling and failure after myocardial infarction in mice. Circulation 113, 1779–1786 [DOI] [PubMed] [Google Scholar]
- 18. Nonn L., Berggren M., Powis G. (2003) Increased expression of mitochondrial peroxiredoxin-3 (thioredoxin peroxidase-2) protects cancer cells against hypoxia and drug-induced hydrogen peroxide-dependent apoptosis. Mol. Cancer Res. 1, 682–689 [PubMed] [Google Scholar]
- 19. Schröder E., Littlechild J. A., Lebedev A. A., Errington N., Vagin A. A., Isupov M. N. (2000) Crystal structure of decameric 2-Cys peroxiredoxin from human erythrocytes at 1.7 Å resolution. Structure 8, 605–615 [DOI] [PubMed] [Google Scholar]
- 20. Chevallet M., Wagner E., Luche S., van Dorsselaer A., Leize-Wagner E., Rabilloud T. (2003) Regeneration of peroxiredoxins during recovery after oxidative stress: only some overoxidized peroxiredoxins can be reduced during recovery after oxidative stress. J. Biol. Chem. 278, 37146–37153 [DOI] [PubMed] [Google Scholar]
- 21. Woo H. A., Chae H. Z., Hwang S. C., Yang K. S., Kang S. W., Kim K., Rhee S. G. (2003) Reversing the inactivation of peroxiredoxins caused by cysteine sulfinic acid formation. Science 300, 653–656 [DOI] [PubMed] [Google Scholar]
- 22. Li Z., Sau A. K., Furdui C. M., Anderson K. S. (2005) Probing the role of tightly bound phosphoenolpyruvate in Escherichia coli 3-deoxy-d-manno-octulosonate 8-phosphate synthase catalysis using quantitative time-resolved electrospray ionization mass spectrometry in the millisecond time range. Anal. Biochem. 343, 35–47 [DOI] [PubMed] [Google Scholar]
- 23. Li Z., Sau A. K., Shen S., Whitehouse C., Baasov T., Anderson K. S. (2003) A snapshot of enzyme catalysis using electrospray ionization mass spectrometry. J. Am. Chem. Soc. 125, 9938–9939 [DOI] [PubMed] [Google Scholar]
- 24. Nagy P., Karton A., Betz A., Peskin A. V., Pace P., O'Reilly R. J., Hampton M. B., Radom L., Winterbourn C. C. (2011) Model for the exceptional reactivity of peroxiredoxins 2 and 3 with hydrogen peroxide: a kinetic and computational study. J. Biol. Chem. 286, 18048–18055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cao Z., Bhella D., Lindsay J. G. (2007) Reconstitution of the mitochondrial PrxIII antioxidant defence pathway: general properties and factors affecting PrxIII activity and oligomeric state. J. Mol. Biol. 372, 1022–1033 [DOI] [PubMed] [Google Scholar]
- 26. Salsbury F. R., Jr., Yuan Y., Knaggs M. H., Poole L. B., Fetrow J. S. (2012) Structural and electrostatic asymmetry at the active site in typical and atypical peroxiredoxin dimers. J. Phys. Chem. B 116, 6832–6843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yuan Y., Knaggs M., Poole L., Fetrow J., Salsbury F., Jr. (2010) Conformational and oligomeric effects on the cysteine pKa of tryparedoxin peroxidase. J. Biomol. Struct. Dyn. 28, 51–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Budde H., Flohe L., Hecht H. J., Hofmann B., Stehr M., Wissing J., Lunsdorf H. (2003) Kinetics and redox-sensitive oligomerisation reveal negative subunit cooperativity in tryparedoxin peroxidase of Trypanosoma brucei. Biol. Chem. 384, 619–633 [DOI] [PubMed] [Google Scholar]
- 29. Halliwell B., Clement M. V., Long L. H. (2000) Hydrogen peroxide in the human body. FEBS Lett. 486, 10–13 [DOI] [PubMed] [Google Scholar]
- 30. Winterbourn C. C., Hampton M. B. (2008) Thiol chemistry and specificity in redox signaling. Free Radic. Biol. Med. 45, 549–561 [DOI] [PubMed] [Google Scholar]
- 31. Claiborne A., Yeh J. I., Mallett T. C., Luba J., Crane E. J., 3rd, Charrier V., Parsonage D. (1999) Protein-sulfenic acids: diverse roles for an unlikely player in enzyme catalysis and redox regulation. Biochemistry 38, 15407–15416 [DOI] [PubMed] [Google Scholar]
- 32. Ellis H. R., Poole L. B. (1997) Novel application of 7-chloro-4-nitrobenzo-2-oxa-1,3-diazole to identify cysteine sulfenic acid in the AhpC component of alkyl hydroperoxide reductase. Biochemistry 36, 15013–15018 [DOI] [PubMed] [Google Scholar]
- 33. Qian J., Wani R., Klomsiri C., Poole L. B., Tsang A. W., Furdui C. M. (2012) A simple and effective strategy for labeling cysteine sulfenic acid in proteins by utilization of β-ketoesters as cleavable probes. Chem. Commun. 48, 4091–4093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wani R., Qian J., Yin L., Bechtold E., King S. B., Poole L. B., Paek E., Tsang A. W., Furdui C. M. (2011) Isoform-specific regulation of Akt by PDGF-induced reactive oxygen species. Proc. Natl. Acad. Sci. U.S.A. 108, 10550–10555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Paulsen C. E., Truong T. H., Garcia F. J., Homann A., Gupta V., Leonard S. E., Carroll K. S. (2012) Peroxide-dependent sulfenylation of the EGFR catalytic site enhances kinase activity. Nat. Chem. Biol. 8, 57–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Klomsiri C., Nelson K. J., Bechtold E., Soito L., Johnson L. C., Lowther W. T., Ryu S. E., King S. B., Furdui C. M., Poole L. B. (2010) Use of dimedone-based chemical probes for sulfenic acid detection evaluation of conditions affecting probe incorporation into redox-sensitive proteins. Methods Enzymol. 473, 77–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cao Z., Tavender T. J., Roszak A. W., Cogdell R. J., Bulleid N. J. (2011) Crystal structure of reduced and of oxidized peroxiredoxin IV enzyme reveals a stable oxidized decamer and a nondisulfide-bonded intermediate in the catalytic cycle. J. Biol. Chem. 286, 42257–42266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yang K. S., Kang S. W., Woo H. A., Hwang S. C., Chae H. Z., Kim K., Rhee S. G. (2002) Inactivation of human peroxiredoxin I during catalysis as the result of the oxidation of the catalytic site cysteine to cysteine-sulfinic acid. J. Biol. Chem. 277, 38029–38036 [DOI] [PubMed] [Google Scholar]
- 39. Jara M., Vivancos A. P., Calvo I. A., Moldón A., Sanso M., Hidalgo E. (2007) The peroxiredoxin Tpx1 is essential as a H2O2 scavenger during aerobic growth in fission yeast. Mol. Biol. Cell 18, 2288–2295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Winterbourn C. C. (2008) Reconciling the chemistry and biology of reactive oxygen species. Nat. Chem. Biol. 4, 278–286 [DOI] [PubMed] [Google Scholar]
- 41. Peskin A. V., Dickerhof N., Poynton R. A., Paton L. N., Pace P. E., Hampton M. B., Winterbourn C. C. (2013) Hyperoxidation of peroxiredoxins 2 and 3: rate constants for the reactions of the sulfenic acid of the peroxidatic cysteine. J. Biol. Chem. 288, 14170–14177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Koo K. H., Lee S., Jeong S. Y., Kim E. T., Kim H. J., Kim K., Song K., Chae H. Z. (2002) Regulation of thioredoxin peroxidase activity by C-terminal truncation. Arch. Biochem. Biophys. 397, 312–318 [DOI] [PubMed] [Google Scholar]
- 43. Sayed A. A., Williams D. L. (2004) Biochemical characterization of 2-Cys peroxiredoxins from Schistosoma mansoni. J. Biol. Chem. 279, 26159–26166 [DOI] [PubMed] [Google Scholar]
- 44. Wang X., Wang L., Wang X., Sun F., Wang C. C. (2012) Structural insights into the peroxidase activity and inactivation of human peroxiredoxin 4. Biochem. J. 441, 113–118 [DOI] [PubMed] [Google Scholar]
- 45. Salmeen A., Andersen J. N., Myers M. P., Meng T. C., Hinks J. A., Tonks N. K., Barford D. (2003) Redox regulation of protein tyrosine phosphatase 1B involves a sulphenyl-amide intermediate. Nature 423, 769–773 [DOI] [PubMed] [Google Scholar]
- 46. van Montfort R. L., Congreve M., Tisi D., Carr R., Jhoti H. (2003) Oxidation state of the active-site cysteine in protein tyrosine phosphatase 1B. Nature 423, 773–777 [DOI] [PubMed] [Google Scholar]
- 47. Lee J. W., Soonsanga S., Helmann J. D. (2007) A complex thiolate switch regulates the Bacillus subtilis organic peroxide sensor OhrR. Proc. Natl. Acad. Sci. U.S.A. 104, 8743–8748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yang J., Groen A., Lemeer S., Jans A., Slijper M., Roe S. M., den Hertog J., Barford D. (2007) Reversible oxidation of the membrane distal domain of receptor PTPα is mediated by a cyclic sulfenamide. Biochemistry 46, 709–719 [DOI] [PubMed] [Google Scholar]
- 49. Fu X., Mueller D. M., Heinecke J. W. (2002) Generation of intramolecular and intermolecular sulfenamides, sulfinamides, and sulfonamides by hypochlorous acid: a potential pathway for oxidative cross-linking of low-density lipoprotein by myeloperoxidase. Biochemistry 41, 1293–1301 [DOI] [PubMed] [Google Scholar]