

Abstract
In the 1960s, I developed methods for directly visualizing DNA and DNA-protein complexes using an electron microscope. This made it possible to examine the shape of DNA and to visualize proteins as they fold and loop DNA. Early applications included the first visualization of true nucleosomes and linkers and the demonstration that repeating tracts of adenines can cause a curvature in DNA. The binding of DNA repair proteins, including p53 and BRCA2, has been visualized at three- and four-way junctions in DNA. The trombone model of DNA replication was directly verified, and the looping of DNA at telomeres was discovered.
Keywords: Chromatin, DNA, Electron Microscopy (EM), Telomeres, Nucleotide
Alaska Was a Territory
Anchorage, Alaska, in the 1950s was a rapidly growing territorial community of 15,000 residents. A single high school provided excellent public education for the town and the two nearby military bases. My family had moved to Anchorage in the late 1940s. With the post-war influx of new residents, Anchorage did not offer ready built housing. We began with a shell of a house lacking indoor plumbing until my father was able to install an electric well pump and a bathroom. The fall moose-hunting season provided meat and a time for my father and me to be together. However, over the years, we both became more interested in photography, laying the basis for a lifelong delight in generating striking photographs from nature and the electron microscope.
The Alaskan mindset of constructing what you need from your collection of scrap parts has served me well over my scientific career. High school science fairs had come to Alaska, and in 1960, during my senior year, I constructed a “nuclear reactor” from a small water heater. This was based on reprints ordered for 25¢ apiece from the Los Alamos National Laboratory. The radiation source was radium obtained from a local aircraft repair shop that used it to paint aircraft dials. I mail-ordered beryllium and mixed it with the radium to generate a neutron source. How strong this source was I never knew, but carrying something like this on a commercial flight from Anchorage to Indianapolis for the National Science Fair would not likely be allowed in these times.
Occidental College in Los Angeles from 1960 to 1964 provided a gentle entry into a much more sophisticated world for a student from “the brush” wishing to study physics. At Christmas time, between my junior and senior years, I happened to meet a scientist from the Oak Ridge National Laboratory as I waited for the fog to clear at the Seattle airport. He arranged a summer internship with his colleague Dick Setlow, who was in the process of discovering how thymine dimers were excised from DNA. I was becoming uneasy that experimental physics was dominated by very large group efforts, and I saw Dick working by himself in the lab, an activity that I still emulate as much as possible. He convinced me that biophysics was a more exciting path, and his recommendation led to me, as a physics student with little genetics or biology background, being admitted to graduate school at the California Institute of Technology (Caltech).
Graduate School and the Development of a Useful Electron Microscopy Technique
James Bonner at Caltech was one of the first to tackle chromatin, and in 1965, I joined his group. Jerry Vinograd and Norman Davidson had built large active labs using the new “Kleinschmidt” method for visualizing DNA (1). This rather unlikely method came from experiments in Germany on how denatured proteins alter surface tension. It was found by accident that when DNA is mixed with a small basic protein, such as cytochrome c, and spread on an air-water interface, the denatured protein binds cooperatively along the DNA to generate a thick protein sheath. This coating makes the DNA thick and stiff, which, together with the surface tension forces, spreads the DNA out. The film with the embedded DNA is picked up with a plastic-coated electron microscope grid and crudely metal-coated in a vacuum prior to examination under the electron microscope. Students and fellows included Ron Davis, Baldomero Olivera, Phil Sharp, and David Clayton and later, Tom Broker and Louise Chow. They became masters of this technique and used it to reveal not only that many plasmid and phage DNAs are circular but that some circles are highly coiled about themselves (supertwisted) (2). Replicating DNAs in the form of rolling circles and bubbles were visualized, and David Clayton began his studies of how human and mouse mitochondrial DNAs replicate (3). The use of the nucleic acid denaturant formamide to open up the secondary structure in single-stranded DNA by Ron Davis, Martha Simon, and Norman Davidson (4) subsequently allowed Louise Chow and Tom Broker to utilize EM in the adenovirus studies carried out by Rich Roberts' group at Cold Spring Harbor Laboratory (5) and Claire Moore (later, my first graduate student) to use similar EM methods with Susan Berget and Phil Sharp at the Massachusetts Institute of Technology (MIT) (6) in their co-discovery of RNA splicing.
With a background in physics, I felt comfortable around vacuum systems and electron microscopes, but the simple Kleinschmidt method was of little use for chromatin work because the structural questions related to how the histones and non-histone proteins interacted with and bound to DNA. The cytochrome c coating was so thick that it obscured any proteins bound along the DNA, and the surface-spreading forces were considerable. My Ph.D. thesis work became an effort to develop methods that would allow the visualization of DNA and DNA-protein complexes directly without cytochrome coating. The solution was to use much thinner and stronger supports provided by thin carbon foils. The granular platinum-palladium metal used for shadow casting was replaced by tungsten after empirical testing and shown later to provide the highest resolution of any metal due to its high melting point. Finally, the samples had to be dehydrated and washed carefully to remove any residual salts that would hide the very thin DNA fiber. A graded series of ethanol/water solutions was settled on for washing and dehydration but had clear drawbacks. Rapid freezing and freeze-drying seemed to offer better three-dimensional structural preservation, and Max Delbrück suggested I shoot the samples into a pot of liquid helium. Although this was far from feasible at that time, his suggestion brings to mind John Heuser's fast freeze-deep etch method, which has revealed such fine structure of cells (7) and which is not that far from Max's idea. My own freeze-drying method failed then due to the lack of clean vacuum systems, something that I have just solved.
Looking back at our images of chromatin from that time, many examples of similarly sized particles strung along bare DNA can be found. However, I was not fixing the material with formaldehyde or glutaraldehyde to protect it from the dehydration steps involving graded ethanol series. As we know now, exposure to ethanol disrupts much of the nucleosomal structure. Furthermore, the long-standing model of a smooth, coiled chromatin fiber was so entrenched that without much more compelling images, combined with a biochemical backup, the particulate nature of the chromatin remained overlooked.
Two Caltech graduate student friends, Regis Kelly and Joel Huberman, had moved to Arthur Kornberg's lab at Stanford University. Searching for a defined DNA-protein complex to test my new EM methods, I heard Joel talk about his work on Escherichia coli DNA polymerase I. Joel was able to make synthetic DNAs containing binding sites (nicks with free 3′ termini) spaced regularly along the DNA by annealing 200-nucleotide polydeoxythymine oligonucleotides to 4000-nucleotide polydeoxyadenine oligonucleotides. This seemed the ideal test sample. It took some tries to find that EDTA was required to keep the polymerase bound to the nicks, after which we obtained what still remains my favorite micrograph (Fig. 1). Fortunately, the binding of polymerase was tenacious and not disturbed by the drying steps. This was the first visualization of a defined DNA-protein complex (8) and showed, as Arthur stated, that “EM could be used to do biochemistry.”
FIGURE 1.
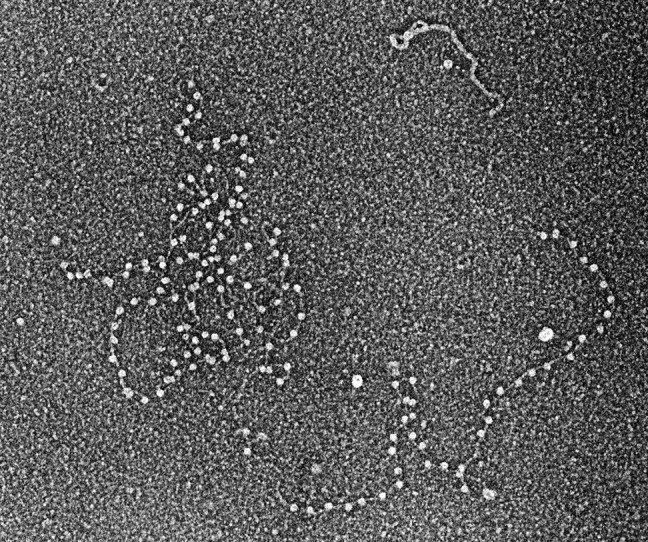
DNA polymerase I bound to DNA. DNA containing evenly spaced nicks to which E. coli DNA polymerase I bound was prepared for EM by rotary shadow casting with tungsten (micrograph circa 1968) (8).
Stanford Biochemistry Department: A Remarkable Time
Following the lead of Joel and Regis, in 1970, I joined Arthur's lab as a postdoctoral fellow and then stayed an additional three years in an independent position working on projects of my own and ones with Arthur and Paul Berg. During this time, I added fixation regimes with formaldehyde and glutaraldehyde and discovered that a mixture of salts, including spermidine, provided very high efficiency binding of the DNA complexes to the carbon foils. These changes, combined with small gel exclusion columns to remove free proteins, established the basic methods we use today, which allow one to visualize duplex DNA as small as 150 bp and up to 20 kb or more. Proteins as small as 30 kDa in mass can be seen bound along the length of a DNA strand.
Arthur's work on E. coli DNA replication had advanced to the stage where EM was able to contribute significantly. The ability to see the major steps in the in vitro replication cycle of the bacteriophage M13 catalyzed by the purified proteins provided a basis for new models that showed that as the nascent single strand of DNA was spun off the circular double-stranded M13 template, the end of the newly synthesized strand was held back at the replication complex as it transited the circle (9). The theme of rolling loops and circles would be reflected in later studies of the trombone model and the discovery of telomere loops (t-loops). These studies also generated a library of images for Arthur's books.
This period was an exciting time to be in the Stanford biochemistry department. DNA enzymology, carried out by members of the department, including Arthur, Paul, Bob Lehman, and Dale Kaiser, had generated a new understanding of how to manipulate DNA. This was used by Jackson, Symons, and Berg (10), and Lobban and Kaiser (11) to show that pieces of DNA from different organisms could be stuck together to create new recombinant molecules. For the students and fellows present at the birth of recombinant DNA, this must remain a singular time in their lives. For me, it was a time to establish a close attachment to Arthur and an active collaboration that lasted unchecked for close to forty years, beginning with the DNA polymerase I work at Caltech and continuing with his love for polyphosphates in his later years. Indeed, at Stanford, I was surprised by how excited Arthur was when I showed him images of polyphosphate bodies in lysed E. coli cells (Fig. 2). I later learned that when he and his wife, Sylvy Kornberg, were at Washington University in St. Louis, she had studied polyphosphate synthesis. Bob Lehman and I established a close friendship that later turned into a series of collaborative projects and that continues today with discussions of herpes virus DNA replication.
FIGURE 2.

The author (left) with Arthur Kornberg (circa 1975).
Paul Berg's lab had moved on to studies of SV40 virus, and it had been noted by Norman Salzman and his group at the National Institutes of Health (NIH) that the viral DNA in the nucleus was bound by cellular histones (12). The DNA itself was a double-stranded circle and, at that time, was one of the few DNAs whose size was well established (5200 bp). Clearly, if this complex could be isolated from infected cells, it would provide a unique miniature chromosome that could be imaged by EM. The long-standing model in which chromatin fibers were considered to be a continuous coil was being questioned by several studies suggesting the presence of small repeating units. Don and Ada Olins obtained EM images of fixed chromatin that showed tiny beads on very long DNA threads (13). However, there was no information as to the amount of DNA in the beads, and due to the centrifugation of the chromatin onto the EM grid, the length of DNA between the beads was often rather long and variable. A paper by Hewish and Burgoyne from Australia on chromatin that had undergone self-digestion revealed a 200-bp DNA repeat (14), but the structural basis for this repeat was unclear.
Working with help from the Berg group, it was possible to isolate the SV40 chromatin complexes on sucrose gradients using a method developed by Hall, Meinke, and Goldstein (15). Imaging, using methods that included aldehyde fixation, revealed a relaxed circular chain of beads joined by short DNA linkers (Fig. 3) (16). The particles were large and of the same size, and the linkers were uniform in length. Each complex contained 21 particles. Simple division showed that the repeating unit of one bead plus one linker was just over 200 bp, with the linkers consisting of ∼40 bp of protein-free DNA. This was the first clear visualization of a nucleosome where the size agreed with the repeat that Hewish and Burgoyne had observed. The relaxed nature of the chain of 21 beads and the subsequent release of supertwisted DNA upon removal of the histones further linked histone assembly with the generation of supertwisting. Subsequent work from Pierre Chambon's lab, now at the National Center for Scientific Research in France, verified this discovery (17).
FIGURE 3.
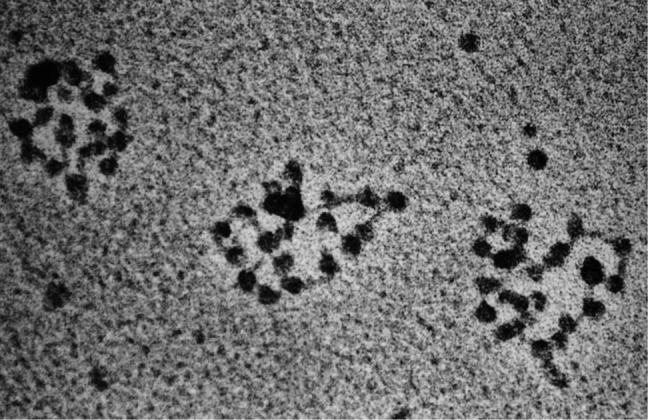
SV40 minichromosomes. SV40 minichromosomes were isolated from cultured monkey cells (16) and prepared for EM (micrograph taken in 1974).
These findings led to an explosion of chromatin studies spurred by Roger Kornberg and Jean Thomas' demonstration that the nucleosome contains two each of the four core histones (18); Luger's x-ray structure of the nucleosome, which showed how the histones are arranged (19); and the recent discovery of histone tail modifications (20). Despite these seminal discoveries, we understand little about the coiling of the nucleosomal chains into higher order structures in the nuclei of actively growing cells and even less about the packaging of DNA in bacterial cells. These questions remain a major challenge for EM in the future.
Importance of a Homologous 3′-End
Chapel Hill, North Carolina, and the Research Triangle Park in the 1970s were just beginning a spectacular growth in molecular biology. Paul Modrich, a close friend and student of Bob Lehman, had moved to Duke University, and three friends from Caltech, Clyde Hutchison III, Marshall Edgell, and John Newbold, were at the University of North Carolina (UNC). Joseph Pagano, whose work on SV40 was well known, offered me a position in his new UNC Lineberger Comprehensive Cancer Center. This environment and the chance to start my own laboratory were opportunities I could not turn down.
Not long after my move to UNC in 1978, the in vitro study of homologous recombination was made possible by Weinstock, McEntee, and Lehman's demonstration that the product of the E. coli recA gene, the RecA protein, could carry out the most fundamental step in recombination, the annealing of complementary DNA strands (21). Furthermore, they showed RecA could catalyze the insertion of a single-stranded DNA into a homologous double-stranded DNA at a site of homology in an ATP-dependent manner (22). These seminal discoveries led to decades of study of the molecular mechanisms of homologous recombination by many groups. Having the proteins available in pure form, now a given in our field, relied on some of the earliest developments in cloning and protein overproduction. In addition to the work in the Lehman lab, the work was done by Aziz Sancar and his colleagues at Yale University, who also cloned the gene for RecA, as well as the E. coli single-strand binding protein (SSB) (23, 24). In some ways, the T4 bacteriophage system was easier to work with because its RecA counterpart (UvsX) and gene 32 protein (the T4 SSB) drive the same reactions and are made in abundance in T4-infected bacteria.
These discoveries presented a virgin field in which EM could be a lead methodology in the analysis of reaction mechanisms. Unlike replication and translation, where single monomers of the polymerases are able to catalyze the synthesis of new RNA or DNA, homologous recombination reactions are driven by very large helical protein assemblies built from hundreds of monomers of RecA and SSB proteins or, in the T4 system, UvsX and gene 32 proteins. These protein machines are easily visualized by EM but present great difficulties to biochemists used to simple assays incorporating radiolabeled nucleoside triphosphates. Furthermore, recombination reactions may reshuffle thousands of base pairs of DNA but do not create new ones. This area provided a wealth of work for eager students and fellows. Our colleague Gunna Christiansen, with whom I had worked at Stanford, returned from Denmark for a sabbatical stay to help in these studies.
Key findings included the demonstration by Jim Register in our lab that RecA polymerizes from 5′ to 3′ on single-stranded DNA (25), an observation easily demonstrated using EM but which would have been difficult to show using only biochemical assays. These reactions were shown to begin with a labile association of two DNAs at sites of homology termed paranemic joints. Here, the single-stranded partner is covered by a helical sheath of the RecA protein. Such joints are unstable when the proteins are removed and thus difficult to demonstrate by simple biochemical means. If a free DNA end is present, the paranemic joints transition to stable plectonemic joints, which can be identified by gel electrophoretic means. Gunna Christiansen's EM study provided the proof of paranemic joining and showed that the double-stranded DNA partner in the joint is topologically unwound (26). It was possible for Gunna and Jim to visualize the process in which a single-stranded DNA with a free 3′-end homologous to a region in a double-stranded DNA invades at the site of homology and then initiates a strand-transfer reaction (27). Lorelei Harris in our lab purified the T4 UvsX and UvsY proteins from T4-infected bacteria and carried out work parallel to the RecA studies, bringing the T4 system into the recombination fold (28). As shown by our studies and parallel EM work by Andrzej Stasiak, Theo Koller, and colleagues (29) in Europe, strand transfer occurs within the large helical filament formed along the single-stranded DNA by the RecA protein. Thus, there was a clear understanding of the importance of single-stranded DNAs with 3′-ends and their ability to catalyze strand invasion at sites of homology, a rule later crucial in thinking about telomeres.
The EM work of Stasiak provided lively competition and confirmation of our mutual findings. Ed Egelman at the University of Virginia developed his approaches of image reconstruction of helical assemblies on the RecA filaments (30), which led to a highly productive career. It is fair to say that EM helped lay a clear understanding of the mechanism of strand transfer by these proteins. This understanding provided a basis for biochemical studies of many groups and laid the foundation for later studies of the mammalian Rad51 protein and its partners. Many questions remain, including the nature of the co-aggregates of RecA and DNA that drive the search for homology over millions of base pairs of sequences.
Nucleotide Triplets
In 1982, Paul Englund's group at The Johns Hopkins University School of Medicine and Don Crothers' group at Yale University reported the discovery of an unusual segment present in the mitochondrial minicircle DNA in trypanosomes (31). This element had the uncomfortable property of appearing to be four times larger when electrophoresed on acrylamide instead of agarose gels. The groups of Englund and Crothers raised the possibility that this seemingly larger size might be caused by a static curvature of the DNA, resulting from the repeating blocks of four to six adenines phased with the helical repeat over the 220-bp length of the DNA. Direct EM visualization, done in with collaboration with Paul, verified this possibility (32). Although there is no question that the elegant phasing experiments of Wu and Crothers (33) had proven the point, images of fields of 223-bp DNA fragments bent into almost perfect circles provided important visual confirmation. We went on to show additional features of these elements, including the fact that they orient domains in supertwisted DNA (34). These studies also provided an entry into our using EM to study DNAs with unusual repeating triplet sequences.
Work with triplet DNAs began with the proposal by Bob Wells, then at the University of Alabama, that we examine DNAs containing very long runs of repeating (CTG)n, which are found in patients with myotonic dystrophy; repeating tracts of (CCG)n, which lead to the fragile X syndrome; and (GAA)n repeats, whose expansion leads to Friedreich ataxia. Although the EM analysis did not show the DNAs to be unusually stiff or curved, we suspected that they might have unusual properties in nucleosome assembly because even small changes in the free energy of assembly would be amplified by their repetitive nature. Yuh-Hwa Wang in our laboratory was engaged in studies with HIV DNA in which she mixed the DNA with purified histones to reconstitute nucleosomes. In parallel, she tested the DNAs containing long tracts of CTG and CCG repeats that Bob and his colleagues had generated. The two repeat elements were found to lie on opposite ends of the spectrum of free energy of histone assembly. Although the CTG repeat turned out to have a very strong propensity to assemble into chromatin in these reconstitution assays (35), repeating CCG DNA assembled very poorly into nucleosomes (36). These conclusions were reached using a combination of EM and gel assays. Indeed, although the repeating CTG DNA remains possibly the strongest nucleosome assembly element known, no clear link between this property and disease pathology has been established. On the other hand, the inability of repeating CCG DNA to assemble into chromatin, a property amplified by methylation of the CpG elements, does have strong biological consequences. Yuh-Hwa observed that as the length of the repeating CCG track increased, its overall ability to assemble into nucleosomes diminished greatly; this was even more pronounced if the CpG elements were methylated (37). In genetic tests for fragile X syndrome, long blocks of uninterrupted CCG repeats and a high level of CpG methylation are considered indicators of a more severe disease state. Poor chromatinization is what would be expected for a chromosomal element prone to breakage. It would also open the door for binding of members of the class of CpG-binding proteins. These proteins are known to inhibit downstream transcription, in this case, the FMR-1 gene. This work provided a wonderful opportunity to apply EM to matters of clinical interest and to engage in lively and fruitful collaborations with Bob Wells and then later with Maurice Swanson at the University of Florida and their colleagues.
Visualizing Proteins That Bind to Unusual DNA Structures
One of the simplest applications of directly imaging proteins bound to DNA has been studying the binding of proteins to DNAs that contain bases that are looped out of the helix (bulged bases), Holliday junctions, and replication forks. Small DNAs containing these structures can be constructed from oligonucleotides and studied by gel shift analysis. However, from gel shift analysis alone, it is difficult to show for certain that a protein binds at the nexus of a Holliday junction instead of along the arms or at the ends of the arms. By constructing DNAs somewhat larger in size (for example, Holliday junctions with 150-bp arms), it is a simple task to examine the affinity of a protein for such structures and to determine where the protein is located along the DNA. Because the DNAs are small and uniform, analysis of hundreds of examples can be made in a short time. The dual approach of EM combined with parallel gel shift analysis is particularly valuable. A useful addition is to include protein mass standards against which the size of the protein on the DNA can be compared. If the standard is close in size to the DNA-bound protein complex, then the mass of the complex can be estimated with assurance (38). Although this does not have the exquisite accuracy of the scanning transmission analyses carried out by Joe Wall and colleagues at the Brookhaven National Laboratory, usually the question at hand is whether the protein is binding as a monomer, dimer, tetramer, and so on; the question can be answered even though the method lacks a high degree of accuracy.
Over the past several decades, we have employed this technology in studies related to DNA damage and repair. A highlight was the demonstration with Arnold Levine at Princeton University that p53 binds to bulged bases and Holliday junctions (39), a possibility that came out of our studies of RecA protein binding to DNA containing bulged bases (40). Work on mismatch repair has involved ongoing collaborations with Paul Modrich at Duke University (41, 42), Richard Kolodner at the University of California, San Diego (43, 44), and Rick Fishel at Ohio State University (45, 46), where these methods have revealed the binding of mismatch repair factors to DNA, as well as DNA looping by MutS (42). These studies paralleled work on the binding of key factors in signal transduction between sites of damage and the damage response pathways with Aziz Sancar at the University of North Carolina (47, 48). Recent work with Steve West and his group at the Cancer Research UK London Research Institute provided images of the BRCA2 and GEN1 proteins bound to branched DNA (49, 50). Although GEN1 can cleave branched DNA, BRCA2 binds and then facilitates loading of the Rad51 protein onto the DNA, setting the stage for subsequent repair events.
These methods have also been valuable in dissecting the structure of complex RNAs, as demonstrated in a series of enjoyable studies of ribozymes with Tom Cech and his group at the University of Colorado (51, 52). Finally, DNA structures far too complex for gel analysis, such as hemicatenated DNAs, can be examined, such as those employed in work with Tao-shih Hsieh at the Duke University Medical Center (53, 54).
Telomeres and t-loops
Few proteins that bind specifically to repeating triplet blocks have been identified. Telomeres in most eukaryotes consist of very long tracts of a short repeating sequence (TTAGGG in vertebrates), followed by a single-stranded 3′-extension of the G-rich strand, usually several hundred nucleotides in length. Work done by Titia de Lange and colleagues at Rockefeller University on a protein she called TRF1, which she found binds with high specificity to double-stranded mammalian telomeric DNA (55), provided a good example when one considers the telomeric repeat to be two tandem triplet repeats. Our EM work with Titia on TRF1 confirmed what the biochemistry had shown: that it binds along the length of telomeric DNA and, as a new twist, can synapse two telomeric DNAs (56). Titia and her group then purified a second human protein with strong specificity for duplex telomeric repeats, TRF2 (57). It was not clear whether TRF2 would show similar binding behavior as TRF1 or perhaps would have other properties at the telomere. Our efforts to visualize its binding to plasmid DNAs containing tracts of TTAGGG repeats were met with complex DNA-protein structures very different from what we had seen with TRF1. Their appearance had a familiar ring, one that suggested that it might have a topological or recombinational activity.
Telomeres contain two canonical elements of the simple in vitro recombination reactions, a single-stranded DNA with a 3′ terminus and a duplex DNA fully homologous to the single-stranded partner. The only difference was that, in this case, the two elements were fused into a single DNA. Action of the RecA or Rad51 protein on this dual element DNA would presumably result in a strand invasion event, in which the very end of the telomere would loop back and form a D-loop structure within the duplex repeats. When I suggested this possibility to Titia, she pointed out the strong biological implications, as it could provide a simple and elegant means of hiding the end of the chromosome and prevent it from being recognized by the omnipresent double-strand break repair machinery, whose action on free double-strand ends would be either to fuse the ends or to trigger an apoptotic response.
Testing this hypothesis in vitro involved generating a model telomere consisting of a linear plasmid DNA with one end containing 1–2 kb of TTAGGG repeats and terminating in a G-rich single-stranded 3′-extension. Rachel Stansel, a graduate student in our laboratory, generated this model DNA. The de Lange group provided a series of telomere factors, including TRF1 and TRF2, for the experiments. By using EM, Rachel observed loops at the telomere end in ∼20% of the molecules (58). The loops were dependent on the presence of TRF2 and a TTAGGG single-stranded 3′-extension. Loops were not observed with TRF1 or if the DNA was blunt or had a 5′-extension. To show that strand invasion had occurred, the complexes were treated with psoralen and UV light to cross-link the putative D-loop structure in place prior to removal of the TRF2 protein. EM analysis showed that the loops remained, confirming the presence of a strand invasion structure. Testing this hypothesis in vivo required combining the psoralen/UV light cross-linking with methods for purifying telomeric DNA that had been worked out by Titia when she was a postdoctoral fellow and cloned human telomeres. The final protocol, refined by our two groups, involved treating nuclei from 3 × 108 cells with psoralen and UV light, followed by purification of the DNA, cleavage by a mixture of restriction enzymes that cleave the genomic DNA into small fragments but leave the telomeric DNA intact, and a final gel filtration step to separate the long telomeric DNA that was present in 1/3000th of the total starting material (58). The demands of the EM methods for enough DNA to visualize and its being spread out in the gel filtration step continued to require large amounts of cells for each experiment. The results showed that long DNAs whose size was consistent with the length of the telomeric restriction fragments (determined by probing in gels) were present. Up to 20% of these long DNAs were found in the form of a circle with a long tail (Fig. 4) (58). The husband of a postdoctoral fellow in the de Lange lab coined the term “t-loops.”
FIGURE 4.
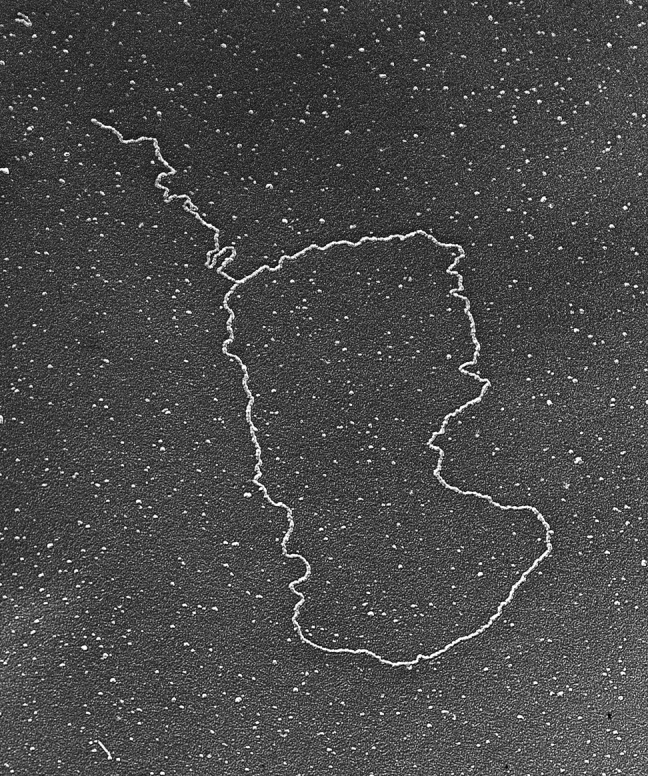
Telomere t-loop. Telomeres photo-cross-linked with psoralen frequently appear as giant duplex loops as seen by surface spreading EM (micrograph taken in 1999) (58).
In addition to the discovery of t-loops in mouse and human cells (58), further work revealed them in trypanosomes (59) and peas (60). Nikitina and Woodcock (61) isolated telomeric chromatin fragments on sucrose gradients and used negative staining and antibodies against TRF1 to show t-loop structures in mouse and chicken samples. Recent work in the de Lange group shows great promise for the visualization of t-loops by super resolution light microscopy (62) and the laboratory of Jerry Shay and Woodring Wright at the University of Texas Southwestern Medical Center has developed an enzyme-based assay for looping.1 Affinity purification methods for isolating telomeric DNA ongoing in our laboratory will greatly facilitate future EM studies by making it possible to scale the experiments down by at least an order of magnitude.
At the time we were first visualizing t-loops, Lubomir Tomaska and Josef Nosek at Comenius University in the Slovak Republic found that the mitochondrial DNA of the yeast Candida parapsilosis was not only linear but contained repeating sequences at its ends (63). However, instead of a 3′-overhang, the DNA contained a single-stranded 5′-protrusion, excluding a possibility of telomerase-dependent telomere maintenance. Rather, they found evidence of small DNA circles consisting of the mitochondrial telomeric repeats and proposed that integration of these circles back to the ends of the mitochondrial DNA could provide a mechanism of telomere maintenance. While visiting our laboratory, Tomaska used EM to directly visualize the small telomeric circles (t-circles) (64). At this time, our student Tony Cesare was isolating t-loops from human cells, which maintain their telomeres in the absence of telomerase, a pathway termed ALT (alternative lengthening of telomeres), and noted a large number of duplex DNA circles lacking tails in the EM spreads and found that these circles were resistant to cutting by four-base cutters, a property of telomeric DNA. Using two-dimensional pulsed-field gel electrophoresis, he confirmed that the presence of free telomeric DNA circles is a marker for the ALT phenotype (65). t-circles may result from the cleavage of t-loops and have now been observed widely and most frequently in cases in which the normal telomere pathways are disrupted. Integration of tagged t-circles back into yeast telomeres was directly demonstrated by Mike McEachern and his group at the University of Georgia (66), further supporting Tomaska and Nosek's hypothesis. Tomaska has recently provided a thoughtful review of the possible functions of t-circles in telomere maintenance (67).
These studies represent an excellent example of what can be accomplished when laboratories combine expertise and technology in very different areas. In this case, it was expertise in recombination and EM with a leading laboratory in the field of telomere biology, genetics, and biochemistry (de Lange) or yeast genetics and biochemistry (Tomaska).
Why It Took Nearly Twenty Years to Visualize Bruce Albert's Trombone Choir
In 1980, Bruce Alberts proposed a structural solution for an architectural dilemma at the replication fork (68, 69) The enzymology dictates that the two strands must be synthesized in the same enzymatic direction, but this requires that replication on the two strands moves in the opposite physical direction. It was known that although the newly synthesized leading strand is continuous, the newly synthesized lagging strand is in the form of short segments termed Okazaki fragments (reviewed in Ref. 70). Bruce proposed that the lagging strand must loop back at the fork, allowing replication to occur in the same physical direction on both strands. Furthermore, he suggested if there are two polymerase molecules, one synthesizing the leading strand and one synthesizing the lagging strand, and if they are in a single replisome at the fork, this would couple leading and lagging strand synthesis.
Bruce envisioned that as the Okazaki fragment cycle proceeds, the lagging strand loop grows out from the replisome in a manner similar to how a trombone slide extends. The loop would then collapse at the end of the Okazaki cycle (the trombone slide retracts). The model was widely accepted, but direct evidence was slow in coming. This was due in large part to the lack of synchrony in these reactions. The complex machinery, involving the helicase, primase, loaders, clamps, and polymerase, must assemble onto a DNA template containing a preformed fork. Once assembled, however, significant time may pass before a cycle of replication begins. Thus, trying to predict events at a single-molecule level by following the bulk ensemble properties of the mixture is nearly impossible. Thus, the key predications of this model remained unproven.
By the mid-1990s, Charles Richardson and his group at Harvard Medical School had characterized the T7 replication system in great detail (71). This simple system uses the T7 DNA polymerase with thioredoxin as a processivity factor, a helicase and primase transcribed from the same gene, and a single-strand binding protein. Using these highly purified proteins and an M13 DNA containing a preformed replication fork, they obtained evidence for robust rolling circle replication generating duplex tails >20 kb and a spectrum of Okazaki fragment lengths from ∼600 to >6000 nucleotides.
In a collaborative effort between our two groups, Zeger Debyser and Kyusung Park used the Kleinschmidt method to directly visualize the M13 template circles with long duplex tails. When the single-strand segments present on the tails were stained with SSB and the complexes were examined via adsorption onto thin carbon foils and tungsten shadow casting, there was a characteristic pattern of single- and double-strand segments along the tails expected for Okazaki fragment synthesis via a trombone cycle (72). Even more exciting, when they fixed the proteins to the DNA prior to EM examination and utilized carbon films and tungsten shadow casting, trombone loops were seen for the first time. Here, the replisome appeared as a single, large nucleoprotein complex at the junction of the circle and long rolling circle tail, and in roughly half of the examples scored, a fully double-stranded loop exited and re-entered the replisome. Measurements of the length of this loop agreed perfectly with the length of the duplex portion of the Okazaki fragments as determined from the analysis of the deproteinized DNAs stained with SSB. Of additional interest, the single-strand segments bound by the T7 SSB were not extended away from the replisome but appeared to be compact and an integral part of the replisome (72).
At the same time, Nancy Nossal at NIH had brought the T4 system to a similar stage. This more complex system utilizes ten different factors, each of which had to be purified free of nucleases and their activity determined. These include a polymerase, clamp, clamp loader, helicase, helicase loader, primase, and single-strand binding protein. Using circular templates with preformed forks, Nancy showed that replication would generate long rolling circle tails. For our EM studies, she drove from NIH to UNC with the proteins and her own glass micropipettes capable of delivering fractions of a microliter. Nancy worked with members of our lab late into the night, carrying out the reactions and preparations for EM. With the T7 studies as a guidepost, the EM analysis revealed trombone loops and distributions of Okazaki fragments (Fig. 5) (73) very similar to those we observed in the studies with Charles Richardson and his group (72). This work extended the visualization of trombone loops to the very system in which Bruce Alberts had been working when he made his proposal. Furthermore, we were poised to test the second part of this model, which was that there would be two polymerase molecules at the fork. We could do this because Paul Chastain II in our laboratory had worked out a very useful nanoscale “biopointer” for EM work (74). The targeted protein is tagged with biotin using cells expressing biotin ligase and then purified over a streptavidin affinity column to ensure that each target protein has a single biotin moiety. The biopointers consist of short (∼200 bp) stiff DNA fragments containing streptavidin at one end and thus provide “molecular arrows” pointing in the micrographs to the location of the tagged protein in a large multiprotein complex. We had first used these in studies of the yeast origin recognition complex with Steve Bell at MIT (74). By using the biopointers, it was possible to show with assurance that the predominant replisome at the fork contained two copies of T4 DNA polymerase (Fig. 5) (75). However, a minor fraction contained three copies, and this fraction corresponded to the fraction of replicating molecules in which there were two trombone loops at the fork. The observation of more than two polymerase molecules in the replisome was subsequently reported by O'Donnell in the E. coli system (76). By using the pointers, it was also possible to count the number of helicase subunits and to show that the helicase loader remained present in the replisome (75). In other studies with Nancy, we used EM to measure the length of successive Okazaki fragments along single rolling circle tails (77), a study ideally suited for an EM approach.
FIGURE 5.
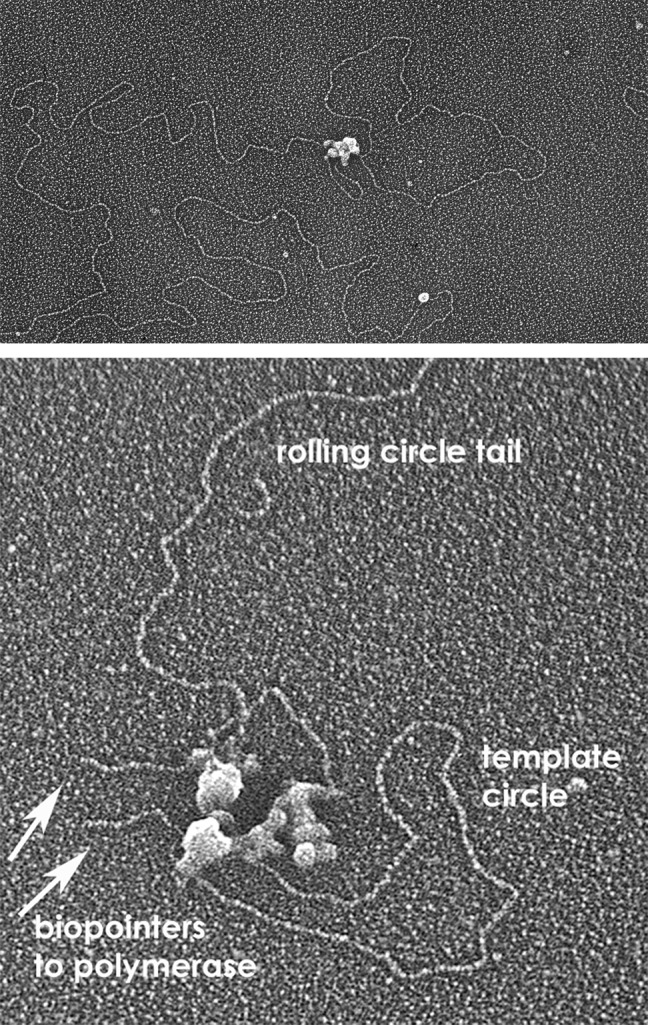
Trombone loops and two polymerase molecules at the fork. Images of a trombone loop at a replication fork driven by the purified T4 proteins (upper) and a fork with biopointers “pointing to” T4 DNA polymerase (lower) (micrograph circa 2007) (74).
The success of the T4 studies is a testament to the biochemical skill of Nancy and her NIH colleague Charles Jones in purifying the T4 replication proteins and optimizing reactions involving many different factors. By working together with members of our lab, she was able to adjust the reaction parameters to the demands set by EM. She sadly passed away just as the 2007 studies were being finalized for publication in this Journal. She will be long remembered.
In collaboration between Charles Richardson's group and Antoine van Oijen's group at Harvard Medical School, single-molecule methods using light microscopy have been employed to follow the trombone cycle catalyzed by the T7 proteins (78, 79). These studies revealed a cyclic movement of the far end of the DNA tagged with a visible bead in perfect agreement with the growth and collapse of single trombone cycles as seen by EM. The verification of the EM studies by a single-molecule approach has provided a strong footing for the use of both EM and the new single-molecule techniques in future work.
For many years, we have enjoyed a collaboration with Bob Lehman, during which we have used EM to examine the activation of the HSV-1 origin (oriS) by the HSV-1 origin-binding protein UL9 and the HSV-1 SSB/recombinase ICP8 (80, 81). Smaranda Willcox and Oya Bermek in our laboratory have purified the five core HSV-1 replication factors: a polymerase and its accessory factor, a three-subunit helicase/primase, and ICP8. Using these proteins, they have used EM to demonstrate a T7/T4 like trombone cycle for the first time in a eukaryotic system.2
The Future of EM in Studies of DNA Transactions
The application of EM to the molecular biology of DNA has advanced greatly in the past four decades. Examination of large, irregular DNA-protein complexes typical of replicating and recombining DNA is not amenable to the image averaging methods that have allowed single-particle reconstructions from negative staining or cryo-EM to achieve subnanometer resolution. Rather, analysis of these large complexes continues to require contrast enhancement by careful metal shadow casting and analysis of a statistically large number of molecules to deduce parameters of the biochemical reactions. Recently, many years after our initial attempts at freeze-drying DNA-protein complexes, we have achieved remarkable success using a new commercial freezing robot instrument and a purpose-built vacuum system of our design with very clean, oil-free pumps. An example of a human macrophage (Fig. 6) prepared this way illustrates the greatly improved structural preservation. With studies of DNA transactions moving steadily toward more and more complex multiprotein reactions, we look forward to a new decade of using this freezing technology to image these structures. Photographing scenery in Alaska (Fig. 7) will continue to provide great personal pleasure.
FIGURE 6.
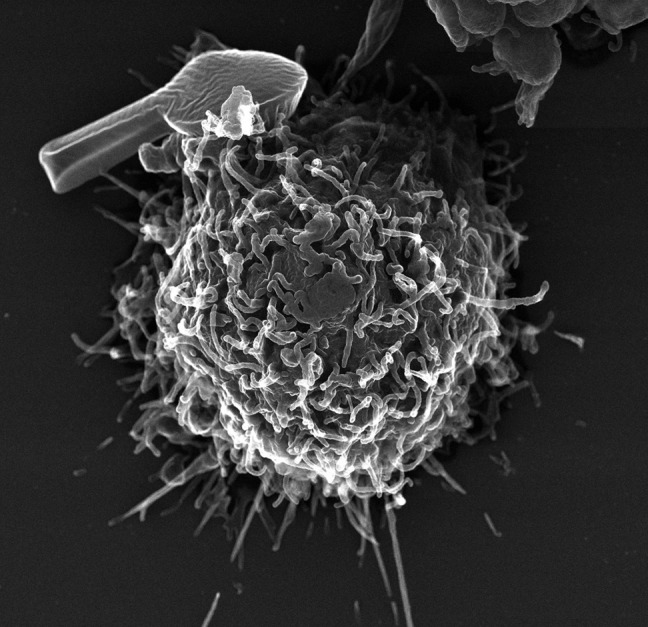
Human lung macrophage with an artificial microparticle bound to the surface. The macrophage was prepared by very fast freezing, freeze-drying, and tungsten shadow casting.
FIGURE 7.

The author on a trip photographing mountains in Alaska in 2013.
Acknowledgments
There are many colleagues who I have not mentioned specifically, yet who have been a strong part of our work for many years, in particular Tom Broker and Louise Chow. Alexander Makhov and Smaranda Willcox have contributed greatly to my success. I thank many colleagues who have critically read this article and provided very helpful suggestions. Much of this work was supported by a long-standing grant from the National Institutes of Health (GM31819).
This article is dedicated to Bob Lehman. In his 32 years as an associate editor of this Journal, he has made a profound impact on the careers of a very large number of younger scientists in our field. Each of us has relied on Bob to help shepherd our papers through to publication and have appreciated his generous time and efforts to improve the manuscripts. Having these papers appear in the Journal of Biological Chemistry has very often made the difference in positive decisions regarding tenure and the funding of our grants.
Footnotes
S. M. Mak, P. G. Smiraldo, T. T. Chow, J. W. Shay, and W. Wright, unpublished results.
O. Bermek, S. Willcox, and J. D. Griffith, unpublished results.
REFERENCES
- 1. Kleinschmidt A. K., Lang D., Jacherts D., Zahn R. K. (1962) Preparation and length measurements of the total desoxyribonucleic acid content of T2 bacteriophages. Biochim. Biophys. Acta 61, 857–864 [PubMed] [Google Scholar]
- 2. Vinograd J., Lebowitz J., Radloff R., Watson R., Laipis P. (1965) The twisted circular form of polyoma viral DNA. Proc. Natl. Acad. Sci. U.S.A. 53, 1104–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Clayton D. A., Vinograd J. (1967) Circular dimer and catenate forms of mitochondrial DNA in human leukaemic leucocytes. Nature 216, 652–657 [DOI] [PubMed] [Google Scholar]
- 4. Davis R. W., Simon M., Davidson N. (1971) Electron microscope heteroduplex methods for mapping regions of base sequence homology in nucleic acids. Methods Enzymol. 21, 413–428 [Google Scholar]
- 5. Chow L. T., Gelinas R. E., Broker T. R., Roberts R. J. (1977) An amazing sequence arrangement at the 5′ ends of adenovirus 2 messenger RNA. Cell 12, 1–8 [DOI] [PubMed] [Google Scholar]
- 6. Berget S. M., Moore C., Sharp P. A. (1977) Spliced segments at the 5′ terminus of adenovirus 2 late mRNA. Proc. Natl. Acad. Sci. U.S.A. 74, 3171–3175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Heuser J. E. (2011) The origins and evolution of freeze-etch electron microscopy. J. Electron Microsc. 60, Suppl. 1, S3–S29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Griffith J., Huberman J. A., Kornberg A. (1971) Electron microscopy of DNA polymerase bound to DNA. J. Mol. Biol. 55, 209–214 [DOI] [PubMed] [Google Scholar]
- 9. Eisenberg S., Griffith J., Kornberg A. (1977) The ϕX174 cistron A protein is a multifunctional enzyme in DNA replication. Proc. Natl. Acad. Sci. U.S.A. 74, 3198–3202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jackson D. A., Symons R. H., Berg P. (1972) Biochemical method for inserting new genetic information into DNA of simian virus 40: circular SV40 DNA molecules containing lambda phage genes an the galactose operon of Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 69, 2904–2909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lobban P. E., Kaiser A. D. (1973) Enzymatic end-to-end joining of DNA molecules. J. Mol. Biol. 78, 453–471 [DOI] [PubMed] [Google Scholar]
- 12. Lake R. S., Barban S., Salzman N. P. (1973) Resolutions and identification of the core deoxynucleoproteins of the simian virus 40. Biochem. Biophys. Res. Commun. 54, 640–647 [DOI] [PubMed] [Google Scholar]
- 13. Olins A. L., Olins D. E. (1974) Spheroid chromatin units (v bodies). Science 183, 330–332 [DOI] [PubMed] [Google Scholar]
- 14. Hewish D. R., Burgoyne L. A. (1973) Chromatin sub-structure. The digestion of chromatin DNA at regularly spaced sites by a nuclear deoxyribonuclease. Biochem. Biophys. Res. Commun. 52, 504–510 [DOI] [PubMed] [Google Scholar]
- 15. Hall M. R., Meinke W., Goldstein D. A. (1973) Nucleoprotein complexes containing replicating simian virus 40 DNA: comparison with polyoma nucleoprotein complexes. J. Virol. 12, 901–908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Griffith J. (1975) Chromatin structure: deduced from a mini-chromosome. Science 187, 1202–1203 [DOI] [PubMed] [Google Scholar]
- 17. Germond J. E., Hirt B., Oudet P., Gross-Bellark M., Chambon P. (1975) Folding of the DNA double helix in chromatin-like structures from simian virus 40. Proc. Natl. Acad. Sci. U.S.A. 72, 1843–1847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Thomas J. O., Kornberg R. D. (1975) An octamer of histones in chromatin and free in solution. Proc. Natl. Acad. Sci. U.S.A. 72, 2626–2630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. White C. L., Suto R. K., Luger K. (2001) Structure of the yeast nucleosome core particle reveals fundamental changes in internucleosome interactions. EMBO J. 20, 5207–5218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gardner K. E., Allis C. D., Strahl B. D. (2011) OPERating ON Chromatin, a colorful language where context matters. J. Mol. Biol. 409, 36–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Weinstock G. M., McEntee K., Lehman I. R. (1979) ATP-dependent renaturation of DNA catalyzed by the RecA protein of Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 76, 126–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. McEntee K., Weinstock G. M., Lehman I. R. (1979) Initiation of general recombination catalyzed in vitro by the RecA protein of Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 76, 2615–2619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sancar A., Rupp W. D. (1979) Cloning of uvrA, lexC, and ssb genes of Escherichia coli. Biochem. Biophys. Res. Commun. 90, 123–129 [DOI] [PubMed] [Google Scholar]
- 24. Chase J. W., Whittier R. F., Auerbach J., Sancar A., Rupp W. D. (1980) Amplification of single-strand DNA binding protein of Escherichia coli. Nucleic Acids Res. 8, 3215–3227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Register J. C., 3rd, Griffith J. (1985) The direction of RecA protein assembly onto single-stranded DNA is the same as the direction of strand assimilation during strand exchange. J. Biol. Chem. 260, 12308–12312 [PubMed] [Google Scholar]
- 26. Christiansen G., Griffith J. (1986) Visualization of the paranemic joining of homologous DNA molecules catalyzed by the RecA protein of Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 83, 2066–2070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Register J. C., 3rd, Christiansen G., Griffith J. (1987) Electron microscopic visualization of the pairing and branch migration phases of strand exchange. J. Biol. Chem. 262, 12812–12820 [PubMed] [Google Scholar]
- 28. Harris L. D., Griffith J. (1987) Visualization of the homologous pairing of DNA catalyzed by the bacteriophage UvsX protein. J. Biol. Chem. 262, 9285–9292 [PubMed] [Google Scholar]
- 29. Stasiak A., Stasiak A. Z., Koller T. (1984) Visualization of RecA-DNA complexes involved in consecutive stages of an in vitro strand exchange reaction. Cold Spring Harb. Symp. Quant. Biol. 49, 561–570 [DOI] [PubMed] [Google Scholar]
- 30. Egelman E. H. (2003) A tale of two polymers: new insights into helical filaments. Nat. Rev. Mol. Cell Biol. 4, 621–630 [DOI] [PubMed] [Google Scholar]
- 31. Marini J. C., Levene S. D., Crothers D. M., Englund P. T. (1982) Bent helical structure in kinetoplast DNA. Proc. Natl. Acad. Sci. U.S.A. 79, 7664–7668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Griffith J., Bleyman M., Rauch C. A., Kitchin P. A., Englund P. T. (1986) Visualization of the bent helix in kinetoplast DNA by electron microscopy. Cell 46, 717–724 [DOI] [PubMed] [Google Scholar]
- 33. Wu H.-M., Crothers D. M. (1984) The locus of sequence-directed and protein-induced bending. Nature 308, 509–513 [DOI] [PubMed] [Google Scholar]
- 34. Laundon C. H., Griffith J. D. (1988) Curved helix segments can uniquely orient the topology of supertwisted DNA. Cell 52, 545–549 [DOI] [PubMed] [Google Scholar]
- 35. Wang Y.-H., Amirhaeri S., Kang S., Wells R. D., Griffith J. D. (1994) Preferential nucleosome assembly at DNA triplet repeats from the myotonic dystrophy gene. Science 265, 669–671 [DOI] [PubMed] [Google Scholar]
- 36. Wang Y.-H., Gellibolian R., Shimizu M., Wells R. D., Griffith J. (1996) Long CCG triplet repeat blocks exclude nucleosomes: a possible mechanism for the nature of fragile sites in chromosomes. J. Mol. Biol. 263, 511–516 [DOI] [PubMed] [Google Scholar]
- 37. Wang Y.-H., Griffith J. (1996) Methylation of expanded CCG triplet repeat DNA from fragile X syndrome patients enhances nucleosome exclusion. (1996) J. Biol. Chem. 271, 22937–22940 [PubMed] [Google Scholar]
- 38. Griffith J. D., Makhov A., Zawel L., Reinberg D. (1995) Visualization of TBP oligomers binding and bending the HIV-1 and adeno promoters. J. Mol. Biol. 246, 576–584 [DOI] [PubMed] [Google Scholar]
- 39. Lee S., Elenbaas B., Levine A., Griffith J. (1995) p53 and its 14 kDa C-terminal domain recognize primary DNA damage in the form of insertion/deletion mismatches. Cell 81, 1013–1020 [DOI] [PubMed] [Google Scholar]
- 40. Wang Y.-H., Bortner C. D., Griffith J. (1993) RecA binding to bulge- and mismatch-containing DNA. Certain single base mismatches provide signals for RecA binding equal to multiple base bulges. J. Biol. Chem. 268, 17571–17577 [PubMed] [Google Scholar]
- 41. Grilley M., Griffith J., Modrich P. (1993) Bidirectional excision in methyl-directed mismatch repair. J. Biol. Chem. 268, 11830–11837 [PubMed] [Google Scholar]
- 42. Allen D. J., Makhov A., Grilley M., Taylor J., Thresher R., Modrich P., Griffith J. D. (1997) MutS mediates heteroduplex formation by a translocation mechanism. EMBO J. 16, 4467–4476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Alani E., Lee S., Kane M. F., Griffith J., Kolodner R. D. (1997) Saccharomyces cerevisiae MSH2, a mispaired base recognition protein also recognizes Holliday junctions in DNA. J. Mol. Biol. 265, 289–301 [DOI] [PubMed] [Google Scholar]
- 44. Marsischky G. T., Lee S., Griffith J., Kolodner R. D. (1999) Saccharomyces cerevisiae MSH2/6 complex interacts with Holliday junctions and facilitates their cleavage by phage resolution enzymes. J. Biol. Chem. 274, 7200–7206 [DOI] [PubMed] [Google Scholar]
- 45. Fishel R., Ewel A., Lee S., Lescoe M. K., Griffith J. (1994) Binding of mismatches microsatellite DNA sequences by the human MSH2 protein. Science 266, 1403–1405 [DOI] [PubMed] [Google Scholar]
- 46. Gradia S., Subramanian D., Wilson T., Acharya S., Makhov A., Griffith J., Fishel R. (1999) hMSH2-hMSH6 forms a hydrolysis-independent sliding clamp on mismatched DNA. Mol. Cell 3, 255–261 [DOI] [PubMed] [Google Scholar]
- 47. Unsal-Kaçmaz K., Makhov A. M., Griffith J. D., Sancar A. (2002) Preferential binding of ATR protein to UV-damaged DNA. Proc. Natl. Acad. Sci. U.S.A. 99, 6673–6678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sar F., Lindsey-Boltz L. A., Subramanian D., Croteau D. L., Hutsell S. Q., Griffith J. D., Sancar A. (2004) Human Claspin is a ring-shaped DNA binding protein with high affinity to branched DNA structures. J. Biol. Chem. 279, 39285–39295 [DOI] [PubMed] [Google Scholar]
- 49. Rass U., Compton S. A., Matos J., Singleton M. R., Ip S. C., Blanco M. G., Griffith J. D., West S. C. (2010) Mechanisms of Holliday junction resolution by the human GEN1 protein. Genes Dev. 24, 1559–1569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Thorslund T., McIllwraith M. J., Compton S. A., Lekomtsev S., Pertonczki M., Griffith J. D., West S. (2010) The breast cancer tumor suppressor BRCA2 promotes the specific targeting of RAD51 to single-stranded DNA. Nat. Mol. Struct. Biol. 17, 1263–1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wang Y.-H., Murphy F. L., Cech T. R., Griffith J. D. (1994) Visualization of a tertiary structural domain of the Tetrahymena group I intron by electron microscopy. J. Mol. Biol. 236, 64–71 [DOI] [PubMed] [Google Scholar]
- 52. Nakamura T. M., Wang Y.-H., Zaug A. J., Griffith J. D., Cech T. R. (1995) Relative orientation of RNA helices in a group I ribozyme determined by helix extension electron microscopy. EMBO J. 14, 4849–4859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chen S. H., Plank J. L., Willcox S., Griffith J. D., Hsieh T.-s. (2013) Improved methods for creating migratable Holliday junctions substrates. Nucleic Acids Res. 41, e60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lee S.-H., Siaw G. E.-L., Willcox S., Griffith J. D., Hsieh T.-s. (2013) Synthesis and dissolution of hemicatenanes by type IA DNA topoisomerases. Proc. Natl. Acad. Sci. U.S.A. 10.1073/pnas.1304103110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chong L., van Steensel B., Broccoli D., Erdjument-Bromage H., Hanish J., Tempst P., de Lange T. (1995) A human telomeric protein. Science 270, 1663–1667 [DOI] [PubMed] [Google Scholar]
- 56. Griffith J., Bianchi A., de Lange T. (1998) TRF1 promotes parallel pairing of telomeric tracts in vitro. J. Mol. Biol. 278, 79–88 [DOI] [PubMed] [Google Scholar]
- 57. Broccoli D., Smogorzewska A., Chong L., de Lange T. (1997) Human telomeres contain two distinct Myb-related proteins, TRF1 and TRF2. Nat. Genet. 17, 231–235 [DOI] [PubMed] [Google Scholar]
- 58. Griffith J. D., Comeau L., Rosenfield S., Stansel R. M., Bianchi A., Moss H., de Lange T. (1999) Mammalian telomeres end in a large duplex loop. Cell 97, 503–514 [DOI] [PubMed] [Google Scholar]
- 59. Muñoz-Jordán J. L., Cross G. A. M., de Lange T., Griffith J. D. (2001) T-loops at trypanosome telomeres. EMBO J. 20, 579–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Cesare A. J., Quinney N., Willcox S., Subramanian D., Griffith J. D. (2003) Telomere looping in P. savitum (common garden pea). Plant J. 36, 271–279 [DOI] [PubMed] [Google Scholar]
- 61. Nikitina T., Woodcock C. L. (2004) Closed chromatin loops at the ends of chromosomes. J. Cell Biol. 166, 161–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Doksani Y., Wu J., de Lange T., Zhuang X. (2013) Super-resolution fluorescence imaging of telomeres reveals TRF2-dependent t-loop formation. Cell 155, in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Nosek J., Dinouël N., Kovac L., Fukuhara H. (1995) Linear mitochondrial DNAs from yeasts: telomeres with large tandem repetitions. Mol. Gen. Genet. 247, 61–72 [DOI] [PubMed] [Google Scholar]
- 64. Tomaska L., Nosek J., Makhov A. M., Pastorakova A., Griffith J. D. (2000) Extragenomic double-stranded DNA circles in yeast with linear mitochondrial genomes: potential involvement in telomere maintenance. Nucleic Acids Res. 28, 4479–4487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Cesare A. J., Griffith J. D. (2004) Telomeric DNA in ALT cells is characterized by free telomeric circles and heterogeneous t-loops. Mol. Cell. Biol. 24, 9948–9957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Natarajan S., McEachern M. J. (2002) Recombinational telomere elongation promoted by DNA circles. Mol. Cell. Biol. 22, 4512–4521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Tomaska L., Nosek J., Kramara J., Griffith J. D. (2009) Telomeric circles: universal players in telomere maintenance. Nat. Struct. Mol. Biol. 16, 1010–1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Sinha N. K., Morris C. F., Alberts B. M. (1980) Efficient in vitro replication of double-stranded DNA templates by a purified T4 bacteriophage replication system. J. Biol. Chem. 255, 4290–4293 [PubMed] [Google Scholar]
- 69. Alberts B. M., Barry J., Bedinger P., Formosa T., Jongeneel C. V., Kreuzer K. N. (1983) Studies on DNA replication in the bacteriophage T4 in vitro system. Cold Spring Harb. Symp. Quant. Biol. 47, 655–668 [DOI] [PubMed] [Google Scholar]
- 70. Kornberg A. (1974) DNA Replication, W. H. Freeman and Co., New York [Google Scholar]
- 71. Lee S. J., Richardson C. C. (2011) Choreography of bacteriophage T7 DNA replication. Curr. Opin. Chem. Biol. 15, 580–586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Park K., Debyser Z., Tabor S., Richardson C. C., Griffith J. D. (1998) Formation of a DNA Loop at the replication fork generated by bacteriophage T7 replication proteins. J. Biol. Chem. 273, 5260–5270 [DOI] [PubMed] [Google Scholar]
- 73. Chastain P. D., 2nd, Makhov A. M., Nossal N. G., Griffith J. (2003) Architecture of the replication complex and DNA loops at the fork generated by the bacteriophage T4 proteins. J. Biol. Chem. 278, 21276–21285 [DOI] [PubMed] [Google Scholar]
- 74. Chastain P. D., 2nd, Bowers J. L., Lee D. G., Bell S. P., Griffith J. D. (2004) Mapping subunit location on the Saccharomyces cerevisiae origin recognition complex free and bound to DNA using a novel nanoscale biopointer. J. Biol. Chem. 279, 36354–36362 [DOI] [PubMed] [Google Scholar]
- 75. Nossal N. G., Makhov A. M., Chastain P. D., 2nd, Jones C. E., Griffith J. D. (2007) Architecture of the bacteriophage T4 replication complex revealed with nanoscale biopointers. J. Biol. Chem. 282, 1098–1108 [DOI] [PubMed] [Google Scholar]
- 76. McInerney P., Johnson A., Katz F., O'Donnell M. (2007) Characterization of a triple DNA polymerase replisome. Mol. Cell 27, 527–538 [DOI] [PubMed] [Google Scholar]
- 77. Chastain P. D., 2nd, Makhov A. M., Nossal N. G., Griffith J. D. (2000) Analysis of the Okazaki fragment distributions along single long DNAs replicated by the bacteriophage T4 proteins. Mol. Cell 6, 803–814 [DOI] [PubMed] [Google Scholar]
- 78. Hamdan S. M., Loparo J. J., Takahashi M., Richardson C. C., van Oijen A. M. (2009) Dynamics of DNA replication loops reveal temporal control of lagging-strand synthesis. Nature 457, 336–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Hamdan S. M., van Oijen A. M. (2010) Timing, coordination, and rhythm: acrobatics at the DNA replication fork. J. Biol. Chem. 285, 18979–18983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Makhov A. M., Boehmer P. E., Lehman I. R., Griffith J. D. (1996) The herpes simplex virus type 1 UL9 protein carries out origin specific DNA unwinding and forms stem/loop structures. EMBO J. 15, 1742–1750 [PMC free article] [PubMed] [Google Scholar]
- 81. Makhov A. M., Lee S. S.-K., Lehman I. R., Griffith J. D. (2003) Origin-specific unwinding of herpes simplex virus type 1 DNA by the viral UL9 and ICP8 proteins: visualization of a specific preunwinding complex. Proc. Natl. Acad. Sci. U.S.A. 100, 898–903 [DOI] [PMC free article] [PubMed] [Google Scholar]