

Background: Contribution of resorption to bone anabolism by PTH receptor signaling in osteocytes is unknown.
Results: Pharmacologic/genetic approaches demonstrated that remodeling- or modeling-based bone formation differentially operate in specific surfaces.
Conclusion: Resorption is critical for anabolism in periosteal/endocortical bone surfaces, but tempers bone gain in cancellous bone.
Significance: Targeting bone compartment-specific actions of PTH receptor signaling could enhance the therapeutic potential of the pathway.
Keywords: Bone, Osteoclast, Osteocyte, Parathyroid Hormone, Wnt Signaling, PTH Receptor, Bone Modeling/Remodeling, Osteocyte, Resorption, Sost/Sclerostin
Abstract
The contribution of remodeling-based bone formation coupled to osteoclast activity versus modeling-based bone formation that occurs independently of resorption, to the anabolic effect of PTH remains unclear. We addressed this question using transgenic mice with activated PTH receptor signaling in osteocytes that exhibit increased bone mass and remodeling, recognized skeletal effects of PTH elevation. Direct inhibition of bone formation was accomplished genetically by overexpressing the Wnt antagonist Sost/sclerostin; and resorption-dependent bone formation was inhibited pharmacologically with the bisphosphonate alendronate. We found that bone formation induced by osteocytic PTH receptor signaling on the periosteal surface depends on Wnt signaling but not on resorption. In contrast, bone formation on the endocortical surface results from a combination of Wnt-driven increased osteoblast number and resorption-dependent osteoblast activity. Moreover, elevated osteoclasts and intracortical/calvarial porosity is exacerbated by overexpressing Sost and reversed by blocking resorption. Furthermore, increased cancellous bone is abolished by Wnt inhibition but further increased by blocking resorption. Thus, resorption induced by PTH receptor signaling in osteocytes is critical for full anabolism in cortical bone, but tempers bone gain in cancellous bone. Dissecting underlying mechanisms of PTH receptor signaling would allow targeting actions in different bone compartments, enhancing the therapeutic potential of the pathway.
Introduction
Parathyroid hormone (PTH)5 has profound effects on the skeleton, and its elevation in the circulation can generate both catabolic and anabolic effects on bone depending on the temporal profile of its increase. Chronic excess of PTH, as in primary hyperparathyroidism or secondary to calcium deficiency, increases the rate of bone remodeling, and can result in loss of bone. In contrast, intermittent PTH elevation, as achieved by daily injections, cause bone gain, and it is the only current bone anabolic therapy. High bone remodeling rates and bone loss with chronic PTH elevation are associated with excessive production of osteoclasts coupled to increased osteoblasts, with a negative balance between formation and resorption within each bone multicellular unit (BMU). Instead, the primary effect of intermittent PTH elevation is a rapid increase in osteoblasts and bone formation, attributed to the ability of PTH to promote proliferation of osteoblast precursors, to inhibit osteoblast apoptosis, to re-activate lining cells to become matrix synthesizing osteoblasts, or to a combination of these effects (1, 2). Daily PTH injections in humans stimulate bone formation by increasing bone remodeling rate and the amount of bone formed by each remodeling unit, named “remodeling-based formation” (3). PTH also stimulates bone formation not coupled to prior resorption, referred to as “modeling-based formation.” However, the relative contribution of each of these mechanisms to the bone gain induced by PTH and the degree at which they operate in different bone surfaces remain unclear.
Earlier work demonstrates that some of actions of PTH on the skeleton are mediated by direct effects of the hormone on osteocytes, the most abundant and highly communicated cells in bone (4). PTH down-regulates the expression of the Sost gene, which encodes the potent inhibitor of bone formation sclerostin, expressed primarily in osteocytes in bone (5, 6). PTH also increases the expression of FGF23, a hormone expressed in osteocytes (and osteoblasts) that regulates phosphate reabsorption in the kidney, contributing to mineral homeostasis (4, 7, 8). Moreover, the major skeletal effects of PTH are recapitulated in transgenic mice expressing a constitutively active PTH receptor in osteocytes, named DMP1–8kb-caPTHR1 mice (4, 9–11). Thus, these mice display a marked increase in bone mineral density and increased bone formation rate (BFR) and osteoblasts in cortical and cancellous bone surfaces. In addition, expression of Sost/sclerostin is reduced and Wnt signaling is activated in DMP1–8kb-caPTHR1 mice. Moreover, induction of Wnt target genes is abolished in double transgenic mice also expressing Sost in osteocytes (10). Furthermore, DMP1–8kb-caPTHR1 mice exhibit high bone resorption, as evidenced by elevated systemic levels of resorption markers, high number of osteoclasts in bone, and increased cortical bone porosity. In addition, circulating osteocalcin, an index of bone formation and remodeling, is also increased in DMP1–8kb-caPTHR1 mice.
We have dissected herein the contribution of modeling-based bone formation and remodeling-based bone formation, to the anabolic effect of PTH receptor signaling in osteocytes. Toward this end, we inhibited bone formation in DMP1–8kb-caPTHR1 mice directly by generating double transgenic mice also overexpressing Sost in osteocytes; and we inhibited remodeling-driven bone formation by blocking resorption with the bisphosphonate alendronate. Our findings demonstrate that PTH receptor signaling in osteocytes regulates bone mass through modeling-based and remodeling-based bone formation mechanisms that operate to a different extent in cortical and cancellous bone surfaces.
EXPERIMENTAL PROCEDURES
Generation of DMP1–8kb-caPTHR1 and DMP-Sost Transgenic Mice
Generation of DMP1–8kb-caPTHR1 and DMP1–8kb-Sost transgenic mice was described previously (9, 10). Mice were born at the expected Mendelian frequency, were fertile, and exhibited normal size and weight. The genetic background of DMP1–8kb-caPTHR1 and DMP1–8kb-Sost mice is C57BL/6. Mouse colonies are maintained by breeding mice hemizygous for the transgene with wild type (WT) C57BL/6 mice. All transgenic mice used in these studies were hemizygous.
DMP1–8kb-caPTHR1 mice were crossed with DMP1–8kb-Sost mice to obtain mice expressing the DMP1–8kb-caPTHR1 transgene mice expressing both the DMP1–8kb-caPTHR1 and the DMP1–8kb-Sost transgenes. Four-week-old mice (n = 6–12 per group) were administered weekly subcutaneous injections of 16.1 μmol/kg/week (5.25 mg/kg/week) of alendronate or the equivalent volume of saline, for 2 weeks. Mice were fed a regular diet (Harlan/Teklad 7001) and water ad libitum and maintained on a 12-h light/dark cycle. Protocols involving genetically modified mice and their WT littermates were approved by the Institutional Animal Care and Use Committees of Indiana University School of Medicine.
Bone Turnover Markers
Plasma osteocalcin (OCN) and C-telopeptide fragments of type I collagen (CTX) were measured using enzyme linked immunoadsorbent assays (Biomedical Technologies, Stoughton, MA, and Immunodiagnostic Systems Inc., Fountain Hills, AZ, respectively) following manufacturer's instructions (10).
Analysis of Skeletal Phenotypes
BMD for the femora and the spine was determined by dual energy x-ray absorptiometry (DXA) using a PIXImus II densitometer (G.E. Medical Systems, Lunar Division, Madison, WI) as previously described (9). Mice were anesthetized via inhalation of 2.5% isoflurane (IsoFlo; Abbott Laboratories, North Chicago, IL) mixed with O2 (1.5 liter/min). Radiographic images were obtained from anesthesized mice using a digital x-ray system, as previously published (9). For micro-CT analysis, bones were dissected, cleaned of soft tissue, stored in 70% ethanol, and scanned at 6 micron resolution (Skyscan 1172, SkyScan, Kontich, Belgium). For histomorphometric analysis, femora and calvariae were dissected, fixed, and embedded in methyl methacrylate. Fluorochrome labeling of the bones was performed by intraperitoneal injections of calcein (30 mg/kg; Sigma) and alizarin (50 mg/kg; Sigma) administered 7 and 2 days before sacrifice, respectively, as previously described (10). Thick cross-sections of undecalcified femora at the mid-diaphysis were prepared using a diamond embedded wire saw (Histosaw, Delaware Diamond Knives, Wilmington, DE) and ground to a final thickness of 30–35 μm. Frontal plane 8 μm-thick calvarial sections were obtained 2 mm anterior to the junction between fronto-parietal and sagittal sutures using an Automated Rotary Microtome Leica RM2255 (Leica Microsystems Inc., Bannockburn, IL). Sections were viewed at 20–40 × magnification on a Leitz DMRXE microscope (Leica Mikroskopie und System GmbH, Wetzlar, Germany). Images were captured using a SPOT digital camera (Diagnostic Instruments, Inc., Sterling Heights, MI). Total, single, and double labeled perimeter, and inter-label width were measured on periosteal and endocortical surfaces of 2 femoral sections per mouse and on outer and inner periosteal surfaces of 1 calvarial section per mouse, using a semiautomatic analysis system (Bioquant OSTEO 7.20.10, Bioquant Image Analysis Co., Nashville, TN) attached to a microscope equipped with an ultraviolet light source (Nikon Optiphot 2 microscope, Nikon Instruments, Melville, NY). A combination of von Kossa followed by enzyme histochemistry for tartrate-resistant acid phosphatase (TRAP) histochemistry was used to visualize mineralized bone and osteoclasts in calvarial sections. TRAP positive multinucleated cells were enumerated and the number was expressed per bone area. The terminology and units used are those recommended by the Histomorphometry Nomenclature Committee of the American Society for Bone and Mineral Research (12).
Quantitative PCR
Total RNA was extracted from ulnae from 6-week-old mice using Ultraspec reagent (Biotecx Laboratories) according to the manufacturer's instructions. Gene expression was analyzed by quantitative PCR as previously described using primer probe sets from Applied Biosystems or from Roche Applied Science (9). Relative mRNA expression levels were normalized to the housekeeping gene ribosomal protein S2 using the ΔCt method.
Immunostaining
Protein detection on paraffin-embedded tibiae was performed as previously described (10). Briefly, sections were deparaffinized, treated with 3% H2O2 to inhibit endogenous peroxidase activity, blocked with mouse, rabbit or goat serum, and then incubated with mouse anti-β-catenin (BD Transduction Laboratories, San Jose, CA) (13, 14) or rabbit anti-phosphoSer133-CREB (16) (Santa Cruz Biotechnology Inc, Santa Cruz, CA) with prior antigen retrieval (DeCal Retrieval Solution, BioGenex, San Ramon, CA) and followed by signal amplification (ABC kit, Vector Laboratories, Burlingame, CA). Corresponding non-immune IgGs were used as negative controls.
Statistical Analysis
Data were analyzed using SigmaStat (SPSS Science, Chicago, IL). Differences between group means were evaluated using two-way ANOVA. All values are reported as the mean ± S.D.
RESULTS
The Increased Bone Remodeling Exhibited by DMP1–8kb-caPTHR1 mice Is Regulated by Sost Overexpression and Alendronate Treatment
DMP1–8kb-caPTHR1 transgenic mice exhibit high bone remodeling indicated by increased circulating levels of the resorption marker CTX and the formation marker osteocalcin (Fig. 1, A and B). Double transgenic mice expressing both the caPTHR1 and Sost transgene in osteocytes exhibited even higher CTX levels than single DMP1–8kb-caPTHR1 transgenic mice (Fig. 1A). And, alendronate decreased CTX levels in both DMP1–8kb-caPTHR1 mice and double transgenic mice. On the other hand, Sost overexpression markedly decreased circulating OCN in DMP1–8kb-caPTHR1 mice. Alendronate also decreased OCN levels in DMP1–8kb-caPTHR1 mice, but did not alter significantly the already low OCN of the double transgenic mice (Fig. 1B). OCN is not only a bone formation marker but also a bone turnover marker because is released from the matrix to the circulation during resorption. However, low circulating OCN in the presence of elevated resorption is indicative of low bone formation rate and decreased osteoblast number. Thus, inhibition of Wnt signaling by Sost overexpression reduces bone formation, whereas alendronate administration blunts both resorption and formation.
FIGURE 1.

The high bone remodeling exhibited by DMP1–8kb-caPTHR1 mice is blunted by alendronate. CTX (A) and OCN (B) were measured in plasma of 6.0-week-old DMP1–8kb-caPTHR1 mice, with and without the DMP1–8kb-Sost transgene and with or without alendronate. C, bone material density (pixels) was measured in cortical bone of the femoral mid-diaphysis, calvaria, and cancellous bone of the distal femur and lumbar vertebrae by μCT. Bars represent the mean ± S.D.; n = 9–12 (A and B) and 4–5 (C) mice per group. *, p < 0.05 versus respective controls not expressing the caPTHR1 transgene and lines connect conditions with p < 0.05, by two-way ANOVA.
Consistent with increased remodeling, activation of the PTH receptor in osteocytes resulted in reduced bone material density, an index of tissue mineralization, measured by micro-CT in cortical and cancellous bone of the femur, in calvaria, as well as in vertebral cancellous bone (Fig. 1C). The effects of Sost overexpression or bisphosphonate treatment were more evident in femoral cortical bone, but did not modify significantly bone material density in the other bones studied. In femoral cortical bone (mid-diaphysis), Sost overexpression further decreased material density as it was lower in DMP1–8kb-caPTHR1; DMP1–8kb-Sost mice compared with DMP1–8kb-caPTHR1 mice, consistent with the increased resorption in the double transgenic mice compared with DMP1–8kb-caPTHR1. In contrast, alendronate treatment led to increased bone material density in both DMP1–8kb-caPTHR1 and in double transgenic mice. These findings validate the genetic and pharmacological interventions chosen to inhibit directly bone formation or bone resorption-driven bone formation, respectively, in DMP1–8kb-caPTHR1 mice.
Molecular Mediators of PTH Receptor Action Are Differentially Regulated by Sost Overexpression and Alendronate
The expression of osteoblast and osteoclast markers, as well as osteoclastogenic cytokines, was elevated in bones from DMP1-caPTHR1 mice (Fig. 2). Alkaline phosphatase, collagen 1, and osteocalcin expression was increased in DMP1-caPTHR1 mice and reversed to wild type levels in DMP1-caPTHR1; DMP1-Sost double transgenic mice. Alendronate markedly blunted the expression of these genes in all mouse genotypes. In contrast, the expression of calcitonin receptor and TRAP was similarly high in single and double transgenic mice. Further, alendronate did not decrease the expression of these osteoclast markers, consistent with the evidence that inhibition of resorption by bisphosphonates results from direct blockade of osteoclast activity without decreasing osteoclast number, and even in some cases inducing accumulation of inactive osteoclasts (17–19). The increased expression of M-CSF exhibited by DMP1-caPTHR1 mice was not altered by Sost overexpression or bisphosphonate treatment. Sost overexpression led to an increase in RANKL expression compared with wild type mice and to a marked reduction in OPG expression in DMP1-caPTHR1 mice. These changes resulted in a RANKL/OPG ratio in double transgenic mice even higher than that of DMP1-caPTHR1 mice. Alendronate treatment increased the expression of RANKL in all mouse genotypes, without inducing major changes in OPG. This resulted in an overall increased RANKL/OPG ratio and in elimination of the effect of DMP1-caPTHR1 transgene.
FIGURE 2.

Molecular mediators of PTH receptor action are differentially regulated by Sost overexpression and alendronate. Gene expression was measured by quantitative PCR. Results are expressed relative to the housekeeping gene ribosomal protein S2. Bars represent mean ± S.D. of 3–7 mice per group. *, p < 0.05 versus respective controls not expressing the caPTHR1 transgene and lines connect conditions with p < 0.05, by two-way ANOVA.
DMP1–8kb-caPTHR1 Mice Exhibit Activation of PTHR/cAMP and PTHR/Wnt Signaling in Osteocytes
Phosphorylated CREB (P-CREB), a direct downstream target of PTHR/cAMP signaling, and β-catenin, the canonical Wnt mediator, accumulated in the nuclei of osteocytes adjacent to both periosteal and endosteal surfaces of cortical bone of DMP1-caPTHR1 mice compared with WT littermates (Fig. 3, B/F and A/E, respectively). Overexpresion of Sost did not affect P-CREB levels in either WT or DMP1–8kb-caPTHR mice (Fig. 3, C and D), whereas it reversed the increased β-catenin accumulation in osteocytes in double transgenic DMP1–8k-caPTHR;DMP1-Sost mice (Fig. 3, H compared with F). In addition, β-catenin staining was also detected within cells covering the periosteal bone surface in bones from DMP1-caPTHR1 mice (Fig. 3F, red arrows). In contrast, β-catenin staining was restricted to the membrane of osteoblasts on the periosteal surface in bone sections from double transgenic mice (Fig. 3H). Thus, PTHR/cAMP and PTHR/Wnt signaling pathways are activated in all osteocytes, regardless of their proximity to periosteal or endosteal bone surfaces in mice with osteocytic activation of the PTHR. In addition, activation of PTHR signaling in osteocytes increases β-catenin accumulation not only in osteocytes but also in cells on the periosteal bone surface; and this effect is specifically prevented by Sost overexpression in double transgenic mice.
FIGURE 3.

DMP1–8kb-caPTHR1 mice exhibit activation of PTHR/cAMP and PTHR/Wnt signaling in osteocytes. Expression of phosphoSer133-CREB (P-CREB, A–D) and β-catenin (E–H) was detected by immunohistochemistry in sections of tibial bone. Bone surfaces facing the periosteum (Periosteal Surface, PS) and the bone marrow (BM) and muscle fibers (M) are indicated. In some sections the bone is detached from the surrounding muscle or bone marrow tissue. Black arrows point to osteocytes labeled with anti-P-CREB (A–D) or anti-β-catenin (E–H) antibodies. Red arrows point to osteoblasts on the periosteal surface of bone labeled with anti-β-catenin antibody. Note that β-catenin is detected within the cells in the bone section from DMP1–8kb-caPTHR1 mice (F), but only in the cell membrane in the bone section from double transgenic DMP1–8kb-caPTHR1; DMP1-Sost mice (H). Bars indicate 10 μm.
Bone Formation Driven by Osteocytic PTH Receptor Signaling on Periosteal and Endocortical Surfaces of Cortical Bone Is Differentially Regulated by Wnt Signaling and Resorption
The increased periosteal bone formation rate displayed by DMP1–8kb-caPTHR1 mice was reversed by Sost overexpression, due to a combination of decreased work of osteoblast teams (MAR) and reduced number of osteoblasts on bone surfaces (MS/BS) (Fig. 4A). On the other hand, inhibition of resorption with alendronate did not affect periosteal bone formation in DMP1–8kb-caPTHR1 mice. In addition, alendronate did not alter significantly bone formation indexes in double transgenic mice, which were already low compared with the single DMP1–8kb-caPTHR1 transgenic mice. Therefore, bone formation induced by PTHR1 signaling in osteocytes on the periosteal surface depends on the Wnt pathway but not on resorption.
FIGURE 4.

The increased periosteal bone formation induced by PTH receptor signaling in osteocytes depends on the Wnt pathway but not on resorption, whereas both Wnt signaling and resorption contribute to the high endocortical bone formation. Dynamic histomorphometric measurements were determined in the femur on the periosteal (A) and endocortical (B) surfaces in 3–5 mice per group. Bars represent the mean ± S.D. *, p < 0.05 versus respective controls not expressing the caPTHR1 transgene and lines connect conditions with p < 0.05, by two-way ANOVA.
On the endocortical surface, Sost overexpression reduced equally BFR in wild type and in DMP1–8kb-caPTHR1 mice. Thus, double transgenic DMP1–8kb-caPTHR1;DMP1–8kb-Sost mice still display higher BFR compared with DMP1–8kb-Sost mice (Fig. 4B). This lack of effect of Wnt inhibition on endocortical bone formation driven by osteocytic PTH receptor signaling resulted from a converse effect on MAR (increase) and on MS/BS (decrease) in the double transgenic mice. Similarly, inhibition of resorption reduced BFR in wild type and in DMP1–8kb-caPTHR1 mice and, as a consequence, in the group of animals treated with alendronate, transgenic mice still exhibited higher BFR than wild type littermates. However, the increase in MAR induced by osteocytic PTH receptor signaling was abolished by alendronate whereas MS/BS was decreased to a similar extent in both DMP1–8kb-caPTHR and wild type littermates. Resulting from the combination of low MS/BS induced by Sost overexpression and low MAR induced by alendronate, double transgenic mice treated with alendronate exhibited a BFR lower than wild type mice receiving vehicle. These results demonstrate that cooperation between Wnt activity and resorption drives bone formation on the endocortical surface of DMP1–8kb-caPTHR1 mice.
Sost Overexpression Exacerbates and Alendronate Blunts Resorption Induced by PTH Receptor Activation in Osteocytes
Sost overexpression further increased the already high intra-cortical remodeling observed in DMP1–8kb-caPTHR1 mice. This is demonstrated by increased porosity in the femoral midshaft and in the calvaria (Fig. 5, A and B). Furthermore, osteoclasts were markedly increased in calvaria from double transgenic mice (Fig. 5, C and D). Alendronate reduced femoral intra-cortical porosity, calvarial porosity, and osteoclast number in both DMP1–8kb-caPTHR and double transgenic mice (Fig. 5, A–D). Intra-cortical bone formation coupled to resorption was high in calvaria from DMP1-caPTHR1 mice and increased further in the double transgenic mice, and decreasing resorption with alendronate blunted these effects (Fig. 5E). The reduction in bone formation is evidenced in the images of fluorochrome labeling, although quantification was not possible due to the intricate nature of the labels.
FIGURE 5.

Sost overexpression exacerbates and alendronate blunts resorption induced by PTH receptor activation in osteocytes. Intracortical porosity in the femoral diaphysis was measured by micro-CT (A) and calvarial marrow space were calculated by measuring histomorphometrically the area occupied by marrow versus bone (B). Osteoclasts were quantified in TRAP stained calvarial sections. n = 3–5 mice/group (C). Bars represent the mean ± S.D. *, p < 0.05 versus respective controls not expressing the caPTHR1 transgene and lines connect conditions with p < 0.05, by two-way ANOVA. Representative images of histologic sections showing TRAP (+) osteoclasts (D) and fluorochome labeling (E) in the calvariae of 6.0-week-old WT and DMP1–8kb-caPTHR1 mice with or without the DMP1–8kb-Sost transgene or alendronate. Bars indicate 100 μm.
Inhibition of Wnt Signaling or Resorption Have Opposite Effects on Bone Mass and Architecture in DMP1–8kb-caPTHR1 Mice
Long bones are more dense and femoral bone mineral density (BMD) is increased in DMP1–8kb-caPTHR1 mice, and overexpression of Sost reverses these effects (Fig. 6) (10). Alendronate increased BMD further in DMP1–8kb-caPTHR mice; and it partially corrected the low BMD of DMP1–8kb-caPTHR1;DMP1–8kb-Sost mice. As a result, double transgenic mice treated with alendronate display bone mass undistinguishable from wild type littermates.
FIGURE 6.

The high femoral bone mass in DMP1–8kb-caPTHR1 mice depends on Wnt activation and is further increased by inhibiting bone resorption. Representative digital x-ray images of the hindlimb (A) and femoral BMD measured by DXA (B) in 6-week-old mice expressing the DMP1–8kb-caPTHR1 and/or the DMP1–8kb-Sost transgenes or alendronate, and WT littermates is shown; n = 9–12 mice per group. Bars represent the mean ± S.D. *, p < 0.05 versus respective controls not expressing the caPTHR1 transgene and lines connect conditions with p < 0.05, by two-way ANOVA.
In cortical bone of the femoral midshaft, DMP1–8kb-caPTHR1 mice exhibit higher bone area and tissue area, increased cortical thickness, and elevated polar moment of inertia (Fig. 7, A and B). This is consistent with the increased periosteal bone formation shown in Fig. 4A. Overexpression of Sost significantly reduced all these architectural parameters in the double transgenic mice. On the other hand, inhibition of bone remodeling with alendronate did not alter bone or tissue area, cortical thickness, or polar moment of inertia in DMP1–8kb-caPTHR1 mice, but reversed the effect of Sost overexpression in DMP1–8kb-caPTHR1;DMP1–8kb-Sost mice.
FIGURE 7.

Changes in cortical bone geometry induced by PTH receptor signaling in osteocytes are abolished by Sost overexpression and this effect is counteracted by alendronate treatment. Representative images (A) and micro-CT cortical bone parameters (B) measured in the femoral mid-diaphysis in the distal femur of 6-week-old mice expressing the DMP1–8kb-caPTHR1 and/or the DMP1–8kb-Sost transgenes or alendronate, and WT littermates. n = 3–6 mice per group. Bars represent the mean ± S.D. *, p < 0.05 versus respective controls not expressing the caPTHR1 transgene and lines connect conditions with p < 0.05, by two-way ANOVA.
In cancellous bone of the distal femur, the increase in bone volume and trabecular thickness and number, and the decrease in trabecular spacing observed in DMP1–8kb-caPTHR1 mice were also prevented by Sost overexpression (Fig. 8, A and B). Alendronate increased bone volume and trabecular number in wild type, DMP1–8kb-caPTHR1 as well as DMP1–8kb-Sost mice. Moreover, the bisphosphonate reversed the decrease in bone volume and trabecular thickness in double transgenic mice, whereas it increased trabecular number to values similar of those of DMP1–8kb-caPTHR1 mice.
FIGURE 8.

Changes in volume and architecture in femoral cancellous bone of DMP1–8kb-caPTHR1 mice are blocked by Sost overexpression and enhanced by alendronate treatment. Representative micro-CT images (A) and the cancellous bone parameters bone volume (BV/TV), and trabecular thickness (Tb. Th.), separation (Tb. Sp.), and number (Tb. N.) (B) measured by micro-CT in the distal femur of 6-week-old mice expressing the DMP1–8kb-caPTHR1 and/or the DMP1–8kb-Sost transgenes or alendronate, and WT littermates. n = 4–5 mice per group. Bars represent the mean ± S.D. *, p < 0.05 versus respective controls not expressing the caPTHR1 transgene and lines connect conditions with p < 0.05, by two-way ANOVA.
Similar changes were found in the cancellous bone of the spine (Fig. 9). The increase in BMD, bone volume, and trabecular thickness and number as well as the decrease in trabecular separation observed in lumbar vertebrae of DMP1–8kb-caPTHR1 mice were reversed in double transgenic mice, to values beyond those of single DMP1–8kb-Sost mice. Alendronate administration further increased spinal BMD and trabecular thickness in DMP1–8kb-caPTHR1 mice; and increased bone mass in wild type littermates (Fig. 9, B and D). Furthermore, alendronate reversed the decrease in BMD, bone volume, trabecular thickness and number, and the increase in trabecular separation in double transgenic DMP1–8kb-caPTHR1;DMP1–8kb-Sost mice.
FIGURE 9.

Increased mass and volume and improved architecture of vertebral cancellous bone of DMP1–8kb-caPTHR1 mice depends on Wnt activation and is further enhanced by inhibiting bone resorption. A, representative micro-CT images of the 4th lumbar vertebrae. Lumbar spine BMD measured by DXA (B) and BV/TV, and Tb. Th., Tb. Sp., and Tb. N. measured by micro-CT (C) in 6-week-old mice expressing the DMP1–8kb-caPTHR1 and/or the DMP1–8kb-Sost transgenes or alendronate, and WT littermates is shown; n = 4–5 mice per group. Bars represent the mean ± S.D. *, p < 0.05 versus respective controls not expressing the caPTHR1 transgene and lines connect conditions with p < 0.05, by two-way ANOVA.
DISCUSSION
This study dissected the contribution to bone gain induced by PTH receptor signaling, of modeling, versus remodeling-dependent mechanisms, and the degree at which they operate in cortical (periosteal and endosteal) and cancellous bone surfaces.
Several aspects of our study are novel. We used DMP1–8kb-caPTHR1 transgenic mice expressing a constitutively active PTH receptor in osteocytes, a unique model that recapitulates the increased bone mass and remodeling induced by PTH elevation. Because bone formation is stimulated in all bone surfaces, this model presents an ideal opportunity to understand the mechanistic bases underlying the intricate actions of PTH in the skeleton. The approach of combining genetic and pharmacological interventions is also innovative and provides a powerful mechanistic insight because these manipulations inhibit bone formation by known mechanisms. Sost overexpression inhibits bone formation directly by antagonizing the Wnt pathway; and the bisphosphonate inhibits bone formation indirectly by stopping bone resorption.
Our report provides mechanistic information of the events that control bone anabolism by systematic analysis at different tissue sublevels. We measured the amount of mineral or BMD, the density of the mineral, the amount of bone, and its architectural distribution by micro-CT, and the rate at which osteoblasts produce bone by dynamic histomorphometry. Further, we have extended our study toward cellular and molecular levels by incorporating molecular analysis of genes that characterize osteoblasts and osteoclasts. We have also investigated which intracellular pathways are activated by PTH signaling in the osteocytes (the cells that send the signals to the osteoblasts) and in the osteoblasts themselves (the cells that execute bone formation). In concert, all this information allowed us to advance our understanding on the mechanisms by which PTH induces bone anabolism in different bone compartments and surfaces. We show that direct inhibition of bone formation blocks completely PTH-induced formation on the periosteal surface of cortical bone and the gain in bone mass in cancellous bone. In contrast, inhibition of bone resorption did not alter bone formation on the periosteal surface of cortical bone, and it even potentiated the increase in bone mass and volume in cancellous bone, in both the appendicular and axial skeletons. Further, elevated bone formation on the endocortical surface was only completely reversed by the combination of inhibiting bone formation directly and indirectly by blocking bone resorption. We conclude that PTH receptor signaling in osteocytes regulates bone mass through modeling-based and remodeling-based bone formation mechanisms that operate to a different extent in cortical and cancellous bone surfaces.
The effects of PTH on the skeleton are complex. Acting on cells of the osteoblastic lineage, PTH activates pathways that increase both bone resorption and formation (1, 3, 4). Depending on the duration and periodicity of the hormonal elevation, the balance between these two effects varies, leading to bone gain or loss on different bone envelops. However, the identity of the osteoblastic cells and the molecular mechanisms underlying PTH effects remain uncertain. Our previous findings that PTH receptor activation in osteocytes is sufficient to elicit both bone formation and bone resorption had pointed to osteocytes as crucial target cells of PTH skeletal actions (4, 9, 10). The current evidence shows that resorption influences bone formation differently depending on the bone compartment. The lack of inhibitory effect of alendronate on the elevated periosteal bone formation exhibited by DMP1–8kb-caPTHR1 mice is consistent with the fact that bone apposition on the periosteal surface is not a consequence of spatially-associated previous bone resorption (20). In contrast, the increased periosteal bone formation is blocked completely by overexpressing Sost, suggesting that the effect depends on activation of Wnt signaling. These findings raise the possibility that the increase in periosteal bone formation associated with intermittent PTH administration to humans (21) or rodents (22) are due to hormonal actions on osteocytes. This stimulatory effect contrasts with the low periosteal bone formation exhibited by mice expressing the same constitutively active PTH receptor not only in osteocytes but also in pre-osteoblasts and osteoblasts (driven by the 2.3-kb fragment of the collagen1a1 promoter, 2.3-col1-caPTHR1 mice) (23). The different outcomes might be a consequence of the purported direct inhibition of osteoblast differentiation in the 2.3-col1-caPTHR1 mice (23, 24), which evidently prevails over the stimulatory effect of activating the receptor in osteocytes. The increased versus decreased periosteal bone formation in mild versus severe chronic PTH elevation in humans (25) might be also a reflection of differential response to PTH by osteocytes versus osteoblasts. Osteocytes appear to express higher levels of PTH receptor than osteoblasts (26).6 Thus, it is possible that lower circulating PTH levels could activate downstream signaling mainly in osteocytes whereas higher hormone levels would be required for activating the receptor also in osteoblasts. This scenario could explain the increase in periosteal bone formation with mild elevations of PTH and the decreased periosteal bone formation with severe elevation of the hormone.
The current study shows that both PTHR/cAMP and PTHR/Wnt pathways are activated in osteocytes juxtaposed to periosteal as well as endosteal surfaces of cortical bone of DMP1-caPTHR1 mice. However, bone formation induced by osteocytic caPTHR is driven by one or the other pathway depending on the bone surface. Our findings underscoring the importance of Wnt signaling for bone formation on the periosteal surface of cortical bone are consistent with evidence from mouse models with genetic manipulations leading to altered levels of Wnt inhibitors/antagonists. Mice expressing low levels (40-fold reduction in calvarial bone) of the Wnt sequestering antagonist secreted frizzle-related protein 4 (SFRP4) exhibit increased periosteal bone formation rate, without changes on the endosteal surface of cortical bone (27). The effect is due exclusively to higher mineralizing surface covered by osteoblasts (MS/BS), indicating that it results from stimulation of osteoblast differentiation and recruitment to the bone surface. Similarly, deletion of the Sost gene leads to marked increased periosteal (as well as endocortical) bone formation rate (28). In this latter case, however, both MS/BS as well as MAR are elevated, demonstrating that osteoblast activity is also stimulated. Consistently, overexpression of the Wnt antagonist Dkk1 in osteoblastic cells reduces cortical bone area and periosteal perimeter (29). Further, we show that DMP1-Sost transgenic mouse exhibit low vertebral cancellous bone, similar to previous reports describing mice overexpressing human Sost under the control of the murine osteocalcin (OG-2) promoter (30) or under the control of the regulatory regions of the Sost gene (31). Cortical bone of the femur is less affected as shown by Winkler et al. (30) and in our own studies (Ref. 32 and the current manuscript). Despite the different effect of the Sost transgene by itself on cancellous versus cortical bone, Sost overexpression was equally effective in reducing the increased bone mass and volume induced by the DMP1-caPTHR1 transgene in both bone envelops. Moreover, although overexpression of Sost does not change the basal rate on bone formation, bone anabolism induced by mechanical stimulation on the periosteal surface is abrogated in mice overexpressing Sost in osteocytes, due to a reduction in both MS/BS and MAR (32). Taken together, these findings establish the requirement of Wnt signaling for bone formation on the periosteal surface of cortical bone.
Remarkably, Wnt activation contributes differently to bone formation on the endosteal surface of cortical bone, as only the increased mineralizing surface is blocked by overexpressing Sost. In contrast, alendronate reduced mineral appositional rate demonstrating the importance of factors released during bone resorption. Complete reversion of bone formation induced by PTH signaling is only accomplished by simultaneous Sost overexpression and alendronate treatment. Therefore, there is cooperation between PTH-induced Wnt activation and bone resorption, and these two effects impact specific functions of osteoblasts on the endosteal surface of cortical bone.
The disorganized and exuberant bone formation exhibited in cancellous bone of DMP1-caPTHR1 mice does not allow to measure bone formation rate in this envelop. Although BMD and micro-CT analyses demonstrate that Sost overexpression abolished bone anabolism, whether this is due to direct inhibition of bone formation (modeling) or inhibition of resorption-dependent bone formation (remodeling) is not possible to distinguish. Decreased bone mass and volume in Sost overexpressing mice could also result from exacerbated bone resorption. Indeed, osteoprotegerin (OPG), a recognized Wnt target gene (33–35), is markedly decreased in double transgenic DMP1-caPTHR1;DMP1–8kb-Sost mice (10). And, RANKL expression is increased in osteoblastic cells derived from mice lacking β-catenin (35) as well as in bones from DMP1–8kb-Sost mice (10). Moreover, circulating CTX is further increased in the double transgenic mice compared with single DMP1-caPTHR1 mice (this report). In addition, although osteoclast number could not be accurately estimated in cancellous bone, the increased calvarial osteoclasts found in single DMP1-caPTHR mice were also further increased in double DMP1-caPTHR1;DMP1–8kb-Sost mice. These pieces of evidence indicate that activation of Wnt signaling by osteocytic PTH receptor restrains bone resorption, thus contributing to increasing bone mass in cancellous bone. Consistent with this notion, blockade of resorption with alendronate leads to further bone gain in the single and double transgenic mice.
In conclusion, crosstalk between modeling-based bone formation and remodeling-based bone formation driven by PTH receptor signaling in osteocytes differentially contribute to bone gain in cortical and cancellous bone surfaces; and resorption is critical for full anabolism in cortical bone but tempers bone gain in cancellous bone (Fig. 10). Understanding the mechanistic bases of PTH receptor action would allow dissecting anabolic versus resorptive effects and specifically trigger one action or the other at will, thus providing new opportunities for the treatment of bone diseases.
FIGURE 10.
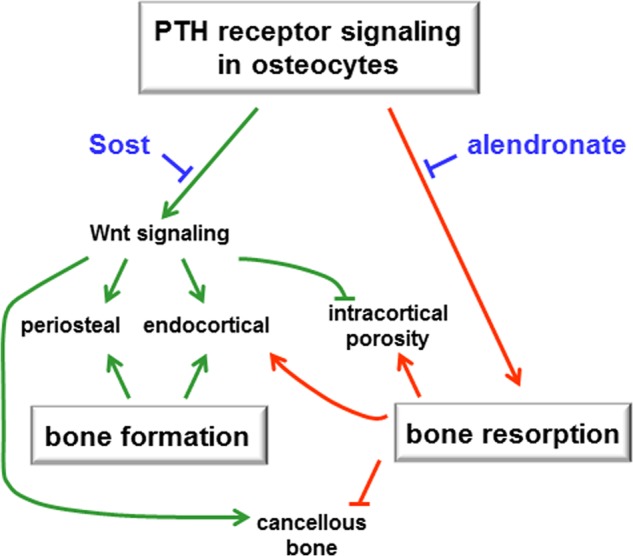
PTH receptor signaling in osteocytes and bone anabolism. Modeling-based bone formation and remodeling-based bone formation driven by PTH receptor signaling in osteocytes differentially contribute to bone gain in cortical and cancellous bone surfaces; and resorption is critical for full anabolism in cortical bone and tempers bone gain in cancellous bone.
Acknowledgments
We thank Dr. N. Bivi for helpful discussions and J. Benson for technical support.
This research was supported by Grants R01AR059357 and R01DK076007 (to T. B.) from the National Institutes of Health and the Dept. of Veterans Affairs (VA Merit Award).
T. Bellido et al., unpublished data.
- PTH
- parathyroid hormone
- BMU
- bone multicellular unit
- BFR
- bone formation rate
- MAR
- mineral apposition rate
- MS/BS
- mineralizing surface
- OCN
- osteocalcin
- CTX
- C-telopeptide fragments of type I collagen
- TRAP
- tartrate-resistant acid phosphatase
- BMD
- bone mineral density.
REFERENCES
- 1. Jilka R. L. (2007) Molecular and cellular mechanisms of the anabolic effect of intermittent PTH. Bone 40, 1434–1446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kim S. W., Pajevic P. D., Selig M., Barry K. J., Yang J. Y., Shin C. S., Baek W. Y., Kim J. E., Kronenberg H. M. (2012) Intermittent parathyroid hormone administration converts quiescent lining cells to active osteoblasts. J. Bone Miner. Res. 27, 2075–2084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hodsman A. B., Bauer D. C., Dempster D. W., Dian L., Hanley D. A., Harris S. T., Kendler D. L., McClung M. R., Miller P. D., Olszynski W. P., Orwoll E., Yuen C. K. (2005) Parathyroid hormone and teriparatide for the treatment of osteoporosis: a review of the evidence and suggested guidelines for its use. Endocr. Rev. 26, 688–703 [DOI] [PubMed] [Google Scholar]
- 4. Bellido T., Saini V., Pajevic P. D. (2013) Effects of PTH on osteocyte function. Bone 54, 250–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bellido T., Ali A. A., Gubrij I., Plotkin L. I., Fu Q., O'Brien C. A., Manolagas S. C., Jilka R. L. (2005) Chronic elevation of parathyroid hormone in mice reduces expression of sclerostin by osteocytes: a novel mechanism for hormonal control of osteoblastogenesis. Endocrinology 146, 4577–4583 [DOI] [PubMed] [Google Scholar]
- 6. Keller H., Kneissel M. (2005) SOST is a target gene for PTH in bone. Bone 37, 148–158 [DOI] [PubMed] [Google Scholar]
- 7. Yang M., Trettel L. B., Adams D. J., Harrison J. R., Canalis E., Kream B. E. (2010) Col3.6-HSD2 transgenic mice: a glucocorticoid loss-of-function model spanning early and late osteoblast differentiation. Bone 47, 573–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lavi-Moshayoff V., Wasserman G., Meir T., Silver J., Naveh-Many T. (2010) PTH increases FGF23 gene expression and mediates the high-FGF23 levels of experimental kidney failure: a bone parathyroid feedback loop. Am. J. Physiol. Renal Physiol. 299, F882–F889 [DOI] [PubMed] [Google Scholar]
- 9. O'Brien C. A., Plotkin L. I., Galli C., Goellner J., Gortazar A. R., Allen M. R., Robling A. G., Bouxsein M., Schipani E., Turner C. H., Jilka R. L., Weinstein R. S., Manolagas S. C., Bellido T. (2008) Control of bone mass and remodeling by PTH receptor signaling in osteocytes. PLoS ONE 3, e2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rhee Y., Allen M. R., Condon K., Lezcano V., Ronda A. C., Galli C., Olivos N., Passeri G., O'Brien C. A., Bivi N., Plotkin L. I., Bellido T. (2011) PTH receptor signaling in osteocytes governs periosteal bone formation and intracortical remodeling. J. Bone Miner. Res. 26, 1035–1046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rhee Y., Bivi N., Farrow E. G., Lezcano V., Plotkin L. I., White K. E., Bellido T. (2011) Parathyroid hormone receptor signaling in osteocytes increases the expression of fibroblast growth factor-23 in vitro and in vivo. Bone 49, 636–643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dempster D. W., Compston J. E., Drezner M. K., Glorieux F. H., Kanis J. A., Malluche H., Meunier P. J., Ott S. M., Recker R. R., Parfitt A. M. (2013) Standardized nomenclature, symbols, and units for bone histomorphometry: a 2012 update of the report of the ASBMR Histomorphometry Nomenclature Committee. J. Bone Miner. Res. 28, 2–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bivi N., Pacheco-Costa R., Brun L. R., Murphy T. R., Farlow N. R., Robling A. G., Bellido T., Plotkin L. I. (2013) Absence of Cx43 selectively from osteocytes enhances responsiveness to mechanical force in mice. J. Orthop. Res. 31, 1075–1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li C., Xing Q., Yu B., Xie H., Wang W., Shi C., Crane J. L., Cao X., Wan M. (2013) Disruption of LRP6 in osteoblasts blunts the bone anabolic activity of PTH. J. Bone Miner. Res. 10.1002/jbmr.1962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deleted in proof
- 16. Signorelli S., Jennings P., Leonard M. O., Pfaller W. (2010) Differential effects of hypoxic stress in alveolar epithelial cells and microvascular endothelial cells. Cell Physiol. Biochem. 25, 135–144 [DOI] [PubMed] [Google Scholar]
- 17. Plotkin L. I., Bivi N., Bellido T. (2011) A bisphosphonate that does not affect osteoclasts prevents osteoblast and osteocyte apoptosis and the loss of bone strength induced by glucocorticoids in mice. Bone 49, 122–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cheung M. S., Glorieux F. H., Rauch F. (2009) Large osteoclasts in pediatric osteogenesis imperfecta patients receiving intravenous pamidronate. J. Bone Miner. Res. 24, 669–674 [DOI] [PubMed] [Google Scholar]
- 19. Weinstein R. S., Roberson P. K., Manolagas S. C. (2009) Giant osteoclast formation and long-term oral bisphosphonate therapy. N. Engl. J. Med. 360, 53–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Martin R. B., Burr D. B., Sharkey N. A. (1998) Skeletal Tissue Mechanics, 1st Ed., Springer-Verlag, New York [Google Scholar]
- 21. Zanchetta J. R., Bogado C. E., Ferretti J. L., Wang O., Wilson M. G., Sato M., Gaich G. A., Dalsky G. P., Myers S. L. (2003) Effects of teriparatide [recombinant human parathyroid hormone (1–34)] on cortical bone in postmenopausal women with osteoporosis. J. Bone Miner. Res. 18, 539–543 [DOI] [PubMed] [Google Scholar]
- 22. Jilka R. L., O'Brien C. A., Ali A. A., Roberson P. K., Weinstein R. S., Manolagas S. C. (2009) Intermittent PTH stimulates periosteal bone formation by actions on post-mitotic preosteoblasts. Bone 44, 275–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Calvi L. M., Sims N. A., Hunzelman J. L., Knight M. C., Giovannetti A., Saxton J. M., Kronenberg H. M., Baron R., Schipani E. (2001) Activated parathyroid hormone/parathyroid hormone-related protein receptor in osteoblastic cells differentially affects cortical and trabecular bone. J. Clin. Invest. 107, 277–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Calvi L. M., Shin H. I., Knight M. C., Weber J. M., Young M. F., Giovannetti A., Schipani E. (2004) Constitutively active PTH/PTHrP receptor in odontoblasts alters odontoblast and ameloblast function and maturation. Mech. Dev. 121, 397–408 [DOI] [PubMed] [Google Scholar]
- 25. Parfitt A. M. (2002) Parathyroid hormone and periosteal bone expansion. J. Bone Miner. Res. 17, 1741–1743 [DOI] [PubMed] [Google Scholar]
- 26. Powell W. F., Jr., Barry K. J., Tulum I., Kobayashi T., Harris S. E., Bringhurst F., Pajevic P. (2011) Targeted ablation of the PTH/PTHrP receptor in osteocytes impairs bone structure and homeostatic calcemic responses. J. Endocrinol. 209, 21–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nakanishi R., Shimizu M., Mori M., Akiyama H., Okudaira S., Otsuki B., Hashimoto M., Higuchi K., Hosokawa M., Tsuboyama T., Nakamura T. (2006) Secreted frizzled-related protein 4 is a negative regulator of peak BMD in SAMP6 mice. J. Bone Miner. Res. 21, 1713–1721 [DOI] [PubMed] [Google Scholar]
- 28. Li X., Ominsky M. S., Niu Q. T., Sun N., Daugherty B., D'Agostin D., Kurahara C., Gao Y., Cao J., Gong J., Asuncion F., Barrero M., Warmington K., Dwyer D., Stolina M., Morony S., Sarosi I., Kostenuik P. J., Lacey D. L., Simonet W. S., Ke H. Z., Paszty C. (2008) Targeted deletion of the sclerostin gene in mice results in increased bone formation and bone strength. J. Bone Miner. Res. 23, 860–869 [DOI] [PubMed] [Google Scholar]
- 29. Li J., Sarosi I., Cattley R. C., Pretorius J., Asuncion F., Grisanti M., Morony S., Adamu S., Geng Z., Qiu W., Kostenuik P., Lacey D. L., Simonet W. S., Bolon B., Qian X., Shalhoub V., Ominsky M. S., Zhu K. H., Li X., Richards W. G. (2006) Dkk1-mediated inhibition of Wnt signaling in bone results in osteopenia. Bone 39, 754–766 [DOI] [PubMed] [Google Scholar]
- 30. Winkler D. G., Sutherland M. K., Geoghegan J. C., Yu C., Hayes T., Skonier J. E., Shpektor D., Jonas M., Kovacevich B. R., Staehling-Hampton K., Appleby M., Brunkow M. E., Latham J. A. (2003) Osteocyte control of bone formation via sclerostin, a novel BMP antagonist. EMBO J. 22, 6267–6276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Loots G. G., Kneissel M., Keller H., Baptist M., Chang J., Collette N. M., Ovcharenko D., Plajzer-Frick I., Rubin E. M. (2005) Genomic deletion of a long-range bone enhancer misregulates sclerostin in Van Buchem disease. Genome Res. 15, 928–935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tu X., Rhee Y., Condon K. W., Bivi N., Allen M. R., Dwyer D., Stolina M., Turner C. H., Robling A. G., Plotkin L. I., Bellido T. (2012) Sost downregulation and local Wnt signaling are required for the osteogenic response to mechanical loading. Bone 50, 209–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kramer I., Halleux C., Keller H., Pegurri M., Gooi J. H., Weber P. B., Feng J. Q., Bonewald L. F., Kneissel M. (2010) Osteocyte Wnt/beta-catenin signaling is required for normal bone homeostasis. Mol. Cell Biol. 30, 3071–3085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Glass D. A., 2nd, Bialek P., Ahn J. D., Starbuck M., Patel M. S., Clevers H., Taketo M. M., Long F., McMahon A. P., Lang R. A., Karsenty G. (2005) Canonical Wnt signaling in differentiated osteoblasts controls osteoclast differentiation. Dev. Cell 8, 751–764 [DOI] [PubMed] [Google Scholar]
- 35. Holmen S. L., Zylstra C. R., Mukherjee A., Sigler R. E., Faugere M. C., Bouxsein M. L., Deng L., Clemens T. L., Williams B. O. (2005) Essential role of beta-catenin in postnatal bone acquisition. J. Biol. Chem. 280, 21162–21168 [DOI] [PubMed] [Google Scholar]