

Background: Leptin expression is induced in lung diseases and lung cancer, but the mechanism of leptin gene expression remains elusive.
Results: Leptin mediates leptin and leptin receptor expression, setting up a feed-forward loop.
Conclusion: DNA elements and intracellular signals activating leptin gene expression were identified.
Significance: Mechanism of leptin/leptin receptor gene regulation will aid in targeting leptin signaling in lung pathologies.
Keywords: Adipokines, Gene Regulation, Inflammation, Lung, Molecular Cell Biology, Electric Cell Substrate Impedance Sensing
Abstract
Elevated levels of systemic and pulmonary leptin are associated with diseases related to lung injury and lung cancer. However, the role of leptin in lung biology and pathology, including the mechanism of leptin gene expression in the pathogenesis of lung diseases, including lung cancer, remains elusive. Here, using conditional deletion of tumor suppressor gene Pten in the lung epithelium in vivo in transgenic mice and human PTEN-null lung epithelial cells, we identify the leptin-driven feed-forward signaling loop in the lung epithelial cells. Leptin-mediated leptin/leptin-receptor gene expression likely amplifies leptin signaling that may contribute to the pathogenesis and severity of lung diseases, resulting in poor clinical outcomes. Loss of Pten in the lung epithelial cells in vivo activated adipokine signaling and induced leptin synthesis as ascertained by genome-wide mRNA profiling and pathway analysis. Leptin gene transcription was mediated by binding of transcription factors NRF-1 and CCAAT/enhancer-binding protein δ (C/EBP) to the proximal promoter regions and STAT3 to the distal promoter regions as revealed by leptin promoter-mutation, chromatin immunoprecipitation, and gain- and loss-of-function studies in lung epithelial cells. Leptin treatment induced expression of the leptin/leptin receptor in the lung epithelial cells via activation of MEK/ERK, PI3K/AKT/mammalian target of rapamycin (mTOR), and JAK2/STAT3 signaling pathways. Expression of constitutively active MEK-1, AKT, and STAT3 proteins increased expression, and treatment with MEK, PI3K, AKT, and mTOR inhibitors decreased LEP expression, indicating that leptin via MAPK/ERK1/2, PI3K/AKT/mTOR, and JAK2/STAT3 pathways, in turn, further induces its own gene expression. Thus, targeted inhibition of the leptin-mediated feed-forward loop provides a novel rationale for pharmacotherapy of disease associated with lung injury and remodeling, including lung cancer.
Introduction
Leptin (LEP)2 is a 16-kDa pleiotropic hormone and a pro-inflammatory adipokine/cytokine. LEP binds to the leptin receptor (LEPR) and activates multiple intracellular signaling pathways (1, 2). Elevated levels of LEP in the lung and serum are associated with, and potentially exacerbate, severity and progression of lung diseases, including acute lung injury (ALI), acute respiratory distress syndrome (ARDS), chronic obstructive pulmonary disease (COPD), airway remodeling associated with asthma, and lung cancer (3–9). In patients, circulating and airway LEP concentrations negatively correlate with lung function (10). Increased LEP expression and secretion following lung injury promotes fibroproliferation, contributing to pulmonary fibrosis (11), particularly in the setting of hyperoxia-induced ALI (11, 12). Pulmonary LEP is also increased in asymptomatic smokers and in mice exposed to cigarette smoke where it modulates innate and adaptive immune cell recruitment (4, 7). In contrast, resistance to the effects of LEP attenuates lung disease pathology, whereas reduction in LEP levels is a strong predictive factor in the improvement of lung function (13, 14). Thus, accumulating evidence indicates that LEP is causally linked to the pathogenesis of many lung diseases associated with injury as well as lung cancer.
ALI/ARDS and COPD cause considerable morbidity and mortality (15), and close to 160,000 people die of lung cancer every year in the United States alone, imposing a major healthcare burden (16). Although clinical interventions do improve alveolar functions marginally in some patients, deterioration of lung function cannot be prevented in these diseases, leading to respiratory failure and death (17). Despite advancements in understanding the pathophysiology of ALI, ARDS, COPD, asthma, and lung cancer, how LEP is induced and contributes to the severity and progression of these lung diseases remains poorly understood (18). LEP and adiponectin (encoded by the ADIPOQ gene) and their respective receptors are expressed by human lung bronchiolar and type II epithelial cells (6). Airway LEP concentrations are high in COPD patients, whereas increased LEP is associated with greater airway inflammation and disease severity in asthma patients; however, these data remain conflicting (6, 10).
LEP signals are pro-angiogenic, pro-inflammatory, and mitogenic and are mediated via multiple cross-regulatory pathways involving oncogenes, cytokines, and growth factors, driving growth of solid tumors (19, 20). LEP activates JAK2/STAT3, MAPK/ERK1/2, and PI3K/AKT signaling pathways (2, 21). As a pro-angiogenic factor, LEP up-regulates VEGF and its receptor VEGF receptor 2 via activation of the IL-1 signaling pathway (1, 22, 23). However, despite the importance of well established LEP signaling pathways outside the adipose tissue and their roles in disease pathology, the mechanisms of regulation and induction of Leptin (LEP) gene expression largely remains limited to adipocytes. Although the LEP gene proximal promoter was defined (24, 25), its role in transcriptional regulation of LEP expression remains limited to adipocytes, including the roles of transcription factors SP1, glucocorticoid receptor, cAMP-response element-binding protein, peroxisome proliferator-activated receptor-γ, C/EBP-α, AP-2, and SREBP1c (26–30). Given the emerging role of LEP in the structural and functional maintenance of the normal and injured lung as well as in the progression of lung cancer (5, 6), it is imperative that a transcriptional regulatory mechanism, especially induction of LEP gene expression in lung epithelial cells, be elucidated.
In this study, we demonstrate that loss of Pten in the lung epithelium in vivo in transgenic mice and in PTEN-null human lung epithelial cells induced LEP signaling in lung epithelial cells. LEP-mediated transcription of LEP and LEPR was mediated by binding of transcription factors NRF-1 and C/EBP-δ to the proximal and STAT3 to the distal LEP gene promoter in lung epithelial cells. Increased LEP expression in PtenΔ/Δ respiratory epithelial cells elicited an autocrine feed-forward loop via up-regulation of LEPR on the lung epithelial cells. LEP/LEPR signaling loop was driven by activation of PI3K/AKT/mTOR, MEK/ERK, and JAK/STAT3 pathways. These three signaling pathways activated expression of both LEP and LEPR, setting up a positive feed-forward LEP/LEPR signaling loop in the lung epithelium. Taken together, aberrant amplification of the LEP-mediated LEP signaling loop potentially deregulates the modulatory role of LEP, likely exacerbating the severity of lung diseases, including cancer, leading to poor clinical outcomes.
MATERIALS AND METHODS
Generation of Transgenic Mouse Lines
Compound transgenic mice harboring the Pten gene with loxP-flanked exon-V (Ptenflox/flox), SP-C-rtTAtg/−, and TetO-Cretg/− were generated and genotyped as described previously (31) with mice harboring SPC-rtTA/Ptenflox/flox, TetO-Cretg/−/Ptenflox/flox, SPC-rtTA, or TetO-Cretg/− as controls. Likewise, doxycycline treatment induced tumors in CCSP-rtTA/TetO-Cretg/−/LSL-KrasG12D/PtenΔ/Δ mice between 10 and 12 weeks of age. Mice expressing rtTA, or bearing TetO-Cretg/− alone, were the normal controls. Animal studies were reviewed and approved by the Institutional Animal Care and Use Committee of the University of South Florida. Mice were housed in humidity- and temperature-controlled rooms on a 12-h light/12-h dark cycle with food and water ad libitum. There was no serologic or histologic evidence of either pulmonary pathogens or infections in sentinel mouse colonies. Gestation was dated E0.5 by vaginal plug. Mice were killed by injection of anesthetic to obtain lung tissue at ∼12 weeks when tachypnea associated with lethargy was observed.
RNA Microarray Analysis
Lung cRNA was hybridized to the murine genome MOE430 chips (Affymetrix) according to the manufacturer's protocol. Affymetrix Microarray Suite 5.0 was used to scan and quantitate the gene chips under default settings. Normalization was performed using the robust multichip average model (32, 33). Data were analyzed using Genespring 7.2 (Silicon Genetics). A volcano plot was used to identify significance (negative log of p values from Welch's approximate t test on y axis) and magnitude of change (log2 of fold change on the x axis) in the expression of a set of genes between PtenΔ/Δ mice and control littermates (34). The selection criteria included a p value of 0.05 or less by two-tailed Student's t test, false discovery rate (35, 36) of no more than 10% (37), and fold change of at least 1.5. Differentially expressed genes were subjected to an additional filter and classified according to Gene Ontology classification on the Biologic Process using the publicly available web-based tool DAVID (38). The Fisher exact test was used to calculate the probability of each gene ontology category that was over-represented in the selected list, using the entire MOE430 mouse genome as a reference data set. Differentially expressed genes (p < 0.05, two-tailed Student's t test; fold change, >1.5) were compared, and correlations of transcript changes among three microarray experiments were measured.
Bioinformatic Analyses of Differentially Regulated Genes
The differentially regulated genes were enriched into different functional clusters using the DAVID Bioinformatic resources 6.7 and quantitatively measured by statistical methods, including χ2, Fisher's exact test, binomial probability, and hypergeometric distribution. Pathway analysis was performed on the enriched clusters using DAVID pathway viewer, GeneGo, and Ingenuity software suites, and top scoring pathways were considered for biologic interpretation. Analysis of the promoter regions of the top 20 PTEN-responsive genes (up and down-regulated) was done using the MatInspector tool (default settings) of the Genomatix software suite.
Cell Culture, Transfection, and Reporter Gene Assays
H1650 cells (ATCC CRL-5883), a gift from Dr. Chellappan (H. Lee Moffitt Cancer Center and Research Institute, University of South Florida, Tampa FL), were cultured in RPMI 1640 medium (Invitrogen) with 10% fetal bovine serum and 5% mixture of penicillin G, streptomycin, and plasmocin (Invitrogen) in a 5% CO2 incubator at 37 °C. A series of LEP promoter-luciferase constructs were used in transient transfection assays using the PEI method (39). Briefly, 6-well plates at 30–50% confluence were transfected with a fixed amount of LEP promoter-luciferase plasmid and various amounts of CMV-based cDNA expressing transactivator plasmids. Total DNA was normalized with corresponding CMV-empty vectors, and transfection efficiency was normalized to β-galactosidase activity using 100 ng/well of pCMV β-galactosidase. Two days after transfection, luciferase and β-galactosidase assays were performed using 50 μl of the supernatant. The light units were assayed by luminometry (MLX, Microtiter Luminometer, DYNEX). Data obtained represent the average of three transfection experiments, each carried out in triplicate and depicted as means ± S.E. unless stated otherwise. Primer sequences for LEP promoter-luciferase constructs were as follows: BGL-HU-LEP-B1-R, cggaacagatcttgcaaccgctggcgctg; 800MluHuLEP-F3, gcgagcacgcgttgacaaaaacgtggctacatctggg; 620MluHuLep-F4, gcgagcacgcgtgaggcttggaactcgattctccg; 399MluHuLep-F5, gcgagcacgcgtcggagcccctcacagcca; 150MluHuLep-F6, gcgagcacgcgtcggcacgtcgctaccctgag; 89MluHuLep-F7, gcgagcacgcgtcggggcagttgcgcaagt; and 52MluHuLep-F8, gcgagcacgcgtagttgtgatcgggccgctataagag.
RNA Isolation and Real Time PCR Assays
Total RNA was isolated from 60 to 80% confluent H1650 cells grown in RPMI 1640 medium using TRIzol reagent (Ambion) as per the manufacturer's instructions. Total RNA was treated with RQ1 RNase-free DNase (Promega) and purified using the RNeasy MinElute cleanup kit (Qiagen). Purified RNA was converted into cDNA using the SuperScript® III reverse transcriptase kit (Invitrogen) and used for real time PCR assays. cDNA samples were mixed with 10 μl of 2× Fast SYBR Green real time PCR master mix containing gene-specific primers. The reaction mixture was denatured at 95 °C for 3 min, followed by 40 cycles of PCRs with the following settings: 95 °C for 15 s, 60 °C for 15 s, and 72 °C for 20 s. The PCR was monitored by the ABI StepOnePlusTM real time PCR system (ABI PRISM 7700; Applied Biosystems, Foster City, CA), and the results were analyzed with the ABI StepOnePlusTM real time PCR version 2.0 software (ABI PRISM 7700). Sequences for primers used were as follows: Lep-R, caccaaaaccctcatcaagaca, and Lep-F, gatagaggcccaggcatttttta; LEPR-F, tagagaaggccagcacgtgaa, and LEPR-R, acaccactctctctctttttgattga; GAPDH-F, tgttgccatcaatgacccctt, and GAPDH-R, ctccacgacgtactcagcg; and NRF-1-F, ccgaggacacctcttacgatg, and NRF-1-R, tacatgaggccgtttccgttt.
RNA Interference Assay
Short hairpin RNAs (shRNAs) specific to human NRF-1 (Hu-SH-29, 29-mer shRNA constructs in retroviral GFP vector) were purchased from OriGene Technologies, Inc. (Rockville, MD). A noneffective 29-mer scrambled shRNA cassette in pGFP-V-RS was used as control. NRF-1 shRNA and scrambled control (3 μg/ml) were transfected into H1650 cells, and RNA was isolated from cells after 48 h. The efficiency of shRNA-based interference of NRF-1 was monitored via real time PCR analysis and gene-specific NRF-1 primers.
Site-directed Mutagenesis
The CEBP-δ and NRF-1 mutant plasmids were generated using site-directed mutagenesis (QuikChange Lightening site-directed mutagenesis kit, Agilent Technologies). Briefly, Lep150 was used as a template with oligonucleotides containing mutations in the CEBP-δ and NRF-1 sites (listed below) to generate PCR products. An annealing temperature of 51 °C was employed for 18 cycles with an extension time of 3 min at 68 °C. This was followed by DpnI digestion of the parental DNA and transformation in XL10-Gold Ultracompetent cells using β-mercaptoethanol provided in the kit. This was followed by routine plating and colony culture procedures. The mutants were confirmed by sequencing. Sequences for primers used are as follows: CEBP-δ (−60/−53): forward primer, 5′-ggcagttcagtacgttgtgatcg-3′, and reverse primer, 5′-acaacgtactgaactgcccg-3′; NRF-1 (−81/−78): forward primer, 5′-tagaaatacaccggggcctg-3′, and reverse primer, 5′-caggccccggtgtatttcta-3′.
Chromatin Immunoprecipitation (ChIP) Assay
ChIP lysates were made using the ChIP-IT Express Magnetic Chromatin Immunoprecipitation kits (Active Motif, Carlsbad, CA). H1650 cells that were 90% confluent were treated with formaldehyde solution, and the chromatin was isolated, digested, and immunoprecipitated as per the manufacturer's instructions. The sheared chromatin was incubated with antibody directed against NRF-1, C/EBP-δ, and STAT-3, and the antibody-bound protein-DNA complexes were precipitated using magnetic protein G-coupled beads. The captured chromatin was eluted and then uncross-linked, and the DNA was recovered. ChIP DNA was subjected to RT-PCR using specific primers flanking the DNA-binding sites for NRF-1, C/EBP-δ, and STAT3. Sequences for primers used are as follows: NRF-1/CEBP-D-ChIP-R, cggaacagatcttgcaaccgctggcgctg, and NRF-1/CEBP-D-ChIP-F, gcgagcacgcgtcggcacgtcgctaccctgag; STAT3-ChIP-R, tcctctctttgtactctctctctttatttctcagc, and STAT3-ChIP-F, ccagatgcagtggctcatgcttgta; GAPDH-ChIP-R, tactagcggttttacgggcg, and GAPDH-ChIP-F, tcgaacaggaggagcagagagcga.
Immunohistochemistry
Lungs from experimental mice PtenΔ/Δ (n = 10 total) and control littermates (n = 8 total) were inflation-fixed by gravity (25 cm of water pressure) with 4% paraformaldehyde in PBS, removed from the chest, and immersed in fixative overnight at 4 °C. The tissue samples were rinsed in PBS, dehydrated, and then embedded in paraffin blocks. Sections were cut at 5-μm intervals, and antigen retrieval was done using pepsin. 3,3′-Diaminobenzidine was used as a substrate, and sections were counter-stained with Mayer's hematoxylin (BioGenex, Fremont, CA) to assess histologic changes.
LEP Treatments and Protein Analysis
PTEN-deficient H1650 lung cancer cell lines were plated at 1 × 106 cells per well of a 6-well plate and allowed to attach overnight. Cells then were serum-deprived for 24 h followed by treatment with 100 ng/ml human recombinant LEP (R&D Systems, Minneapolis, MN) for 48 and 72 h in serum-free medium. For measurements of ERK/MAPK activity after LEP treatment, cells were treated with 100 ng/ml human recombinant LEP (R&D Systems, Minneapolis, MN) for 15 and 30 min. Proteins were isolated post-LEP treatment and separated by SDS-PAGE on 10% gel and electroblotted to nitrocellulose membranes (0.1 μm; Invitrogen). Blots were blocked with 5% nonfat dry milk in TBST (10 mm Tris, pH 8, 150 mm NaCl, 0.1% Tween 20) and incubated with 1:1000 diluted specific primary antibodies to P-mTOR (catalog no. 2971), P-AKT (catalog no. 9271) and phospho-p44/42 ERK1/2 (Thr-202/Tyr-204, catalog no. 4370) from Cell Signaling Technology. p-STAT3 (Y-705, catalog no. 2236-1) was from EPITOMICS, and ERK1/2 (Abcam; catalog no. AB17942) and β-actin (catalog no. A5060; Sigma) peroxidase-conjugated AffiniPure goat anti-rabbit IgG secondary antibody (Jackson ImmunoResearch) were used at a 1:1000 concentration. Peroxidase-conjugated AffiniPure goat anti-rabbit IgG secondary antibody (Jackson ImmunoResearch) was used at 1:10,000 concentration. Blots were developed by chemiluminescence (Pierce) and autoradiographed.
Cell Proliferation Assay
Cell proliferation assays were performed using Cell Counting kit-8 (Fluka, BioChemika). Cells were plated in 96-well plates at increasing density ranging from 5 × 103 to 1.2 × 104 cells/well and cultured in RPMI 1640 growth medium as described above. The cells were transferred to serum-free medium for 16 h and replenished with serum-free medium containing increasing concentrations (50–200 ng/ml) of human recombinant LEP (R&D Systems Minneapolis, MN). At the indicated time points, the cell numbers in triplicate wells were measured as a function of absorbance (450 nm) of reduced WST-8 (2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium, monosodium salt).
Inhibitor Studies
Dose-response studies for pathway-specific inhibitors of PI3K/mTOR (BEZ-235, LY294002), MEK1/2 (U0126), and AKT 1/2/3 (MK-2206) (Selleck Chemicals) were carried out on 30–40% confluent H1650 cells grown in RPMI 1640 medium for 24 h in the presence of LEP promoter fragment (150 bp). Post-transfection, luciferase and β-galactosidase assays were performed using 50 μl of the supernatant on MLX, Microtiter Luminometer, (DYNEX). Data obtained represent the average of three transfection experiments, each carried out in triplicate and depicted as means ± S.E. unless stated otherwise.
Electric Cell Substrate Impedance Sensing Wounding (Migration) Assay
H1650 cells were grown on electric cell substrate impedance sensing 8-well plate arrays (8W1E; Applied Biophysics, Troy, NY) in growth media with serum until fully confluent, after which the media were replaced with serum-free media for 24 h. Serum-deprived cells were treated with 100 ng/ml of human recombinant LEP (R&D Systems, Minneapolis, MN) for 2 h prior to wounding. Cells were wounded using an elevated field pulse of 1400 mA at 32,000 Hz applied for 20 s, producing a uniform circular lesion of 250 mm in size, and wounds were tracked over a period of 24 h. The impedance (Z) was measured at 4000 Hz, normalized to its value at the initiation of data acquisition, and plotted as a function of time. Assays were performed in triplicate and reported as mean ± S.E. unless stated otherwise (p value of ≤0.05).
RESULTS
Loss of Pten Induces LEP Signaling in the Lung Epithelial Cells in Vivo and in Vitro
Triple transgenic mice harboring a conditional Pten allele were developed (Fig. 1A), and Pten was conditionally deleted in vivo from the lung epithelial cells using the doxycycline-dependent Cre/LoxP approach (Fig. 1A(i)) as described previously (31). Pten was selectively deleted in the respiratory epithelial cells after administration of doxycycline to the dam (Fig. 1A(ii)). At birth, the transmission of all of the genes followed Mendelian inheritance as confirmed by genotyping (Fig. 1A(iii)). Selective deletion of Pten gene in the lung resulted in epithelial hyperplasia at 20 weeks as compared with control littermates, and the bronchial epithelium in PtenΔ/Δ mice was hypercellular (Fig. 1A(iv), red arrow). Microarray analysis of lung RNA isolated from Pten-deleted lung epithelial cells (PtenΔ/Δ) and control mice revealed that expression of 1389 genes was altered significantly (≥2-fold change, p value of ≤0.05) (Fig. 1B). Network enrichment analysis of the Pten-responsive genes using the Metacore software suite (40, 41) (Metacore from GeneGo Inc. New York) revealed that the LEP signal transduction is among the most significantly perturbed networks (Fig. 2C). This was independently confirmed by the disease enrichment analysis using the Ingenuity Pathway Analysis (IPA) suite (42), which identified LEP signaling in obesity as one of the most significantly altered pathways (Fig. 2D). Loss of Pten in the lung epithelial cells up-regulated the adipocyte signaling pathway, including a number of genes involved in the LEP pathway as assessed by the KEGG signaling pathway database (Fig. 2A) (43). Robust expression of LEP expression was observed after deletion of Pten (PtenΔ/Δ) in lung adenocarcinomas (Fig. 2B) developed in mice. To identify a PTEN null cancer cell line, we compared endogenous PTEN protein expression in A549, H292, and H1650 human lung epithelial cell lines (Fig. 3A). Because H1650 did not show any expression of PTEN as opposed to the other two cell lines, we used it for all further experiments. Significant expression of LEP and LEPR mRNAs (Fig. 3B) and LEP/LEPR proteins (Fig. 3C) was observed in PTEN-deficient human lung cancer cells (NCI-H1650), consistent with the concept that the LEP/LEPR signaling pathway is operational and may be up-regulated following loss of Pten in lung epithelial cells, including in lung cancer.
FIGURE 1.

Generation of PtenΔ/Δ mice and microarray profiling of PtenΔ/Δ lung mRNA identifies 1389 Pten-responsive genes. A, generation of the PtenΔ/Δ mice. Triple-transgenic mice containing three alleles, loxP-flanked exon V (Ptenflox/flox), SP-C-rtTAtg/−, and OTet-Cretg/− were produced (i and ii) and selected by genotyping (iii). Mice harboring SPC-rtTAtg/−/Ptenflox/flox, OTetCretg/−/Ptenflox/flox, or Ptenflox/flox were used as controls. Lungs were harvested from 5-month-old mice. Hematoxylin/eosin staining of lung sections from PtenΔ/Δ mice demonstrated normal branching morphogenesis (indicated by blue arrows) and postnatal lung formation with increased hyperplasia (indicated by red arrows) (iv). B, microarray analysis of lung RNA isolated from Pten-deleted lung epithelial cells (PtenΔ/Δ) (orange oval) and control mice (blue oval) revealed that expression of 1389 genes was altered significantly (≥2-fold change, p value ≤ 0.05).
FIGURE 2.

Activation of the adipocytokine signaling pathway in PtenΔ/Δ mouse lung and expression of lep and lepr receptor in lung epithelial cells. A, KEGG pathway analysis of Pten-responsive genes identified adipocytokine signaling as being significantly up-regulated. Lep, Lepr, and adiponectin (Adipoq) were up-regulated in the PtenΔ/Δ mice and overlapped (red stars) on the canonical KEGG adipocyte signaling pathway, suggesting that loss of Pten activated adipokine synthesis and signaling in the lung epithelium. B, immunostaining with LEP antibodies confirmed exuberant induction and secretion of Lep in K-rasG12D (89)-driven PtenΔ/Δ lung tumors in mice. C, enrichment for gene networks in PtenΔ/Δ mice. Biologic networks were enriched from the Pten-responsive gene set using standard software tools from MetaCoreTM. Lep signaling was identified as a significant metabolic pathway activated in the PtenΔ/Δ enriched list (p value = 0.007). D, likewise, application of Ingenuity Systems software tool called Intelligent Pathway Analysis (IPATM) independently enriched the LEP signaling pathway (p value = 0.04).
FIGURE 3.

H1650 human lung adenocarcinoma cells are PTEN-deficient and express high leptin and leptin receptor mRNA. Comparison of different lung cancer cell lines reveals H1650 cells are PTEN-deficient (A) and express the highest levels of LEP and its receptor LEPR mRNA (B). C, leptin and leptin receptor protein levels as a result of PTEN deletion as assessed in H1650 cells, suggesting the likely presence of a functional LEP signaling pathway in lung epithelial cells. D, dose-dependent increase in H1650 cell proliferation (∼3-fold) was observed when cells were treated for 48 h with increasing concentrations (50, 100, and 200 ng/ml; lane 2–4) of human recombinant LEP. E, continuous impedance sensing measurements identify greater wound closure efficiency in cells treated with 100 ng/ml leptin prior to wounding as compared with control cells without leptin. F, identification of the LEP gene core promoter region in H1650 lung cancer cell line revealed a proximal enhancer containing NRF-1 and δ-binding sites. Subconfluent cultures were transiently transfected with various promoter-reporter deletion constructs derived from 5′-upstream regulatory sequence of the LEP gene. Luciferase activity was expressed relative to the base-line luciferase activity of a promoter-less luciferase reporter construct (pGL3-Basic) set to unity. Data are represented from four independent experiments performed in triplicate (± S.E.; *, p value ≤ 0.05). G, diagrammatic representation of the most active promoter region −149 to +21 bp of the LEP gene (Luc-150), including proximal enhancers depicting the positions of NRF-1 and C/EBP sites.
LEP Directly Influences Lung Epithelial Cell Physiology by Inducing Cell Proliferation and Wound Healing
Pathophysiologic alterations in the lung after Pten loss in the respiratory epithelium have been characterized in great detail (31, 44–47). We hypothesized that an increase in LEP signaling will likely alter lung epithelial cell physiology and behavior, and as a hallmark of LEP signaling, treatment with LEP should enhance proliferation of H1650 cells. To test this premise, we treated exponentially growing H1650 cells with increasing concentrations of LEP. There was an ∼3-fold increase in cell proliferation in a dose-dependent manner (Fig. 3D), indicating a physiologic response to LEP in H1650 lung epithelial cells, consistent with studies on other cancer cells (48). Using the electric cell-substrate impedance sensing method (49–52), we assessed the role of leptin in wound healing in PTEN-null H1650 cells. For the wound-healing assays, confluent H1650 cells were serum-starved for 24 h on ECIS 8W1E plates treated with 100 ng/ml leptin, and impedance values were measured. Real time measurements of impedance values prior to wounding clearly indicated that cells treated with leptin reached confluence earlier than the ones without treatment (Fig. 3E). As shown in Fig. 3E, the application of the high field pulse led to a drastic drop of cell impedance. Post-wounding (as represented by the dotted line) on control H1650 cells (no leptin treatment) had a lower impedance value as compared with leptin-treated H1650 cells (Fig. 3E), indicating that PTEN-null H1650 cells showed a physiologic response to leptin in the medium. Taken together, leptin treatment increased cell proliferation (Fig. 3D) and accelerated wound healing due to increased cell migration (Fig. 3E). Consequently, it is highly likely that pathologic conditions associated with increased LEP synthesis and secretion may accompany LEP binding to the LEP receptor on lung epithelial cells, activating its own synthesis and setting up a self-sustaining feed-forward auto-regulatory loop that may further drive pathogenic events in the lung following injury or in lung cancer. Therefore, we sought to define the molecular mechanisms underlying LEP-mediated LEP gene expression.
LEP Gene Expression Is Regulated by a Proximal Enhancer via Transcription Factors NRF-1 and CEBP-δ
To define potential proximal enhancer elements that drive LEP gene expression in PTEN-null lung epithelial cells, luciferase reporter plasmids comprising various lengths of the 5′-upstream regulatory regions of the LEP gene promoter were subcloned. Transcription activity of each promoter-deletion plasmid was evaluated in transient transfection assays using H1650 PTEN-null lung epithelial cells (Fig. 3F). The promoter region spanning −149 to +21 (Luc-150) was found to be transcriptionally the most active, although further deletion of the promoter up to −52 bp (Luc-50) decreased the activity to basal levels, indicating the presence of a proximal enhancer element in the region between −149 and −50 bp (Fig. 3F). Consensus sites for NRF-1 (pink solid box) and C/EBP (green solid box) were identified in the region spanning −83 to −78 bp and −60 to −53 bp from the transcription start site, respectively (Fig. 3G), using MatInspector 7.0 (53), a transcription factor-binding site identification software derived from the Genomatix Suite (Genomatix Software GmbH, Munich, Germany).
To ascertain the role of transcription factors NRF-1 and C/EBP in transcriptional regulation of the LEP gene via the proximal enhancer, constitutively active NRF-1 (CA-NRF-1) was co-transfected with Luc-150 in H1650 cells. Dose-dependent expression of CA-NRF-1 increased the transcriptional activity from Luc-150 (Fig. 4A, lanes 3–5). In contrast, co-transfection of Luc-150 with increasing amounts of dominant negative NRF-1 (DN-NRF-1) in H1650 cells decreased Luc-150 activity (Fig. 4B, lanes 3–5), indicating that NRF-1 was able to transcriptionally activate the LEP gene via the proximal enhancer that contains the NRF-1-binding element. The role of the C/EBP site within the proximal enhancer (−60/−53 bp) was examined by co-transfection of Luc-150 together with either C/EBP-α-, C/EBP-β-, or C/EBP-δ-expressing plasmids because all three forms bind similar DNA consensus sites and are highly expressed in lung epithelial cells (54, 55). Plasmids expressing C/EBP-δ selectively up-regulated Luc-150 activity by ∼20-fold (Fig. 4C, lane 5) as opposed to C/EBP-α and C/EBP-β, which showed 4–5-fold activation (Fig. 4C, lanes 3 and 4), indicating that C/EBP-δ likely plays a major regulatory role in transcriptional activation of the LEP gene. Indeed, dose-dependent co-transfection of C/EBP-δ and fixed amounts of Luc-150 activated Luc-150 activity (Fig. 4D, lanes 3–5), whereas co-transfection of dominant negative C/EBP-δ (DN-C/EBP-δ) abrogated Luc-150 activity (Fig. 4D, lanes 6 and 7).
FIGURE 4.

NRF-1 and CEBP-δ activate LEP gene proximal promoter. A, NRF-1 increased LEP promoter activity as assessed after co-transfection of a fixed amount of Luc-150 (0.1 μg) together with increasing amounts of CA-NRF-1 expression plasmid (1, 2, and 4 μg/per well). All wells were normalized with empty pCDNA control vector as empty vector had no significant activity. B, expression of dominant negative NRF-1 (DN-NRF-1) repressed LEP gene promoter activity as assessed after co-transfection of a fixed amount of Luc-150 (0.1 μg), with increasing amounts of DN-NRF-1 expression plasmid (1, 2, and 4 μg/per well). C, CEBP-δ selectively activated LEP gene proximal promoter after co-transfection of a fixed amount of Luc-150 (0.1 μg) and 4 μg of CEBP-α, -β, and -δ expression plasmids. D, expression of increasing amounts of CEBP-δ (0.25, 0.5, and 1 μg/per well) stimulated (lanes 3–5), whereas the dominant negative form (0.5 and 1 μg) of CEBP-δ (DN-CEBP-δ) repressed LEP gene promoter activity (lanes 6 and 7) as assessed after co-transfection of a fixed amount of Luc-150 (0.1 μg). E, additive effect of NRF-1 and CEBP-δ expression on the proximal enhancer as assessed on Luc-150. Independent expression of CA-NRF-1 and CEBP-δ (1 μg/per well) stimulated LEP gene promoter activity (lanes 3 and 4) as assessed after co-transfection with a fixed amount of Luc-150 (0.1 μg) per well in 6-well plates containing H1650 cells, whereas co-expression of CA-NRF-1 and CEBP-δ (1 μg each/per well) showed an additive effect. F, expression of CA-STAT3 did not affect Luc-150 activity. Co-transfection of Luc-150 (1 μg) with CA-STAT3 expression plasmid (1 μg per well) in 6-well plates containing H1650 cells was performed. All transfections were done in H1650 lung epithelial cells in 6-well plates. Values represent three independent experiments carried out in triplicate ± S.E. (**, p value ≤0.001).
Because NRF-1 and C/EBP-δ both up-regulated LEP gene promoter activity, we tested the possibility that NRF-1 and C/EBP-δ might have potential synergistic or additive effect on the proximal enhancer in the induction of LEP transcription. Co-transfection of Luc-150 in combination with CA-NRF-1 and C/EBP-δ-expressing plasmids showed an ∼8-fold increase in Luc-150 activity (Fig. 4E, lane 5) as compared with a 4-fold increase after expression of CA-NRF-1 (Fig. 4E, lane 3) and a 2.5-fold increase after expression of C/EBP-δ alone (Fig. 4E, lane 4), indicating the additive effect of the two transcription factors. Because canonical LEP signaling is mediated via binding of the transcription factor STAT3 to various gene promoter elements (56, 57), we tested the hypothesis whether STAT3 itself may play a critical role in transcriptional activation of Luc-150. However, co-transfection of Luc-150 with plasmids expressing constitutively active STAT3 (CA-STAT3) did not activate transcription from Luc-150 (Fig. 4F), suggesting that the proximal LEP gene enhancer functions independently of STAT3 and that LEP transcription may be regulated by STAT3-binding elements present further upstream in the LEP gene promoter. Together, NRF-1 and C/EBP-δ activated LEP gene transcription utilizing the LEP gene proximal enhancer element.
LEP Gene Proximal Enhancer Binds Transcription Factors NRF-1 and CEBP-δ
The NRF-1 and C/EBP-δ DNA-binding elements that were identified by MatInspector were matched with true consensus elements defined by the JASPAR database (53, 58). Although the C/EBP site showed complete conservation, the NRF-1 site deviated by one nucleotide at position 2. Site-directed mutagenesis of the NRF-1 and C/EBP-δ-binding sites within the proximal enhancer abrogated transcription activity, indicating that both these transcription factors directly bind to the enhancer element and transcriptionally regulate LEP gene expression (Fig. 5A, lane 3, and Fig. 5B, lane 3). Indeed, ChIP of NRF-1 and C/EBP-δ using the DNA primers spanning the 5′-regulatory region containing the proximal LEP gene enhancer (Fig. 5, D and E) readily detected the bound form of NRF-1 and C/EBP-δ in the chromatin in vivo in H1650 cells (Fig. 5, D, lane 4, and E, lane 4). In contrast, NRF-1 and C/EBP-δ failed to bind the GAPDH proximal promoter (Fig. 5, D, lane 3, and E, lane 3), implicating NRF-1 and C/EBP-δ as direct transcriptional activators of the LEP gene. Taken together, these experiments confirm that LEP gene transcription is mediated by binding of NRF-1 and C/EBP-δ to the LEP gene proximal enhancer in the chromatin context in lung epithelial cells.
FIGURE 5.
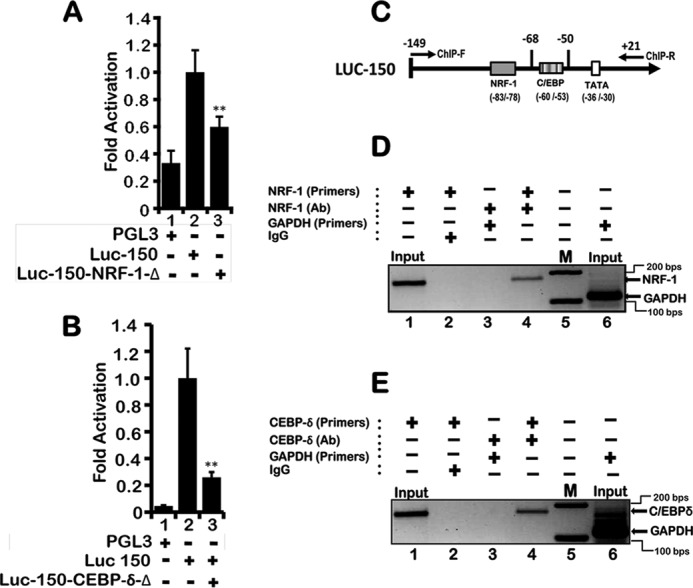
Site-directed mutations to the NRF-1 and CEBP-δ consensus binding sites abrogate LEP gene proximal promoter activity. A, as compared with wild-type LEP promoter (Luc-150), mutation in the NRF-1 site (Luc-150-NRF-1-Δ) luciferase promoter-reporter plasmid (1 μg each) decreased the activity by >50% as assessed after transfection in H1650 cells. Values represent three independent experiments carried out in triplicate ± S.E. (**, p value ≤0.001). B, site-directed mutagenesis of CEBP-δ-binding site (Luc-150-CEBP-δ-Δ) abrogated LEP gene proximal promoter activity following transient transfection in H1650 cells. Values represent three independent experiments carried out in triplicate ± S.E. (**, p value ≤0.001). C, primer locations that spanned the proximal enhancer containing NRF-1 and CEBP-δ sites for identification of PCR product following ChIP. D, ChIP analysis reveals NRF-1 occupancy on the LEP proximal promoter (lane 4). ChIP was performed with antibodies to NRF-1. IgG was used as a control (lane 2). Primers flanking the NRF-1 site were used for PCR analyses, and GAPDH primers used as an internal control did not show amplification of specific product (lane 3). E, ChIP analysis reveals C/EBP-δ occupancy on the LEP proximal promoter (lane 4). ChIP was performed with antibodies to C/EBP-δ. IgG was used as a control (lane 2). Primers flanking the C/EBP-δ site were used for PCR analyses, whereas GAPDH primers used as an internal control did not show amplification of specific product (lane 3) as compared with lane 4 and DNA marker M showing 100- and 200-bp reference DNA sizes (lane 5).
PI3K/AKT and MEK Pathways Regulate LEP Gene Transcription via a Proximal Enhancer
When LEP binds to its receptor LEPR, it triggers the activation of PI3K/AKT, MEK/ERK, and JAK2/STAT3 pathways in many cell types (21). However, whether LEP expression is controlled and induced by its own signaling via these three signaling pathways remains unknown. It is plausible that to maintain continuous LEP signaling, mainly in an autocrine loop, LEP itself may regulate its own expression via up-regulating LEPR and its downstream signaling pathways. To test this hypothesis, we transfected H1650 cells with Luc-150 and subsequently subjected these cells to PI3K/AKT and MEK pathway-specific inhibitors. MK-2206, an AKT inhibitor, U0126 a MEK inhibitor, LY-294002 a pan-PI3K inhibitor, and BEZ-235 a dual-PI3K/mTOR kinase inhibitor reduced LEP promoter activity in a dose-dependent manner, respectively (Fig. 6, A–C, lanes 3–5, and D, lanes 3 and 4). A combination of PI3K/AKT, PI3K/MEK, and MEK/mTOR pathway inhibitors further reduced LEP promoter activity, indicating that signals from these pathways independently regulate the LEP gene promoter (Fig. 6, E–G, lane 5). In all these experiments, using trypan blue staining, we made sure that cell viability was not compromised. The highest concentrations used in our experiments were consistent or lower than previously reported studies (59–62) without any observable toxicity.
FIGURE 6.

LEP gene transcription is regulated by PI3K/AKT/mTOR and MEK/ERK signaling pathways. Effect of inhibitors of AKT (MK-2206), MEK (U0126), PI3K (LY294), and PI3K/mTOR dual inhibitor (BEZ-235) on LEP promoter activity. H1650 cells transfected with Luc-150 (1 μg) were either treated with increasing concentrations of AKT inhibitor MK-2206 (50, 100, and 250 nm) (A), MEK-inhibitor U0126 (50, 100, and 250 nm) (B), PI3K/mTOR dual inhibitor BEZ-235 (50, 75 and 250 nm) (C), or a potent PI3K inhibitor LY294002 (5 and 10 nm) (D), and a dose-dependent decrease in luciferase activity was observed at 16 h after transfection (lanes 3–5 for A–C, and lanes 3 and 4 for D). Combination of MK-2206 (50 nm) and LY294002 (10 nm) (E), U0126 (50 nm) and LY294002 (10 nm) (F), or BEZ-235 (50 nm) and LY294002 (10 nm) (G) completely abrogated Luc-150 luciferase activity, indicating that PI3K/AKT/mTOR and MEK/ERK pathways independently modulate the LEP proximal promoter activity, likely via post-translational modification of NRF-1 and CEBP-δ, influencing their transcription activity. Throughout, values represent three independent experiments carried out in triplicate. Data are presented as ± S.E. (*, p value ≤0.05).
To further support the role of PI3K/AKT/mTOR and MEK/ERK signaling pathways, we performed co-transfection of Luc-150 with plasmids expressing constitutively active AKT (CA-AKT) and constitutively active MEK (CA-MEK) in lung epithelial cells H1650. Indeed, expression of CA-AKT and CA-MEK induced LEP gene promoter activation in H1650 cells (Fig. 9C). In summary, the LEP gene proximal promoter was regulated by PI3K/AKT/mTOR and MEK/ERK signaling pathways in lung epithelial cells. These observations raise the possibility that LEP via PI3K/AKT/mTOR and MEK/ERK signaling likely mediates its own transcription followed by increased LEP synthesis in lung epithelial cells that are exposed to extracellular LEP, reinforcing an auto-regulatory LEP signaling loop.
FIGURE 9.

LEP treatment increases endogenous LEP and LEPR mRNA levels. A, H1650 cells were serum-starved for 16 h and then replenished with either 10% fetal calf serum (FCS), or 10% FCS containing LEP (100 ng/ml) for 24 and 72 h (2nd and 3rd lanes). Endogenous levels of LEP mRNA were measured using RT-qPCR analysis as described under “Materials and Methods.” B, when LEP treatment was performed on H1650 cells and expression of LEPR mRNA was analyzed by RT-qPCR, a modest but significant increase in LEPR mRNA was detected. Exon-specific primers for LEP and LEPR were used as described under “Materials and Methods.” Values represent three independent experiments carried out in triplicate. Data are presented as ± S.E. (*, p value ≤0.05). C, co-transfection of Luc-150 (1 μg) together with expression plasmids CA-AKT (6 μg) or CA-MEK (6 μg) in 6-well plates containing H1650 cells stimulated with LEP promoter activity by ∼3- and 4-fold, respectively (3rd and 4th lanes), indicating a direct role for AKT and MEK pathways in the modulation of the LEP proximal promoter activity. Values represent three independent experiments carried out in triplicate. Data are presented as ± S.E. (**, p value ≤0.001). D, When H1650 cells growing in 10-cm plates were transfected with plasmid (8 μg) expressing constitutively active STAT3 (CA-STAT3), LEP, and LEPR (E), mRNA expression was induced by ∼2.5-fold as measured by RT-qPCR using gene-specific primers. Values represent three independent experiments carried out in triplicate. Data are presented as ± S.E. (*, p value ≤ 0.05). F, location of primers spanning the distal promoter region containing STAT3 consensus site for identification of PCR product following ChIP using STAT3 antibodies. G, ChIP analysis reveals STAT3 occupancy on the LEP distal promoter (lane 5). ChIP was performed with antibodies to STAT3. IgG was used as a control (lane 3). Primers flanking the STAT3 site were used for PCR analyses, whereas GAPDH primers used as an internal control did not show amplification of specific product (lane 4) as compared with lane 5 and DNA marker M showing 100 and 200 bp reference DNA sizes (lane 1).
NRF-1, C/EBP-δ, and STAT3 Up-regulates Endogenous LEP and LEP Receptor Gene Transcripts
Although the potential role of NRF-1 and C/EBP-δ in the regulation of LEP gene transcription was established in transient transfection assays using the Luc-150 as a reporter, whether NRF-1 and C/EBP-δ can directly activate endogenous LEP gene transcription in the native chromatin context in H1650 lung epithelial cells was not determined. Therefore, following expression of CA-NRF-1 and CA-C/EBP-δ in H1650 cells, induction of LEP mRNA transcription was evaluated by quantitative real time PCR (RT-qPCR). Indeed, expression of CA-NRF-1 and C/EBP-δ significantly increased LEP and its receptor LEPR mRNA expression (Fig. 7, A and B, lanes 3 and 4, and 2nd and 3rd lanes of insets). To further confirm that endogenous NRF-1 does activate LEP and its receptor LEPR transcription in lung epithelial cells, plasmid vectors expressing shRNA targeting the NRF-1 mRNA were expressed in H1650 cells. NRF-1 shRNA expression vectors 1A and 1B resulted in ∼60–80% decrease in levels of NRF-1 transcripts as measured by RT-qPCR (Fig. 7C, lanes 2 and 3 and inset, lanes 3 and 4). Expression of shRNA-1B in H1650 cells significantly down-regulated endogenous LEP and LEPR gene mRNA transcripts as measured by RT-qPCR analysis (Fig. 7, D and E), thereby validating the role of NRF-1 in the transcriptional regulation of LEP and LEP receptor expression in lung epithelial cells.
FIGURE 7.

LEP and LEPR mRNA levels are up-regulated by NRF-1 and CEBP-δ. A, when H1650 lung epithelial cells growing in 10-cm plates were transfected with expression plasmids (8 μg) containing CA-NRF-1 or CEBP-δ, endogenous expression of LEP mRNA was increased by 8- and 4-fold, respectively, after 48 h (lanes 3 and 4, and inset, 2nd and 3rd lanes). mRNA expression was quantitated by real time qPCR using LEP exon-specific primers as described under “Materials and Methods.” B, likewise, transfection of H1650 cells with CA-NRF-1 and CEBP-δ increased endogenous LEPR mRNA by ∼3.5- and 2.5-fold, respectively, after 48 h, suggesting a direct role for CA-NRF-1 and CEBP-δ in modulating LEP and LEPR gene expression. C, robust expression of endogenous NRF-1 mRNA was detected in H1650 cells. Two distinct shRNAs abrogated NRF-1 mRNA, with shRNA-1B being more effective (lane 3). Overexpression of shNRF-1B significantly decreased LEP (D), and LEPR mRNA expression in H1650 (E), confirming a critical transcriptional role for NRF-1 in LEP and LEPR expression in lung epithelial cells. Throughout, the values represent three independent experiments carried out in triplicate. Data are presented as ± S.E. (*, p value ≤0.05).
LEP-mediated LEP and LEPR Gene Expression
LEP as a cytokine and a paracrine factor activates JAK2/STAT3, PI3K/AKT/mTOR, and MEK/ERK signaling pathways that are directly involved in cancer progression (21). LEP also activates expression of several gene targets that participate in cancer progression, including pro-inflammatory cytokines and factors promoting angiogenesis (63). However, whether LEP signaling modulates the transcription of its own gene (LEP) and receptor (LEPR), amplifying its function has not been studied. When H1650 cells were treated with recombinant LEP for 48 and 72 h, JAK2/STAT3 and PI3K/AKT/mTOR signaling pathways were activated as assessed by increase in phosphorylation of STAT3 AKT and mTOR (Fig. 8, A–C). Within 15 min after treatment with leptin, an increase in phosphorylation of ERK was observed (Fig. 8D). Likewise, 30 min of treatment with leptin-regulated p38 and JNK signaling pathways as assessed by an increase in P-P38, P-P54 SNP/JNK, and P-P46 SNP/JNK (Fig. 8, E–G). These results clearly indicate that the LEP/LEPR signaling pathway was operational in the lung epithelial cells. Furthermore, treatment of LEP significantly increased LEP and LEP receptor gene expression as assessed by measurement of their mRNA transcripts by RT-qPCR (Fig. 9, A and B). Taken together, LEP via the LEP receptor up-regulated the expression of LEP and LEPR genes in lung epithelial cells, likely driving a feed-forward LEP-signaling loop that may potentially be required for sustained LEP signaling as observed in chronic lung injury diseases and lung cancer.
FIGURE 8.

Leptin-mediated activation of AKT/mTOR, MEK/ERK, and JAK2/STAT3 signaling pathways. H1650 cells were serum-starved for 16 h and then replenished with either 10% FCS or 10% FCS with LEP (100 ng/ml). After 48 and 72 h, total protein was isolated and immunoblotted to detect activation of PI3K/AKT/mTOR, MEK/ERK, and JAK2/STAT3 pathways via phosphorylation of AKT, mTOR, and STAT3. An increase in P-STAT3 (A), P-AKT (B), and P-mTOR (C) was readily observed at 48 and 72 h after treatment with LEP (lanes 2 and 4), suggesting an active LEP signaling pathway in the lung epithelial cells that operates via downstream activation of AKT, MEK, and STAT3 pathways. β-Actin was used as a biologic and loading control. All data are representative of at least three independent experiments. Likewise, following serum starvation, when H1650 cells were treated with LEP (100 ng/ml), activation of p-ERK (D), P-P38 (E), P-P54 SNP/JNK (F), and P-P46 SNP/JNK (G) was transient but detectable at 15 min, for p-ERK, and at 30 min, for P-P38, P-P54 SNP/JNK, and P-P46 SNP/JNK, respectively. Throughout, values represent three independent experiments carried out in triplicate. Data are presented as ± S.E. (*, p value ≤0.05).
STAT3 Binds to a Distal Enhancer and Activates LEP Gene Transcription
When CA-STAT3 was expressed in H1650 cells, transcription of LEP and its receptor LEPR mRNA were significantly induced (Fig. 9, D and E) as assessed by RT-qPCR. This result indicated that although STAT3 did not function via the proximal enhancer, it certainly regulated LEP and LEP gene transcription from the endogenous promoter in H1650 cells, likely via an upstream STAT3-responsive elements located distally. To ascertain whether the distal enhancer element comprising STAT3-binding sites is present within the LEP gene promoter, we scanned up to −2 kb of the human LEP gene 5′-upstream regulatory promoter sequence. The STAT3-binding sites were identified by homology search using softwares such as JASPAR and GENOMATIX and the published STAT3 consensus DNA-binding sequence (64). Although the STAT3 site was not completely conserved at every nucleotide, it did show the classical TT-N3–6-GG STAT3 binding sequence (64). ChIP of STAT3 using DNA primers spanning the 5′-regulatory region (−1610 to −1493 bp) containing the STAT3 site (Fig. 9F) readily detected the bound form of STAT3 on the chromatin in vivo in H1650 cells (Fig. 9G, lane 5). In contrast, STAT3 antibodies failed to bind the GAPDH proximal promoter (Fig. 9G, lane 4), implicating STAT3 as a direct transcriptional activator of LEP gene expression. Taken together, these experiments confirm that transcriptional regulation of LEP and its receptor LEPR gene expression is mediated by STAT3 in lung epithelial cells.
DISCUSSION
LEP and LEP receptor are synthesized by several nonadipose tissues, wherein LEP functions as a pleiotropic cytokine, modulating a variety of physiologic and pathologic functions (65). Increased pulmonary and circulating LEP levels are observed with several lung diseases associated with injury/repair and remodeling, including lung cancer (5, 66–68). LEP is also involved in fetal lung development and pulmonary homeostasis (69, 70). Emerging evidence indicates that LEP as a pro-inflammatory and pro-angiogenic cytokine may play critical roles in exacerbating acute and chronic pulmonary pathologies and drive lung cancer as an inflammatory molecule (6, 11, 71). However, the molecular mechanism of LEP gene expression in lung diseases and lung cancer remains elusive.
In this study, we demonstrate that Cre/LoxP-mediated conditional deletion of Pten (PtenΔ/Δ) activated adipocyte signaling in the respiratory epithelium that was associated with increased expression of LEP and its receptor. Using PTEN-null lung epithelial cells, we show that LEP gene was transcriptionally activated by a proximal enhancer element via binding of NRF-1 and C/EBP-δ transcription factors, although STAT3 bound a distal promoter element in the LEP gene and activated its expression. Transcription of the active form of LEPR was also induced by NRF-1, C/EBP-δ, and STAT3, suggesting that these three factors concertedly activate the LEP/LEPR signaling pathway in the lung epithelial cells. C/EBP-δ and STAT3 play critical roles during inflammatory responses in the lung (54, 72, 73). Because lung epithelial cells, particularly type II alveolar epithelial cells, have high lipid metabolic activity and turnover that are required for surfactant synthesis (74), it is plausible that LEP may play a critical role in surfactant homeostasis following lung epithelial injury. Indeed, several studies have demonstrated that LEP directly stimulates proliferation of alveolar epithelial cells (75) up-regulating type IV collagen synthesis, which reinforce the alveolar walls (76, 77).
Extra lipid accumulation in type II pneumocytes may lead to lipotoxicity as observed in many nonadipose tissues (78, 79). Because LEP stimulates fatty acid oxidation via activation of AMP-activated protein kinase (80), it participates in reducing lipid stores, thereby reducing lipotoxicity (80–82). Consistent with this concept, sustained LEP signaling, as seen in many inflammatory conditions, causes chronic activation of AMP-activated protein kinase (80), which activates the transcription factor NRF-1 (83). Activated NRF-1 binds gene promoters involved in enhancing oxidative capacity and mitochondrial biogenesis, including LEP as demonstrated in this study, increasing energy metabolism via known LEP signaling pathways (83, 84). Thus, our results presented here are consistent with the role of NRF-1 in PTEN-null H1650 lung epithelial cells. In addition, loss of PTEN in lung epithelial cells drives rapid cell proliferation that would indeed be associated with increased mitochondriogenesis (31). Supporting these observations, our LEP promoter-reporter deletion analysis, site-directed mutagenesis, ChIP, and NRF-1 overexpression studies demonstrated increased activity of NRF-1 on the LEP promoter itself, indicating that LEP-mediated activation of NRF-1 contributes to transcriptional activation and expression of the LEP gene in lung epithelial cells.
In adipocytes, LEP gene transcription is regulated by C/EBP-α (27, 30); however, transcriptional regulation of the LEP gene and the roles of C/EBP isoforms, including C/EBP-β and C/EBP-δ in nonadipocyte tissues, remain unclear. This study revealed that C/EBP-δ, but not C/EBP-α or C/EBP-β, strongly activated LEP gene transcription in lung epithelial cells, suggesting a critical and selective role for C/EBP-δ in LEP gene regulation. Given the important role of LEP in inflammatory processes, it is highly likely that concerted modulation of pro-inflammatory genes, including LEP, is under the control of C/EBP-δ in the lung epithelium (55). Recent studies demonstrate that C/EBP-β and C/EBP-δ participate in inflammatory responses following lung injury and infection (85), consistent with the identified role of C/EBP-δ in the transcriptional activation of LEP gene expression in the lung epithelial cells.
LEP signaling is transduced via the activation of canonical PI3K/AKT, MEK/ERK, and JAK2/STAT3 pathways in many cell types (21). However, whether LEP itself is activated by these three signaling pathways and regulates its own expression has not been explored.
Tumors maintain continuous LEP signaling that facilitate cancer progression and metastasis, whereas inhibition of LEP signaling results in efficient anti-tumor activity (86); therefore, it is likely that LEP itself may regulate its own gene expression. When H1650 PTEN-null lung epithelial cells were treated with LEP, activation of PI3K/AKT/mTOR, MEK/ERK, JAK2/STAT3, p38, and JNK signaling pathways were detected by immunoblotting for P-mTOR, P-AKT, P-STAT, p-ERK, and P38 MAPK as well as the active and inactive forms of JNK (p54 JNK and p46 JNK). This is consistent with previous findings where leptin-mediated activation of canonical (PI3K and ERK) and noncanonical (p38 MAPK, JNK, and PKC) signaling pathways has been observed (87, 88) Together, our results support the concept that loss of PTEN in lung adenocarcinoma would activate PI3K/AKT/mTOR, MEK/ERK, and the p38/JNK MAPK signaling pathways, which would in turn contribute to the induction of LEP gene expression and subsequent secretion.
To test this hypothesis in vivo, we developed a mouse model with conditional deletion of Pten in an oncogenic Kras background (89). Exuberant LEP secretion was indeed detected in tumors generated after Pten loss in the oncogenic Kras background (Fig. 2B), indicating an important role for LEP signaling in lung cancer progression. Because treatment of H1650 cells with inhibitors of PI3K, AKT, mTOR, and MEK abrogated LEP gene transcription, whereas constitutive expression of AKT and MEK activated LEP gene promoter activity, we proposed in our conceptual model that LEP-mediated LEP gene expression is directly influenced by PI3K/AKT/mTOR, MEK/ERK, and p38 JNK signaling pathways (Fig. 10). The major hallmarks of LEP signaling are activation of STAT3 via JAK2 phosphorylation and increased cell proliferation. Overexpression of constitutively active STAT3 induced LEP gene expression, whereas treatment with LEP enhanced proliferation and wound healing of H1650 cells, consistent with the promotion of invasion and migration in cancer cells (90). Taken together, our findings demonstrate that LEP itself regulates its own expression via the LEP receptor-mediated downstream signaling in lung epithelial cells. This study supports the concept that therapies that can abrogate LEP signaling in lung pathologies may reduce disease morbidity.
FIGURE 10.
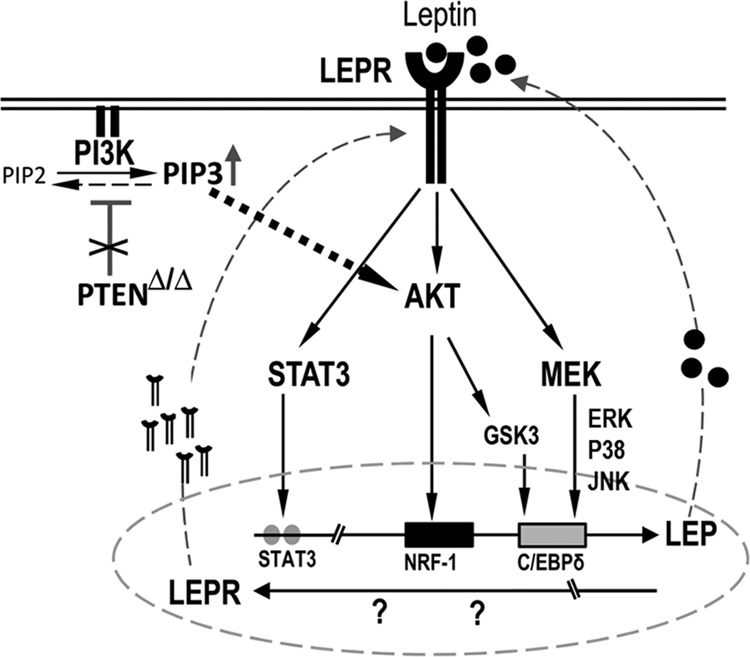
Proposed model of LEP mediated LEP gene expression. Proposed model of LEP mediated LEP gene expression indicates that loss of Pten in the lung epithelial cells activates AKT, which in turn induces LEP expression and LEP secretion into the extracellular space. Such AKT mediated increases in extracellular LEP (LEP) concentration may occur in lung cancer and many lung diseases associated with increased PI3K activity. Because lung epithelial cells express LEP receptor (LEPR) that readily bind to LEP, multiple kinase pathways are activated, including AKT/mTOR, GSK3, JAK2/STAT3 and MEK/ERK that influence NRF-1, CEBP-δ and STAT3 binding, activating LEP and LEPR expression. Thus, a continuous LEP/LEPR feed-forward loop is set-up, further deteriorating lung function. Activation of these anti-apoptotic pathways also provides survival benefit to lung tumor cells, contributing to chemo-radio- and targeted therapy resistance, leading to poor clinical outcomes.
Acknowledgments
We thank Dr. Todd Gulick (Sanford-Burnham Medical Research Institute, Lake Nano Orlando, FL) for the NRF-1 plasmids; Dr. Steven McKnight (University of Texas-Southwestern Medical Center, Dallas) for C/EBP-δ plasmid, and Dr. Jim Darnell (Rockefeller University, New York) for CA-STAT3 expression plasmid. We also thank Dr. Jeffrey Whitsett (Cincinnati Children's Hospital, Cincinnati OH) for the use of microarray core facility.
This work was supported, in whole or in part, by National Institutes of Health-Lung SPORE Career Development Grant (to V. D.). This work was supported by American Heart Association Scientist Development Grant SDG-0830101N (to V. D.).
- LEP
- leptin
- PTEN
- phosphatase and tensin homolog
- LEPR
- leptin receptor
- AKT
- protein kinase B
- mTOR
- mammalian target of rapamycin
- STAT3
- signal transducer and activator of transcription 3
- C/EBP
- CCAAT/enhancer-binding protein
- KEGG
- Kyoto Encyclopedia of Genes and Genomes
- ALI
- acute lung injury
- COPD
- chronic obstructive pulmonary disease
- ARDS
- acute respiratory distress syndrome
- CA
- constitutively active
- qPCR
- quantitative PCR
- F
- forward
- R
- reverse.
REFERENCES
- 1. Procaccini C., Jirillo E., Matarese G. (2012) Leptin as an immunomodulator. Mol. Aspects Med. 33, 35–45 [DOI] [PubMed] [Google Scholar]
- 2. Morris D. L., Rui L. (2009) Recent advances in understanding leptin signaling and leptin resistance. Am. J. Physiol. Endocrinol. Metab. 297, E1247–E1259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bruno A., Chanez P., Chiappara G., Siena L., Giammanco S., Gjomarkaj M., Bonsignore G., Bousquet J., Vignola A. M. (2005) Does leptin play a cytokine-like role within the airways of COPD patients? Eur. Respir. J. 26, 398–405 [DOI] [PubMed] [Google Scholar]
- 4. Vernooy J. H., Drummen N. E., van Suylen R. J., Cloots R. H., Möller G. M., Bracke K. R., Zuyderduyn S., Dentener M. A., Brusselle G. G., Hiemstra P. S., Wouters E. F. (2009) Enhanced pulmonary leptin expression in patients with severe COPD and asymptomatic smokers. Thorax 64, 26–32 [DOI] [PubMed] [Google Scholar]
- 5. Ribeiro R., Araujo A., Lopes C., Medeiros R. (2007) Immunoinflammatory mechanisms in lung cancer development: is leptin a mediator? J. Thoracic Oncol. 2, 105–108 [DOI] [PubMed] [Google Scholar]
- 6. Vernooy J. H., Ubags N. D., Brusselle G. G., Tavernier J., Suratt B. T., Joos G. F., Wouters E. F., Bracke K. R. (2013) Leptin as regulator of pulmonary immune responses: Involvement in respiratory diseases. Pulm. Pharmacol. Ther. 26, 464–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Vernooy J. H., Bracke K. R., Drummen N. E., Pauwels N. S., Zabeau L., van Suylen R. J., Tavernier J., Joos G. F., Wouters E. F., Brusselle G. G. (2010) Leptin modulates innate and adaptive immune cell recruitment after cigarette smoke exposure in mice. J. Immunol. 184, 7169–7177 [DOI] [PubMed] [Google Scholar]
- 8. Broekhuizen R., Vernooy J. H., Schols A. M., Dentener M. A., Wouters E. F. (2005) Leptin as local inflammatory marker in COPD. Respir. Med. 99, 70–74 [DOI] [PubMed] [Google Scholar]
- 9. Alemán M. R., Santolaria F., Batista N., de La Vega M., González-Reimers E., Milena A., Llanos M., Gómez-Sirvent J. L. (2002) Leptin role in advanced lung cancer. A mediator of the acute phase response or a marker of the status of nutrition? Cytokine 19, 21–26 [DOI] [PubMed] [Google Scholar]
- 10. Sood A. (2010) Obesity, adipokines, and lung disease. J. Appl. Physiol. 108, 744–753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jain M., Budinger G. R., Lo A., Urich D., Rivera S. E., Ghosh A. K., Gonzalez A., Chiarella S. E., Marks K., Donnelly H. K., Soberanes S., Varga J., Radigan K. A., Chandel N. S., Mutlu G. M. (2011) Leptin promotes fibroproliferative acute respiratory distress syndrome by inhibiting peroxisome proliferator-activated receptor-γ. Am. J. Respir. Crit. Care Med. 183, 1490–1498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Barazzone-Argiroffo C., Muzzin P., Donati Y. R., Kan C. D., Aubert M. L., Piguet P. F. (2001) Hyperoxia increases leptin production: a mechanism mediated through endogenous elevation of corticosterone. Am. J. Physiol. Lung Cell. Mol. Physiol. 281, L1150–L1156 [DOI] [PubMed] [Google Scholar]
- 13. Bellmeyer A., Martino J. M., Chandel N. S., Scott Budinger G. R., Dean D. A., Mutlu G. M. (2007) Leptin resistance protects mice from hyperoxia-induced acute lung injury. Am. J. Respir. Crit. Care Med. 175, 587–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Leão da Silva P., de Mello M. T., Cheik N. C., Sanches P. L., Munhoz da Silveira Campos R., Carnier J., Inoue D., do Nascimento C. M., Oyama L. M., Tock L., Tufik S., Dâmaso A. R. (2012) Reduction in the leptin concentration as a predictor of improvement in lung function in obese adolescents. Obes. Facts 5, 806–820 [DOI] [PubMed] [Google Scholar]
- 15. Malli F., Papaioannou A. I., Gourgoulianis K. I., Daniil Z. (2010) The role of leptin in the respiratory system: an overview. Respir. Res. 11, 152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. MacCallum N. S., Evans T. W. (2005) Epidemiology of acute lung injury. Curr. Opin. Crit. Care 11, 43–49 [DOI] [PubMed] [Google Scholar]
- 17. Matthay M. A., Zimmerman G. A. (2005) Acute lung injury and the acute respiratory distress syndrome: four decades of inquiry into pathogenesis and rational management. Am. J. Respir. Cell Mol. Biol. 33, 319–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Calfee C. S., Matthay M. A., Eisner M. D., Benowitz N., Call M., Pittet J. F., Cohen M. J. (2011) Active and passive cigarette smoking and acute lung injury after severe blunt trauma. Am. J. Respir. Crit. Care Med. 183, 1660–1665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sierra-Honigmann M. R., Nath A. K., Murakami C., García-Cardeña G., Papapetropoulos A., Sessa W. C., Madge L. A., Schechner J. S., Schwabb M. B., Polverini P. J., Flores-Riveros J. R. (1998) Biological action of leptin as an angiogenic factor. Science 281, 1683–1686 [DOI] [PubMed] [Google Scholar]
- 20. Vansaun M. N. (2013) Molecular pathways: adiponectin and leptin signaling in cancer. Clin. Cancer Res. 19, 1926–1932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sweeney G. (2002) Leptin signalling. Cell. Signal. 14, 655–663 [DOI] [PubMed] [Google Scholar]
- 22. Zhou W., Guo S., Gonzalez-Perez R. R. (2011) Leptin pro-angiogenic signature in breast cancer is linked to IL-1 signalling. Br. J. Cancer 104, 128–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gonzalez-Perez R. R., Xu Y., Guo S., Watters A., Zhou W., Leibovich S. J. (2010) Leptin upregulates VEGF in breast cancer via canonic and non-canonical signalling pathways and NFκB/HIF-1α activation. Cell. Signal. 22, 1350–1362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. de la Brousse F. C., Shan B., Chen J. L. (1996) Identification of the promoter of the mouse obese gene. Proc. Natl. Acad. Sci. U.S.A. 93, 4096–4101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gong D. W., Bi S., Pratley R. E., Weintraub B. D. (1996) Genomic structure and promoter analysis of the human obese gene. J. Biol. Chem. 271, 3971–3974 [DOI] [PubMed] [Google Scholar]
- 26. He Y., Chen H., Quon M. J., Reitman M. (1995) The mouse obese gene. J. Biol. Chem. 270, 28887–28891 [DOI] [PubMed] [Google Scholar]
- 27. Miller S. G., De Vos P., Guerre-Millo M., Wong K., Hermann T., Staels B., Briggs M. R., Auwerx J. (1996) The adipocyte-specific transcription factor C/EBPα modulates human ob gene expression. Proc. Natl. Acad. Sci. U.S.A. 93, 5507–5511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fuke T., Yoshizaki T., Kondo M., Morino K., Obata T., Ugi S., Nishio Y., Maeda S., Kashiwagi A., Maegawa H. (2010) Transcription factor AP-2β inhibits expression and secretion of leptin, an insulin-sensitizing hormone, in 3T3-L1 adipocytes. Int. J. Obes. 34, 670–678 [DOI] [PubMed] [Google Scholar]
- 29. Kim J. B., Sarraf P., Wright M., Yao K. M., Mueller E., Solanes G., Lowell B. B., Spiegelman B. M. (1998) Nutritional and insulin regulation of fatty acid synthetase and leptin gene expression through ADD1/SREBP1. J. Clin. Invest. 101, 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hwang C. S., Mandrup S., MacDougald O. A., Geiman D. E., Lane M. D. (1996) Transcriptional activation of the mouse obese (ob) gene by CCAAT/enhancer binding protein α. Proc. Natl. Acad. Sci. U.S.A. 93, 873–877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Davé V., Wert S. E., Tanner T., Thitoff A. R., Loudy D. E., Whitsett J. A. (2008) Conditional deletion of Pten causes bronchiolar hyperplasia. Am. J. Respir. Cell Mol. Biol. 38, 337–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dranoff G., Crawford A. D., Sadelain M., Ream B., Rashid A., Bronson R. T., Dickersin G. R., Bachurski C. J., Mark E. L., Whitsett J. A. (1994) Involvement of granulocyte-macrophage colony-stimulating factor in pulmonary homeostasis. Science 264, 713–716 [DOI] [PubMed] [Google Scholar]
- 33. Irizarry R. A., Bolstad B. M., Collin F., Cope L. M., Hobbs B., Speed T. P. (2003) Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 31, e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dinu I., Potter J. D., Mueller T., Liu Q., Adewale A. J., Jhangri G. S., Einecke G., Famulski K. S., Halloran P., Yasui Y. (2007) Improving gene set analysis of microarray data by SAM-GS. BMC Bioinformatics 8, 242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Verhoeven K. J., Simonsen K. L., McIntyre L. M. (2005) Implementing false discovery rate control: increasing your power. Oikos 108, 643–647 [Google Scholar]
- 36. Storey J. D. (2002) A direct approach to false discovery rates. J. R. Stat. Soc. B 64, 479–498 [Google Scholar]
- 37. Benjamini Y., Drai D., Elmer G., Kafkafi N., Golani I. (2001) Controlling the false discovery rate in behavior genetics research. Behav. Brain Res. 125, 279–284 [DOI] [PubMed] [Google Scholar]
- 38. Dennis G., Jr., Sherman B. T., Hosack D. A., Yang J., Gao W., Lane H. C., Lempicki R. A. (2003) DAVID: Database for annotation, visualization, and integrated discovery. Genome Biol. 4, P3. [PubMed] [Google Scholar]
- 39. Boussif O., Lezoualc'h F., Zanta M. A., Mergny M. D., Scherman D., Demeneix B., Behr J. P. (1995) A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethyleneimine. Proc. Natl. Acad. Sci. U.S.A. 92, 7297–7301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ekins S., Nikolsky Y., Bugrim A., Kirillov E., Nikolskaya T. (2007) Pathway mapping tools for analysis of high content data. Methods Mol. Biol. 356, 319–350 [DOI] [PubMed] [Google Scholar]
- 41. Ekins S., Nikolsky Y., Bugrim A., Kirillov E., Nikolskaya T. (2006) in High Content Screening (Taylor D. L., Haskins J., Giuliano K., eds) pp. 319–350, Humana Press Inc., Totowa, NJ [Google Scholar]
- 42. Jimenez-Marin A., Collado-Romero M., Ramirez-Boo M., Arce C., Garrido J. J. (2009) Biological pathway analysis by ArrayUnlock and ingenuity pathway analysis. BMC Proc. 3, S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kanehisa M., Goto S., Sato Y., Furumichi M., Tanabe M. (2012) KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 40, D109–D114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yanagi S., Kishimoto H., Kawahara K., Sasaki T., Sasaki M., Nishio M., Yajima N., Hamada K., Horie Y., Kubo H., Whitsett J. A., Mak T. W., Nakano T., Nakazato M., Suzuki A. (2007) Pten controls lung morphogenesis, bronchioalveolar stem cells, and onset of lung adenocarcinomas in mice. J. Clin. Invest. 117, 2929–2940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Miyoshi K., Yanagi S., Kawahara K., Nishio M., Tsubouchi H., Imazu Y., Koshida R., Matsumoto N., Taguchi A., Yamashita S., Suzuki A., Nakazato M. (2013) Epithelial Pten controls acute lung injury and fibrosis by regulating alveolar epithelial cell integrity. Am. J. Respir. Crit. Care Med. 187, 262–275 [DOI] [PubMed] [Google Scholar]
- 46. Mihai C., Bao S., Lai J. P., Ghadiali S. N., Knoell D. L. (2012) PTEN inhibition improves wound healing in lung epithelia through changes in cellular mechanics that enhance migration. Am. J. Physiol. Lung Cell. Mol. Physiol. 302, L287–L299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tiozzo C., De Langhe S., Yu M., Londhe V. A., Carraro G., Li M., Li C., Xing Y., Anderson S., Borok Z., Bellusci S., Minoo P. (2009) Deletion of Pten expands lung epithelial progenitor pools and confers resistance to airway injury. Am. J. Respir. Crit. Care Med. 180, 701–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Okumura M., Yamamoto M., Sakuma H., Kojima T., Maruyama T., Jamali M., Cooper D. R., Yasuda K. (2002) Leptin and high glucose stimulate cell proliferation in MCF-7 human breast cancer cells: reciprocal involvement of PKC-α and PPAR expression. Biochim. Biophys. Acta 1592, 107–116 [DOI] [PubMed] [Google Scholar]
- 49. Keese C. R., Wegener J., Walker S. R., Giaever I. (2004) Electrical wound-healing assay for cells in vitro. Proc. Natl. Acad. Sci. U.S.A. 101, 1554–1559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Schiller K. R., Maniak P. J., O'Grady S. M. (2010) Cystic fibrosis transmembrane conductance regulator is involved in airway epithelial wound repair. Am. J. Physiol. Cell Physiol. 299, C912–C921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wu N. L., Chiang Y. C., Huang C. C., Fang J. Y., Chen D. F., Hung C. F. (2010) Zeaxanthin inhibits PDGF-BB-induced migration in human dermal fibroblasts. Exp. Dermatol. 19, e173–e181 [DOI] [PubMed] [Google Scholar]
- 52. Gorshkova I., He D., Berdyshev E., Usatuyk P., Burns M., Kalari S., Zhao Y., Pendyala S., Garcia J. G., Pyne N. J., Brindley D. N., Natarajan V. (2008) Protein kinase C-ϵ regulates sphingosine 1-phosphate-mediated migration of human lung endothelial cells through activation of phospholipase D2, protein kinase C-ζ, and Rac1. J. Biol. Chem. 283, 11794–11806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Quandt K., Frech K., Karas H., Wingender E., Werner T. (1995) MatInd and MatInspector: new fast and versatile tools for detection of consensus matches in nucleotide sequence data. Nucleic Acids Res. 23, 4878–4884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Roos A. B., Nord M. (2012) The emerging role of C/EBP in glucocorticoid signaling: lessons from the lung. J. Endocrinol. 212, 291–305 [DOI] [PubMed] [Google Scholar]
- 55. Cassel T. N., Nord M. (2003) C/EBP transcription factors in the lung epithelium. Am. J. Physiol. Lung Cell. Mol. Physiol. 285, L773–L781 [DOI] [PubMed] [Google Scholar]
- 56. Buettner C., Pocai A., Muse E. D., Etgen A. M., Myers M. G., Jr., Rossetti L. (2006) Critical role of STAT3 in leptin's metabolic actions. Cell Metab. 4, 49–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Myers M. G., Cowley M. A., Münzberg H. (2008) Mechanisms of leptin action and leptin resistance. Annu. Rev. Physiol. 70, 537–556 [DOI] [PubMed] [Google Scholar]
- 58. Sandelin A., Alkema W., Engström P., Wasserman W. W., Lenhard B. (2004) JASPAR: an open-access database for eukaryotic transcription factor binding profiles. Nucleic Acids Res. 32, D91–D94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Maira S. M., Stauffer F., Brueggen J., Furet P., Schnell C., Fritsch C., Brachmann S., Chène P., De Pover A., Schoemaker K., Fabbro D., Gabriel D., Simonen M., Murphy L., Finan P., Sellers W., García-Echeverría C. (2008) Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol. Cancer Ther. 7, 1851–1863 [DOI] [PubMed] [Google Scholar]
- 60. DeSilva D. R., Jones E. A., Favata M. F., Jaffee B. D., Magolda R. L., Trzaskos J. M., Scherle P. A. (1998) Inhibition of mitogen-activated protein kinase kinase blocks T cell proliferation but does not induce or prevent anergy. J. Immunol. 160, 4175–4181 [PubMed] [Google Scholar]
- 61. Hirai H., Sootome H., Nakatsuru Y., Miyama K., Taguchi S., Tsujioka K., Ueno Y., Hatch H., Majumder P. K., Pan B.-S., Kotani H. (2010) MK-2206, an allosteric akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol. Cancer Ther. 9, 1956–1967 [DOI] [PubMed] [Google Scholar]
- 62. Semba S., Itoh N., Ito M., Harada M., Yamakawa M. (2002) The in vitro and in vivo effects of 2-(4-morpholinyl)-8-phenyl-chromone (LY294002), a specific inhibitor of phosphatidylinositol 3′-kinase, in human colon cancer cells. Clin. Cancer Res. 8, 1957–1963 [PubMed] [Google Scholar]
- 63. Aleffi S., Petrai I., Bertolani C., Parola M., Colombatto S., Novo E., Vizzutti F., Anania F. A., Milani S., Rombouts K., Laffi G., Pinzani M., Marra F. (2005) Upregulation of proinflammatory and proangiogenic cytokines by leptin in human hepatic stellate cells. Hepatology 42, 1339–1348 [DOI] [PubMed] [Google Scholar]
- 64. Seidel H. M., Milocco L. H., Lamb P., Darnell J. E., Jr., Stein R. B., Rosen J. (1995) Spacing of palindromic half sites as a determinant of selective STAT (signal transducers and activators of transcription) DNA binding and transcriptional activity. Proc. Natl. Acad. Sci. U.S.A. 92, 3041–3045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Margetic S., Gazzola C., Pegg G. G., Hill R. A. (2002) Leptin: a review of its peripheral actions and interactions. Int. J. Obes. Relat. Metab. Disord. 26, 1407–1433 [DOI] [PubMed] [Google Scholar]
- 66. Karakas S., Karadag F., Karul A. B., Gurgey O., Gurel S., Guney E., Cildag O. (2005) Circulating leptin and body composition in chronic obstructive pulmonary disease. Int. J. Clin. Pract. 59, 1167–1170 [DOI] [PubMed] [Google Scholar]
- 67. Shen Y., Wang Q., Zhao Q., Zhou J. (2009) Leptin promotes the immune escape of lung cancer by inducing proinflammatory cytokines and resistance to apoptosis. Mol. Med. Rep. 2, 295–299 [DOI] [PubMed] [Google Scholar]
- 68. Carpagnano G. E., Spanevello A., Curci C., Salerno F., Palladino G. P., Resta O., Di Gioia G., Carpagnano F., Foschino Barbaro M. P. (2007) IL-2, TNF-α, and leptin: local versus systemic concentrations in NSCLC patients. Oncol. Res. 16, 375–381 [DOI] [PubMed] [Google Scholar]
- 69. Henson M. C., Swan K. F., Edwards D. E., Hoyle G. W., Purcell J., Castracane V. D. (2004) Leptin receptor expression in fetal lung increases in late gestation in the baboon: a model for human pregnancy. Reproduction 127, 87–94 [DOI] [PubMed] [Google Scholar]
- 70. Bergen H. T., Cherlet T. C., Manuel P., Scott J. E. (2002) Identification of leptin receptors in lung and isolated fetal type II cells. Am. J. Respir. Cell Mol. Biol. 27, 71–77 [DOI] [PubMed] [Google Scholar]
- 71. Xu Y. J., Shao Y. F., Zhao X., Geng Y. T., Wang K., Yin Y. M. (2011) Expression and clinical significance of leptin, the functional receptor of leptin (OB-Rb) and HER-2 in non-small-cell lung cancer: a retrospective analysis. J. Cancer Res. Clin. Oncol. 137, 1841–1848 [DOI] [PubMed] [Google Scholar]
- 72. Gao H., Guo R. F., Speyer C. L., Reuben J., Neff T. A., Hoesel L. M., Riedemann N. C., McClintock S. D., Sarma J. V., Van Rooijen N., Zetoune F. S., Ward P. A. (2004) Stat3 activation in acute lung injury. J. Immunol. 172, 7703–7712 [DOI] [PubMed] [Google Scholar]
- 73. Hokuto I., Ikegami M., Yoshida M., Takeda K., Akira S., Perl A. K., Hull W. M., Wert S. E., Whitsett J. A. (2004) Stat-3 is required for pulmonary homeostasis during hyperoxia. J. Clin. Invest. 113, 28–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Dobbs L. G. (1989) Pulmonary surfactant. Annu. Rev. Med. 40, 431–446 [DOI] [PubMed] [Google Scholar]
- 75. Torday J. S., Sun H., Wang L., Torres E., Sunday M. E., Rubin L. P. (2002) Leptin mediates the parathyroid hormone-related protein paracrine stimulation of fetal lung maturation. Am. J. Physiol. Lung Cell. Mol. Physiol. 282, L405–L410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Wolf G., Ziyadeh F. N. (2006) Leptin and renal fibrosis. Contrib. Nephrol. 151, 175–183 [DOI] [PubMed] [Google Scholar]
- 77. Maina J. N., West J. B. (2005) Thin and strong! The bioengineering dilemma in the structural and functional design of the blood-gas barrier. Physiol. Rev. 85, 811–844 [DOI] [PubMed] [Google Scholar]
- 78. Unger R. H., Zhou Y. T., Orci L. (1999) Regulation of fatty acid homeostasis in cells: novel role of leptin. Proc. Natl. Acad. Sci. U.S.A. 96, 2327–2332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. van Herpen N. A., Schrauwen-Hinderling V. B. (2008) Lipid accumulation in non-adipose tissue and lipotoxicity. Physiol. Behav. 94, 231–241 [DOI] [PubMed] [Google Scholar]
- 80. Minokoshi Y., Kim Y. B., Peroni O. D., Fryer L. G., Müller C., Carling D., Kahn B. B. (2002) Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase. Nature 415, 339–343 [DOI] [PubMed] [Google Scholar]
- 81. Muoio D. M., Dohm G. L., Fiedorek F. T., Jr., Tapscott E. B., Coleman R. A. (1997) Leptin directly alters lipid partitioning in skeletal muscle. Diabetes 46, 1360–1363 [DOI] [PubMed] [Google Scholar]
- 82. Minokoshi Y., Kahn B. B. (2003) Role of AMP-activated protein kinase in leptin-induced fatty acid oxidation in muscle. Biochem. Soc. Trans. 31, 196–201 [DOI] [PubMed] [Google Scholar]
- 83. Bergeron R., Ren J. M., Cadman K. S., Moore I. K., Perret P., Pypaert M., Young L. H., Semenkovich C. F., Shulman G. I. (2001) Chronic activation of AMP kinase results in NRF-1 activation and mitochondrial biogenesis. Am. J. Physiol. Endocrinol. Metab. 281, E1340–E1346 [DOI] [PubMed] [Google Scholar]
- 84. Porter R. K., Andrews J. F. (1998) Effects of leptin on mitochondrial 'proton leak' and uncoupling proteins: implications for mammalian energy metabolism. Proc. Nutr. Soc. 57, 455–460 [DOI] [PubMed] [Google Scholar]
- 85. Yan C., Johnson P. F., Tang H., Ye Y., Wu M., Gao H. (2013) CCAAT/enhancer-binding protein δ is a critical mediator of lipopolysaccharide-induced acute lung injury. Am. J. Pathol. 182, 420–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Rene Gonzalez R., Watters A., Xu Y., Singh U. P., Mann D. R., Rueda B. R., Penichet M. L. (2009) Leptin-signaling inhibition results in efficient anti-tumor activity in estrogen receptor positive or negative breast cancer. Breast Cancer Res. 11, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Andò S., Catalano S. (2012) The multifactorial role of leptin in driving the breast cancer microenvironment. Nat. Rev. Endocrinol. 8, 263–275 [DOI] [PubMed] [Google Scholar]
- 88. Onuma M., Bub J. D., Rummel T. L., Iwamoto Y. (2003) Prostate cancer cell-adipocyte interaction: leptin mediates androgen-independent prostate cancer cell proliferation through c-Jun NH2-terminal kinase. J. Biol. Chem. 278, 42660–42667 [DOI] [PubMed] [Google Scholar]
- 89. Johnson L., Mercer K., Greenbaum D., Bronson R. T., Crowley D., Tuveson D. A., Jacks T. (2001) Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature 410, 1111–1116 [DOI] [PubMed] [Google Scholar]
- 90. Saxena N. K., Sharma D., Ding X., Lin S., Marra F., Merlin D., Anania F. A. (2007) Concomitant activation of the JAK/STAT, PI3K/AKT, and ERK signaling is involved in leptin-mediated promotion of invasion and migration of hepatocellular carcinoma cells. Cancer Res. 67, 2497–2507 [DOI] [PMC free article] [PubMed] [Google Scholar]