

Background: Mechanism of prion adaptation and evolution has not been fully elucidated.
Results: Distinct human prion particles co-exist and undergo competitive selection during replication.
Conclusion: The process is governed by preferential replication of the least stable pathogenic conformers.
Significance: The spectrum of conformers in wild human prion isolates enables adaptation and evolution by selection of the progressively less stable and faster replicating subset.
Keywords: Molecular Evolution, Neurobiology, Neurodegeneration, Prions, Protein Conformation, Sporadic Creutzfeldt-Jakob Disease
Abstract
The unique phenotypic characteristics of mammalian prions are thought to be encoded in the conformation of pathogenic prion proteins (PrPSc). The molecular mechanism responsible for the adaptation, mutation, and evolution of prions observed in cloned cells and upon crossing the species barrier remains unsolved. Using biophysical techniques and conformation-dependent immunoassays in tandem, we isolated two distinct populations of PrPSc particles with different conformational stabilities and aggregate sizes, which frequently co-exist in the most common human prion disease, sporadic Creutzfeldt-Jakob disease. The protein misfolding cyclic amplification replicates each of the PrPSc particle types independently and leads to the competitive selection of those with lower initial conformational stability. In serial propagation with a nonglycosylated mutant PrPC substrate, the dominant PrPSc conformers are subject to further evolution by natural selection of the subpopulation with the highest replication rate due to its lowest stability. Cumulatively, the data show that sporadic Creutzfeldt-Jakob disease PrPSc is not a single conformational entity but a dynamic collection of two distinct populations of particles. This implies the co-existence of different prions, whose adaptation and evolution are governed by the selection of progressively less stable, faster replicating PrPSc conformers.
Introduction
Ample genetic, transgenetic, and biophysical data, and ultimately the generation of infectious prions from recombinant prion protein (PrP)2 in vitro (1–3) all provide compelling evidence that prion diseases are caused by the accumulation of an aberrantly folded isoform of the prion protein termed PrPSc (4). Variations in prions, which cause remarkably different disease phenotypes in the same host, are referred to as strains (5, 6). For several decades, the existence of distinct prion strains that can be passaged indefinitely has polarized the scientific community and was offered as an argument for the existence of a prion-specific genome. Subsequently, extraordinary progress in the past decade has produced convincing experimental evidence indicating that the species of prion is encoded in the primary amino acid sequence of PrPSc (6) and that prion strain characteristics are encoded in the self-replicating conformation of PrPSc (7–10). These phenotypic characteristics may undergo mutation in cloned cells, but the molecular mechanism responsible for this phenomenon remained elusive in the absence of informative nucleic acid (10). Although recent important experiments with synthetic and rodent-adapted laboratory prions suggest that structural plasticity of PrPSc is a key factor in adaptation and evolution, the exact conformational mechanism and relevancy of these observations to wild prions causing natural human prion diseases have not been established (11–13).
The extensive phenotypic heterogeneity of the most frequent human prion disease, sporadic Creutzfeldt-Jakob disease (sCJD) (14), is currently understood as a complex interplay between polymorphisms in the PRNP gene and different PrPSc conformers (6, 14). Because the conformations of PrPSc vary in different prion strains, the broad spectrum of distinct PrPSc conformers recently found in different cases of sCJD using sensitive biophysical techniques implies that sCJD is caused by a broad array of distinct prions (5, 15, 16). Furthermore, the frequent, and perhaps universal, presence of both the 21-kDa (type 1) and 19-kDa (type 2) unglycosylated fragments of protease-resistant (r) PrPSc in sCJD (17–21) indicates the co-occurrence of markedly different PrPSc conformers, often in the same anatomical structure in the same brain. Apart from challenging the validity of the clinicopathological classification of sCJD based on PRNP gene polymorphism and Western blot patterns of type 1 or type 2 rPrPSc (14, 22), these findings raise some fundamental questions. (a) Do the co-existent type 1 and type 2 rPrPSc form distinct or hybrid particles composed of both types of PrPSc? (b) Do they replicate independently and thus imply co-existence of different sCJD prions? (c) What is the impact of co-existence of distinct PrPSc conformers on prion adaptation and evolution?
In our earlier experiments, we found a remarkable inter-individual conformational heterogeneity of sCJD PrPSc, and we established that progression rates of the disease correlate with replication rate of human prions in vitro, which is in turn governed by the size and instability of aggregates formed by this protein (6, 15, 16). To extend these observations with the aim to advance our understanding of the molecular mechanism of human prion co-existence, adaptation, and evolution, we applied complementary biophysical techniques to a set of representative sCJD cases with the co-occurrence of type 1 and type 2 rPrPSc (type 1 + 2) in the same cortical location (20) in the same brain. First, we investigated whether sCJD with mixed type 1 + 2 sCJD PrPSc would contain a single particle composed of conformers that generate two different fragments after proteolytic digestion or whether they represent the distinct prion particles expected for different prions (15). We isolated two distinct prion particle populations that display differing conformational stabilities and different sedimentation velocities in the sucrose gradient, which argues that the co-occurrence of type 1 and 2 PrPSc in sCJD is due to the presence of two distinct aggregates, each containing a subpopulation of similar conformers of PrPSc. The independent replication of type 1 and type 2 PrPSc populations with markedly different replication rates in sPMCA argues for the co-existence of different prions in the same sCJD brain. The invariable and progressive dominance of type 1 PrPSc and the subsequent disappearance of type 2 from the mixture during serial amplification suggest a competition between different conformers and ongoing conformational evolution through the natural selection of faster replicating PrPSc conformers.
MATERIALS AND METHODS
Ethics Statement
All procedures were performed under protocols approved by the Institutional Review Board at Case Western Reserve University. In all cases, written informed consent for research was obtained from the patient or legal guardian, and the material used had appropriate ethical approval for use in this project. All patients' data and samples were coded and handled according to National Institutes of Health guidelines to protect the patients' identities.
sCJD Cases
We selected six representative subjects with a mixed pattern of rPrPSc on Western blots of brain tissue from a group of 36 patients with a definitive diagnosis of sCJD. The criteria for inclusion were as follows: (a) availability of clinical diagnosis of CJD according to World Health Organization criteria (23–25); (b) methionine homozygous at codon 129 of the human prion protein (PrP) gene (PRNP); (c) unequivocal WB classification; (d) unequivocal classification of pathology as sCJD at the National Prion Disease Pathology Surveillance Center in Cleveland, OH; (e) demographic data distribution within the 95% confidence interval of the whole sCJD group, resulting in no difference between selected cases and the whole group in any of the statistically followed parameters.
Brain Samples and PRNP Gene Sequencing
DNA was extracted from frozen brain tissues in all cases, and genotypic analysis of the PRNP coding region was performed as described (26–28). Patients lacked pathogenic mutations in the PRNP and had no history of familial diseases or known exposure to prion agents. These cases underwent additional detailed WB analyses of the PrPSc so that we could ascertain the accuracy of their original classification and confirm that the same brain homogenate analyzed by CDI contained mixed type 1 + 2 PrPSc(129M). Coronal sections of human brain tissues were obtained at autopsy and stored at −80 °C. Three 200–350-mg cuts of frontal (superior and more posterior middle gyri) or occipital cortex were taken from each brain and used for molecular analyses.
Brain Homogenates
Slices of tissues weighing 200–350 mg were first homogenized to a final 15% (w/v) concentration in calcium- and magnesium-free PBS, pH 7.4, by three 75-s cycles with Mini-beadbeater 16 Cell Disrupter (Biospec, Bartlesville, OK). The homogenates were then diluted to a final 5% (w/v) in 1% (v/v) Sarkosyl in PBS, pH 7.4, and rehomogenized. After clarification at 500 × g for 5 min, 1 aliquot of the supernatant was treated with protease inhibitors (0.5 mm PMSF and aprotinin and leupeptin at 5 μg/ml, respectively). The second aliquot was treated with 50 μg/ml of proteinase K (Amresco, Solon, OH) for 1 h at 37 °C and shaking 600 rpm on an Eppendorf Thermomixer (Eppendorf, Hauppauge, NY), and PK was blocked with PMSF and aprotinin/leupeptin mixture.
Western Blots
Both PK-treated and -untreated samples were diluted 9-fold in 1× Laemmli Buffer (Bio-Rad) containing 5% (v/v) β-mercaptoethanol and a final 115 mm Tris-HCl, pH 6.8. Samples were heated for 5 min at 100 °C, and ∼2 ng of PrP per lane was loaded onto 15% Tris-HCl, SDS-polyacrylamide gels (Bio-Rad) mounted in Western blot apparatus (Bio-Rad). After electrotransfer to Immobilon-P transfer membranes (Millipore, Bedford, MA), the membranes were blocked with 2% (w/v) BSA in TBS containing 0.1% of Tween 20 (v/v) and 0.05% (v/v) Kathon CG/ICP (Sigma). The PVDF membranes were developed with 0.05 μg/ml biotinylated mAb 3F4 (Covance, Princeton, NJ) followed by 0.0175 μg/ml streptavidin-peroxidase conjugate (Fisher) or with ascitic fluid containing mAb 3F4 (kindly supplied by Richard Kascsak) diluted 1:20,000 followed by peroxidase-labeled sheep anti-mouse IgG Ab (Amersham Biosciences) and diluted 1:3000. Alternatively, the membranes were developed with mAb 12B2 (29) for detection of type 1 rPrPSc or mAb 1E4 (30) for detection of type 2 rPrPSc, respectively. The membranes were developed with the ECL Plus detection system (Amersham Biosciences) and exposed to Kodak BioMax MR films (Fisher) or Kodak BioMax XAR films (Fisher).
Conformation-dependent Immunoassay
The CDI for human PrP was performed as described previously (28, 31), with several modifications. First, we used white Lumitrac 600 High Binding Plates (E&K Scientific, Santa Clara, CA) coated with mAb 8H4 (epitope 175–185) (32) in 200 mm NaH2PO4 containing 0.03% (w/v) NaN3, pH 7.5. Second, aliquots of 20 μl from each fraction containing 0.007% (v/v) of Patent Blue V (Sigma) were directly loaded into wells of white strip plates prefilled with 200 μl of Assay Buffer (PerkinElmer Life Sciences). Finally, the captured PrP was detected by a europium-conjugated (9) anti-PrP mAb 3F4 (epitope 108–112) (33) or europium-labeled mAb 12B2 (29), and the time-resolved fluorescence (TRF) signals were measured by the multimode microplate reader PHERAstar Plus (BMG LabTech, Durham, NC). The recHuPrP(90–231,129M) and PrP(23–231,129M) used as a calibrant in the CDI was a gift from Witold Surewicz, and preparation and purification have been described previously (34). The initial concentration of recombinant human PrP(23–231) and PrP(90–231) was calculated from absorbance at 280 nm and molar extinction coefficients 56,650 and 21,640 m−1cm−1, respectively. The purified recombinant proteins were dissolved in 4 m GdnHCl and 50% Stabilcoat (SurModics, Eden Prairie, MN) and stored at −80 °C. The concentration of PrP was calculated from the CDI signal of denatured samples using calibration curve prepared with either recPrP(23–231) for samples containing full-length PrPSc or recPrP(90–231) for samples containing truncated rPrPSc (PrP(27–30)) after proteinase K treatment.
The calibration and validation of CDI has been published extensively by us and others (15, 16, 28, 35–41). Briefly, the PK-untreated sample containing PrP was divided into native and denatured aliquots, and the latter was denatured with 4 m GdnHCl for 5 min at 80 °C. Using europium-labeled mAb 3F4 for detection, the TRF signal of the native sample corresponded to epitope 107–112 that was exposed in α-helical PrPC and hidden in PrPSc and is proportional to the concentration of PrPC (37). The signal of the denatured aliquot corresponded to the total PrP in a sample, and the concentration of PrPSc was then calculated according to the following: [PrPSc] = [PrPD] − [PrPN]. Next, the concentration of protease-resistant rPrPSc was calculated in samples subjected to the protease K treatment followed by complete denaturation using the PrP(90–231) calibration curve. The concentration of sPrPSc was calculated according to the following: [sPrPSc] = [PrPSc] − [rPrPSc]. The separate calibration for PK-treated and -untreated samples was critical for correct results due to the ∼3.5-fold lower affinity of mAb 3F4 with denatured full-length human PrP(23–231,129M) compared with PrP(90–231,129M) (15, 16).
Monitoring Dissociation and Unfolding of PrPSc by CDI
The denaturation of human PrPSc was performed as described previously (9), with several modifications. Frozen aliquots of PrPSc were thawed, sonicated three times for 5 s at 60% power with Sonicator 4000 (Qsonica, Newtown, CT), and the concentration was adjusted to a constant ∼50 ng/ml PrPSc. The 15-μl aliquots in 15 tubes were treated with increasing concentrations of 8 m GdnHCl containing 0.007% (v/v) Patent Blue V (Sigma) in 0.25 m or 0.5 m increments. After a 30-min incubation at room temperature, individual samples were rapidly diluted with Assay Buffer (PerkinElmer Life Sciences) containing diminishing concentrations of 8 m GdnHCl, so that the final concentration in all samples was 0.411 m. Each individual aliquot was immediately loaded in triplicate to dry white Lumitrac 600, High Binding Plates (E&K Scientific, Santa Clara, CA), coated with mAb 8H4, and developed in accordance with CDI protocol using europium-labeled mAb 3F4 or 12B2 for detection (9, 28, 36, 42).
The raw TRF signal was converted into the apparent fractional change of unfolding (Fapp) as follows: F = (TRFOBS − TRFN)/(TRFU − TRFN), where TRFOBS is the observed TRF value, and TRFN and TRFU are the TRF values for native and unfolded forms, respectively, at the given GdnHCl concentrations (43). To determine the concentration of GdnHCl where 50% of PrPSc is unfolded ([GdnHCl]½), the data were fitted by the least square method with a sigmoidal transition model (Equation 1),
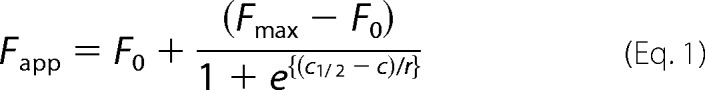 |
The apparent fractional change (F) in the TRF signal is the function of GdnHCl concentration (c); c½ is the concentration of GdnHCl at which 50% of PrPSc is dissociated/unfolded, and r is the slope constant. To determine the impact of protease treatment on the conformational stability of PrPSc, the values of fractional change after PK were subtracted from Fapp values obtained before PK (ΔFapp = F0 − FPK) and then fitted with a Gaussian model to estimate the proportion and average stability of sPrPSc conformers (Equation 2),
In this model, the PK-induced fractional change is ΔF; F0 is the fractional change at 0 concentration of GdnHCl, and c0 is the GdnHCl concentration at the maximum height A of the peak.
Sucrose Gradient Ultracentrifugation
The 400-μl aliquots of 10% brain homogenate in PBS, pH 7.4, containing 2% Sarkosyl were clarified by centrifugation at 500 × g for 5 min and carefully layered onto the top of the 10–45% sucrose gradient. The sucrose gradient was prepared in PBS, pH 7.4, containing 1% Sarkosyl in thin wall polyallomer (13 × 51 mm) tubes (Beckman Instruments, Palo Alto, CA). Alternatively, the brain homogenates and sucrose gradients destined for PMCA were prepared in 1% Triton X-100. The ultracentrifugation was performed at 50,000 rpm for 73 min at 5 °C in an Optima TL ultracentrifuge (Beckman Instruments) equipped with an SW 55 Ti rotor (Beckman Instruments). After the centrifugation, the 200- or 400-μl fractions of gradients were collected from the bottom of the tube and assayed for PrP by CDI and WBs. The densitometry of WBs was performed with ImageJ software. To compare the sucrose gradient profiles of different PrPs developed with different antibodies, the raw data were converted to apparent fractional change according to the following: Fapp = (DOBS − Dmin)/(Dmax − Dmin) where DOBS is the observed D value, and Dmax and Dmin are the density values for fractions with maximum and minimum values, respectively, at the given sucrose fraction.
Nonglycosylated Mutated Human PrPC(129M) Substrate Used in PMCA of sCJD PrPSc
The human PrPN181Q/N197Q ORF, which contains two point mutations that change Asn to Gln at positions 181 and 197, thereby eliminating the two N-linked glycosylation sites on human PrP, was generated by PCR mutagenesis from the human PrP-129M transgene construct used for the generation of the Tg40 mice and described previously (44). The human PrPN181Q/N197Q transgene construct was then generated by inserting the human PrPN181Q/N197Q ORF into the NruI site of the pHGD3 plasmid that was made by replacing the mouse PrP ORF in the half-genomic PrP clone (pHGPRP) with the restriction sites for ClaI and NruI (44). The transgene construct was microinjected into fertilized FVB/NJ eggs and planted into the oviducts of pseudo-pregnant CD-1 mice to obtain TgPrPN181Q/N197Q founder pups. All TgPrPN181Q/N197Q founder mice were bred with FVB/Prnp0/0 mice (45) to obtain TgPrPN181Q/N197Q mice in the mouse PrP-null background. Several TgPrPN181Q/N197Q lines were obtained, and the TgPrPN181Q/N197Q line was bred to homozygosity to generate the TgNN6h mice, whose human PrPN181Q/N197Q expression level in the brain is about 60% of the PrP level found in wild type FVB mice based on CDI analysis using monoclonal antibody 12B2 in CDI (Fig. 6, A and B). Perfused brain tissues from the TgPrPN181Q/N197Q mice homogenized in PBS, pH 7.4, containing 1% Triton X-100, 5 mm EDTA, and protease inhibitor mixture (Roche Applied Science) were used as substrates for PMCA.
FIGURE 6.

Preferential amplification of type 1 PrPSc in sPMCA of mixed type 1 + 2 cases followed by WBs and CDI. The sPMCA was performed with homologous human PrPC as described previously (15) with limited 2-fold dilution between rounds. A, representative WB was developed with mAb 3F4 for both type 1 and type 2, mAb 12B2 for type 1, and mAb 1E4 for type 2 PrPSc. B, amplification index of different types of PrPSc followed by WB densitometry. C–E, amplifications of different mixed cases with type 1 + 2 PrPSc in sPMCA followed by CDI with europium-labeled mAb 3F4 for total PrPSc (purple diamonds) and 12B2 selective for type 1 PrPSc (red circles) and expressed as an amplification index.
Protein Misfolding Cyclic Amplification (PMCA)
Sonication-driven PMCA was performed as described previously (15, 46) with the following modifications. The PrPSc was replicated using brains of transgenic mice expressing unglycosylated human PrPN181Q/N197Q with methionine at position 129 (44). Brain homogenates from infected mice or the human type MM1 sCJD case were diluted as described in the specific experiments to attain a final 10% brain homogenate, and 60 μl was transferred into 0.2 ml of PCR tubes equipped with 2.38 mm diameter PTFE ball (K-mac Plastics, Wyoming, MI). The buffer in all PMCA reactions was PBS, pH 7.4, containing a final 1% Triton X-100, 500 mm NaCl, 0.1% Sarkosyl, and 1 mm of an antioxidant α-tocopherol. Tubes were positioned on an adaptor placed on the plate holder of a microsonicator (Misonix Model 3000, Farmingdale, NY) and programmed to perform cycles of 60 min of incubation at 35 °C followed by a 30-s pulse of sonication set at 80% power. Samples were incubated, without shaking, and immersed in the water of the sonicator bath for 48 cycles. The 30 μl of the amplified materials were transferred to the next tube prefilled with 30 μl of substrate brain homogenate for the next round, and the remaining 30-μl aliquot was analyzed with CDI and WBs.
RESULTS
Measurement of Type 1 + 2 and Type 1 PrPSc in sCJD Cortex with CDI
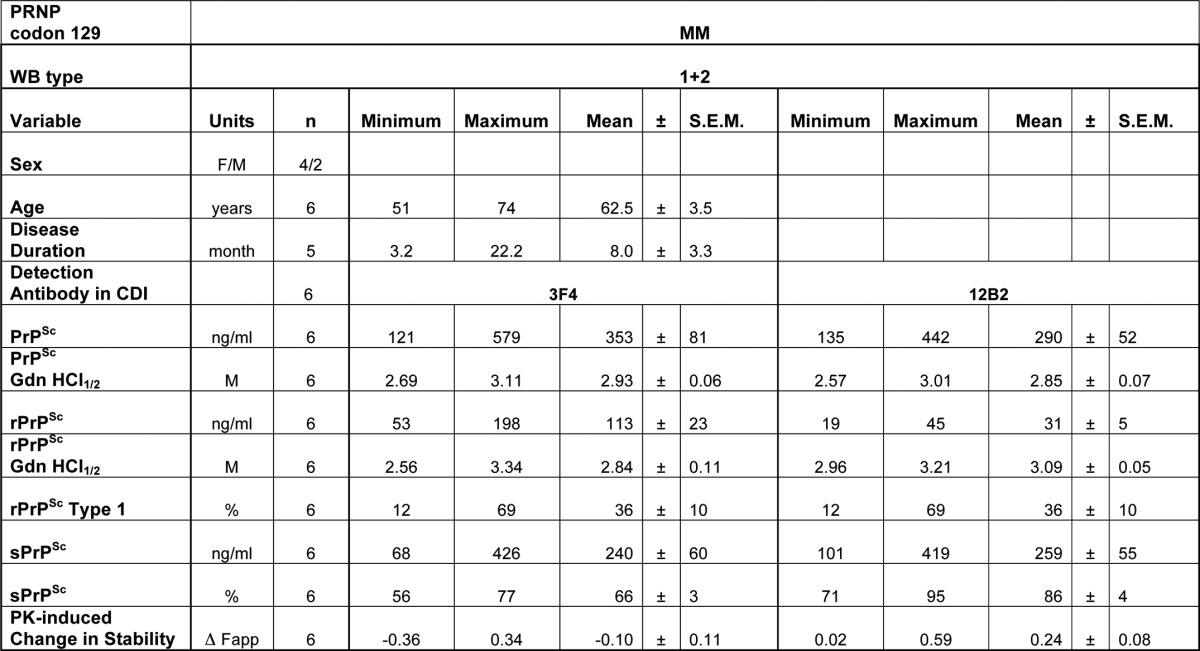
Based on a previous study of 36 cases with mixed type 1 + 2 rPrPSc (20), we selected six cases with a co-occurrence of type 1 + 2 rPrPSc in the same cortical location for detailed investigation. All cases were methionine homozygous at codon 129 (MM) of the human PrP gene (PRNP) and had a definitive diagnosis of sCJD at the National Prion Disease Pathology Surveillance Centre in Cleveland, OH. The mixed type 1 + 2 rPrPSc were found either in the frontal (n = 3) or occipital (n = 3) cortex. All cases lacked pathogenic mutations in the PRNP and had no history of familial diseases or known exposure to prions. The presence of mixed type 1 + 2 rPrPSc was verified using the recommended conditions (20, 47) and high resolution gels, to verify the accuracy of their original classification as sCJD type 1 + 2 (Fig. 1A). The descriptive demographics indicate that these cases are representative of the whole mixed type 1 + 2 group reported previously (20) and are similar to mixed type 1 + 2 cases reported by others (Table 1) (20, 48, 49).
FIGURE 1.

Western blot specificity and calibration of CDI using mAb 3F4 and 12B2. A, WB pattern of typical sCJD with mixed type 1 + 2 PrPSc before and after proteinase K (PK) treatment. The WB was developed with monoclonal antibody 3F4 (epitope 107–112) reacting with the doublet of both 21- and 19-kDa bands of unglycosylated rPrPSc, 12B2 (epitope 89–93) reacting preferentially with 21-kDa band (type 1), and 1E4 (epitope 97–108) reacting preferentially with type 2 rPrPSc. The molecular mass of the standard proteins is in kDa. B and C, calibration of CDI with (blue squares) full-length (PrP(23–231,129M)) or (red circles) truncated (PrP(90–231,129M)) human prion protein and detected with europium-labeled mAb 3F4 (B) or with Europium-labeled mAb 12B2 (C). D and E, sensitivity and specificity of type 1 rPrPSc detection with CDI using europium-labeled mAb 12B2. The different cases of sCJD that were classified pure after diagnostic WBs and a case of other neurological disorders (OND) were serially diluted and tested with CDI using europium-labeled mAb 3F4 (D) or europium-labeled mAb 12B2 (E). To obtain optimum discrimination of type 1 from type 2 rPrPSc, samples were treated with PK at a concentration equivalent to 3 IU/ml (100 μg/ml) of 10% brain homogenate for 1 h at 37 °C and precipitated with phosphotungstic acid after blocking PK with the protease inhibitor mixture. The 8H4 mAb was used for capture of denatured PrPSc. Data points and bars represent average ± S.D. obtained from three or four independent measurements.
TABLE 1.
Descriptive statistics of the data and demographics of sCJD cases
To measure the concentration of total type 1 + 2 PrPSc, protease-resistant fraction (rPrPSc), and protease-sensitive fraction (sPrPSc) in the brain cortex, we used europium-labeled mAb 3F4 (epitope 107–112) (33) for detection, and 8H4 mAb (epitope residues 175–185) (32) to capture human PrPSc in a sandwich CDI format (Fig. 1B) (28, 31). The concentration of PrP was calculated from the CDI signal of completely GdnHCl-denatured samples using a calibration curve prepared with either denatured human recPrP(23–231) for samples containing full-length PrPSc or recPrP(90–231) for samples containing truncated rPrPSc (PrP(27–30)) after proteinase K treatment. As we reported previously, this separate calibration is necessary due to the ∼3.5-fold lower affinity of mAb 3F4 with full-length human PrP(23–231,129M) compared with PrP(90–231,129M) (15, 16). The analytical sensitivity and specificity of the optimized CDI for the detection of both sPrPSc and rPrPSc using europium-labeled mAb 3F4 were previously reported by us and others (9, 28, 36, 39).
For the selective measurement of denatured type 1 rPrPSc after PK treatment, we developed a sandwich CDI using europium-labeled mAb 12B2 (epitope 89–93) to detect and 8H4 mAb (epitope residues 175–185) (32) to capture human rPrPSc type 1. The sandwich CDI with europium-labeled mAb 12B2 has a detection sensitivity of full-length denatured recombinant human PrP(23–231) comparable with europium-labeled 3F4 (Fig. 1C) (16, 28). In contrast to mAb 3F4, the mAb 12B2 does not detect shorter fragments of denatured PrP starting at residue ∼90 (Fig. 1C). The serial dilutions of brain homogenates obtained from “pure” sCJD with MM1, MM2, and VV2 PrPSc demonstrate sensitivity and specificity of detection of denatured full-length PrPSc similar to europium-labeled 3F4 (Fig. 1D). After protease K treatment, the type 1 PrPSc present in MM1 sCJD was detected with the same end point sensitivity as with mAb 3F4 (16, 28). Even though the MM2 and VV2 cases did not show any residual type 1 PrP on Western blots, and were considered pure type 2, the CDI with mAb 12B2 demonstrated low levels of type 1 PrPSc that are equivalent to ∼1% of total rPrPSc (Fig. 1E).
To expand these findings, we tested 36 sCJD cases that were classified with WBs as pure MM1 (n = 10), mixed type 1 + 2 cases (n = 6), MM2 (n = 10), and VV2 (n = 10) sCJD. Based on CDI monitoring rPrPSc with both 3F4 and 12B2 antibodies, we detected in each group a variable proportion of both type 1 and type 2 components (Fig. 2A). The content of type 1 rPrPSc in the MM2 WB group varied between 0.6 and 20% and in the VV2 WB group between 4 and 20% (Fig. 2B). From these experiments, we concluded that the sandwich CDI with europium-labeled mAb 12B2 detects type 1 rPrPSc with high sensitivity and specificity. The low but reproducible levels of type 1 PrPSc we observed in type 2 sCJD cases with different polymorphisms in the PRNP gene confirmed the data obtained with different techniques and antibodies by others (17–19, 21) and indicated that the presence of both types of rPrPSc in the same sCJD case is a universal phenomenon.
FIGURE 2.

Concentration and dichotomous impact of PK treatment on the conformational stability of PrPSc isoforms in the cortex of sCJD with mixed type 1 + 2 PrPSc in the same location. A, concentration of total and type 1 rPrPSc in the cortex of 36 cases of sCJD homozygous for different codon 129 polymorphisms and with WB classification of MM type 1 (n = 10), MM type 2 (n = 10), VV type 2 (n = 10), and MM type 1 + 2 (n = 6) rPrPSc in the same location. B, relative proportion of type 1 rPrPSc in each codon 129 polymorphism and WB classification group; the sCJD cases MM type 1 + 2 (n = 6) studied in detail in this paper are depicted as brown circles. C, concentrations of total PrPSc (purple circles), rPrPSc (black circles), sPrPSc (green diamonds). D, relative proportion of sPrPSc (green diamonds), type 1 (red triangles), and type 2 (blue triangles) in each sCJD case (n = 6). E, distinct conformational stability of total rPrPSc, type 1 rPrPSc, and incongruent impact of proteinase K treatment (−/+). The same color symbols and links indicate data obtained in the same mixed case of sCJD. F, differential stability curves of type 2 rPrPSc obtained after subtracting stability curves of type 1 rPrPSc obtained with mAb 12B2 from stability curves of total rPrPSc obtained with mAb 3F4 after PK treatment. The CDI with europium-labeled mAb 3F4 was used to determine the concentration and stability of total PrPSc and mAb 12B2 to measure the concentration and stability of type 1 rPrPSc. Each data point represents a unique patient measured in triplicate, and the concentration of PrPSc in 10% brain homogenate was calculated; the percentage of rPrPSc or type 1 is expressed over total rPrPSc. The horizontal line represents mean for each parameter.
Protease Sensitivity and Conformational Stability of Type 1 + 2 rPrPSc
The CDI allows a differentiation of PrPSc from PrPC without PK treatment, and after calibration with human recPrP(23–231), a reliable measurement of total PrPSc (15, 16, 28, 35–37). The CDI has been now introduced in numerous other laboratories (38–41), and by measuring the concentration of completely denatured PrPSc before and after PK treatment, this allowed us to determine the levels of the prion strain-specific protease-sensitive (see under “Materials and Methods” for detail explanation) PrPSc (6, 15, 16, 37). Measured with CDI, the sCJD cases with mixed type 1 + 2 rPrPSc in the same location, and homozygous for methionine in codon 129 of the PRNP gene, had variable levels of PrPSc, and the levels of sPrPSc varied from 55 to 80% in individual cases (Table 1 and Fig. 2, C and D). The type 1 rPrPSc content varied in individual cases from 12 to 69% of the total rPrPSc, with complementary type 2 levels between 31 and 88% (Fig. 2D). We observed wide inter-individual variations in stabilities of PrPSc, and the stabilities of total PrPSc measured with either mAb 3F4 or mAb 12B2 (Fig. 2E) were similar. In contrast to the variable response of PrPSc stability to PK treatment when followed with mAb 3F4, we observed a remarkably uniform increase in stability of all cases with mAb 12B2. In general, the stabilities of rPrPSc obtained with mAb 12B2 were significantly higher than those obtained with mAb 3F4. Because the unfolding curves obtained with mAb 3F4 reflect the unfolding of both type 1 and type 2 rPrPSc, and unfolding curves obtained with mAb 12B2 report on the unfolding of type 1 only, we constructed differential curves of the stability of rPrPSc for each sCJD case. These differential unfolding curves of rPrPSc correspond to the stability profiles of type 2 rPrPSc (Fig. 2F).
Cumulatively, CDI data demonstrate two populations of conformers with distinctly different stabilities and responses to PK treatment. The stabilities of type 2 rPrPSc co-occurring in the same sCJD host and in the same location are invariably lower after the PK treatment than the stability of type 1 rPrPSc. These findings are in accord with our previous observations demonstrating the lower stability of undigested oligomers of type 1 sCJD PrPSc than type 2 sCJD PrPSc and opposite effects of PK treatments leading to the lower stability of residual pure type 2 sCJD rPrPSc and the higher stability of residual pure type 1 rPrPSc (15, 16).
Prion Particle Size and Composition in sCJD Cases with Mixed Type 1 + 2 PrPSc
To investigate sCJD prion particle size and composition, we separated the prion particles according to sedimentation velocity using high speed centrifugation in sucrose gradient. The sCJD prions present in the brain homogenates of six sCJD patients with mixed type 1 + 2 PrPSc in the same location were separated in 10–45% sucrose gradients, and the collected fractions were analyzed before and after PK treatment by WBs and CDI. WBs of the fractions developed with mAb 12B2 specific for type 1 rPrPSc (17, 20) and mAb 1E4 preferential for type 2 rPrPSc (20) indicate that most of the PrPSc collected in fractions 3–5 were composed of type 1 rPrPSc (Fig. 3, A and B). In contrast, type 2 was present predominantly in fraction 1 in all six cases examined (Fig. 3, A and B). The PrPSc present in pure type 1 sCJD brain homogenate, and mixed in vitro with type 2 sCJD brain homogenate, separated in a similar manner; type 1 was collected in fractions 3–5, and type 2 sedimented mostly to fraction 1 (Fig. 3, C and D).
FIGURE 3.
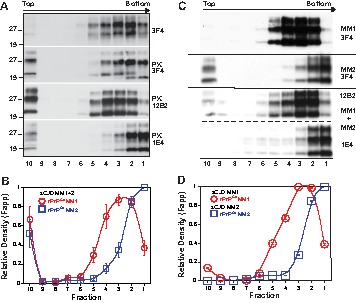
Mixed type 1 + 2 sCJD PrPSc present in the same cortical location of sCJD separated by sedimentation velocity fractionation in sucrose gradient into type 1 and type 2 sCJD PrPSc. A, representative distribution of total PrPSc before or after PK was monitored with mAb 3F4, type 1 with mAb 12B2, and type 2 with mAb 1E4 after PK in a typical mixed type 1 + 2 case of sCJD. B, cumulative plot of relative distribution of type 1 (red circles) and type 2 rPrPSc (blue squares) in sucrose gradients of six sCJD patients with mixed type 1 + 2 PrPSc present in the same cortical location. C, representative distribution of type 1 rPrPSc of a typical pure MM1 sCJD, pure type 2 rPrPSc in typical MM2 sCJD, and after mixing both types before in sucrose gradient. D, relative distribution of type 1 (red circles) and type 2 rPrPSc (blue squares) in sucrose gradient after densitometry of WBs shown in C. The total PrPs were monitored with mAb 3F4, type 1 with mAb 12B2, and type 2 with mAb 1E4 after PK. The Western blots were developed before or after PK digestion was performed with 3 IU/ml (∼100 μg/ml) of 10% brain homogenate for 1 h at 37 °C and scanned using ImageJ. The data points are mean ± S.E. from six sCJD cases each scanned in triplicate.
Next, we performed a CDI using sucrose gradient fractions obtained from six mixed type 1 + 2 cases. The parallel CDI conducted with mAb 3F4 (Fig. 4A) and with mAb 12B2 (Fig. 4B) allowed for the measurement of different PrPSc isoforms, type 1 rPrPSc directly and type 2 rPrPSc by subtracting the concentration of type 1 rPrPSc from total rPrPSc. The results expressed as a relative fractional distribution (Fapp) confirmed the data obtained with WBs and showed that the maximum level of type 1 rPrPSc was present in fractions 3–5, whereas type 2 sedimented to the bottom of the tube (Fig. 4C). We concluded from these experiments that co-existent type 1 + 2 PrPSc is composed of a mixture of distinct type 1 and type 2 particles, which can be separated according to their different sedimentation velocities. Additionally, the mixing of both PrPSc types in vitro did not induce changes in sedimentation that would indicate an interaction of type 1 and type 2 PrPSc particles.
FIGURE 4.

Sedimentation velocity fractionation and conformational stability of PrPs monitored with CDI in sCJD cases with mixed type 1 + 2 rPrPSc. A, concentrations of PrPC (green bars), total PrPSc (brown bars), and rPrPSc (black bars) determined with europium-labeled mAb 3F4. B, concentration of type 1 rPrPSc (red bars), PrPC (green bars), and total PrPSc (brown bars) determined with europium-labeled mAb 12B2. C, relative proportion of type 1 (red circles) and type 2 (blue squares) rPrPSc. D, representative conformational stability curve of type 1 rPrPSc obtained in fraction 4 (red circles) and type 2 rPrPSc (blue squares) obtained in fraction 1 of the sucrose gradient of mixed type 1 + 2 sCJD. E, typical conformational stability curves of pure type 1 rPrPSc of MM1 sCJD and type 2 rPrPSc of MM2 sCJD that were mixed in vitro. The original MM1 sCJD rPrPSc before (red circles) and fraction 4 after mixing and sucrose gradient separation (red pentagons) are shown; the original MM2 sCJD rPrPSc before (blue squares) and fraction 1 after mixing and sucrose gradient separation (blue diamonds) are shown. The fractions were obtained from the frontal (n = 3) or occipital (n = 3) cortex of six mixed type 1 + 2 sCJD cases. The total rPrPSc was measured with europium-labeled mAb 3F4, type 1, with europium-labeled mAb 12B2, and the difference is type 2 rPrPSc. The bars and data points are an average ± S.E. obtained in six cases each measured in triplicate with CDI before and after PK treatment as described in the legend for Fig. 3.
Conformational Stability of Mixed Type 1 + 2 PrPSc Fractionated in Sucrose Gradient
The conformational stability of rPrPSc collected from mixed type 1 + 2 cases in fraction 4 of the sucrose gradient was markedly different from the stability obtained in fraction 1 (Fig. 4D). The stability of rPrPSc collected in fraction 4 and fraction 1 was similar to the stability of type 1 PrPSc and type 2 PrPSc, respectively, collected from pure MM1 or MM2 cases of sCJD (Fig. 4E). Mixing brain homogenates of pure type 1 and type 2 sCJD in vitro did not change the sedimentation velocity of rPrPSc, and the stability of each type of rPrPSc remained unchanged. We concluded that brain homogenates of mixed type 1 + 2 sCJD contain particles of different sizes in the same cortical location, with distinctly different stabilities, corresponding in general to the range we have seen in pure type 1 or pure type 2 sCJD, as observed previously after PK treatment (15, 16). The experiments also confirmed our earlier observation of a paradoxically decreased stability of type 2 sCJD PrPSc after PK treatment (15). The mixing experiments performed in vitro did not indicate any interaction between type 1 and type 2 particles or the formation of hybrid aggregates.
PMCA of Mixed Type 1 + 2 sCJD PrPSc
In the next experiments, we addressed the question whether the type 1 and type 2 particles in the mixture replicate independently as would be expected from a mixture of different prions. The WBs performed on the reaction product generated in sPMCA with homologous human PrPC substrate expressed in the brains of Tg(HuPrP,129M) mice (15, 50) were developed with mAb 3F4 to detect total rPrPSc, and with mAb 12B2 for type 1 rPrPSc (Fig. 5, A–C). The impact of amplification on the type 1 and type 2 doublet was difficult to investigate directly due to the limited amplification rate and low concentrations of unglycosylated PrPSc in the reaction product. The enzymatic deglycosylation revealed a sudden change in the reaction product with a dominant band of type 1 and absent type 2 PrPSc (Fig. 5C). The second band with an apparent mass of ∼27 kDa corresponds to the deglycosylated PrPSc that remained incompletely digested even after 1 h of incubation with up to 100 μg/ml of PK at 37 °C in the presence of 1% Sarkosyl; this highly PK-resistant subfraction was observed after PMCA by us and others previously (11, 15). To investigate the abrupt transition in detail and to “slow down” the conformational transformation of PrPSc from original human brain homogenate seed, we decided to use only a 2-fold dilution between sequential PMCA rounds, in contrast to the usual 10-fold dilution of the reaction product, into fresh substrate Tg(HuPrP) brain homogenate. The WB data confirmed previous observations (Fig. 6, A and B) and show preferential amplification of type 1 over type 2 rPrPSc; the same trends were observed in the experiments followed with CDI (Fig. 6, C–E). These data indicate that mixed sCJD cases are composed of two distinct populations of conformers that replicate in PMCA independently, with preferential amplification of the type 1 PrPSc particles. The data extend our previous observations of generally more efficient amplification of type 1 PrPSc due to the lower conformational stability (15, 16).
FIGURE 5.
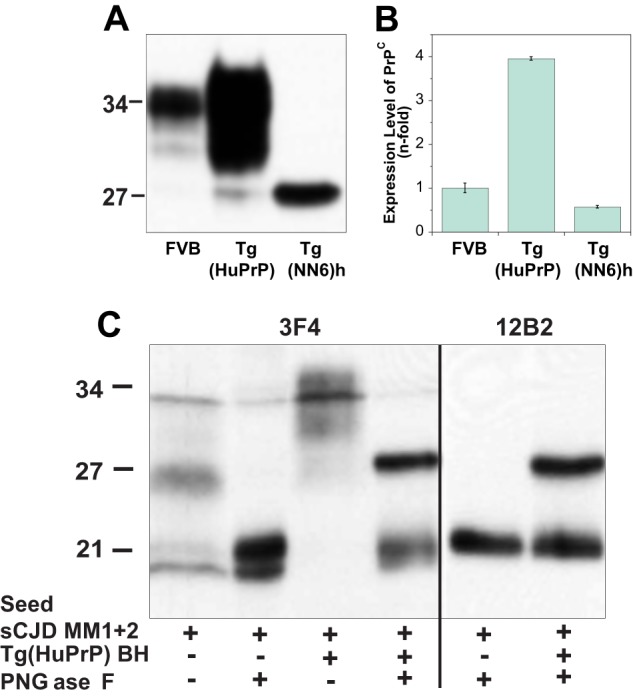
Expression levels of PrPC in Tg(HuPrP) and Tg(NN6)h mice and sPMCA of sCJD MM1 + 2 PrPSc with human PrPC substrate. A, Western blot pattern and levels of PrPC and nonglycosylated PrPC(N181Q/N197Q) in the brain homogenate of Tg(HuPrP) and Tg(NN6)h mice, respectively, developed with mAb 12B2. B, relative expression levels of PrPC and PrPC(N181Q/N197Q) in Tg(HuPrP) and in Tg(NN6)h mouse brains, respectively, were measured with europium-labeled mAb 12B2 by CDI and compared with the levels in FVB mice. C, preferential amplification of type 1 PrPSc in sPMCA of mixed type 1 + 2 cases using Tg(HuPrP) and followed by WBs. The sPMCA was performed with homologous human PrPC as described previously (15) and 10-fold dilution of type 1 + 2 sCJD brain homogenate. The reaction product was after sPMCA and PK treatment deglycosylated with peptide:N-glycosidase F (PNGase F).
To investigate the impact of an alternative substrate on the amplification of mixed type 1 and type 2 PrPSc particles and the adaptation process, we introduced as a substrate brain homogenates from transgenics that express unglycosylated PrPC (Tg(HuPrPN181Q,N197Q,129M)). Even though the PrPC levels in the Tg(HuPrPN181,197Q,129M) are 60% of the level in FVB mice (Fig. 6, A and B), this substrate amplifies both pure type 1 and type 2 PrPSc and offers the critical advantage of an unequivocal high intensity band of unglycosylated PrPSc without enzymatic deglycosylation. The WBs performed on the reaction product generated in nine rounds of sPMCA were developed with mAb 3F4 to detect total rPrPSc, with mAb 12B2 for type 1 rPrPSc and mAb 1E4 for type 2 rPrPSc (Fig. 7A). The WBs indicate a gradual loss of type 2 rPrPSc bands from the original doublet in the mixed type 1 + 2 sCJD samples, and after an adaptation period in rounds 4–6, the dominant band became type 1 rPrPSc. The experiments performed with pure type 1 sCJD (Fig. 8A) or pure type 2 sCJD (Fig. 8B) show within nine rounds of sPMCA a faithful replication of the unglycosylated rPrPSc from the original sCJD brain homogenate.
FIGURE 7.
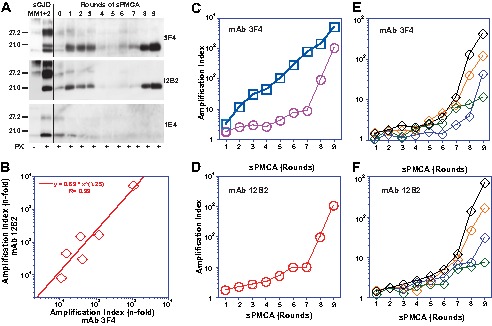
PMCA of mixed type 1 + 2 PrPSc with PrPC(N181Q,N197Q) substrate. The sPMCA was performed with initial 10-fold dilution of type 1 + 2 sCJD brain homogenate followed by limited 2-fold dilution between rounds. A, representative WB was developed with mAb 3F4 for both type 1 and type 2, mAb 12B2 for type 1, and mAb 1E4 for type 2 PrPSc. B, amplification index of total PrPSc and type 1 PrPSc was determined with CDI in six cases of type 1 + 2 sCJD using europium-labeled mAb 3F4 and europium-labeled mAb 12B2, respectively, after 9 rounds of PMCA. C and D, amplification of pure type 1 (red circles) PrPSc and pure type 2 (blue squares) in sPMCA followed by CDI with europium-labeled mAb (C) 3F4 or (D) 12B2 and expressed as an amplification index. E and F, amplification indexes of mixed cases of type 1 + 2 sCJD (diamonds) followed with mAb 3F4 (E) or 12B2 (F).
FIGURE 8.
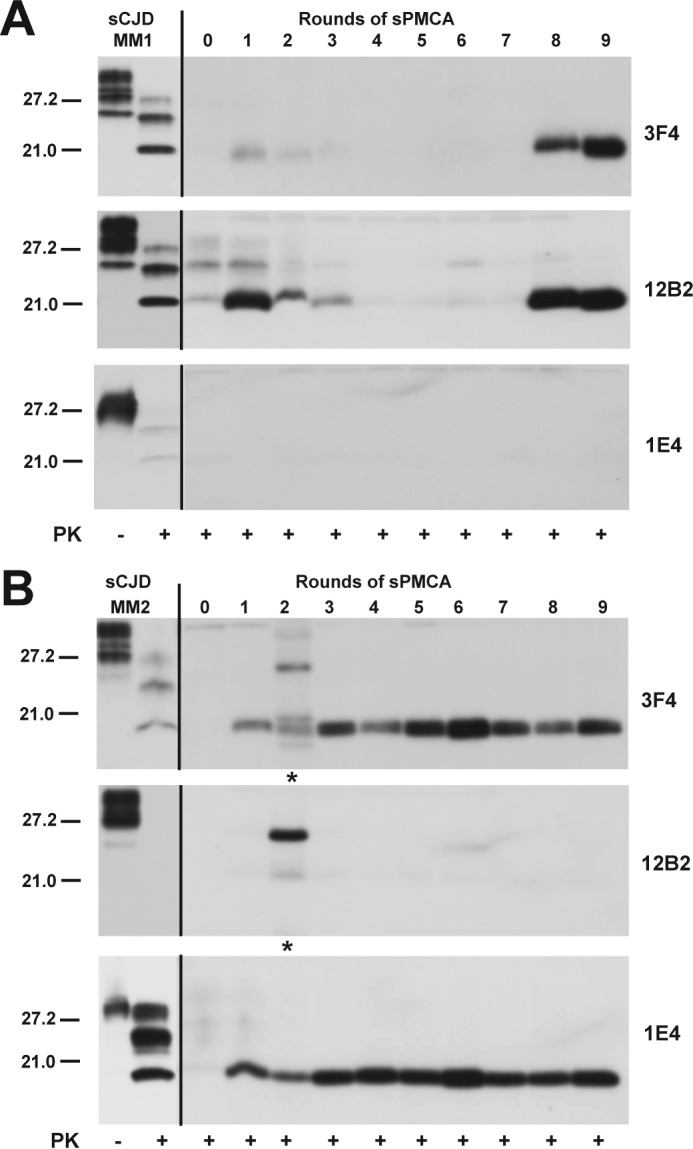
Selective amplification of pure type 1 or pure type 2 sCJD PrPSc using PrPC(N181Q/N197Q) substrate. The sPMCA was performed as described with initial 10-fold dilution of pure sCJD type 1 (A) and pure type 2 sCJD(B). The total rPrPSc was after PK treatment detected with mAb 3F4, type 1 rPrPSc with mAb 12B2, and type 2 with mAb 1E4. The incompletely PK-digested PrPSc in B is indicated by an asterisk.
The sPMCA of mixed type 1 + 2 sCJD cases was also followed by CDI. We used europium-labeled mAb 3F4 for the detection of total rPrPSc and europium-labeled mAb 12B2 for the selective detection of type 1 rPrPSc (Fig. 7B). The amplification index was calculated in each round using the following: (PMCA rPrPSc)/((seed rPrPSc)/2n), taking into account the number (n) of PMCA rounds and sequential 2-fold dilution of the seed between rounds. The regression analysis of amplification indexes obtained with six mixed sCJD cases after nine rounds of PMCA indicated consistently higher amplification rates of the type 1 rPrPSc in comparison with the total rPrPSc (Fig. 7B). To understand the dynamics of changes and inter-individual differences, we performed new experiments with sequential CDI after each round and with both antibodies. The control experiments with pure type 1 and type 2 sCJD replicated both rPrPSc conformers efficiently, with type 2 replicating faster and with a less prominent adaptation phase (Fig. 7, C and D). Even though the amplification index varied significantly between different cases of mixed type 1 + 2 sCJD, the data obtained with mAb 12B2 are in agreement with the data obtained using mAb 3F4, which indicate that the primary amplifying species from the mixture was type 1 rPrPSc (Fig. 7, E and F). Thus, the more sensitive and specific CDI confirmed qualitative WB findings, and both methods support the conclusion that type 1 conformers present in a mixture of type 1 + 2 rPrPSc replicate independently and more efficiently and as a result dominate the later rounds of sPMCA. In contrast, the type 2 conformers initially present in the mixture gradually disappeared. This adaptation phase, which apparently differs between different sCJD PrPSc, is intriguing and may suggest distinct optimum PrPSc/PrPC stoichiometry for the conversion of different PrPSc conformers or an adaptation to different auxiliary molecules that may be species-specific.
The conformational stability assay of rPrPSc performed with CDI using mAb 12B2 showed a dramatic shift of type 1 rPrPSc to the less stable conformers after nine rounds of PMCA with unglycosylated PrPC(N181,197Q) substrate (Fig. 9A). In control experiments performed with pure type 2 sCJD, the unfolding curves of rPrPSc collected before and after PMCA were superimposable (Fig. 9B). These experiments indicate that PMCA performed with unglycosylated PrPC(N181,197Q) substrate and sCJD PrPSc harboring predominantly type 1 or type 2 propagate these conformers with high fidelity. The data are in accord with our previous observations showing that the faster replication of type 1 PrPSc in vitro is due to the higher proportion of less stable protease-sensitive oligomers than in type 2 PrPSc (5, 15). Consequently, in “mixed” cases of sCJD, type 1 rPrPSc replicated preferentially, and its stability significantly decreased further below the stability of type 1 that was observed in the original brain homogenate. Cumulatively, these data indicate that mixed sCJD cases are composed of two populations of conformers that replicate in PMCA independently and, under favorable conditions, may undergo further conformational selection in favor of faster replicating, less stable conformers.
FIGURE 9.
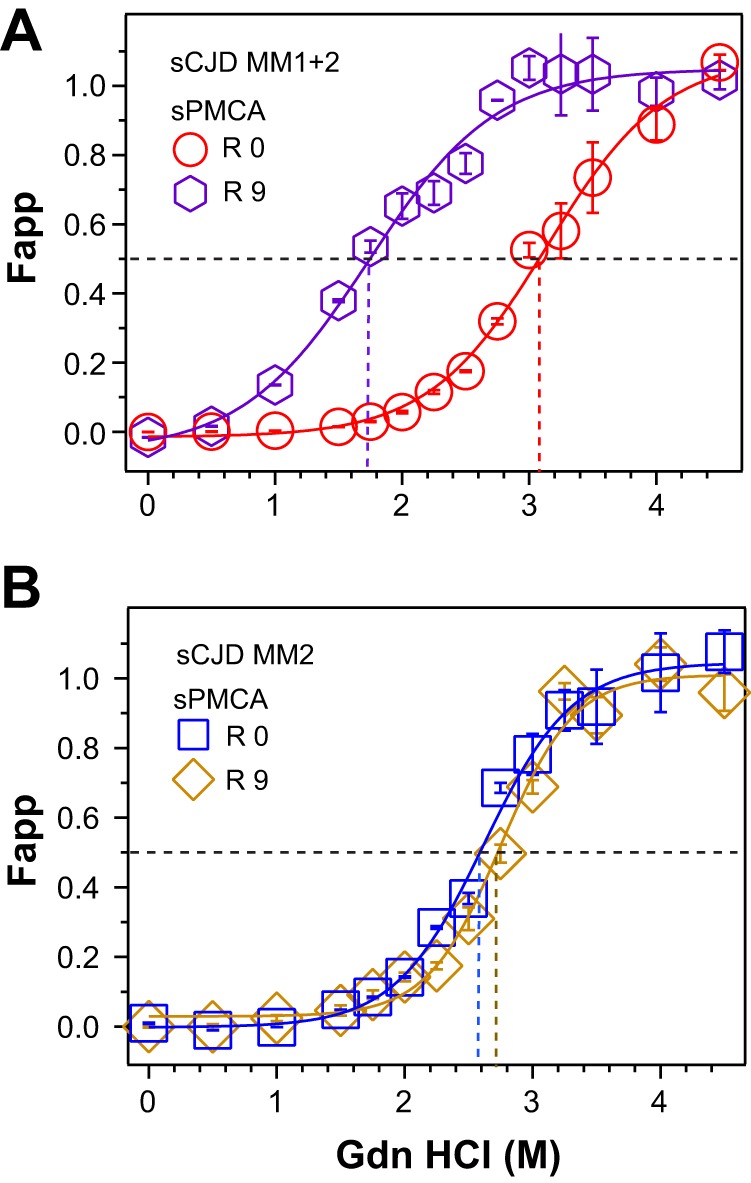
Selection of type 1 rPrPSc and further shift in conformational stability after PMCA of mixed type 1 + 2 sCJD with unglycosylated PrPC substrate. A, conformational stability curve of type 1 rPrPSc obtained with europium-labeled mAb 12B2 before (red circles) and after 9 rounds of PMCA (purple hexagons). B, conformational stability curve of a typical pure type 2 rPrPSc obtained with europium-labeled mAb 3F4 before (blue squares) and after 9 rounds of PMCA (yellow diamonds).
DISCUSSION
Here, we disclose two major phenomena defining sporadic human prion diseases as follows: (a) the co-existence of two distinct populations of human PrPSc particles in the same host; and (b) a conformational evolution of human PrPSc by natural selection. Using sedimentation velocity separation by high speed centrifugation in sucrose gradient, we show that PrPSc in mixed type 1 + 2 sCJD is a blend of distinct type 1 and type 2 particles, each composed of conformers with unique stability and replication rates in vitro. Each of these features offer evidence of a distinct prion strain and suggest that distinct prions frequently co-exist in the most prevalent human prion disease, sCJD. Remarkably, the independent replication of type 1 PrPSc with novel PrPC substrate leads to gradual evolution by natural selection of the conformational subset with the highest replication rate due to the lowest stability.
Co-existence of Distinct PrPSc Conformers in sCJD
Our CDI data obtained with mAb 12B2 (Fig. 2, A and B) are in accord with WB observations obtained with more sensitive and specific antibodies that uncovered frequent, and probably ubiquitous, co-existence of 21-kDa (type 1) and 19-kDa (type 2) fragments of unglycosylated rPrPSc in the same human prion-infected brain (17–21). Additionally, the growing body of evidence accumulated with sensitive conformational methods, including CDI, indicates extensive conformational heterogeneity of sCJD rPrPSc fragments that otherwise show a single band (type) on WBs (5, 15–17). Thus, the remarkably variable phenotypes of all human prion diseases, including sporadic CJD, apparently stems from the PRNP gene polymorphisms in codon 129 of the PRNP gene and a complex interaction with different conformers of PrPSc (5, 6, 14, 22). Taken together, the pure type 1 or type 2 cases may frequently contain low levels of the second minor component (type 2 or type 1, respectively), which is not detected by regular WBs.
We used the measurement of sedimentation velocity (51) to investigate whether co-existent type 1 and type 2 rPrPSc form distinct particles in the brains of patients with mixed type 1 + 2 sCJD in the same cortical location. We previously observed a broad range of sedimentation velocities using ultracentrifugation of sCJD brain homogenates in sucrose gradient, which indicates that sCJD PrPSc proteins exist in the continuum of aggregates composed from <20 to >600 PrPSc molecules (15). However, the major fraction of type 2 rPrPSc conformers showed strikingly high sedimentation velocity, in contrast to the lower sedimentation velocity of MM1 PrPSc, indicating that type 2 PrPSc formed aggregates with different sizes, shapes, or both (15). We used the differences in sedimentation velocity here to isolate type 1 and type 2 rPrPSc from the mixture extracted from sCJD brains and to characterize them conformationally. The data indicate that type 1 and type 2 rPrPSc co-existing in the same cortical location form distinct particles, which are composed of homogeneous populations of conformers that have uniform N-terminal proteolytic cleavage sites and conformational stability. Because both types of particles had Gaussian distribution and overlapped in some fractions, we asked whether a minor fraction of these conformers may interact or form hybrid particles. This is apparently not the case, because we observed no changes in the sedimentation velocity of aggregates nor any formation of hybrids after mixing different types in vitro. Taken together with the growing number of independent studies indicating that type 1 and type 2 rPrPSc co-exist in high proportion, and likely in the majority of sCJD cases, our findings suggest that the co-existence of distinct prion particles is a common feature of sCJD. The different quaternary structure, or packing of the monomers of PrPSc with different conformations in distinct particles, is responsible for the peptide fragmentation pattern, consisting of predominantly 19-kDa fragments in type 2 rPrPSc or 21 kDa in type 1 rPrPSc, after PK treatment.
Implications for Adaptation, Competition, Evolution, and Selection of Prions
Although the possible co-existence of different prions in naturally prion-infected sheep, goat, and mink has been suspected early on (7, 52–56), these experiments could not discriminate between two possibilities as follows: (a) selection of strains from a co-existing pool in the natural host, or (b) strain adaptation (mutation) caused by the switch from the primary sequence of the original host's PrPSc to the different PrP sequence in the new host (57, 58). We used a modified PMCA with homologous as well as unglycosylated PrPC(N181Q,N197Q) substrate to test whether the type 1 and type 2 particles of PrPSc we isolated in sucrose gradients replicate independently as is expected for different prions (46). The unglycosylated PrPC(N181Q,N197Q) expressed in the brains of transgenic mice that carry the PrP construct with double N181Q and N197Q mutations, allowed for direct monitoring of unglycosylated bands of type 1 and type 2 rPrPSc in PMCA with WBs, and thus the detection of subtle changes in their mobility and ratio without additional enzymatic deglycosylation.
In contrast to pure type 2 sCJD seeds, the serial PMCA of pure type 1 and mixed type 1 + 2 seeds underwent two distinct phases. In the first adaptation phase, the amplification was limited, and as a result, the bands of unglycosylated rPrPSc gradually disappeared on WBs; the amplification was detectable only with CDI. In the second phase, we observed an abrupt increase in the amplification rate from 64- to 128-fold dilution of the original seed in rounds 6 and 7. Within the type 1 + 2 mixture, type 2 rPrPSc gradually disappeared, even though pure type 2 sCJD amplified very well. This resulted in the uniform selection of type 1 rPrPSc in mixed type 1 + 2 cases (Fig. 7, A and B). Surprisingly, even though the N-terminal PK cleavage sites and mobility on WBs corresponded to the type 1 rPrPSc from the original brain type 1 + 2 mixture, the stability of the PMCA rPrPSc significantly dropped in comparison (Fig. 9, A and B). These findings provide experimental evidence for an evolutionary process within the type 1 + 2 mixture, with selection that favors type 1 conformers with the lowest stability. The initial preferential amplification rate of type 1 PrPSc is not surprising due to the usually higher percentage of less stable protease-sensitive oligomers and may explain the predominant presence of type 1 PrPSc in ∼70% of all sCJD cases (5, 15). These data correlate with the superior transmissibility and short incubation times of MM1 sCJD prions and with incomplete transmissions and extended incubations times of MM2 sCJD prions observed in transgenic mice overexpressing homologous or chimeric human PrPC (59, 60). Moreover, the dramatic difference in the amplification rate of pure type 2 compared with type 2 present in the type 1 + 2 mixture suggests an interference between type 1 and type 2 conformers. The prion interference has been observed in vivo in mice (61) and Syrian hamsters (62) that were inoculated simultaneously or sequentially with two distinct strains of prions. The exact molecular mechanism of this phenomenon remains to be fully elucidated, but because we observed no direct interaction between different conformers of PrPSc, the most likely explanation is a competition for PrPC substrate or auxiliary molecule implied from experiments with Syrian hamster-adapted prions (63).
Cumulatively, the distinct particle size, conformational stability, and amplification rate all argue for the frequent co-existence of different sCJD prions in the same host. Under favorable conditions with compatible PrPC substrate, the mixture of human PrPSc conformers may undergo an evolution that selects a subset with the highest replication rate, due to their lowest stability. Notably, the adaptation phase and prion strain evolution inferred from experiments with clone cells (10) and Tg mice (57, 58), has been shown here to be a conformational process. It remains to be established, however, whether the first adaptation phase is due to the requirement for the critical threshold stoichiometry of PrPSc/PrPC(N181Q,N197Q) needed for optimum replication (adaptation due to the absence of sugar chains on PrPC(N181Q,N197Q) substrate and double Asn to Gln mutation) or whether this first phase is caused by the difference between mouse and human auxiliary molecules. Even though the enzymatic deglycosylation had no impact on the biological characteristics of prions replicated in PMCA (64), and the conservative Asn to Gln mutations are outside the central PrP region between residues 96 and 167, which has a major role in determining the species barrier (57, 58), the evaluation of biological characteristics of nonglycosylated sCJD prions generated with PrPC(N181Q,N197Q) must await the results of ongoing bioassays.
Concluding Remarks and New Directions
Several explanations have been proposed for the etiology of sporadic prion diseases, including spontaneous somatic mutations in the PRNP gene or rare stochastic conformational changes in the structure of PrPC (65). Whether the co-existent type 1 and type 2 PrPSc in sCJD is the result of such a primordial event or evolution in the passage through different cells expressing different post-translationally modified PrPC remains to be established. When using new PrPC substrate that may differ in primary sequence, post-translational modifications, or both, co-existent prions may undergo progressive conformational shifts due to the natural selection of the least stable conformers with the highest replication rate. Thus, the evolutionary conformational selection mechanism of PrPSc presented in this study may explain the recently observed acquisition of drug resistance by mammalian prions (66) and calls for the reevaluation of different therapeutic strategies targeting PrPSc. The high resolution structural tools and the study of the role of PrPSc ligands must address the apparent conformational plasticity of PrPSc, which is likely responsible for the natural selection process of prions and results in prion evolution and phenotypic diversity.
Acknowledgments
We are grateful to the patients' families, the CJD Foundation, and all the members of the National Prion Disease Pathology Surveillance Center. We thank Witold Surewicz for providing recombinant PrPs for CDI calibration and Earl Poptic from Cleveland Clinic Hybridoma Facility for scaled up production of mAb 8H4.
This work was supported, in whole or in part, by National Institutes of Health Grant NS074317. This work was also supported by Centers for Disease Control and Prevention Grant UR8/CCU515004 and the Charles S. Britton Fund.
- PrP
- prion protein
- CDI
- conformation-dependent immunoassay
- MM1 sCJD
- sporadic Creutzfeldt-Jakob disease homozygous for methionine in polymorphic codon 129 of PRNP gene and 21-kDa fragment (type 1) of unglycosylated rPrPSc on WB
- MM2 sCJD
- sporadic Creutzfeldt-Jakob disease homozygous for methionine in polymorphic codon 129 of PRNP gene and 19-kDa fragment (type 2) of unglycosylated rPrPSc on WB
- PRNP
- prion protein gene
- PrPC
- normal or cellular prion protein
- PrPSc
- misfolded pathogenic prion protein
- rPrPSc
- protease-resistant conformers of pathogenic prion protein (PrP(27–30))
- sPMCA
- serial protein misfolding cyclic amplification
- sPrPSc
- protease-sensitive conformers of pathogenic prion protein
- PMCA
- protein misfolding cyclic amplification
- WB
- Western blot
- PK
- proteinase K
- TRF
- time-resolved fluorescence
- Tg
- transgenic.
REFERENCES
- 1. Legname G., Baskakov I. V., Nguyen H.-O., Riesner D., Cohen F. E., DeArmond S. J., Prusiner S. B. (2004) Synthetic mammalian prions. Science 305, 673–676 [DOI] [PubMed] [Google Scholar]
- 2. Deleault N. R., Harris B. T., Rees J. R., Supattapone S. (2007) Formation of native prions from minimal components in vitro. Proc. Natl. Acad. Sci. U.S.A. 104, 9741–9746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wang F., Wang X., Yuan C. G., Ma J. (2010) Generating a prion with bacterially expressed recombinant prion protein. Science 327, 1132–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Prusiner S. B. (1982) Novel proteinaceous infectious particles cause scrapie. Science 216, 136–144 [DOI] [PubMed] [Google Scholar]
- 5. Safar J. G. (2012) Molecular pathogenesis of sporadic prion diseases in man. Prion 6, 108–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Safar J. G. (2012) in Prions and Diseases (Gambetti P., ed) pp. 161–179, Springer-Verlag, New York [Google Scholar]
- 7. Bessen R. A., Marsh R. F. (1994) Distinct PrP properties suggest the molecular basis of strain variation in transmissible mink encephalopathy. J. Virol. 68, 7859–7868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Telling G. C., Parchi P., DeArmond S. J., Cortelli P., Montagna P., Gabizon R., Mastrianni J., Lugaresi E., Gambetti P., Prusiner S. B. (1996) Evidence for the conformation of the pathologic isoform of the prion protein enciphering and propagating prion diversity. Science 274, 2079–2082 [DOI] [PubMed] [Google Scholar]
- 9. Safar J., Prusiner S. B. (1998) Molecular studies of prion diseases. Prog. Brain Res. 117, 421–434 [DOI] [PubMed] [Google Scholar]
- 10. Li J., Browning S., Mahal S. P., Oelschlegel A. M., Weissmann C. (2010) Darwinian evolution of prions in cell culture. Science 327, 869–872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Makarava N., Savtchenko R., Baskakov I. V. (2013) Selective amplification of classical and atypical prions using modified protein misfolding cyclic amplification. J. Biol. Chem. 288, 33–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ghaemmaghami S., Colby D. W., Nguyen H. O., Hayashi S., Oehler A., DeArmond S. J., Prusiner S. B. (2013) Convergent replication of mouse synthetic prion strains. Am. J. Pathol. 182, 866–874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Colby D. W., Wain R., Baskakov I. V., Legname G., Palmer C. G., Nguyen H. O., Lemus A., Cohen F. E., DeArmond S. J., Prusiner S. B. (2010) Protease-sensitive synthetic prions. PLoS Pathog. 6, e1000736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Puoti G., Bizzi A., Forloni G., Safar J. G., Tagliavini F., Gambetti P. (2012) Sporadic human prion diseases: molecular insights and diagnosis. Lancet Neurol. 11, 618–628 [DOI] [PubMed] [Google Scholar]
- 15. Kim C., Haldiman T., Surewicz K., Cohen Y., Chen W., Blevins J., Sy M. S., Cohen M., Kong Q., Telling G. C., Surewicz W. K., Safar J. G. (2012) Small protease-sensitive oligomers of PrP(Sc) in distinct human prions determine conversion rate of PrP(C). PLoS Pathog. 8, e1002835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kim C., Haldiman T., Cohen Y., Chen W., Blevins J., Sy M. S., Cohen M., Safar J. G. (2011) Protease-sensitive conformers in broad spectrum of distinct PrP structures in sporadic Creutzfeldt-Jakob disease are indicators of progression rate. PLoS Pathog. 7, e1002242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Uro-Coste E., Cassard H., Simon S., Lugan S., Bilheude J. M., Perret-Liaudet A., Ironside J. W., Haik S., Basset-Leobon C., Lacroux C., Peoch K., Streichenberger N., Langeveld J., Head M. W., Grassi J., Hauw J. J., Schelcher F., Delisle M. B., Andreoletti O. (2008) Beyond PrP9res type 1/type 2 dichotomy in Creutzfeldt-Jakob disease. PLoS Pathog. 4, e1000029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yull H. M., Ritchie D. L., Langeveld J. P., van Zijderveld F. G., Bruce M. E., Ironside J. W., Head M. W. (2006) Detection of type 1 prion protein in variant Creutzfeldt-Jakob disease. Am. J. Pathol. 168, 151–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schoch G., Seeger H., Bogousslavsky J., Tolnay M., Janzer R. C., Aguzzi A., Glatzel M. (2006) Analysis of prion strains by PrPSc profiling in sporadic Creutzfeldt-Jakob disease. PLoS Med. 3, e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cali I., Castellani R., Alshekhlee A., Cohen Y., Blevins J., Yuan J., Langeveld J. P., Parchi P., Safar J. G., Zou W. Q., Gambetti P. (2009) Co-existence of scrapie prion protein types 1 and 2 in sporadic Creutzfeldt-Jakob disease: its effect on the phenotype and prion-type characteristics. Brain 132, 2643–2658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kobayashi A., Mizukoshi K., Iwasaki Y., Miyata H., Yoshida Y., Kitamoto T. (2011) Co-occurrence of types 1 and 2 PrP(res) in sporadic Creutzfeldt-Jakob disease MM1. Am. J. Pathol. 178, 1309–1315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Parchi P., Giese A., Capellari S., Brown P., Schulz-Schaeffer W., Windl O., Zerr I., Budka H., Kopp N., Piccardo P., Poser S., Rojiani A., Streichemberger N., Julien J., Vital C., Ghetti B., Gambetti P., Kretzschmar H. (1999) Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann. Neurol. 46, 224–233 [PubMed] [Google Scholar]
- 23. World Health Organization (1999) in WHO Infection Control Guidelines for Transmissible Spongiform Encephalopathies (Asher D., Pocchiari M., eds) pp. 1–38, World Health Organization Emerging and Other Communicable Diseases, Geneva [Google Scholar]
- 24. Collins S. J., Sanchez-Juan P., Masters C. L., Klug G. M., van Duijn C., Poleggi A., Pocchiari M., Almonti S., Cuadrado-Corrales N., de Pedro-Cuesta J., Budka H., Gelpi E., Glatzel M., Tolnay M., Hewer E., Zerr I., Heinemann U., Kretszchmar H. A., Jansen G. H., Olsen E., Mitrova E., Alpérovitch A., Brandel J. P., Mackenzie J., Murray K., Will R. G. (2006) Determinants of diagnostic investigation sensitivities across the clinical spectrum of sporadic Creutzfeldt-Jakob disease. Brain 129, 2278–2287 [DOI] [PubMed] [Google Scholar]
- 25. Geschwind M. D., Shu H., Haman A., Sejvar J. J., Miller B. L. (2008) Rapidly progressive dementia. Ann. Neurol. 64, 97–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Parchi P., Zou W., Wang W., Brown P., Capellari S., Ghetti B., Kopp N., Schulz-Schaeffer W. J., Kretzschmar H. A., Head M. W., Ironside J. W., Gambetti P., Chen S. G. (2000) Genetic influence on the structural variations of the abnormal prion protein. Proc. Natl. Acad. Sci. U.S.A. 97, 10168–10172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Parchi P., Castellani R., Capellari S., Ghetti B., Young K., Chen S. G., Farlow M., Dickson D. W., Sima A. A., Trojanowski J. Q., Petersen R. B., Gambetti P. (1996) Molecular basis of phenotypic variability in sporadic Creutzfeldt-Jakob disease. Ann. Neurol. 39, 767–778 [DOI] [PubMed] [Google Scholar]
- 28. Safar J. G., Geschwind M. D., Deering C., Didorenko S., Sattavat M., Sanchez H., Serban A., Vey M., Baron H., Giles K., Miller B. L., Dearmond S. J., Prusiner S. B. (2005) Diagnosis of human prion disease. Proc. Natl. Acad. Sci. U.S.A. 102, 3501–3506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Langeveld J. P., Jacobs J. G., Erkens J. H., Bossers A., van Zijderveld F. G., van Keulen L. J. (2006) Rapid and discriminatory diagnosis of scrapie and BSE in retro-pharyngeal lymph nodes of sheep. BMC Vet. Res. 2, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yuan J., Xiao X., McGeehan J., Dong Z., Cali I., Fujioka H., Kong Q., Kneale G., Gambetti P., Zou W. Q. (2006) Insoluble aggregates and protease-resistant conformers of prion protein in uninfected human brains. J. Biol. Chem. 281, 34848–34858 [DOI] [PubMed] [Google Scholar]
- 31. Choi E. M., Geschwind M. D., Deering C., Pomeroy K., Kuo A., Miller B. L., Safar J. G., Prusiner S. B. (2009) Prion proteins in subpopulations of white blood cells from patients with sporadic Creutzfeldt-Jakob disease. Lab. Invest. 89, 624–635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zanusso G., Liu D., Ferrari S., Hegyi I., Yin X., Aguzzi A., Hornemann S., Liemann S., Glockshuber R., Manson J. C., Brown P., Petersen R. B., Gambetti P., Sy M.-S. (1998) Prion protein expression in different species: Analysis with a panel of new mAbs. Proc. Natl. Acad. Sci. U.S.A. 95, 8812–8816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kascsak R. J., Rubenstein R., Merz P. A., Tonna-DeMasi M., Fersko R., Carp R. I., Wisniewski H. M., Diringer H. (1987) Mouse polyclonal and monoclonal antibody to scrapie-associated fibril proteins. J. Virol. 61, 3688–3693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Swietnicki W., Morillas M., Chen S. G., Gambetti P., Surewicz W. K. (2000) Aggregation and fibrillization of the recombinant human prion protein huPrP90–231. Biochemistry 39, 424–431 [DOI] [PubMed] [Google Scholar]
- 35. Safar J. G., Lessard P., Tamgüney G., Freyman Y., Deering C., Letessier F., Dearmond S. J., Prusiner S. B. (2008) Transmission and detection of prions in feces. J. Infect. Dis. 198, 81–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Safar J. G., Scott M., Monaghan J., Deering C., Didorenko S., Vergara J., Ball H., Legname G., Leclerc E., Solforosi L., Serban H., Groth D., Burton D. R., Prusiner S. B., Williamson R. A. (2002) Measuring prions causing bovine spongiform encephalopathy or chronic wasting disease by immunoassays and transgenic mice. Nat. Biotechnol. 20, 1147–1150 [DOI] [PubMed] [Google Scholar]
- 37. Safar J., Wille H., Itri V., Groth D., Serban H., Torchia M., Cohen F. E., Prusiner S. B. (1998) Eight prion strains have PrPSc molecules with different conformations. Nat. Med. 4, 1157–1165 [DOI] [PubMed] [Google Scholar]
- 38. Thackray A. M., Hopkins L., Bujdoso R. (2007) Proteinase K-sensitive disease-associated ovine prion protein revealed by conformation-dependent immunoassay. Biochem. J. 401, 475–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bellon A., Seyfert-Brandt W., Lang W., Baron H., Gröner A., Vey M. (2003) Improved conformation-dependent immunoassay: suitability for human prion detection with enhanced sensitivity. J. Gen. Virol. 84, 1921–1925 [DOI] [PubMed] [Google Scholar]
- 40. Choi Y. P., Gröner A., Ironside J. W., Head M. W. (2011) Comparison of the level, distribution and form of disease-associated prion protein in variant and sporadic Creutzfeldt-Jakob diseased brain using conformation-dependent immunoassay and Western blot. J. Gen. Virol. 92, 727–732 [DOI] [PubMed] [Google Scholar]
- 41. McCutcheon S., Hunter N., Houston F. (2005) Use of a new immunoassay to measure PrP Sc levels in scrapie-infected sheep brains reveals PrP genotype-specific differences. J. Immunol. Methods 298, 119–128 [DOI] [PubMed] [Google Scholar]
- 42. Safar J. G., Wille H., Geschwind M. D., Deering C., Latawiec D., Serban A., King D. J., Legname G., Weisgraber K. H., Mahley R. W., Miller B. L., Dearmond S. J., Prusiner S. B. (2006) Human prions and plasma lipoproteins. Proc. Natl. Acad. Sci. U.S.A. 103, 11312–11317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Safar J., Roller P. P., Gajdusek D. C., Gibbs C. J., Jr. (1993) Conformational transitions, dissociation, and unfolding of scrapie amyloid (prion) protein. J. Biol. Chem. 268, 20276–20284 [PubMed] [Google Scholar]
- 44. Kong Q., Huang S., Zou W., Vanegas D., Wang M., Wu D., Yuan J., Zheng M., Bai H., Deng H., Chen K., Jenny A. L., O'Rourke K., Belay E. D., Schonberger L. B., Petersen R. B., Sy M. S., Chen S. G., Gambetti P. (2005) Chronic wasting disease of elk: transmissibility to humans examined by transgenic mouse models. J. Neurosci. 25, 7944–7999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Fischer M., Rülicke T., Raeber A., Sailer A., Moser M., Oesch B., Brandner S., Aguzzi A., Weissmann C. (1996) Prion protein (PrP) with amino-proximal deletions restoring susceptibility of PrP knockout mice to scrapie. EMBO J. 15, 1255–1264 [PMC free article] [PubMed] [Google Scholar]
- 46. Castilla J., Morales R., Saá P., Barria M., Gambetti P., Soto C. (2008) Cell-free propagation of prion strains. EMBO J. 27, 2557–2566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Notari S., Capellari S., Langeveld J., Giese A., Strammiello R., Gambetti P., Kretzschmar H. A., Parchi P. (2007) A refined method for molecular typing reveals that co-occurrence of PrP(Sc) types in Creutzfeldt-Jakob disease is not the rule. Lab. Invest. 87, 1103–1112 [DOI] [PubMed] [Google Scholar]
- 48. Pocchiari M., Puopolo M., Croes E. A., Budka H., Gelpi E., Collins S., Lewis V., Sutcliffe T., Guilivi A., Delasnerie-Laupretre N., Brandel J. P., Alperovitch A., Zerr I., Poser S., Kretzschmar H. A., Ladogana A., Rietvald I., Mitrova E., Martinez-Martin P., de Pedro-Cuesta J., Glatzel M., Aguzzi A., Cooper S., Mackenzie J., van Duijn C. M., Will R. G. (2004) Predictors of survival in sporadic Creutzfeldt-Jakob disease and other human transmissible spongiform encephalopathies. Brain 127, 2348–2359 [DOI] [PubMed] [Google Scholar]
- 49. Gambetti P., Kong Q., Zou W., Parchi P., Chen S. G. (2003) Sporadic and familial CJD: classification and characterisation. Br. Med. Bull. 66, 213–239 [DOI] [PubMed] [Google Scholar]
- 50. Telling G. C., Scott M., Mastrianni J., Gabizon R., Torchia M., Cohen F. E., DeArmond S. J., Prusiner S. B. (1995) Prion propagation in mice expressing human and chimeric PrP transgenes implicates the interaction of cellular PrP with another protein. Cell 83, 79–90 [DOI] [PubMed] [Google Scholar]
- 51. Steensgaard J., Humphries S., Spragg S. P. (1992) in Preparative Centrifugation: A Practical Approach (Rickwood D., ed) pp. 187–232, IRL Press at Oxford University Press, Oxford [Google Scholar]
- 52. Kimberlin R. H., Walker C. A. (1978) Evidence that the transmission of one source of scrapie agent to hamsters involves separation of agent strains from a mixture. J. Gen. Virol. 39, 487–496 [DOI] [PubMed] [Google Scholar]
- 53. Bruce M. E., Fraser H. (1991) Scrapie strain variation and its implications. Curr. Top. Microbiol. Immunol. 172, 125–138 [DOI] [PubMed] [Google Scholar]
- 54. Bruce M. E. (1993) Scrapie strain variation and mutation. Br. Med. Bull. 49, 822–838 [DOI] [PubMed] [Google Scholar]
- 55. Bessen R. A., Marsh R. F. (1992) Identification of two biologically distinct strains of transmissible mink encephalopathy in hamsters. J. Gen. Virol. 73, 329–334 [DOI] [PubMed] [Google Scholar]
- 56. Bessen R. A., Marsh R. F. (1992) Biochemical and physical properties of the prion protein from two strains of the transmissible mink encephalopathy agent. J. Virol. 66, 2096–2101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Scott M. R., Peretz D., Nguyen H.-O., Dearmond S. J., Prusiner S. B. (2005) Transmission barriers for bovine, ovine, and human prions in transgenic mice. J. Virol. 79, 5259–5271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Scott M., Peretz D., Ridley R. M., Baker H. F., DeArmond S. J., Prusiner S. B. (2004) in Prion Biology and Diseases (Prusiner S. B., ed) 2nd Ed., pp. 435–482, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 59. Bishop M. T., Will R. G., Manson J. C. (2010) Defining sporadic Creutzfeldt-Jakob disease strains and their transmission properties. Proc. Natl. Acad. Sci. U.S.A. 107, 12005–12010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Korth C., Kaneko K., Groth D., Heye N., Telling G., Mastrianni J., Parchi P., Gambetti P., Will R., Ironside J., Heinrich C., Tremblay P., DeArmond S. J., Prusiner S. B. (2003) Abbreviated incubation times for human prions in mice expressing a chimeric mouse—human prion protein transgene. Proc. Natl. Acad. Sci. U.S.A. 100, 4784–4789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Manuelidis L., Lu Z. Y. (2003) Virus-like interference in the latency and prevention of Creutzfeldt-Jakob disease. Proc. Natl. Acad. Sci. U.S.A. 100, 5360–5365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Bartz J. C., Kramer M. L., Sheehan M. H., Hutter J. A., Ayers J. I., Bessen R. A., Kincaid A. E. (2007) Prion interference is due to a reduction in strain-specific PrPSc levels. J. Virol. 81, 689–697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Shikiya R. A., Ayers J. I., Schutt C. R., Kincaid A. E., Bartz J. C. (2010) Coinfecting prion strains compete for a limiting cellular resource. J. Virol. 84, 5706–5714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Piro J. R., Harris B. T., Nishina K., Soto C., Morales R., Rees J. R., Supattapone S. (2009) Prion protein glycosylation is not required for strain-specific neurotropism. J. Virol. 83, 5321–5328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Prusiner S. B. (2001) Shattuck Lecture—Neurodegenerative diseases and prions. N. Engl. J. Med. 344, 1516–1526 [DOI] [PubMed] [Google Scholar]
- 66. Oelschlegel A. M., Weissmann C. (2013) Acquisition of drug resistance and dependence by prions. PLoS Pathog. 9, e1003158. [DOI] [PMC free article] [PubMed] [Google Scholar]