

Background: As well as inhibiting insulin-like growth factor (IGF) actions, IGF-binding protein (IGFBP)-6 also promotes rhabdomyosarcoma cell migration.
Results: IGFBP-6 binds to prohibitin-2 on the cell membrane, and knockdown of the latter abrogates IGFBP-6-induced migration.
Conclusion: Prohibitin-2 is required for IGFBP-6-induced rhabdomyosarcoma cell migration.
Significance: Prohibitin-2 may be a novel target to inhibit cancer cell migration.
Keywords: Cell Migration, Insulin-like Growth Factor (IGF), Protein Phosphorylation, Protein-Protein Interactions, Rhabdomyosarcoma, Insulin-like Growth Factor-binding Protein-6, Prohibitin-2
Abstract
Insulin-like growth factor (IGF)-binding protein (IGFBP)-6 decreases cancer cell proliferation and survival by inhibiting the effects of IGF-II. More recently, IGFBP-6 was found to promote the migration of rhabdomyosarcoma (RMS) cells in an IGF-independent manner, and MAPK pathways were involved in this process. However, the precise molecular mechanisms of these IGF-independent migratory actions of IGFBP-6 are largely unknown. Here, we report that prohibitin-2 (PHB2), a single-span membrane protein, is a key regulator of IGFBP-6-induced RMS cell migration. PHB2 and IGFBP-6 co-localize on the RMS cell surface, and they specifically interact, as demonstrated by affinity chromatography, co-immunoprecipitation, biosensor analysis, and confocal microscopy. Binding affinities for PHB2 are 9.0 ± 1.0 nm for IGFBP-6 and 10.2 ± 0.5 nm for mIGFBP-6, a non-IGF-binding mutant of IGFBP-6. The C-domain but not the N-domain of IGFBP-6 is involved in PHB2 binding. In addition, IGFBP-6 indirectly increases PHB2 tyrosine phosphorylation on RMS membranes. Importantly, PHB2 knockdown completely abolished IGFBP-6-mediated RMS cell migration. In contrast, IGFBP-6-induced MAPK pathway activation was not affected, suggesting that PHB2 may act as a downstream effector of these pathways. These results indicate that PHB2 plays a key role in this IGF-independent action of IGFBP-6 and suggest a possible therapeutic target for RMS.
Introduction
Rhabdomyosarcoma (RMS)2 arises from primitive skeletal myoblasts and is the most common soft tissue malignancy of childhood and adolescence. Embryonal (ERMS) and alveolar (ARMS) are the two principal subtypes of RMS. ERMS mainly affects children less than 10 years old (1), whereas ARMS is predominantly found in adolescents and young adults, is more aggressive, and has a poorer prognosis (2).
Insulin-like growth factor (IGF)-II has been implicated as an autocrine growth factor for RMS and plays an important role in its development and progression (3). IGF actions are modulated by a family of six high affinity IGF-binding proteins (IGFBPs). Of these, IGFBP-6 is distinctive for its ∼50-fold higher binding affinity for IGF-II over IGF-I, and it inhibits IGF-II actions but has little effect on IGF-I (3). This specificity makes IGFBP-6 an attractive potential therapeutic candidate for IGF-II-dependent pediatric malignancies, such as RMS, where it may be possible to inhibit IGF-II in the tumor environment with minimal effect on IGF-I-induced normal growth. However, IGFBP-6 also promotes RMS cell migration by an IGF-independent mechanism that involves MAPK pathway activation (4, 5), raising the possibility that a receptor or membrane protein may be involved.
Prohibitins (PHB1 and PHB2) are highly conserved and ubiquitously expressed proteins. They are primarily found in mitochondria but are also present in several other cellular compartments, including the cytosol, nucleus, and plasma membrane (6, 7). They have diverse functions. For example, PHB1 and PHB2 form large ring complexes in the inner mitochondrial membrane and function as chaperone proteins that stabilize mitochondrial respiratory enzymes and maintain mitochondrial integrity (8, 9). In the nucleus, PHBs serve as transcriptional regulators, thereby repressing cell proliferation (10). Cytoplasmic PHB1 plays an important role in Ras-mediated epithelial cell migration (11), whereas cytoplasmic PHB2 has been linked to phosphatidylinositol 3-kinase (PI3K)/protein kinase B (Akt) signaling, thereby modulating skeletal muscle differentiation (12). Cell surface PHB1·PHB2 complexes interact with the C-terminal cytoplasmic domain of the HIV-1 glycoprotein (13) and the Vi polysaccharide of Salmonella typhi in intestinal epithelial cells (14), with the latter interaction resulting in suppression of early inflammatory responses. Plasma membrane PHB2 has also been identified as a receptor for insect dengue serotype 2 (15). However, it is unknown whether there is a native mammalian ligand for cell surface PHBs.
In the present study, we report the identification and characterization of PHB2 as a binding partner of IGFBP-6 on the RMS cell surface. IGFBP-6 indirectly increases its tyrosine phosphorylation, and PHB2 knockdown completely prevents IGFBP-6-induced RMS cell migration. These results suggest that PHB2 plays a key role in this IGF-independent action of IGFBP-6.
EXPERIMENTAL PROCEDURES
Cells, Reagents, and Antibodies
Human Rh30 cells, derived from an alveolar RMS, and RD cells, derived from an embryonal RMS, were obtained from ATCC. The Mem-Per membrane protein extraction kit and SuperSignal West Pico chemiluminescent substrate were from Thermo Fisher Scientific. Sequencing grade trypsin was from Promega. Affi-Gel 10 was from Bio-Rad. PCR primers for Phb2 expression were obtained from Sigma. DharmaFECT3 transfection reagent, On-Target plus SMARTpool siRNA for human PHB2, On-Target Plus non-targeting siRNA 1, and siGLO RISC-Free control siRNA were from Millennium Science (Mulgrave, Australia). Phosphatase inhibitor mixtures 1 and 2, streptavidin-agarose, and protein G-agarose were from Sigma-Aldrich. Complete, EDTA-free protease inhibitor mixture tablets were from Roche Applied Science. Antibodies to phospho-ERK1/2 (Thr-204/Tyr-204), phospho-JNK (Thr-181/Tyr-185), phospho-p38 (Thr-180/Tyr-182), ERK1/2, JNK, p38, PHB1, and horseradish peroxidase-conjugated donkey anti-rabbit IgG were from Genesearch (Arundel, Australia). β-Actin, phosphotyrosine, and PHB2 antibodies were from Millipore (North Ryde, Australia). IGFBP-6 antibody was from Gropep (Adelaide, Australia). Alexa Fluor 568 goat anti-rabbit IgG, Alexa Fluor 488 streptavidin, TRIzol, and the Superscript II reverse transcriptase kit were from Invitrogen. Disposable PD-10 desalting columns, protein A-Sepharose CL-4B, sheep anti-mouse IgG horseradish peroxidase-linked whole antibody, the CM5 chip, and the amine coupling kit were from GE Healthcare. Ni-NTA resin was from Qiagen (Melbourne, Australia). Polycarbonate membranes (12-μm pores) were from Neuro Probe, Inc. (Gaithersburg, MD). SeeBlue Plus2 molecular weight markers were obtained from Invitrogen.
RMS Cell Culture and IGFBP-6 Stimulation
Rh30 and RD cells were routinely cultured in RPMI 1640 and DMEM, respectively. Both media were supplemented with 10% bovine calf serum, 2 mm glutamine, 100 units/ml penicillin, 100 μg/ml streptomycin, and 0.25 μg/ml amphotericin B.
Cells were serum-starved in serum-free RPMI 1640 or DMEM (SFM) containing 0.05% BSA at 37 °C for 16 h, followed by incubation with SFM with or without IGFBP-6 or mIGFBP-6 (1 μg/ml) for various times as described.
Isolation of RD RMS Cell Membrane Proteins Using mIGFBP-6 Affinity Chromatography
RD cells (20 175-cm2 flasks) were detached with PBS (pH 7.4), 0.5 mm EDTA, 0.1% BSA. Cell membrane proteins were extracted using the Mem-Per membrane protein extraction kit according to the manufacturer's instructions. An mIGFBP-6 affinity column was prepared using 1 ml of Affi-Gel 10 and 5 mg of recombinant mIGFBP-6. The mIGFBP-6 affinity column was loaded with isolated membrane proteins diluted with column binding buffer (20 mm Tris, pH 7.4, 50 mm NaCl, 0.1% Triton X-100). The column was washed with column binding buffer, and bound proteins were eluted with 0.5 m and then 1 m NaCl in column binding buffer, followed by 0.5 m acetic acid. Eluted fractions were subjected to SDS-10% PAGE.
Trypsin Digestion and Mass Spectrometry
Coomassie Blue staining bands from the acetic acid eluate were cut out and washed extensively with 100 mm ammonium bicarbonate (pH 8.0) and then ammonium bicarbonate/acetonitrile (1:1) prior to drying. Sequencing grade trypsin (5 μl) was added directly at 40 °C for 4 h. Tryptic peptides were then mixed with α-cyano-2,5-dihydroxycinnamic acid matrix and dried. Samples were analyzed by MALDI-TOF-MS (Bruker Daltonics, Bremen, Germany), and spectra were analyzed using the Mascot search engine (Matrixscience) against the SwissProt protein database.
PHB2 Expression Plasmid
Total RNA was extracted from RD cells using TRIzol followed by DNase I digestion. RNA (10 μg) was reverse-transcribed using Superscript II reverse transcriptase. The PHB2 cDNA was amplified, and 5′ NcoI and 3′ EcoRI sites were introduced by PCR (94 °C for 3 min; 32 cycles of 94 °C for 30 s or 60 or 68 °C for 30 s, depending on the primer pair; 72 °C for 30 s; and then extension at 72 °C for 10 min). Primers (5′-TCAGCCATGGCCCAGAACTTGAAGG-3′ and 5′-CTGGGTGATCAGCTGTGAGGC-3′) were used to generate a 474-bp 5′-PHB2 cDNA, and primers (5′-TGTGGTGGCCAAGTTCAATGC-3′ and 5′-GGTGGAATTCTTGGTGACTAGGCTCATTTC-3′) were used to obtain a 496-bp 3′-PHB2 cDNA. These PCR products were digested by NcoI/BclI and BclI/EcoRI, respectively, and ligated into the pProExHTb expression vector in-frame with an N-terminal His6 tag. The PCR product was sequenced to confirm that it was correct.
Expression and Purification of Recombinant Proteins
Expression and Ni-NTA chromatographic purification of recombinant His6-tagged IGFBP-6, mIGFBP-6, and N- and C-domains of IGFBP-6 have been described previously (5). Following purification, recombinant proteins were dialyzed against RPMI 1640 and stored in aliquots (1–2 μg/ml) at −80 °C.
The PHB2 expression plasmid was transformed into Escherichia coli strain JM109, and expression was induced with 1.5 mm isopropyl 1-thio-β-d-galactopyranoside at 37 °C for 4–5 h. Recombinant PHB2 was purified by Ni-NTA affinity chromatography and eluted with 200 mm imidazole, 6 m guanidine hydrochloride. The protein was refolded by dialysis against 10 mm sodium acetate (pH 5.5) and stored at −80 °C.
PHB2 Pull-down
Recombinant IGFBP-6, mIGFBP-6, and RNase (1 mg) were mixed with sulfo-NHS-biotin (2.5- and 20-fold molar excess), respectively, at room temperature for 1 h, followed by desalting. Biotinylated proteins (4.5 μm) were incubated with Rh30 cells (10-cm dish, 80% confluent) at 4 °C for 1 h. Cell lysates were prepared and incubated with streptavidin-agarose (50 μl) at 4 °C for 2 h. Biotin solution (250 μl, 2 mm) was added, and the mixture was incubated at room temperature for 5 min. Beads were washed five times with 250 μl of TBS (20 mm Tris, pH 7.4, 140 mm NaCl), and bound proteins were eluted with sample loading dye (2% SDS), followed by SDS-10% PAGE and PHB2 or PHB1 Western blotting.
Co-immunoprecipitation
Recombinant IGFBP-6 or mIGFBP-6 (200 ng) was incubated with recombinant PHB2 (300 ng) at room temperature for 3 h. PHB2 antibody or IgG (2.5 μg), protein A-Sepharose, and protein G-agarose beads (15 μl each) were added for 1 h at 4 °C. Beads were washed three times with 1 ml of TBS, and bound proteins were eluted by boiling in sample loading buffer for 5 min, followed by IGFBP-6 Western blotting.
To detect PHB phosphorylation, Rh30 cells were incubated with mIGFBP-6 (1 μg/ml) at 37 °C for 20 min. After preclearance of cell lysates with protein A/G bead mixture (60 μl), anti-phosphotyrosine antibody or IgG (3 μg) was added with protein A/G beads (60 μl) for 3 h at 4 °C. Beads were washed, and bound proteins were eluted as above prior to PHB2 or PHB1 Western blotting.
Surface Plasmon Resonance
Surface plasmon resonance analysis was performed using the Biacore T100. Recombinant PHB2 in 10 mm sodium acetate (pH 5.5) was immobilized on a CM5 sensor chip using the manufacturer's amine coupling kit, followed by blocking with 1 m ethanolamine (pH 6.0). The increase in signal of recombinant PHB2 was 1784 response units. The control flow cell was activated and blocked the same way. IGFBP-6, mIGFBP-6, or isolated N- and C-domains of IGFBP-6 (8–512 nm) in HBS-EP buffer (10 mm HEPES, pH 7.4, 150 mm NaCl, 3 mm EDTA, 0.05% Surfactant p20 (GE Healthcare)) were passed over control and PHB2 flow cells at 10 μl/min for 70 s at 25 °C. Following injection, dissociation was evaluated by passing HBS-EP buffer alone over the chip at 10 μl/min for 10 min. After each run, the chip was regenerated with 0.045% SDS for 30 s. Data were processed using BIAevaluation software (version 1.1.1).
Biotinylation and Pull-down of RMS Cell Surface Proteins
Rh30 or RD cells from four 10-cm dishes each were incubated with IGFBP-6 (1 μg/ml) at 37 °C for 20 min. Cells were washed twice with cold PBS and surface-biotinylated with sulfo-NHS-S-S-biotin (0.5 mg/ml in 0.25% DMSO/PBS) at 4 °C for 40 min. Cells were then incubated with RPMI or DMEM at 4 °C for 10 min, followed by a 20-ml cold PBS wash. Cell lysates (3 mg) were incubated with 160 μl of streptavidin-agarose (50%) in TBS containing protease inhibitors and phosphatase inhibitors for 3 h. Beads were washed three times with 1 ml of cold TBS (pH 7.4). Biotinylated cell surface proteins were eluted with sample loading buffer containing DTT and analyzed by phosphotyrosine, PHB2, and PHB1 Western blotting. Band intensities were quantitated using ImageJ software (version 1.47).
In Vitro Phosphorylation
PHB2 (5 μg) was incubated with WT or mIGFBP-6 (5 μg) in 20 μl of Hepes buffer, pH 7.4, containing 1 mm DTT, 5 mm MgCl2, and 5 mm MnCl2 with or without 100 μm ATP, for 30 min at 30 °C. Phosphorylation of N-ezrin (5 μg) by the EGF receptor (1.62 units) was used as a positive control as described previously (16). The reaction was stopped by the addition of SDS-PAGE sample buffer and boiling, and the products were analyzed by SDS-10% PAGE and phosphotyrosine Western blotting.
Confocal Microscopy
Rh30 cells were detached and incubated with biotinylated IGFBP-6 or RNase (50 μg/ml) in 200 μl of PBS at 4 °C for 20 min and then with anti-PHB2 antiserum (150 μl, 1:100) at 4 °C for 20 min, followed by fixation with 4% paraformaldehyde at 4 °C for 30 min. After PBS washing (1.5 ml), cells were incubated with a mixture of Alexa Fluor 568 donkey anti-rabbit IgG (1:100) and Alexa Fluor 488 streptavidin (1:100) in 1% BSA at room temperature for 1 h and then stained with DAPI (600 nm) for 1 min. Cells were washed with 1.5 ml of PBS and examined using a Nikon Alr confocal microscope with a ×40 water lens. Confocal images were generated using Fuji ImageJA 1.45b.
PHB2 Knockdown
Human PHB-2 siRNAs SMARTpool and an On-TARGETplus non-targeting siRNA 1 were used for knockdown experiments. PHB2 or control siRNA (final concentration 300 nm) was incubated at room temperature for 20 min in 100 μl of serum-free RPMI medium with 0.05% BSA and 5.3 μl of DharmaFECT3 transfection reagent. Rh30 cells (1.5 × 105 cells/well in 12-well plates) were grown for 24 h and then incubated for 72 h at 37 °C with the siRNA mixture diluted with 400 μl of fresh 10% serum-containing RPMI. Western blotting and cell migration assays were then performed.
Cell Migration Assay
Rh30 cell migration studies were performed using a 48-well chemotaxis chamber (Neuroprobe, Cabin John, MD) as described previously (4, 5). WT or mIGFBP-6 (1 μg/ml) or IGF-II (100 ng/ml) in SFM was added to the bottom wells of the chamber. Polycarbonate filters (12-μm pore size, coated with gelatin (100 μg/ml) in 10 mm acetic acid at room temperature for 30 min) were placed over the bottom wells, and Rh30 cells (5.6 × 104 cells/well) in SFM were seeded in the top wells. After 24 h, the filter was removed, fixed in 100% methanol for 5 min, and stained with 0.5% crystal violet, 50% methanol solution for 20 min. Membranes were washed in water to remove excess dye and unbound cells and mounted on glass slides. Three random fields/well were photographed, and migrating cells were counted using ImageJ software (version 1.44o). Each data point represents 4–6 replicates.
Statistical Analysis
Results are shown as mean ± S.E. of 3–5 independent experiments, as indicated. Data were log-transformed to stabilize variance if required and analyzed by one-way analysis of variance, followed by Fisher's protected least significant difference test to compare individual treatments.
RESULTS
PHB2 Binds IGFBP-6 on RMS Cell Membranes
We previously reported that IGFBP-6 induces RMS cell migration in an IGF-independent manner (4, 5), suggesting that IGFBP-6 may bind to a cell surface protein or receptor. To identify membrane proteins that interact with IGFBP-6, we carried out a large scale preparation of RD membrane proteins, and IGFBP-6-binding membrane proteins were isolated using an mIGFBP-6 (a mutant of IGFBP-6 that does not bind IGFs) affinity column. A 35 kDa Coomassie Blue-stained band was isolated and subjected to tryptic peptide mapping followed by mass spectrometry. Six peptides corresponding to sequences within PHB2 (17), also called B-cell receptor-associated protein 37 (BAP 37) and repressor of estrogen receptor activity (REA), were identified (Table 1).
TABLE 1.
Tryptic peptides of PHB2 identified by mass spectrometry
| Amino acid position | Tryptic peptide sequences |
|---|---|
| 25–48 | LLLGAGAVAYGVRESVFTVEGGHR |
| 55–84 | IGGVQQDTILAEGLHFRIPWFQYPIIYDIR |
| 98–123 | DLQMVNISLRVLSRPNAQELPSMYQR |
| 148–165 | FNASQLITQRAQVSLLIR |
| 201–209 | QVAQQEAQR |
| 290–299 | GSDSLIKGK |
IGFBP-6 Specifically Binds to PHB2
To further confirm the interaction between IGFBP-6 and PHB2, co-immunoprecipitation and Biacore biosensor experiments were performed. Biotinylated WT and mIGFBP-6 both pulled down PHB2 from Rh30 cell lysates, whereas biotinylated RNase, an unrelated control protein, did not (Fig. 1A). Furthermore, a 3-fold excess of unlabeled mIGFBP-6 competed for the interaction between biotinylated mIGFBP-6 and PHB2. In contrast, PHB1 was not found in the pulled down fraction (not shown). These results suggest that IGFBP-6 physically interacts with PHB2 and that the interaction is reversible.
FIGURE 1.

IGFBP-6 binds to PHB2. A, Rh30 cells were incubated with biotinylated IGFBP-6, mIGFBP-6, or RNase. Proteins in cell lysates were pulled down by streptavidin-agarose and analyzed by PHB2 Western blotting. The blot is representative of three independent experiments. B, Coomassie Blue staining of purified recombinant PHB2 after SDS-10% PAGE. C, co-immunoprecipitation of recombinant IGFBP-6 or mIGFBP-6 with recombinant PHB2 using a PHB2 antiserum for immunoprecipitation and an IGFBP-6 antiserum for Western blotting. D and E, binding of IGFBP-6 or mIGFBP-6 (8–512 nm) to immobilized PHB2. Sensorgrams were recorded using a Biacore T100 and are representative of three independent experiments.
Recombinant human PHB2 was next expressed in E. coli and purified (Fig. 1B). Co-immunoprecipitation using recombinant IGFBP-6 or mIGFBP-6 and PHB2 was performed. The PHB2 antibody, but not control IgG, successfully pulled down PHB2·IGFBP-6 and PHB2·mIGFBP-6 complexes (Fig. 1C).
The kinetics of the IGFBP-6/PHB2 interaction was characterized by surface plasmon resonance. Both WT and mIGFBP-6 bound to immobilized PHB2 in a concentration-dependent fashion (Fig. 1, D and E). Calculated KD values for the interactions were 9.0 ± 1.0 nm for IGFBP-6 and 10.2 ± 0.5 nm for mIGFBP-6. In contrast, immobilized PHB-2 did not bind to unrelated proteins, including 500 nm BSA or γ-globulin (not shown).
The C-domain of IGFBP-6 Binds to PHB2
IGFBP-6 shares a three-domain structure with the other IGFBPs, and the structured N- and C-domains are involved in binding to IGFs and other biomolecules. To further analyze the PHB2 interaction with IGFBP-6, we prepared recombinant N- and C-domains of IGFBP-6 as described previously (5, 18). The isolated C-domain of IGFBP-6 bound PHB2 with a KD of 68.8 ± 10.2 nm (Fig. 2A). In contrast, the N-domain of IGFBP-6 did not bind (Fig. 2B).
FIGURE 2.
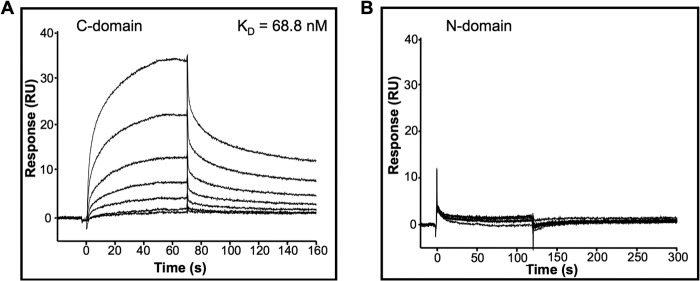
The C-domain, but not the N-domain, of IGFBP-6 binds to PHB2. Biosensor analysis of the C-domain (A) and the N-domain (B) of IGFBP-6 (8–512 nm) binding to immobilized PHB2. Sensorgrams are representative of three independent experiments.
PHB2 and IGFBP-6 Are Co-localized on the RMS Cell Membrane
To determine whether PHB2 is located on RMS cell membranes, cell surface proteins were biotinylated and then pulled down using streptavidin-agarose beads. Western blotting showed that both PHB2 and PHB1 were found in the pulled-down fraction of RD (Fig. 3A) and Rh30 cells (Fig. 3B). These results suggest that both PHB2 and PHB1 are located on RMS cell membranes. Importantly, incubation of biotinylated IGFBP-6 with Rh30 cells resulted in its co-localization with membrane-associated PHB2 (Fig. 3C).
FIGURE 3.
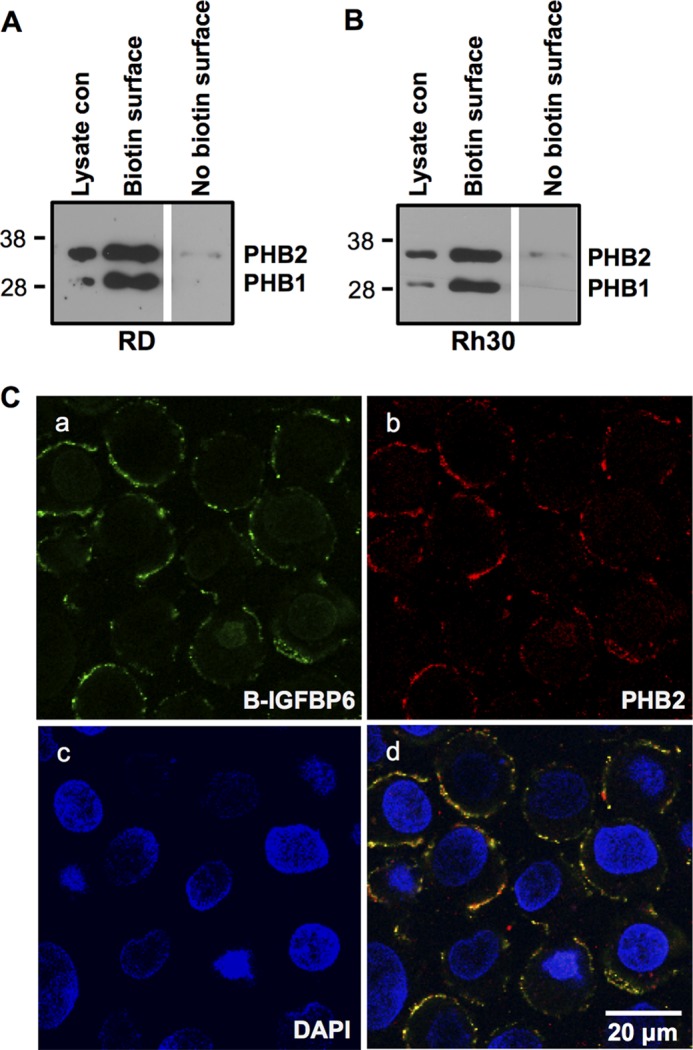
PHB2 and IGFBP-6 colocalize to the plasma membrane in RMS cells. RD (A) or Rh30 (B) cells were subjected to cell surface biotinylation, followed by streptavidin-agarose pull-down and PHB1 and PHB2 Western blotting. Whole lysate control (con) is shown. The blots are representative of three experiments. C, Rh30 cells were incubated with biotinylated IGFBP-6 and PHB2 antiserum, followed by incubation with streptavidin-Alexa Fluro 488 nm and anti-rabbit antibody-Alexa Fluor 567 nm, and then stained with DAPI. Staining is shown for IGFBP-6 (a), PHB2 (b), and DAPI (c). d, merge of a–c.
IGFBP-6 Indirectly Stimulates Tyrosine Phosphorylation of PHB2
PHB phosphorylation may contribute to its biological actions (19). Rh30 cells were incubated with mIGFBP-6 for 20 min, after which cell lysates were immunoprecipitated with a phosphotyrosine antibody followed by PHB2 Western blotting. This showed that mIGFBP-6 increased tyrosine phosphorylation of PHB2 in Rh30 cells (Fig. 4A). In contrast, PHB1 was not pulled down (not shown), indicating that it is not tyrosine-phosphorylated in these cells. Furthermore, PHB2 was not Ser/Thr-phosphorylated in the absence or presence of mIGFBP-6 (not shown). These results suggest that mIGFBP-6 specifically increases PHB2 tyrosine phosphorylation in these cells.
FIGURE 4.
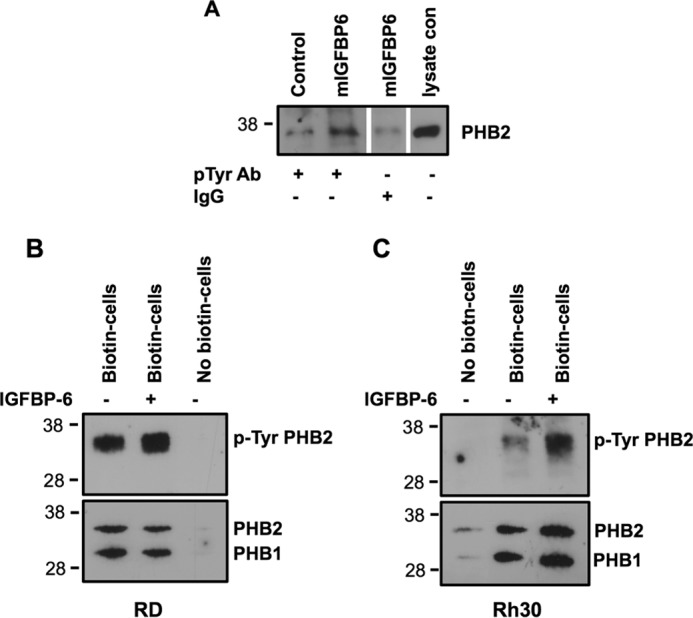
IGFBP-6 increases tyrosine phosphorylation of PHB2 in Rh30 cells. A, Rh30 cells were incubated with mIGFBP-6 (1 μg/ml) at 37 °C for 20 min. Cell lysates were immunoprecipitated with a phosphotyrosine antibody (pTyr Ab) or IgG, followed by PHB2 Western blotting. The blot is representative of four independent experiments. RD (B) or Rh30 (C) cells were incubated with mIGFBP-6 (1 μg/ml) for 20 min. Cell surface proteins were biotinylated and pulled down with streptavidin-agarose, followed by phospho-Tyr, PHB1, and PHB2 Western blotting. Blots are representative of 2–4 independent experiments.
To further investigate if IGFBP-6 increases tyrosine phosphorylation of PHB2 on RMS cell surfaces, cells were treated with IGFBP-6 (1 μg/ml) for 20 min, and cell surface biotinylation was performed, followed by streptavidin-agarose pull-down, SDS-10% PAGE, and then phosphotyrosine, PHB2, and PHB1 Western blotting. Tyrosine phosphorylation of PHB2, but not PHB1, was increased by IGFBP-6 on both RD and Rh30 cell surfaces, whereas it had no effect on PHB2 or PHB1 abundance (Fig. 4, B and C). After correcting for total PHB2, IGFBP-6 significantly increased phosphorylation 2.5 ± 0.5- and 1.4 ± 0.2-fold, respectively, in Rh30 and RD cells (p = 0.04 for each).
In vitro assays using purified proteins were next performed to determine whether IGFBP-6 directly stimulates PHB2 phosphorylation. Although the EGF receptor stimulated tyrosine phosphorylation of ezrin as described previously (16), PHB2 phosphorylation was not detected in the absence or presence of WT or mIGFBP-6 (Fig. 5), indicating that IGFBP-6 does not directly phosphorylate PHB2.
FIGURE 5.
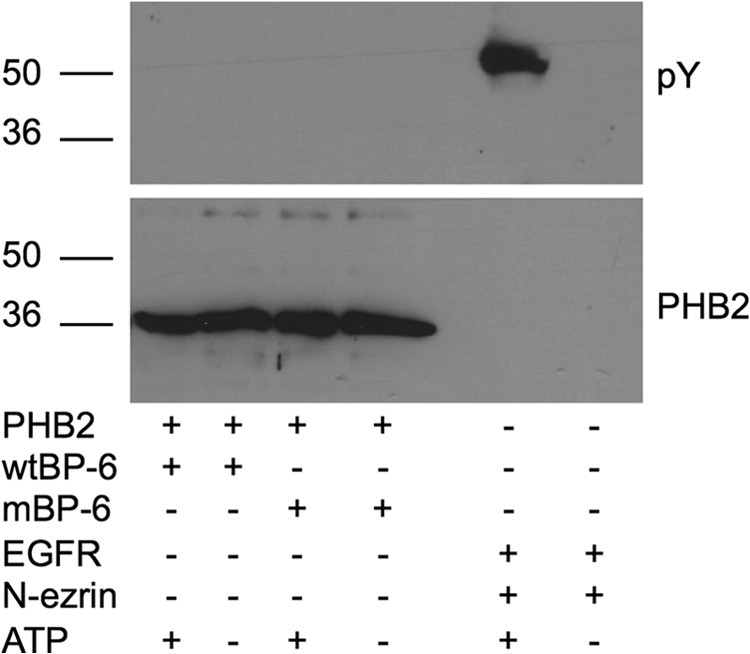
IGFBP-6 does not directly phosphorylate PHB2 in vitro. Recombinant PHB2 (5 μg) was incubated with WT or mIGFBP-6 (5 μg) in the presence or absence of 100 μm ATP. Samples were analyzed by phosphotyrosine (pY) Western blotting. Phosphorylation of N-ezrin (5 μg) by the EGF receptor (EGFR; 1.62 units) was used as a positive control. The blot is representative of two independent experiments.
Knockdown of PHB2 Abolishes IGFBP-6-induced Rh30 Cell Migration
siRNA was used to transiently knock down PHB2 expression to determine whether it plays a role in IGFBP-6-induced RMS cell migration. PHB2 levels were reduced to 10 ± 3% of control levels in the presence of PHB2-specific pooled siRNAs, whereas levels were unaffected by control siRNA 72 h after transfection (Fig. 6A). Furthermore, disruption of PHB2 expression also reduced PHB1 levels to 6 ± 2% of control levels, as reported previously in other cells (20–22). PHB2 siRNA had no effect on PHB1 mRNA levels, indicating that the effect on protein levels was post-transcriptional. PHB2 siRNA also had no effect on IGFBP-6 mRNA levels, further supporting the specificity of the siRNA. mIGFBP-6 had no effect on PHB2 and PHB1 levels in the absence or presence of PHB2 siRNA.
FIGURE 6.
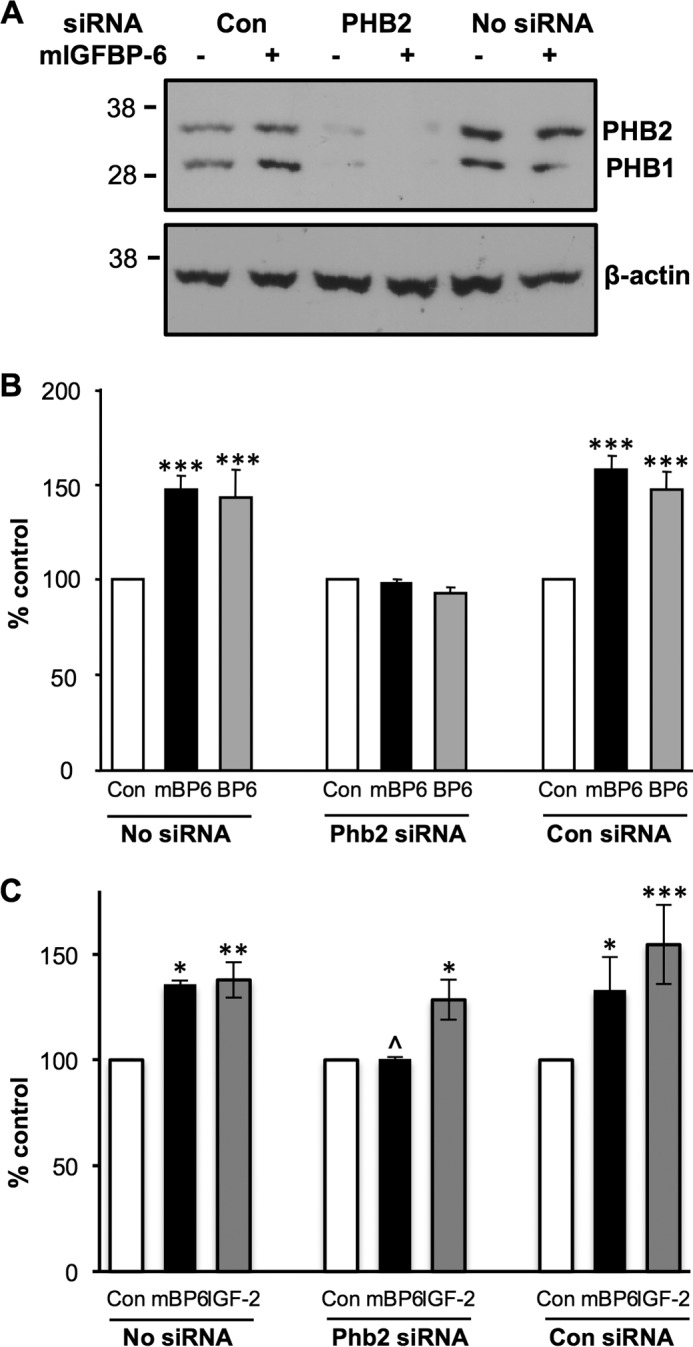
Knockdown of PHB2 abolishes IGFBP-6-induced Rh30 cell migration. A, Rh30 cells were treated with PHB2 or control (Con) siRNA for 72 h, followed by PHB2 and PHB1 Western blotting. Control cells without siRNA are shown for comparison. β-Actin was used as a protein loading control. The blot is representative of five independent experiments. B, migration assay was performed in the absence (Con) and presence of 1 μg/ml WT (BP6) or mIGFBP-6 (mBP6) for 24 h. Results are expressed as a percentage of the relevant control and shown as mean ± S.E. (error bars) of five independent experiments (***, p < 0.001 versus control). C, migration assay was performed as above in the absence (Con) and presence of mIGFBP-6 (mBP6; 1 μg/ml) or IGF-II (100 ng/ml) for 24 h. Results are expressed as a percentage of the relevant control and shown as mean ± S.E. of three independent experiments (*, p < 0.05; **, p < 0.01; ***, p < 0.001 versus control; ^, p < 0.05 mBP6/Phb2 siRNA versus mBP6 with no or control siRNA).
To investigate the effects of PHB2 knockdown on IGFBP-6-induced Rh30 cell migration, assays were performed 72 h after transfection with PHB2 or control siRNA. IGFBP-6 and mIGFBP-6 both significantly increased cell migration in the absence of siRNA or in the presence of control siRNA (p < 0.001; Fig. 6B). In contrast, the IGFBP-6-induced increase in Rh30 cell migration was completely abolished in PHB2 knockdown cells. These results clearly demonstrate that PHB2 plays an important role in IGFBP-6-dependent Rh30 cell migration. Basal and IGF-II-stimulated Rh30 cell migration were not affected by PHB2 knockdown (Fig. 6C), suggesting that PHB2 is specifically involved in IGFBP-6-induced migration.
Knockdown of PHB2 Does Not Affect Basal or IGFBP-6-induced MAPK Activation
PHB1 has been reported to regulate the ERK MAPK pathway in both normal and cancer cells (11, 19, 23), and we have shown that IGFBP-6-induced ERK and JNK MAPK phosphorylation contributes to IGFBP-6-induced Rh30 cell migration (4). We therefore investigated if PHB2 knockdown affected MAPK signaling. mIGFBP-6 significantly increased ERK (Fig. 7A) and JNK1 (Fig. 7B) in the presence or absence of PHB2 knockdown, and knockdown had no effect on the magnitude of this increase. In addition, basal levels of phospho-p38 MAPK were not changed by IGFBP-6 or PHB2 knockdown (Fig. 6C). These results suggest that neither PHB is involved in IGFBP-6-induced or basal MAPK activation in Rh30 cells.
FIGURE 7.

Knockdown of PHB2 does not affect basal or IGFBP-6-induced MAPK activation in Rh30 cells. A, Western blot analysis of phospho-ERK (pERK) and ERK; B, phospho-JNK (pJNK) and JNK; C, phospho-p38 (pp38) and p38 of Rh30 cells treated with PHB2 and control siRNA for 72 h, followed by incubation with mIGFBP-6 (1 μg/ml) for 5 min. β-Actin was used as a protein loading control. Blots from 3–4 independent experiments were scanned, and the ratio of phosphoprotein to total protein abundance was quantified. Results are shown as mean ± S.E. (error bars), with mIGFBP-6-stimulated levels designated as 1.0. **, p < 0.01; ***, p < 0.001 versus mIGFBP-6.
DISCUSSION
In the present study, we describe five novel findings: (a) IGFBP-6 co-localizes with PHB2 on RMS cell plasma membranes, and the affinity of the IGFBP-6/PHB2 interaction is within the plausible biological range; (b) the C-domain, but not the N-domain, of IGFBP-6, is directly involved in binding; (c) IGFBP-6 does not alter PHB2 levels but indirectly induces tyrosine phosphorylation of plasma membrane PHB2 in RMS cells; (d) disruption of PHB2 expression abolishes IGFBP-6-induced Rh30 cell migration; (e) PHBs are not involved in basal or IGFBP-6-induced MAPK activation in Rh30 cells.
Many studies have shown that IGFBP-6 inhibits IGF-II actions in a range of cell types (3, 24). IGF-II is an autocrine growth factor for RMS (3, 25), and IGFBP-6 inhibited proliferation and survival of RMS cells in vitro (26). Further, IGFBP-6 overexpression inhibited RMS xenograft growth in vivo (26). IGFBP-6 is strongly down-regulated in spontaneously metastatic RMS cells compared with non-metastatic cells (additional data in Ref. 27). IGFBP-6 may also localize to the nucleus in RMS cells, and this has been associated with increased apoptosis (28, 29).
Although most literature suggests that IGFBP-6 is an IGF-II inhibitor and is therefore anti-tumorigenic, there is circumstantial evidence that it may have additional actions. For example, MNU-induced glial carcinogenesis in the rat is associated with increased IGFBP-6 expression (30). IGFBP-6 levels are increased in pancreatic (31) and adrenocortical cancer (32), and IGFBP-6 levels correlate with meningioma invasiveness in vivo (33). These examples may represent a compensatory response to increased IGF-II activity, or they may reflect IGF-independent actions of IGFBP-6. Indeed, we have recently shown that IGFBP-6 promotes RMS cell migration in an IGF-independent manner (4, 5).
PHB2 Is a Cell Surface Protein That Binds IGFBP-6
Although a number of IGF-independent actions of IGFBPs have been described via distinct mechanisms (34), there is limited information regarding cell surface proteins (“IGFBP receptors”) that are responsible for these actions (35, 36). IGFBP-1 and -2 interact with cell surface integrins to modulate cell migration and adhesion via Arg-Gly-Asp (RGD) sequences in their C-terminal domains (37, 38). IGFBP-3 binds to a cell death receptor, IGFBP-3R, which mediates anti-tumor effects in breast and prostate cancer (36). IGFBP-4 binds to a Wnt receptor (Frizzled 8) and low density lipoprotein receptor-related protein 6, a Wnt co-receptor, to enhance cardiomyocyte differentiation (35). In this study, we show for the first time that cell surface PHB2 binds to IGFBP-6 and modulates its IGF-independent promigratory effect in RMS cells.
PHB1 and -2 are highly conserved membrane proteins that act interdependently (20–22, 39) by forming ring complexes that serve as protein scaffolds and chaperones in mitochondria (8, 21). However, some actions and protein interactions are specific to PHB1 or PHB2. PHBs modulate signaling pathways (19), including MAPK pathways, and affect apoptosis, proliferation, adhesion, and migration in a cell- and context-specific manner. They are found in mitochondria, nuclei, and cell membranes and translocate between these sites (8). In particular, PHBs may be functional on the cell surface. In human intestinal epithelial cells, PHB1 and PHB2 are components of a cell surface-associated complex that binds to the Vi capsular polysaccharide of Salmonella typhi, resulting in decreased IL-8 production (14). Cell surface PHB1 and -2 are involved in the T cell receptor-mediated signaling cascade in activated T cells (40) and also interact with the IgM receptor on B lymphocyte membranes (41). PHB2 was identified as a receptor mediating dengue serotype 2 entry into insect cells (15). However, all of these ligands are either small molecules or viral proteins. Whether there is a eukaryotic ligand for membrane-associated PHB1 and -2 was not known. In this study, however, IGFBP-6 was identified as a high affinity mammalian ligand.
The IGFBP-6/PHB2 Interaction
PHB2 interacts with various proteins in different subcellular compartments. In mitochondria, PHB2 interacts with Mdm12p (20) and sphingosine-1-phosphate (42), thereby regulating mitochondrial functions. In the nucleus, PHB2 physically interacts with the estrogen receptor (43) and brefeldin A-inhibited guanine nucleotide-exchange protein 3 (BIG3) (44), resulting in suppression of gene transcription and contributing to tumor growth inhibition (10). In the cytosol, PHB2 but not PHB1 binds Akt2 and reciprocally regulates Akt2 levels and muscle differentiation (12, 45). PHB2 also interacts with α-actinin and annexin A2 (46). However, the in vitro binding affinities of PHB2 for these ligands are higher than 500 nm (46), which raises the question of their biological significance. We found that IGFBP-6 binds to PHB2 with high affinity (9 nm), which is within the biological range for multiprotein complex formation and is the highest affinity so far reported.
In most cases, the C-domains of IGFBPs are responsible for binding with non-IGF ligands, and these interactions may contribute to their IGF-independent actions (3). Consistent with this, the C-domain, but not the N-domain, of IGFBP-6 binds PHB2. RMS cell migration is also mediated by the C-domain of IGFBP-6 (5), further supporting the notion that PHB2 is involved in IGFBP-6-dependent actions.
IGFBP-6 Indirectly Induces PHB2 Tyrosine Phosphorylation
PHB phosphorylation at different residues has been suggested as a mechanism to regulate its subcellular localization and actions, particularly with respect to cell signaling and cross-talk between pathways (19). The PHB1·PHB2 phosphocomplex is required for mitochondrial homeostasis and survival of human T cells (47). Insulin-induced phosphorylation of cell membrane PHB1 at tyrosine 114 and 259 by insulin receptor kinase facilitates its interaction with Shp1, which in turn negatively regulates PIP3/Akt signaling in myoblasts (48) and breast cancer cells (48, 49). On the other hand, Akt-induced threonine 258 phosphorylation of PHB1 inhibits its interactions with PIP3 and Shp2, which may facilitate insulin-induced PIP3/Akt signaling in 3T3-L1 adipocytes (50). Increased threonine 258 phosphorylation of PHB1 in lipid rafts is associated with enhanced cancer cell migration/invasion (23). Consistent with this, viral capsid protein VP1 decreased both cervical cancer migration/invasion and threonine 258 phosphorylation of PHB1 in lipid rafts (51). Calcium/calmodulin-dependent kinase IV phosphorylates serine 91 of PHB2, resulting in decreased PHB2-mediated repression of MEF2-dependent gene expression (52). In the current study, we found that IGFBP-6 increased tyrosine phosphorylation of PHB2, but not PHB1, in plasma membranes of RMS cells. In keeping with its lack of a kinase catalytic domain, IGFBP-6 did not directly phosphorylate PHB2, suggesting that it may induce a structural change that facilitates phosphorylation by another yet to be determined kinase. Nevertheless, this finding is consistent with an emerging role of PHB phosphorylation in its actions (19) and suggests that IGFBP-6-induced tyrosine phosphorylation of PHB2 may play a role in IGFBP-6-induced RMS cell migration.
PHB2 Plays a Critical Role in IGFBP-6-induced RMS Cell Migration
In this study, we showed that knockdown of PHB2 completely abolished IGFBP-6-induced Rh30 RMS cell migration. This is the first report that links PHB2 with cancer cell migration. However, PHB1 plays an important role in Ras-mediated epithelial cell migration (53) and regulates prostate cancer cell migration mediated by TGF-β (54). This may be relevant because PHB1 and PHB2 expression are interdependent, so that depletion of one protein leads to loss of the other by post-transcriptional mechanisms (20–22, 39), as was observed in our study. This finding has been attributed to enhanced degradation of one PHB in the absence of the other (21). Although the effects of reduced PHB2 or PHB1 in the present study could not be definitively distinguished, they are likely to be due to PHB2 because IGFBP-6 binds to it and increases its phosphorylation. Although we localized the binding interaction to the cell membrane, it remains possible that PHB2 in other cellular compartments may also contribute to its actions.
PHBs Are Not Required for IGFBP-6-induced MAPK Signaling in Rh30 Cells
PHB1 has been shown to be indispensable for Ras-induced Raf-1/MEK/ERK activation in several cell types (11, 19), including human epithelial cells (14, 53) and human prostate (55) and cervical cancer cells (51). PHB1 (51, 53), PHB2 (56), and PHB1·PHB2 complexes (14, 57) associate with plasma membrane lipid rafts that are believed to modulate many intracellular signaling events. Overexpression of PHB1 in 3T3-L1 fibroblasts up-regulates basal ERK MAPK signaling, leading to adipogenesis (58). In contrast, increased PHB1 phosphorylation, rather than changed PHB1 protein levels in lipid rafts, plays an important role in the Ras-driven activation of PIP3/AKT and Raf-1/ERK signaling cascades, resulting in the promotion of cancer cell metastasis (23). Our results differ from these findings, showing that reduced PHB levels in RMS cells had no effect on basal or IGFBP-6-induced ERK MAPK activation. This suggests that the PHBs may modulate cell migration as downstream effectors of ERK signaling (55), as shown in Fig. 8, pathway 3. Alternatively, PHB1 and PHB2 may regulate RMS cell migration independently of IGFBP-6-induced ERK and JNK MAPK activation (Fig. 8, pathway 4). Further studies are required to investigate these possibilities.
FIGURE 8.
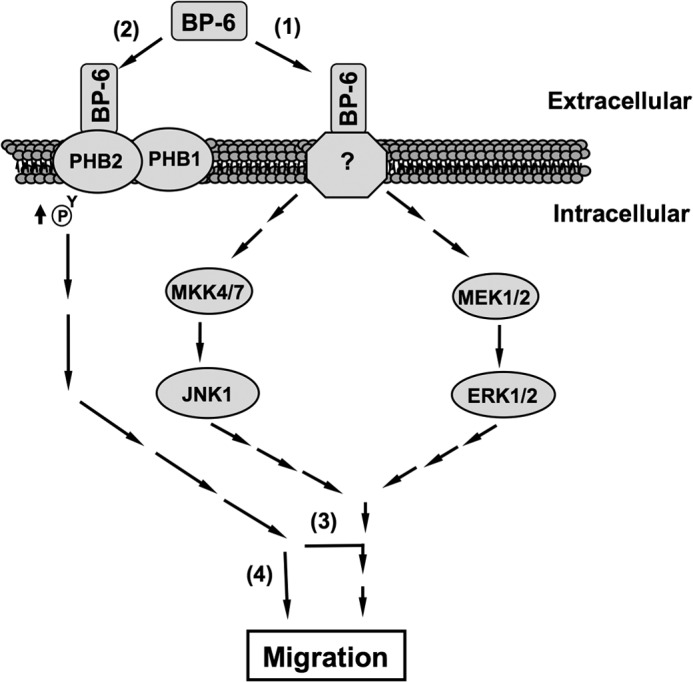
Model of the potential roles of PHB2 in IGFBP-6-induced Rh30 cell migration. IGFBP-6 binds to an unknown cell surface receptor (1), leading to activation of MAPK pathways (2). IGFBP-6 also binds to PHB2 on the cell surface and indirectly increases its tyrosine phosphorylation. PHB2 is essential for IGFBP-6-induced migration either by acting as a downstream effector of MAPKs (3) or regulating migration independently of MAPK activation (4).
In summary, we demonstrate for the first time that PHB2 is a cell surface protein that binds IGFBP-6 in RMS cells. The interaction between PHB2 and IGFBP-6 on RMS cell membranes results in increased tyrosine phosphorylation of PHB2 and leads to cancer cell migration. Although PHB2 is indispensable for IGFBP-6-induced RMS cell migration, this action is not dependent on MAPK activation. IGFBP-6 has been shown to have anti-tumorigenic properties by inhibiting IGF-II actions and, more recently, by inhibiting angiogenesis via an IGF-independent mechanism (59). However, IGFBP-6-induced migration may favor cell invasion, and targeting PHB2 may ameliorate this potentially adverse effect of an IGFBP-6-based therapeutic for IGF-II-dependent tumors.
Acknowledgments
We thank Dr. Stephen Cody for confocal microscope assistance and Dr. Anne McRobert for critical reading of the manuscript.
This work was supported by grants from the National Health and Medical Research Council of Australia, Cancer Council Victoria, and the Alfred Research Trust.
- RMS
- rhabdomyosarcoma
- ERMS and ARMS
- embryonal and alveolar RMS, respectively
- PHB
- prohibitin
- IGF
- insulin-like growth factor
- IGFBP
- IGF-binding protein
- mIGFBP-6
- mutant IGFBP-6 (P93A/L94A/L97A/L98A)
- SFM
- serum-free medium
- Ni-NTA
- nickel-nitrilotriacetic acid.
REFERENCES
- 1. Visser M., Sijmons C., Bras J., Arceci R. J., Godfried M., Valentijn L. J., Voûte P. A., Baas F. (1997) Allelotype of pediatric rhabdomyosarcoma. Oncogene 15, 1309–1314 [DOI] [PubMed] [Google Scholar]
- 2. Barr F. G. (2001) Gene fusions involving PAX and FOX family members in alveolar rhabdomyosarcoma. Oncogene 20, 5736–5746 [DOI] [PubMed] [Google Scholar]
- 3. Bach L. A. (2005) IGFBP-6 five years on; not so “forgotten”? Growth Horm. IGF Res. 15, 185–192 [DOI] [PubMed] [Google Scholar]
- 4. Fu P., Liang G. J., Khot S. S., Phan R., Bach L. A. (2010) Cross-talk between MAP kinase pathways is involved in IGF-independent, IGFBP-6-induced Rh30 rhabdomyosarcoma cell migration. J. Cell Physiol. 224, 636–643 [DOI] [PubMed] [Google Scholar]
- 5. Fu P., Thompson J. A., Bach L. A. (2007) Promotion of cancer cell migration. An insulin-like growth factor (IGF)-independent action of IGF-binding protein-6. J. Biol. Chem. 282, 22298–22306 [DOI] [PubMed] [Google Scholar]
- 6. Mishra S., Murphy L. C., Nyomba B. L., Murphy L. J. (2005) Prohibitin. A potential target for new therapeutics. Trends Mol. Med. 11, 192–197 [DOI] [PubMed] [Google Scholar]
- 7. Theiss A. L., Sitaraman S. V. (2011) The role and therapeutic potential of prohibitin in disease. Biochim. Biophys. Acta 1813, 1137–1143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Osman C., Merkwirth C., Langer T. (2009) Prohibitins and the functional compartmentalization of mitochondrial membranes. J. Cell Sci. 122, 3823–3830 [DOI] [PubMed] [Google Scholar]
- 9. Artal-Sanz M., Tavernarakis N. (2009) Prohibitin and mitochondrial biology. Trends Endocrinol. Metab. 20, 394–401 [DOI] [PubMed] [Google Scholar]
- 10. Wang S., Faller D. V. (2008) Roles of prohibitin in growth control and tumor suppression in human cancers. Transl. Oncogenomics 3, 23–37 [PMC free article] [PubMed] [Google Scholar]
- 11. Rajalingam K., Rudel T. (2005) Ras-Raf signaling needs prohibitin. Cell Cycle 4, 1503–1505 [DOI] [PubMed] [Google Scholar]
- 12. Héron-Milhavet L., Mamaeva D., Rochat A., Lamb N. J., Fernandez A. (2008) Akt2 is implicated in skeletal muscle differentiation and specifically binds Prohibitin2/REA. J. Cell Physiol. 214, 158–165 [DOI] [PubMed] [Google Scholar]
- 13. Emerson V., Holtkotte D., Pfeiffer T., Wang I. H., Schnölzer M., Kempf T., Bosch V. (2010) Identification of the cellular prohibitin 1/prohibitin 2 heterodimer as an interaction partner of the C-terminal cytoplasmic domain of the HIV-1 glycoprotein. J. Virol. 84, 1355–1365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sharma A., Qadri A. (2004) Vi polysaccharide of Salmonella typhi targets the prohibitin family of molecules in intestinal epithelial cells and suppresses early inflammatory responses. Proc. Natl. Acad. Sci. U.S.A. 101, 17492–17497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kuadkitkan A., Wikan N., Fongsaran C., Smith D. R. (2010) Identification and characterization of prohibitin as a receptor protein mediating DENV-2 entry into insect cells. Virology 406, 149–161 [DOI] [PubMed] [Google Scholar]
- 16. McRobert E. A., Gallicchio M., Jerums G., Cooper M. E., Bach L. A. (2003) The amino-terminal domains of the ezrin, radixin, and moesin (ERM) proteins bind advanced glycation end products, an interaction that may play a role in the development of diabetic complications. J. Biol. Chem. 278, 25783–25789 [DOI] [PubMed] [Google Scholar]
- 17. Strausberg R. L., Feingold E. A., Grouse L. H., Derge J. G., Klausner R. D., Collins F. S., Wagner L., Shenmen C. M., Schuler G. D., Altschul S. F., Zeeberg B., Buetow K. H., Schaefer C. F., Bhat N. K., Hopkins R. F., Jordan H., Moore T., Max S. I., Wang J., Hsieh F., Diatchenko L., Marusina K., Farmer A. A., Rubin G. M., Hong L., Stapleton M., Soares M. B., Bonaldo M. F., Casavant T. L., Scheetz T. E., Brownstein M. J., Usdin T. B., Toshiyuki S., Carninci P., Prange C., Raha S. S., Loquellano N. A., Peters G. J., Abramson R. D., Mullahy S. J., Bosak S. A., McEwan P. J., McKernan K. J., Malek J. A., Gunaratne P. H., Richards S., Worley K. C., Hale S., Garcia A. M., Gay L. J., Hulyk S. W., Villalon D. K., Muzny D. M., Sodergren E. J., Lu X., Gibbs R. A., Fahey J., Helton E., Ketteman M., Madan A., Rodrigues S., Sanchez A., Whiting M., Young A. C., Shevchenko Y., Bouffard G. G., Blakesley R. W., Touchman J. W., Green E. D., Dickson M. C., Rodriguez A. C., Grimwood J., Schmutz J., Myers R. M., Butterfield Y. S., Krzywinski M. I., Skalska U., Smailus D. E., Schnerch A., Schein J. E., Jones S. J., Marra M. A. (2002) Generation and initial analysis of more than 15,000 full-length human and mouse cDNA sequences. Proc. Natl. Acad. Sci. U.S.A. 99, 16899–16903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Headey S. J., Leeding K. S., Norton R. S., Bach L. A. (2004) Contributions of the N- and C-terminal domains of IGF binding protein-6 to IGF binding. J. Mol. Endocrinol. 33, 377–386 [DOI] [PubMed] [Google Scholar]
- 19. Mishra S., Ande S. R., Nyomba B. L. (2010) The role of prohibitin in cell signaling. FEBS J. 277, 3937–3946 [DOI] [PubMed] [Google Scholar]
- 20. Berger K. H., Yaffe M. P. (1998) Prohibitin family members interact genetically with mitochondrial inheritance components in Saccharomyces cerevisiae. Mol. Cell Biol. 18, 4043–4052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Merkwirth C., Langer T. (2009) Prohibitin function within mitochondria. Essential roles for cell proliferation and cristae morphogenesis. Biochim. Biophys. Acta 1793, 27–32 [DOI] [PubMed] [Google Scholar]
- 22. Sievers C., Billig G., Gottschalk K., Rudel T. (2010) Prohibitins are required for cancer cell proliferation and adhesion. PLoS One 5, e12735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chiu C. F., Ho M. Y., Peng J. M., Hung S. W., Lee W. H., Liang C. M., Liang S. M. (2013) Raf activation by Ras and promotion of cellular metastasis require phosphorylation of prohibitin in the raft domain of the plasma membrane. Oncogene 32, 777–787 [DOI] [PubMed] [Google Scholar]
- 24. Bach L. A. (1999) Insulin-like growth factor binding protein-6. The “forgotten” binding protein? Horm. Metab. Res. 31, 226–234 [DOI] [PubMed] [Google Scholar]
- 25. Martins A. S., Olmos D., Missiaglia E., Shipley J. (2011) Targeting the insulin-like growth factor pathway in rhabdomyosarcomas. Rationale and future perspectives. Sarcoma 2011, 209736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gallicchio M. A., Kneen M., Hall C., Scott A. M., Bach L. A. (2001) Overexpression of insulin-like growth factor binding protein-6 inhibits rhabdomyosarcoma growth in vivo. Int. J. Cancer 94, 645–651 [DOI] [PubMed] [Google Scholar]
- 27. Scholl F. A., Betts D. R., Niggli F. K., Schäfer B. W. (2000) Molecular features of a human rhabdomyosarcoma cell line with spontaneous metastatic progression. Br. J. Cancer 82, 1239–1245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Iosef C., Gkourasas T., Jia C. Y., Li S. S., Han V. K. (2008) A functional nuclear localization signal in insulin-like growth factor binding protein-6 mediates its nuclear import. Endocrinology 149, 1214–1226 [DOI] [PubMed] [Google Scholar]
- 29. Iosef C., Vilk G., Gkourasas T., Lee K. J., Chen B. P., Fu P., Bach L. A., Lajoie G., Gupta M. B., Li S. S., Han V. K. (2010) Insulin-like growth factor binding protein-6 (IGFBP-6) interacts with DNA-end binding protein Ku80 to regulate cell fate. Cell. Signal. 22, 1033–1043 [DOI] [PubMed] [Google Scholar]
- 30. Kokkinakis D. M., Rushing E. J., Shareef M. M., Ahmed M. M., Yang S., Singha U. K., Luo J. (2004) Physiology and gene expression characteristics of carcinogen-initiated and tumor-transformed glial progenitor cells derived from the CNS of methylnitrosourea (MNU)-treated Sprague-Dawley rats. J. Neuropathol. Exp. Neurol. 63, 1182–1199 [DOI] [PubMed] [Google Scholar]
- 31. Xu X. F., Guo C. Y., Liu J., Yang W. J., Xia Y. J., Xu L., Yu Y. C., Wang X. P. (2009) Gli1 maintains cell survival by up-regulating IGFBP6 and Bcl-2 through promoter regions in parallel manner in pancreatic cancer cells. J. Carcinog. 8, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Velázquez-Fernandez D., Laurell C., Geli J., Höög A., Odeberg J., Kjellman M., Lundeberg J., Hamberger B., Nilsson P., Bäckdahl M. (2005) Expression profiling of adrenocortical neoplasms suggests a molecular signature of malignancy. Surgery 138, 1087–1094 [DOI] [PubMed] [Google Scholar]
- 33. Nordqvist A. C., Mathiesen T. (2002) Expression of IGF-II, IGFBP-2, -5, and -6 in meningiomas with different brain invasiveness. J. Neurooncol. 57, 19–26 [DOI] [PubMed] [Google Scholar]
- 34. Firth S. M., Baxter R. C. (2002) Cellular actions of the insulin-like growth factor binding proteins. Endocr. Rev. 23, 824–854 [DOI] [PubMed] [Google Scholar]
- 35. Zhu W., Shiojima I., Ito Y., Li Z., Ikeda H., Yoshida M., Naito A. T., Nishi J., Ueno H., Umezawa A., Minamino T., Nagai T., Kikuchi A., Asashima M., Komuro I. (2008) IGFBP-4 is an inhibitor of canonical Wnt signalling required for cardiogenesis. Nature 454, 345–349 [DOI] [PubMed] [Google Scholar]
- 36. Ingermann A. R., Yang Y. F., Han J., Mikami A., Garza A. E., Mohanraj L., Fan L., Idowu M., Ware J. L., Kim H. S., Lee D. Y., Oh Y. (2010) Identification of a novel cell death receptor mediating IGFBP-3-induced anti-tumor effects in breast and prostate cancer. J. Biol. Chem. 285, 30233–30246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jones J. I., Gockerman A., Busby W. H., Jr., Wright G., Clemmons D. R. (1993) Insulin-like growth factor binding protein 1 stimulates cell migration and binds to the α5β1 integrin by means of its Arg-Gly-Asp sequence. Proc. Natl. Acad. Sci. U.S.A. 90, 10553–10557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Schütt B. S., Langkamp M., Rauschnabel U., Ranke M. B., Elmlinger M. W. (2004) Integrin-mediated action of insulin-like growth factor binding protein-2 in tumor cells. J. Mol. Endocrinol. 32, 859–868 [DOI] [PubMed] [Google Scholar]
- 39. Merkwirth C., Dargazanli S., Tatsuta T., Geimer S., Löwer B., Wunderlich F. T., von Kleist-Retzow J. C., Waisman A., Westermann B., Langer T. (2008) Prohibitins control cell proliferation and apoptosis by regulating OPA1-dependent cristae morphogenesis in mitochondria. Genes Dev. 22, 476–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yurugi H., Tanida S., Ishida A., Akita K., Toda M., Inoue M., Nakada H. (2012) Expression of prohibitins on the surface of activated T cells. Biochem. Biophys. Res. Commun. 420, 275–280 [DOI] [PubMed] [Google Scholar]
- 41. Terashima M., Kim K. M., Adachi T., Nielsen P. J., Reth M., Köhler G., Lamers M. C. (1994) The IgM antigen receptor of B lymphocytes is associated with prohibitin and a prohibitin-related protein. EMBO J. 13, 3782–3792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Strub G. M., Paillard M., Liang J., Gomez L., Allegood J. C., Hait N. C., Maceyka M., Price M. M., Chen Q., Simpson D. C., Kordula T., Milstien S., Lesnefsky E. J., Spiegel S. (2011) Sphingosine-1-phosphate produced by sphingosine kinase 2 in mitochondria interacts with prohibitin 2 to regulate complex IV assembly and respiration. FASEB J. 25, 600–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kasashima K., Ohta E., Kagawa Y., Endo H. (2006) Mitochondrial functions and estrogen receptor-dependent nuclear translocation of pleiotropic human prohibitin 2. J. Biol. Chem. 281, 36401–36410 [DOI] [PubMed] [Google Scholar]
- 44. Kim J. W., Akiyama M., Park J. H., Lin M. L., Shimo A., Ueki T., Daigo Y., Tsunoda T., Nishidate T., Nakamura Y., Katagiri T. (2009) Activation of an estrogen/estrogen receptor signaling by BIG3 through its inhibitory effect on nuclear transport of PHB2/REA in breast cancer. Cancer Sci. 100, 1468–1478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sun L., Liu L., Yang X. J., Wu Z. (2004) Akt binds prohibitin 2 and relieves its repression of MyoD and muscle differentiation. J. Cell Sci. 117, 3021–3029 [DOI] [PubMed] [Google Scholar]
- 46. Bacher S., Achatz G., Schmitz M. L., Lamers M. C. (2002) Prohibitin and prohibitone are contained in high-molecular weight complexes and interact with α-actinin and annexin A2. Biochimie 84, 1207–1220 [DOI] [PubMed] [Google Scholar]
- 47. Ross J. A., Nagy Z. S., Kirken R. A. (2008) The PHB1/2 phosphocomplex is required for mitochondrial homeostasis and survival of human T cells. J. Biol. Chem. 283, 4699–4713 [DOI] [PubMed] [Google Scholar]
- 48. Ande S. R., Gu Y., Nyomba B. L., Mishra S. (2009) Insulin induced phosphorylation of prohibitin at tyrosine 114 recruits Shp1. Biochim. Biophys. Acta 1793, 1372–1378 [DOI] [PubMed] [Google Scholar]
- 49. Ande S. R., Moulik S., Mishra S. (2009) Interaction between O-GlcNAc modification and tyrosine phosphorylation of prohibitin. Implication for a novel binary switch. PLoS One 4, e4586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ande S. R., Mishra S. (2009) Prohibitin interacts with phosphatidylinositol 3,4,5-triphosphate (PIP3) and modulates insulin signaling. Biochem. Biophys. Res. Commun. 390, 1023–1028 [DOI] [PubMed] [Google Scholar]
- 51. Chiu C. F., Peng J. M., Hung S. W., Liang C. M., Liang S. M. (2012) Recombinant viral capsid protein VP1 suppresses migration and invasion of human cervical cancer by modulating phosphorylated prohibitin in lipid rafts. Cancer Lett. 320, 205–214 [DOI] [PubMed] [Google Scholar]
- 52. Fukasawa H., Obayashi H., Schmieder S., Lee J., Ghosh P., Farquhar M. G. (2011) Phosphorylation of podocalyxin (Ser415) Prevents RhoA and ezrin activation and disrupts its interaction with the actin cytoskeleton. Am. J. Pathol. 179, 2254–2265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Rajalingam K., Wunder C., Brinkmann V., Churin Y., Hekman M., Sievers C., Rapp U. R., Rudel T. (2005) Prohibitin is required for Ras-induced Raf-MEK-ERK activation and epithelial cell migration. Nat. Cell Biol. 7, 837–843 [DOI] [PubMed] [Google Scholar]
- 54. Zhu B., Fukada K., Zhu H., Kyprianou N. (2006) Prohibitin and cofilin are intracellular effectors of transforming growth factor β signaling in human prostate cancer cells. Cancer Res. 66, 8640–8647 [DOI] [PubMed] [Google Scholar]
- 55. Zhu B., Zhai J., Zhu H., Kyprianou N. (2010) Prohibitin regulates TGF-β induced apoptosis as a downstream effector of Smad-dependent and -independent signaling. Prostate 70, 17–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ponce J., Brea D., Carrascal M., Guirao V., Degregorio-Rocasolano N., Sobrino T., Castillo J., Dávalos A., Gasull T. (2010) The effect of simvastatin on the proteome of detergent-resistant membrane domains. Decreases of specific proteins previously related to cytoskeleton regulation, calcium homeostasis and cell fate. Proteomics 10, 1954–1965 [DOI] [PubMed] [Google Scholar]
- 57. Mengwasser J., Piau A., Schlag P., Sleeman J. P. (2004) Differential immunization identifies PHB1/PHB2 as blood-borne tumor antigens. Oncogene 23, 7430–7435 [DOI] [PubMed] [Google Scholar]
- 58. Ande S. R., Xu Z., Gu Y., Mishra S. (2012) Prohibitin has an important role in adipocyte differentiation. Int. J. Obes. 36, 1236–1244 [DOI] [PubMed] [Google Scholar]
- 59. Zhang C., Lu L., Li Y., Wang X., Zhou J., Liu Y., Fu P., Gallicchio M. A., Bach L. A., Duan C. (2012) IGF binding protein-6 expression in vascular endothelial cells is induced by hypoxia and plays a negative role in tumor angiogenesis. Int. J. Cancer 130, 2003–2012 [DOI] [PMC free article] [PubMed] [Google Scholar]