

Background: The non-methyl-CpG DNA-binding CXXC domain is critical for MLL leukemia.
Results: Only the DNMT1 CXXC domain substitutes in an MLL leukemia model because of similar capacity to protect from CpG DNA methylation and H3K9 methylation.
Conclusion: Differential protection from specific CpG DNA methylation mechanistically distinguishes similar CXXC domains.
Significance: CXXC domains have specific nonredundant activities that impact downstream regulatory functions.
Keywords: Chromatin Histone Modification, Chromatin Regulation, DNA Methyltransferase, Epigenetics, Leukemia, CXXC Domain, MLL
Abstract
The MLL CXXC domain binds nonmethylated CpG-containing DNA and is essential for the oncogenic properties of MLL fusion proteins. To determine potential functional promiscuity of similar DNA binding domains, we replaced the MLL CXXC domain in the context of the leukemogenic MLL-AF9 fusion with CXXC domains from DNMT1, CGBP (CFP1), and MBD1, or with a methyl-CpG-binding domain (MBD) from MBD1. MLL(DNMT1 CXXC)-AF9 shows robust in vitro colony forming activity and in vivo leukemogenesis, similar to MLL-AF9. However, colony forming ability and leukemogenicity are abrogated in MLL-AF9 containing either the CGBP or MBD1 CXXC domains or the MBD1 MBD domain. Direct comparison of in vitro DNA binding affinity of the isolated CXXC or MBD domains demonstrated that MLL, DNMT1, and CGBP CXXC domains could each bind to unmethylated DNA but with differing affinity. In contrast, the isolated MBD1 CXXC and MBD1 MBD domains were unable to bind to the same DNA. However, all substituted domains still allowed targeting of the MLL fusions to the functionally important Hoxa9 locus in primary bone marrow progenitor cells. In addition to DNA binding activity, it was critical that specific CpG residues in the Hoxa9 locus were protected from methylation for leukemia development. This ultimately prevented histone 3 lysine 9 trimethylation (H3K9me3) of the locus and enabled Hoxa9 expression. These were properties shared by MLL and DNMT1 CXXC domains but not by CGBP CXXC or the other swapped fusions tested. We demonstrate that similar CXXC domains can be mechanistically distinguished by specificity of CpG nucleotides preferentially protected from DNA methylation.
Introduction
The mixed lineage leukemia (MLL)2 gene was initially identified through its involvement in chromosome translocations that cause acute leukemia (1, 2). MLL leukemia patients have a relatively poor prognosis, so the development of specific targeted therapies would be beneficial (3, 4). MLL encodes a large multi-domain protein that is involved in positively maintaining Hox gene expression during development and hematopoiesis (5–7). MLL translocations encode leukemogenic MLL fusion proteins in which the amino-terminal domains of MLL are retained. These include a menin-LEDGF interacting motif (8), AT hooks which bind to AT-rich DNA (9), nuclear localization motifs, and the DNA-binding CXXC domain (10, 11).
DNA methylation occurs on position five on the cytosine ring of CpG dinucleotides (12). DNA methyl marks can inhibit the binding of transcriptional activators while recruiting co-repressors. Methyl-CpG binding domains (MBDs) are protein domains that specifically bind to methylated CpG DNA. They are present in the MBD family of repressor proteins, which include MeCP2 and MBD1, -2, -3, and -4 (13). One family member, MBD1, is present with the maintenance DNA methyltransferase enzyme DNMT1 at the replication fork during S phase of the cell cycle (14, 15). When DNMT1 adds a DNA methyl mark on newly replicated, hemimethylated DNA, MBD1 can bind directly to the newly methylated DNA. MBD1 then recruits the repressive histone H3K9 methyltransferase enzyme SETDB1 (16), thus coupling DNA methylation to histone modifications and chromatin remodeling.
Although promoter DNA methylation is most often associated with repressive chromatin states, unmethylated CpG DNA found in the promoters of genes is often associated with active transcription. The CXXC protein domain, similar to the MBD domain, also specifically recognizes CpG DNA, but only in the alternate, unmethylated state (17, 18). CXXC domains have highly conserved spacing of eight cysteine residues, which function to coordinate two zinc ions. This domain folds into a saddle-like structure that centers over an unmethylated CpG residue when bound to DNA (19, 20). CXXC domains are present in several chromatin-associated proteins, including MLL, DNMT1, MBD1, and the transcriptional activator CpG binding protein CGBP/CFP1/CXXC-1. Most CXXC domains that have been studied show binding specificity for unmethylated CpG DNA, including those from CGBP (21, 22), MLL (17, 19, 20, 23), and DNMT1 (24). However, two of the three CXXC domains from MBD1 are unable to bind DNA in any state (25). Similar to MBD domains, CXXC domains have also been implicated in bridging DNA and histone epigenetic states. The MLL protein contains both the CXXC domain, which recognizes unmethylated DNA, and the histone methyltransferase SET domain, which methylates histone H3 tails at lysine 4 (H3K4), a mark of active transcription. Similarly, the CXXC domain-containing CGBP protein also helps direct H3K4 methylation patterns set by Setd1, another histone methyltransferase (26). It has recently been shown that the CXXC domain in DNMT1 acts as an auto-inhibitory domain that binds to unmethylated DNA, protecting it from DNMT1 DNA methyltransferase activity (24).
The CXXC domain of MLL was initially identified by its homology to DNMT1 (27) and was first shown to bind unmethylated DNA through electrophoretic mobility shift assays (17). Through deletion and point mutation experiments, the presence of the MLL CXXC domain was shown to be essential to MLL fusion protein function in colony formation assays (23). Further structural and functional work by our lab and others has shown the specific interaction between the MLL CXXC domain and unmethylated CpG DNA (19, 20). By mutating specific residues that are critical for the DNA binding activity of the MLL CXXC domain while keeping the structure of the domain intact, we showed that the DNA binding function of the MLL CXXC domain is essential for the ability of the MLL fusion protein to transform bone marrow cells and promote leukemia development (19). Additionally, MLL and MLL fusion proteins bind to the Hoxa9 locus and protect specific CpG DNA in this region from becoming methylated (28). This protection against DNA methylation function of MLL contributes to keeping the chromatin of target genes in the open state to allow for active gene expression.
Although CXXC domains are defined by a particular clustering of cysteine residues, multiple amino acid differences exist between CXXC domains of different proteins. We tested our hypothesis that not all CXXC domains possess the necessary properties required for an MLL fusion protein to function as an oncogene and delineate the mechanism underlying CXXC domain functional specificity.
EXPERIMENTAL PROCEDURES
Cloning and Purification of GST-tagged Proteins
Isolated CXXC or MBD domains were cloned into the pGex-4T1 vector. The designs of the domains were based off of our previously published structure using 57 amino acids of the MLL CXXC domain (1147–1203) (19). Primers for the CXXC domains were designed to include the same number of amino acids before and after the CXXC cysteines as were used from MLL CXXC and 75 amino acids for the MBD domain, as was used previously to solve the MBD domain structure (29). DNMT1 (NM_001379) CXXC includes amino acids 645–700; CGBP (NM_001101654) CXXC includes amino acids 161–217; MBD1 transcript variant 1 (NM_015846) CXXC-1 includes amino acids 168–224; MBD1 (NM_015846) MBD includes amino acids 1–75. Cloning details provided upon request. Briefly, lysates were sonicated, cell debris was pelleted, and soluble GST-tagged CXXC or MBD proteins were purified using glutathione affinity chromatography (Fluka 49739). The GST-tagged proteins were eluted in 50 mm Tris, pH 7.2, 400 mm sodium chloride, 50 μm zinc chloride, 1 mm DTT, and 10 mm glutathione (G4251, Sigma). Protein purity was verified by Coomassie Blue staining.
DNA Binding Experiments using Fluorescence Polarization
Unmethylated C-terminal fluorescein labeled DNA (5′ GGGTCGCGGGAG 3′, Integrated DNA Technologies) and the purified GST-tagged CXXC or MBD domains were dialyzed into fluorescence polarization buffer (50 mm Tris-HCl, 150 mm KCl, 1 mm DTT, 10 μm ZnCl2). Fluorescein-labeled DNA was added to 96-well black COSTAR (Corning Life Sciences, Lowell, MA) plates. The proteins were separately mixed with fluorescein-labeled DNA and serially diluted 1:2 onto the DNA-containing COSTAR plates. A PHERAstar microplate reader (BMG Labtech, Durham, NC) was used to measure fluorescence polarization with excitation at 494 nm and emission at 525 nm. The DNA binding experiments were performed three times. To calculate Kd for each protein, anisotropy values (mA) were plotted versus the log of protein concentration (μm), and the resulting plots were fit to a one-site sigmoidal binding curve using Origin software (version 7.0, MicroCal, Northampton, MA).
Cloning of MSCVneo-MLL-AF9-FLAG CXXC Domain Swap Constructs
Modified versions of our MSCVneo-MLL-AF9-FLAG construct were generated, which substituted the same domains used for the DNA binding experiments in place of the MLL CXXC domain. Cloning details will be provided upon request.
Cell Lines Used
A Phoenix-Eco cell line was obtained from Orbigen, Inc. (San Diego, CA) where it was tested for appropriate resistance to hygromycin and diphtheria toxin and negative for helper virus production. Stocks of early passage cells were frozen, and when used for experiments, were passaged less than three months.
Bone Marrow Colony Replating Assays and in Vivo Leukemia Experiments
Bone marrow colony assays and in vivo murine leukemia experiments were performed as described previously (19). All experiments using mice in this study were performed with the approval of and in accordance with the Loyola University Medical Center Institutional Animal Care and Use Committee, in accordance with federal guidelines.
Quantitative RT-PCR
RNA was isolated from week 1 methylcellulose colony assay cells, and cDNA was prepared. Real time PCR using Taqman probes or SYBR green was performed to measure Hoxa9 and MLL fusion expression, respectively. Further details will be provided upon request.
Chromatin Immunoprecipitation
Chromatin immunoprecipitation was performed using EZ-Magna ChIP A+G (Upstate/Millipore, Temecula, CA) with anti-FLAG, anti-H3K9me3 antibody, or control IgG on primary murine bone marrow progenitor cells expressing retroviral constructs. Results were analyzed by quantitative PCR in triplicate on an ABI 7300 real time PCR machine using iTaq SYBR Green Supermix with Rox (Bio-Rad). Enrichment was calculated as percent input chromatin. Primer sequences for Hoxa9 DNA were as follows: 5′-CGGTGATTTAGGTAGTTTCCTGTTG-3′ and 5′-CACAGCGCCGAGGAAGAC-3′.
DNA Methylation Analysis
DNA was isolated from primary mouse bone marrow progenitor cells expressing MLL fusion constructs using DNeasy blood and tissue kit (Qiagen). DNA was bisulfite-treated and analyzed as described previously (19). Primers used for amplification were as follows: forward (outside), TYGAAATTYGYGGAGGAGGGTTTA; reverse (outside), CCCTACRATTATACCCAATCRAACCC; forward (inside), GTTAGGTTAYGYGTTTTTTGTT; reverse (inside), CCAACRATATAAAACRAATTCC.
RESULTS
To determine whether the functions of the MLL CXXC domain could be replaced by other CXXC domains or by the MBD meCpG-DNA binding domain, domain swap experiments were designed in the context of an oncogenic MLL-AF9 fusion protein to test for leukemogenic activity. As seen in the alignment, all CXXC domains contain the eight conserved cysteines, but there are various degrees of amino acid conservation between the domains at other positions (Fig. 1A). Furthermore, the MBD methyl CpG DNA-binding domain does not have any apparent amino acid sequence or structural similarity to the CXXC domains (29) (Fig. 1A).
FIGURE 1.

Relative binding affinities of isolated CXXC domains to unmethylated CpG-containing DNA. A, CXXC domains are aligned to show amino acid sequence conservation: those identical in all or present in two or more are shaded in dark and light gray, respectively. The sequence of the MBD1 MBD is also shown. B, representative binding curves for fluorescein labeled DNA titrated with increasing concentrations of GST-tagged CXXC domains and measured by fluorescence polarization. C, absolute and relative Kd values for each of the GST-tagged CXXC domains. Absolute Kd values were determined from the fluorescence polarization titration curves. Relative Kd values were determined by comparison to GST-MLL CXXC (set to 1).
Direct Comparison of DNA Binding Affinities for Isolated CXXC and MBD Domains
The DNA binding affinities of multiple CpG DNA binding domains to a specific DNA sequence have not been compared previously. Fluorescence polarization was used to measure the DNA binding affinities of several isolated CpG DNA binding domains (Fig. 1A). The DNA sequence used to measure the DNA binding affinities is derived from an MLL binding site in the Hoxa9 locus, which contains two central CpG motifs and is protected from DNA methylation by MLL (28). We previously used isothermal titration calorimetry to demonstrate that the MLL CXXC domain binds to this specific nonmethylated DNA sequence (28). The other domains chosen for comparison with MLL vary in their CpG DNA binding capacity (Fig. 1A). Proteins were expressed as GST fusions in Escherichia coli and purified using glutathione agarose affinity chromatography (data not shown). Protein-DNA binding curves were generated using fluorescence polarization and show the approach of saturation of protein binding to DNA (Fig. 1B). From these curves, absolute and relative binding constants (Kd) were calculated (Fig. 1C). These results indicate that the MLL CXXC domain has the highest DNA binding affinity (0.64 μm) of the domains tested to this unmethylated DNA target. The CGBP CXXC domain has the second highest affinity at ∼7-fold lower than MLL CXXC. DNMT1 CXXC could bind to this unmethylated DNA, but with a lower affinity: 34.4-fold lower affinity than the MLL CXXC domain. We tested binding of the MBD1 CXXC domain that has the least amino acid identity to MLL CXXC. It was shown previously that the mouse isoform of this Mbd1 CXXC domain was unable to bind to DNA regardless of methylation status (25), and our current data with the human orthologous region also shows no DNA binding activity (Fig. 1, B and C). As expected, the MBD1 MBD was unable to bind to the unmethylated DNA. Thus, most of the CXXC domains were able to bind nonmethylated CpG-containing DNA but with significant differences in binding affinity.
Construction of MLL-AF9 Retroviral Constructs with Substituted CXXC or MBD Domain
Retroviral constructs were generated to produce FLAG-tagged MLL-AF9 fusion proteins with various CpG DNA binding domains replacing the MLL CXXC domain (Fig. 2A). Amino acids 1150 through 1201 of MLL, which contain the minimal CXXC domain, were deleted and replaced with other CpG-recognizing domains analyzed above (Fig. 1). The CXXC domain from DNA methyltransferase 1 (DNMT1 amino acids 649–697), CpG binding protein (CGBP/CFP1 amino acids 163–215), and methyl-CpG binding domain protein 1 (MBD1 amino acids 172–221), as well as the MBD from MBD1 (amino acids 1–75) were swapped into the MLL-AF9 fusion. Stable expression of all the MLL-AF9 fusion proteins was confirmed by Western blot and quantitative RT-PCR (data not shown). We predicted that the different CpG DNA binding domains would change the strength or specificity of MLL protection of DNA against methylation, which would affect the ability of MLL-AF9 to cause leukemia.
FIGURE 2.

Myeloid colony forming assays with bone marrow progenitor cells. A, schematic of CXXC domain swaps into MLL-AF9-FLAG. B, average colony numbers for weeks 1–4 after plating primary mouse bone c-Kit+ progenitor cells expressing MLL-AF9 or MLL-AF9 with indicated CXXC domain swaps in methylcellulose, with error bars representing S.E. from 6–8 independent biological replicates. C, digital photographs (original magnification, 4×) showing colony morphologies at week 4 of the colony assay. Scale bar represents 250 μm. Pictures were taken through air on a Leica model DMIL microscope (Wetzlar, Germany), through a 4×/0.10 numerical aperture lens, with a Canon PowerShot S40 digital camera. Images were acquired with Canon ZoomBrowser EX (version 8).
Ability of Other CXXC and MBD Domains to Functionally Replace MLL CXXC Domain in MLL-AF9 in Vitro Immortalization Assay
MLL fusion proteins, including MLL-AF9, confer increased proliferative capacity and immortalization when exogenously expressed in murine bone marrow progenitor cells, such that they can be serially replated in methylcellulose (19, 30). Vector-infected or non-leukemogenic mutant-infected progenitor cells typically form colonies only in the first week and then differentiate and die, similar to normal bone marrow progenitors. To determine whether the CpG binding domains from other proteins would affect this ability of MLL-AF9 to give an enhanced proliferative capacity to bone marrow progenitor cells, we performed in vitro colony assays. Murine bone marrow c-Kit+ progenitor cells were isolated and infected with domain swap or wild type MLL-AF9 retroviral fusion genes (Fig. 2A). We observed significant differences between the ability of various CpG DNA binding domains to function in the context of an MLL-AF9 fusion protein (Fig. 2B). As expected, bone marrow cells expressing MLL-AF9 continue to proliferate and form many colonies through four sequential platings of the colony assay, whereas MSCVneo vector infected cells have very few, if any, colonies remaining. MLL(DNMT1 CXXC)-AF9 shows robust in vitro colony forming activity, similar to the oncogenic MLL-AF9 fusion. However, MLL(CGBP CXXC)-AF9, MLL(MBD1 CXXC)-AF9, and MLL(MBD1 MBD)-AF9 almost completely abrogated colony forming ability. Those few colonies that persisted for these constructs were often diffuse in nature and typical of differentiated bone marrow cells, with few cell numbers (Fig. 2C). The MLL-AF9 and MLL(DNMT1 CXXC)-AF9 cells formed dense compact round colonies typical of bone marrow progenitor cells. Cell counts over the course of the replating experiment also reinforced this observation. MLL-AF9 and MLL(DNMT1 CXXC)-AF9 both conferred an ∼870-fold expansion of cell numbers at each replating, whereas for the remaining constructs, the cell expansion ranged from 3- to 7-fold (data not shown). Cytospins of the bone marrow cells taken from week 3 or 4 of the colony assays also confirm that the transformed MLL-AF9 and MLL(DNMT1 CXXC)-AF9 cells primarily resemble leukemic blasts, whereas the other constructs produce a more heterogeneous differentiated bone marrow cell population (data not shown).
Because the domains from MBD1 were not able to function in MLL-AF9, our hypothesis that MLL CXXC must maintain the ability to bind to unmethylated DNA to transform bone marrow cells was supported. However, we expected that both CGBP and DNMT1 CXXC domains would replace MLL CXXC domain in the context of the MLL-AF9 fusion because the isolated domains bound to the nonmethylated CpG-containing DNA in vitro. This capability did not strictly align with DNA binding affinity of the isolated CXXC domain, however, because the CXXC domain from CGBP had a higher affinity for DNA binding as compared with the DNMT1 CXXC domain (Fig. 1B). These results suggest that CXXC DNA binding affinity is not the only function of this domain necessary for MLL-AF9 transformation.
Ability of Other CXXC and MBD Domains to Functionally Replace MLL CXXC Domain in MLL-AF9 to Cause Leukemia in Vivo
In vivo murine studies were performed to determine whether the different CpG DNA binding domains would alter the ability of MLL fusion proteins to cause leukemia. Murine c-Kit+ progenitor cells isolated from B6/SJL CD45.1+ mice were infected with MLL-AF9 or domain swap constructs and then transplanted into irradiated C57Bl/6 recipient mice as described previously (19). The results of the leukemia assay correlate with the colony assay data in that all MLL-AF9 mice and all but one of the MLL(DNMT1 CXXC)-AF9 mice developed MLL acute myeloid leukemia, whereas none of the MSCVneo, MLL(CGBP CXXC)-AF9, MLL(MBD1 CXXC)-AF9, or MLL(MBD1 MBD)-AF9 mice developed leukemia (Fig. 3A). A small number of the mice that did not develop leukemia (one or two mice from each group, excluding MLL-AF9 and MLL (CGBP CXXC)-AF9) died from other cancers during the course of the experiment. However, these tumor cells did not express the CD45.1 marker from the donor cells; therefore, they were unrelated to MLL-AF9 and were likely a side effect of the radiation that the recipients received prior to transplant. Peripheral blood and bone marrow from leukemic mice show increased blast cells (Fig. 3B). Wright-Giemsa stained organ sections also show infiltration of leukemia cells into the spleen, liver, lungs, and kidneys of the MLL-AF9 and MLL(DNMT1 CXXC)-AF9 mice (data not shown). Bone marrow cells taken upon sacrifice from the leukemic or healthy mice were subjected to surface marker staining for multiple murine hematopoietic cell markers followed by flow cytometric analysis. All MLL-AF9 and MLL(DNMT1 CXXC)-AF9 mice with leukemia showed a high percentage of CD45.1+ cells, indicating that the leukemic cells had replaced the normal bone marrow cells of the mice (Fig. 3C), whereas the healthy mice only had a small population of CD45.1+ cells in their bone marrow. Nearly all of the CD45.1+ leukemia cells expressed the myeloid markers CD11b and Gr-1. Smaller proportions were positive for the progenitor markers Sca-1 and c-Kit (CD117). The cells were negative for lymphocyte and other hematopoietic cell markers (data not shown). The results of this leukemia assay corroborate the in vitro colony assay results and again emphasize that the DNMT1 CXXC domain can functionally replace the MLL CXXC domain to enable an MLL-AF9 fusion to cause leukemia. However, although the nonmethyl-CpG DNA binding function of MLL CXXC domain remains essential, the CGBP CXXC domain with similar DNA binding function is not sufficient for MLL-AF9 oncogenic activity.
FIGURE 3.

Incidence of leukemia in mice expressing MLL-AF9 or MLL-AF9 with substituted CXXC domains. A, survival curve of mice transplanted with bone marrow progenitor cells expressing MLL-AF9 or MLL-AF9 CXXC domain swap fusion proteins. B, peripheral blood smears and bone marrow samples taken from indicated mice at time of sacrifice (original magnification, 100×). Scale bar represents 12 μm. Pictures were taken on an Olympus BH-2 microscope (Tokyo, Japan), under a 100×/1.25 Numerical aperture oil immersion lens, with a Sony 3CCD camera (model DXC-76OMD). Images were acquired with Adobe Premier software (version 4.2.1) and processed using Adobe Photoshop CS3 (version 10.0). C, FACS profiles of bone marrow cells from leukemic mice. CD45.1 indicates transplanted cell population expressing the MLL-AF9 or domain swap construct. The second and third panels show the percentage of CD45.1 positive cells that express myeloid markers CD11b and Gr-1 and progenitor markers CD117 and Sca-1.
Hoxa9 Expression in MLL-AF9 and Domain-swapped MLL-AF9-transduced Bone Marrow Progenitors
The MLL target gene Hoxa9 is frequently up-regulated in MLL leukemias, and its overexpression is involved in disease progression (31, 32). To determine whether the MLL-AF9 CXXC domain swap proteins cause increased Hoxa9 expression, RNA was isolated from bone marrow cells expressing the domain swap fusion proteins. Hoxa9 transcript levels were assessed by quantitative RT-PCR (Fig. 4A). The MLL-AF9 and MLL(DNMT1 CXXC)-AF9 proteins cause overexpression of Hoxa9, whereas the other swap constructs that were non-transforming did not increase expression over the levels of MSCVneo-infected bone marrow cells. The lower levels of Hoxa9 expression help explain why the MLL-AF9 fusion proteins with the CGBP CXXC, MBD1 CXXC, or MBD1 MBD domain were unable to transform bone marrow cells.
FIGURE 4.
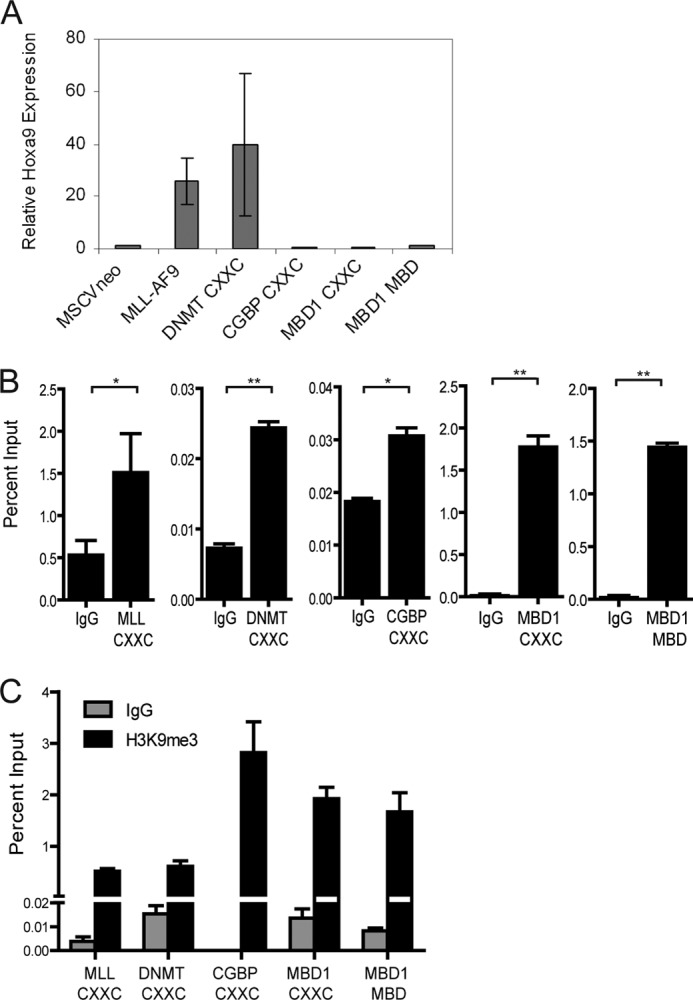
Hoxa9 expression, fusion protein binding to the Hoxa9 locus, and H3K9 methylation in bone marrow progenitor cells expressing MLL-AF9 or MLL-AF9 with substituted CXXC domains. A, quantitative RT-PCR for Hoxa9 in bone marrow cells expressing the indicated constructs. Shown are mean relative expression levels compared with Hprt from two independent experiments each done in triplicate, with error bars indicating S.D. B and C, chromatin immunoprecipitation assay from primary murine progenitor cells expressing the indicated constructs. Anti-FLAG or IgG (B) or anti-H3K9me3 or IgG (C) were used to immunoprecipitate chromatin, and quantitative PCR was performed with primers that amplify an MLL-binding region in the Hoxa9 locus. Primer location indicated by arrows in the Hoxa9 locus schematic shown in Fig. 5. Results are shown as average percent input from triplicate experiments with error bars representing S.E.
Chromatin Localization in MLL-AF9 and Domain-swapped MLL-AF9 Fusion Proteins
To determine whether MLL-AF9 domain swap constructs which do not cause overexpression of Hoxa9 still bind to the locus, chromatin immunoprecipitation assays were performed using bone marrow cells expressing the MLL-AF9 domain swap constructs. All of the MLL-AF9 CXXC domain swap proteins show significant binding to the Hoxa9 locus, regardless of their ability to activate transcription of the locus (Fig. 4B). This suggests that, similar to what we found previously for MLL-AF9 (19), the MLL CXXC domain is not essential for MLL-AF9 proteins to localize to the Hoxa9 locus.
Transforming MLL Fusions Protect a Specific Subset of CpG Sequences from DNA Methylation
Although both DNMT- and CGBP-CXXC domain swapped MLL-AF9 fusion proteins were able to bind the Hoxa9 locus, only DNMT CXXC functioned to cause leukemia. Our previous studies demonstrated that MLL and MLL fusion proteins protect specific CpG sequences from methylation. We hypothesized that differential ability to protect CpG DNA methylation may be the mechanism underlying functional differences. DNA was isolated from bone marrow progenitor cells expressing the MLL fusions and assessed for DNA methylation status using bisulfite sequencing. Different DNA methylation patterns were observed when comparing transforming fusions with the non-transforming fusion (Fig. 5). In particular, the CGBP CXXC domain-containing fusion did not efficiently protect CpG-5 and CpG-8 from methylation, whereas both MLL-AF9 and the DNMT1 CXXC-containing MLL-AF9 abrogated methylation of this region. Conversely, CGBP more effectively protected CpG-6 and -7 from methylation than the transforming fusion proteins. This suggests that transforming ability may depend on the methylation status of specific CpG nucleotides.
FIGURE 5.

Differential DNA methylation in bone marrow progenitor cells expressing MLL-AF9 or MLL-AF9 with substituted CXXC domains. Top, schematic representation of the Hoxa9 locus. CpG island (CGI) containing eight CpG residues dependent on Mll for protection from DNA methylation are located upstream of mir196b and Hoxa9 transcription start sites. Location of primers used for ChIP is indicated by arrows. Middle, heat map depicting DNA methylation levels in primary bone marrow progenitor cells expressing the MLL CXXC, DNMT CXXC, or CGBP CXXC domains swapped into MLL-AF9 or control MSCVneo vector. Methylation levels represent the average of five experiments. CpG 1–8 correspond to the CpGs shown in schematic. Bottom, representative chromatograms of sequenced PCR products from bisulfite-treated genomic DNA isolated from primary MLL-AF9 bone marrow cells with substituted CXXC domains. CpGs correspond to those in schematic and heat map.
Transforming MLL Fusions Protect from Histone 3 Lysine 9 Trimethylation
CpG methylation has been shown to enable recruitment of protein complexes that ultimately cause histone modifications associated with silent chromatin such as HeK9me3 (16). To determine whether MLL-AF9 domain swap fusion proteins that do not cause leukemia enable increased H3K9me3, ChIP assays were performed (Fig. 4C). We found that an increased level of H3K9me3 at the Hoxa9 locus correlated with inability of the swapped fusion proteins to activate Hoxa9 expression, to transform bone marrow progenitors in vitro, and cause leukemia in vivo.
DISCUSSION
We have explored the functional specificity of the CXXC domain in MLL leukemia. For the first time, the DNA binding affinities of CXXC domains from multiple proteins have been directly measured and compared with each other for binding to the same CpG-containing DNA sequence. MLL CXXC binds with highest affinity to the unmethylated CpG DNA, followed by CGBP CXXC, and then DNMT1 CXXC. MBD1 CXXC and MBD domains were unable to bind to unmethylated DNA. MBD1 MBD domain sequence is unrelated to CXXC domains and was previously shown to bind only methylated CpG-containing DNA; therefore, its lack of binding was expected (33). One of the MBD1 splice variants contains three CXXC domains (34). Previous DNA binding studies of the murine Mbd1 CXXC domains demonstrated that the first two CXXC domains from Mbd1 were unable to bind DNA regardless of its methylation status (25), which we also found to be true of the first CXXC domain from human MBD1, which has the least identity to MLL of the CXXC domains studied here. These different CXXC or MBD domains were substituted in place of the MLL CXXC domain in the context of an MLL-AF9 fusion, and the mutant fusion proteins were tested for leukemogenic activities in vitro and in vivo. The CXXC and MBD domains from MBD1, which are unable to interact with unmethylated CpG DNA, were incapable of replacing the MLL CXXC domain in promoting MLL-AF9 oncogenicity. This confirmed our hypothesis that DNA binding is an essential function of the MLL CXXC domain in MLL-AF9 promotion of Hoxa9 overexpression and development of acute leukemia. However, an intriguing finding from this study is that the DNMT1 CXXC domain, which has a lower DNA binding affinity compared with that of the MLL CXXC domain, is able to replace MLL CXXC in MLL-AF9, whereas the CGBP CXXC domain, which has a higher DNA binding affinity compared with the DNMT1 CXXC domain, is not functional in MLL-AF9. Overall, these results suggest that for a CXXC domain to function in the context of an MLL-AF9 fusion protein, DNA binding activity to relevant target DNA sequence is required, but a high DNA binding affinity alone is not sufficient. Additional properties of the CXXC domain must also contribute to function.
Our ChIP studies show that MLL-AF9 is able to localize to Hoxa9 regardless of which CpG-binding domain is present. Additional MLL targeting domains, including menin/LEDGF binding domains and the AT hooks, would likely contribute to the chromatin binding ability of the fusion protein. Although all domain swapped proteins bound to the Hoxa9 locus, they differed in the specific CpG residues protected from DNA methylation. CGBP cells had demethylated CpG-6 and -7, which agrees with the previous finding that CGBP CXXC preferentially binds a CpGG motif (22). Arginine 213 of CGBP (highlighted in Fig. 6A) forms two hydrogen bonds with the guanosine nucleotide base following the CpG dinucleotide. In contrast, MLL and DNMT contain nonpolar amino acids in the same site (Fig. 6A) and are associated with preferentially demethylated CpG-5 and -8. Therefore, it may be of critical importance that CpG-5 and -8 remain unmethylated for transformation of hematopoietic progenitors by MLL fusion proteins. CXXC domain specificity likely determines preferential gene targets affected by proteins containing these structurally similar domains. Our data also support the ability of the non-transforming fusions to enable proteins with H3K9 methyltransferase activity access to this critical locus. It has been previously shown that the MBD1 MBD domain binds to co-repressors HP1 and Suv39h1, a histone H3K9 methyltransferase. This would act to further enforce transcriptional repression of gene targets by linking DNA and histone modifications (35). In addition, a region of MBD1 that includes the MBD domain and its first two CXXC domains is able to bind the chromatin assembly factor-1 and the histone methyltransferase SETDB1 to provide histone H3K9 trimethylation on newly replicated, DNA-methylated chromatin (16). Our data also suggest that CGBP CXXC allows recruitment of repressive H3K9 methyltransferase activity to Hoxa9, whereas MLL and DNMT CXXC domains protect against this effect.
FIGURE 6.

Model of MLL-AF9 CXXC domain functional specificity in leukemogenesis. A, amino acid alignment to show sequence conservation between different CXXC domains. Identical residues to MLL are shaded in gray, whereas boxed residues highlight differences. Red residues are identical in MLL and DNMT1 but not present in CGBP or MBD1 CXXC. Green residues are conserved in CGBP and MBD1 but not present in MLL CXXC. B, solution structure of MLL CXXC domain in complex with CpG DNA (Protein Data Bank code 2KKF). The MLL CXXC domain is shown as a space-filling model in contact with the ball and stick structure of DNA, in four orientations. The same color scheme is used as described in A. C, model of functional specificity of CpG DNA binding domains in the context of MLL fusion proteins, adapted from Ref. 19.
Eight structural cysteines are conserved among all CXXC domains. Additional residues are identical between the two transforming CXXC domains from MLL and DNMT1 (Fig. 6, A and B, highlighted in red) but not the other CXXC domains, suggesting they may be essential to MLL CXXC function. Green residues are identical between CGBP and MBD1 CXXC domains but are not present in MLL or DNMT1, which suggests that the amino acids at these positions in CGBP or MBD1 may be inhibitory to CXXC function in MLL-AF9. These differences could influence DNA binding affinity or protein-protein interactions.
The MLL CXXC domain is a critical functional domain in MLL fusion proteins. The CXXC DNA binding function is essential to promote an unmethylated DNA state, which helps to keep target genes activated (19). In this study, we have shown that of the domains tested, only the DNMT1 CXXC domain can functionally replace MLL CXXC to provide critical functions required of the CXXC domain in a leukemogenic MLL fusion protein (Fig. 6C). CXXC domains from different proteins share some but not all functional characteristics. MLL fusion proteins require a CXXC domain that can both bind to unmethylated DNA and provide the ability to prevent both specific CpG DNA methylation and histone H3K9 methylation. This allows critical target loci such as Hoxa9, to remain active. The MLL CXXC domain remains a potentially tractable region for targeted therapies to be developed to treat MLL leukemia.
Acknowledgments
We thank Drs. Adrian Bird, Moshe Szyf, and David Skalnik for providing DNA constructs and Dr. Pieter De Tombe for equipment use.
This work was supported, in whole or in part, by National Institutes of Health Grants CA105049 and HL087188 (to N. J. Z.-L.); the Dr. Ralph and Marian Falk Medical Research Trust (to N. J. Z.-L.); the Arthur J. Schmitt Foundation (to L. E. R.); National Institutes of Health Experimental Immunology Training Grant T32 AI007508-11A1 (to L. E. R.); Leukemia and Lymphoma Society SCOR Grant 7006-05 (to J. H. B.); National Institutes of Health, NHLBI 1F30HL103208-01 (to N. W. B.); and the Stritch School of Medicine M.D./Ph.D. program (to N. W. B.).
- MLL
- mixed lineage leukemia
- MBD
- methyl-CpG binding domain
- CGBP
- CpG binding protein
- DNMT1
- DNA methyltransferase 1.
REFERENCES
- 1. Rowley J. D. (1993) Rearrangements involving chromosome band 11Q23 in acute leukaemia. Semin. Cancer Biol. 4, 377–385 [PubMed] [Google Scholar]
- 2. Bernard O. A., Berger R. (1995) Molecular basis of 11q23 rearrangements in hematopoietic malignant proliferations. Genes Chromosomes Cancer 13, 75–85 [DOI] [PubMed] [Google Scholar]
- 3. Mann G., Attarbaschi A., Schrappe M., De Lorenzo P., Peters C., Hann I., De Rossi G., Felice M., Lausen B., Leblanc T., Szczepanski T., Ferster A., Janka-Schaub G., Rubnitz J., Silverman L. B., Stary J., Campbell M., Li C. K., Suppiah R., Biondi A., Vora A., Valsecchi M. G., Pieters R., and Interfant-99 Study Group (2010) Improved outcome with hematopoietic stem cell transplantation in a poor prognostic subgroup of infants with mixed-lineage-leukemia (MLL)-rearranged acute lymphoblastic leukemia: results from the Interfant-99 Study. Blood 116, 2644–2650 [DOI] [PubMed] [Google Scholar]
- 4. Chen C. S., Hilden J. M., Frestedt J., Domer P. H., Moore R., Korsmeyer S. J., Kersey J. H. (1993) The chromosome 4q21 gene (AF-4/FEL) is widely expressed in normal tissues and shows breakpoint diversity in t(4;11)(q21;q23) acute leukemia. Blood 82, 1080–1085 [PubMed] [Google Scholar]
- 5. Hanson R. D., Hess J.L., Yu B. D., Ernst P., van Lohuizen M., Berns A., van der Lugt N. M., Shashikant C. S., Ruddle F. H., Seto M., Korsmeyer S. J. (1999) Mammalian Trithorax and polycomb-group homologues are antagonistic regulators of homeotic development. Proc. Natl. Acad. Sci. U.S.A. 96, 14372–14377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tkachuk D. C., Kohler S., Cleary M. L. (1992) Involvement of a homolog of Drosophila trithorax by 11q23 chromosomal translocations in acute leukemias. Cell 71, 691–700 [DOI] [PubMed] [Google Scholar]
- 7. Yu B. D., Hess J. L., Horning S. E., Brown G. A., Korsmeyer S. J. (1995) Altered Hox expression and segmental identity in Mll-mutant mice. Nature 378, 505–508 [DOI] [PubMed] [Google Scholar]
- 8. Yokoyama A., Cleary M. L. (2008) Menin critically links MLL proteins with LEDGF on cancer-associated target genes. Cancer Cell 14, 36–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zeleznik-Le N. J., Harden A. M., Rowley J. D. (1994) 11q23 translocations split the “AT-hook” cruciform DNA-binding region and the transcriptional repression domain from the activation domain of the mixed-lineage leukemia (MLL) gene. Proc. Natl. Acad. Sci. U.S.A. 91, 10610–10614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Muntean A. G., Tan J., Sitwala K., Huang Y., Bronstein J., Connelly J. A., Basrur V., Elenitoba-Johnson K. S., Hess J. L. (2010) The PAF complex synergizes with MLL fusion proteins at HOX loci to promote leukemogenesis. Cancer Cell 17, 609–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Milne T. A., Kim J., Wang G. G., Stadler S. C., Basrur V., Whitcomb S. J., Wang Z., Ruthenburg A. J., Elenitoba-Johnson K. S., Roeder R. G., Allis C. D. (2010) Multiple interactions recruit MLL1 and MLL1 fusion proteins to the HOXA9 locus in leukemogenesis. Mol. Cell 38, 853–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ehrlich M., Wang R. Y. (1981) 5-Methylcytosine in eukaryotic DNA. Science 212, 1350–1357 [DOI] [PubMed] [Google Scholar]
- 13. Wade P. A. (2001) Methyl CpG-binding proteins and transcriptional repression. Bioessays 23, 1131–1137 [DOI] [PubMed] [Google Scholar]
- 14. Reese B. E., Bachman K. E., Baylin S. B., Rountree M. R. (2003) The methyl-CpG binding protein MBD1 interacts with the p150 subunit of chromatin assembly factor 1. Mol. Cell Biol. 23, 3226–3236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Iida T., Suetake I., Tajima S., Morioka H., Ohta S., Obuse C., Tsurimoto T. (2002) PCNA clamp facilitates action of DNA cytosine methyltransferase 1 on hemimethylated DNA. Genes Cells 7, 997–1007 [DOI] [PubMed] [Google Scholar]
- 16. Sarraf S. A., Stancheva I. (2004) Methyl-CpG binding protein MBD1 couples histone H3 methylation at lysine 9 by SETDB1 to DNA replication and chromatin assembly. Mol. Cell 15, 595–605 [DOI] [PubMed] [Google Scholar]
- 17. Birke M., Schreiner S., García-Cuéllar M. P., Mahr K., Titgemeyer F., Slany R. K. (2002) The MT domain of the proto-oncoprotein MLL binds to CpG-containing DNA and discriminates against methylation. Nucleic Acids Res. 30, 958–965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lee J. H., Voo K. S., Skalnik D. G. (2001) Identification and characterization of the DNA binding domain of CpG-binding protein. J. Biol. Chem. 276, 44669–44676 [DOI] [PubMed] [Google Scholar]
- 19. Cierpicki T., Risner L. E., Grembecka J., Lukasik S. M., Popovic R., Omonkowska M., Shultis D. D., Zeleznik-Le N. J., Bushweller J. H. (2010) Structure of the MLL CXXC domain-DNA complex and its functional role in MLL-AF9 leukemia. Nat. Struct. Mol. Biol. 17, 62–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Allen M. D., Grummitt C. G., Hilcenko C., Min S. Y., Tonkin L. M., Johnson C. M., Freund S. M., Bycroft M., Warren A. J. (2006) Solution structure of the nonmethyl-CpG-binding CXXC domain of the leukaemia-associated MLL histone methyltransferase. EMBO J. 25, 4503–4512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Voo K. S., Carlone D. L., Jacobsen B. M., Flodin A., Skalnik D. G. (2000) Cloning of a mammalian transcriptional activator that binds unmethylated CpG motifs and shares a CXXC domain with DNA methyltransferase, human trithorax, and methyl-CpG binding domain protein 1. Mol. Cell Biol. 20, 2108–2121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Xu C., Bian C., Lam R., Dong A., Min J. (2011) The structural basis for selective binding of non-methylated CpG islands by the CFP1 CXXC domain. Nat. Commun. 2, 227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ayton P. M., Chen E. H., Cleary M. L. (2004) Binding to nonmethylated CpG DNA is essential for target recognition, transactivation, and myeloid transformation by an MLL oncoprotein. Mol. Cell Biol. 24, 10470–10478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Song J., Rechkoblit O., Bestor T. H., Patel D. J. (2011) Structure of DNMT1-DNA complex reveals a role for autoinhibition in maintenance DNA methylation. Science 331, 1036–1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jørgensen H. F., Ben-Porath I., Bird A. P. (2004) Mbd1 is recruited to both methylated and nonmethylated CpGs via distinct DNA binding domains. Mol. Cell Biol. 24, 3387–3395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Thomson J. P., Skene P. J., Selfridge J., Clouaire T., Guy J., Webb S., Kerr A. R., Deaton A., Andrews R., James K. D., Turner D. J., Illingworth R., Bird A. (2010) CpG islands influence chromatin structure via the CpG-binding protein Cfp1. Nature 464, 1082–1086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ma Q., Alder H., Nelson K. K., Chatterjee D., Gu Y., Nakamura T., Canaani E., Croce C. M., Siracusa L. D., Buchberg A. M. (1993) Analysis of the murine All-1 gene reveals conserved domains with human ALL-1 and identifies a motif shared with DNA methyltransferases. Proc. Natl. Acad. Sci. U.S.A. 90, 6350–6354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Erfurth F. E., Popovic R., Grembecka J., Cierpicki T., Theisler C., Xia Z. B., Stuart T., Diaz M. O., Bushweller J. H., Zeleznik-Le N. J. (2008) MLL protects CpG clusters from methylation within the Hoxa9 gene, maintaining transcript expression. Proc. Natl. Acad. Sci. U.S.A. 105, 7517–7522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ohki I., Shimotake N., Fujita N., Jee J., Ikegami T., Nakao M., Shirakawa M. (2001) Solution structure of the methyl-CpG binding domain of human MBD1 in complex with methylated DNA. Cell 105, 487–497 [DOI] [PubMed] [Google Scholar]
- 30. Lavau C., Szilvassy S. J., Slany R., Cleary M. L. (1997) Immortalization and leukemic transformation of a myelomonocytic precursor by retrovirally transduced HRX-ENL. EMBO J. 16, 4226–4237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Armstrong S. A., Staunton J. E., Silverman L. B., Pieters R., den Boer M. L., Minden M. D., Sallan S. E., Lander E. S., Golub T. R., Korsmeyer S. J. (2002) MLL translocations specify a distinct gene expression profile that distinguishes a unique leukemia. Nat. Genet. 30, 41–47 [DOI] [PubMed] [Google Scholar]
- 32. Ayton P. M., Cleary M. L. (2003) Transformation of myeloid progenitors by MLL oncoproteins is dependent on Hoxa7 and Hoxa9. Genes Dev. 17, 2298–2307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ng H. H., Jeppesen P., Bird A. (2000) Active repression of methylated genes by the chromosomal protein MBD1. Mol. Cell Biol. 20, 1394–1406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fujita N., Takebayashi S., Okumura K., Kudo S., Chiba T., Saya H., Nakao M. (1999) Methylation-mediated transcriptional silencing in euchromatin by methyl-CpG binding protein MBD1 isoforms. Mol. Cell Biol. 19, 6415–6426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fujita N., Watanabe S., Ichimura T., Tsuruzoe S., Shinkai Y., Tachibana M., Chiba T., Nakao M. (2003) Methyl-CpG binding domain 1 (MBD1) interacts with the Suv39h1-HP1 heterochromatic complex for DNA methylation-based transcriptional repression. J. Biol. Chem. 278, 24132–24138 [DOI] [PubMed] [Google Scholar]