

Background: Leishmania donovani salvage all pyrimidines through uracil phosphoribosyltransferase (LdUPRT).
Results: LdUPRT phosphoribosylates uracil, 5-fluorouracil, and 4-thiouracil and is susceptible to substrate inhibition.
Conclusion: LdUPRT recognizes pyrimidine analogs, and substrate inhibition by LdUPRT explains the supersensitivity of pyrimidine auxotrophs to uracil.
Significance: Substrate inhibition of LdUPRT provides a mechanism for uracil susceptibility and offers a protective function for the parasite.
Keywords: Enzyme Kinetics, Leishmania, Parasite, Parasite Metabolism, Pyrimidine, Leishmania donovani, Pyrimidine Salvage, Pyrimidines, Substrate Inhibition, Uracil Phosphoribosyltransferase
Abstract
The pathogenic protozoan parasite Leishmania donovani is capable of both de novo pyrimidine biosynthesis and salvage of pyrimidines from the host milieu. Genetic analysis has authenticated L. donovani uracil phosphoribosyltransferase (LdUPRT), an enzyme not found in mammalian cells, as the focal enzyme of pyrimidine salvage because all exogenous pyrimidines that can satisfy the requirement of the parasite for pyrimidine nucleotides are funneled to uracil and then phosphoribosylated to UMP in the parasite by LdUPRT. To characterize this unique parasite enzyme, LdUPRT was expressed in Escherichia coli, and the recombinant enzyme was purified to homogeneity. Kinetic analysis revealed apparent Km values of 20 and 99 μm for the natural substrates uracil and phosphoribosylpyrophosphate, respectively, as well as apparent Km values 6 and 7 μm for the pyrimidine analogs 5-fluorouracil and 4-thiouracil, respectively. Size exclusion chromatography revealed the native LdUPRT to be tetrameric and retained partial structure and activity in high concentrations of urea. L. donovani mutants deficient in de novo pyrimidine biosynthesis, which require functional LdUPRT for growth, are hypersensitive to high concentrations of uracil, 5-fluorouracil, and 4-thiouracil in the growth medium. This hypersensitivity can be explained by the observation that LdUPRT is substrate-inhibited by uracil and 4-thiouracil, but 5-fluorouracil toxicity transpires via an alternative mechanism. This substrate inhibition of LdUPRT provides a protective mechanism for the parasite by facilitating purine and pyrimidine nucleotide pool balance and by sparing phosphoribosylpyrophosphate for consumption by the nutritionally indispensable purine salvage process.
Introduction
Leishmania donovani is a protozoan parasite and etiologic agent of visceral leishmaniasis, a disease that is ultimately fatal if untreated. Leishmania are digenetic parasites subsisting as the motile, extracellular promastigote in the female Phlebotomine sandfly vector and as the nonmotile, intracellular amastigote within the phagolysosome of macrophages inside the infected mammalian host. There is no vaccine against leishmaniasis, and the current assortment of drugs used to treat leishmaniasis is far from ideal. These drugs are toxic to the host, require invasive means of administration, and trigger resistance in the field. Thus, the need to discover new drugs and validate new drug targets for the treatment of leishmaniasis—or for that matter any disease of parasitic origin—is imperative.
Among the pathways that have been touted as potential antiparasitic targets are those for purines and pyrimidines, the building blocks for nucleic acid synthesis. Leishmania, like all protozoan parasites studied to date, are incapable of synthesizing purine nucleotides de novo, and therefore, each genus must obligatorily scavenge purines from its hosts (1). In contrast, most, but not all, protozoan parasites, including Leishmania, are prototrophic for pyrimidines (1). The de novo pathway for pyrimidine biosynthesis is conserved among eukaryotes and prokaryotes and consists of six enzymes that generate UMP from CO2, amino acids, and 5-phosphoribosyl-1-pyrophosphate (PRPP)2 (Fig. 1). UMP is then distributed into ribonucleotides via nucleotide kinases and into deoxyribonucleotides by ribonucleotide reductase and thymidylate synthase. Gene sequencing, supported by biochemical studies, has revealed a number of significant differences between the pyrimidine biosynthetic pathways of Leishmania and mammals: 1) the genes encoding the pyrimidine pathway of Leishmania are syntenic (2–5), whereas the mammalian pyrimidine genes are not; 2) the genes encoding the first three enzymes in Leishmania are discrete, unlike the mammalian pathway in which there is a single gene encoding a trifunctional protein (6, 7); 3) the last two enzymes of the pyrimidine biosynthetic pathway are expressed as a single bifunctional protein, designated UMP synthase (UMPS), although the domain order in Leishmania and mammalian cells is reversed (3, 8); and 4) the UMP synthase of Leishmania is localized within the glycosome (3), a unique peroxisomal-like organelle that is found uniquely among Leishmania and related parasites (9, 10). Genetic ablation of either carbamoyl phosphate synthetase (CPS), the first enzyme of pyrimidine biosynthesis, or UMPS in L. donovani confer pyrimidine auxotrophy that can be circumvented by supplementation of the defined growth medium with uracil, uridine, deoxyuridine, cytidine, or deoxycytidine (3, 5). Furthermore, both the Δcps and the Δumps null mutants exhibit a striking collateral supersensitivity to uracil, which is innocuous to wild type parasites, that is not observed with any of the ribonucleosides (5). A comparable growth susceptibility toward uracil is also observed in other species of protozoan parasites in which the de novo pyrimidine pathway has been genetically disrupted. These uracil-sensitive mutants include Δcps strains of Toxoplasma gondii (11) and Trypanosoma cruzi (12), the causative agents of toxoplasmosis and Chagas disease in humans, respectively, as well as a Δumps null mutant in T. brucei (13), which causes African sleeping sickness. Furthermore, repressing expression of dihydroorotate dehydrogenase, the fourth enzyme in the pyrimidine biosynthesis pathway, by RNA interference elicits susceptibility to 5-fluorouracil in Trypanosoma brucei (14).
FIGURE 1.
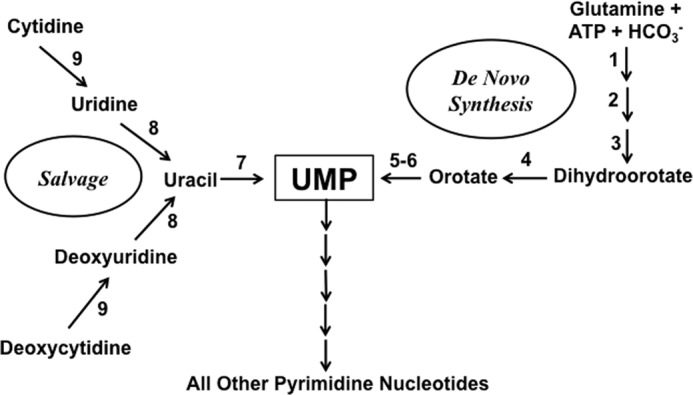
Schematic diagram of the pyrimidine biosynthetic and salvage pathways of L. donovani. The portions of the pyrimidine biosynthetic and pyrimidine salvage pathways that are pertinent to this manuscript are depicted. The following enzymes are depicted: CPS (1), aspartate transcarbamoylase (2), dihydroorotase (3), dihydroorotate dehydrogenase (4), orotate phosphoribosyltransferase (5), orotidylate decarboxylase (6), LdUPRT (7), uridine nucleosidase (8), and cytidine deaminase (9). Orotate phosphoribosyltransferase and orotidylate decarboxylase are the enzymatic activities encoded by UMPS.
Despite the pyrimidine auxotrophy observed for Δcps and Δumps L. donovani promastigotes in culture, both knock-out lines sustain relatively robust infections in mice (Ref. 5 and data not shown). These findings imply that the null mutants within the macrophage phagolysosome can access a source of host pyrimidines that can satisfy the pyrimidine nucleotide requirements of the parasite. Thus, L. donovani, in contrast to the purine pathway, has two routes for pyrimidine nucleotide synthesis, biosynthesis, and salvage. Genetic analysis has also authenticated that this salvage of preformed pyrimidines in both promastigotes and amastigotes of L. donovani is mediated through uracil phosphoribosyltransferase (LdUPRT) and that pyrimidine nucleosides (other than thymidine) are converted to uracil within the parasite (5). Uridine and deoxyuridine are cleaved to form uracil via nucleoside hydrolase enzymes, whereas a cytidine deaminase converts cytidine and deoxycytidine to their uracil-containing counterparts (15, 16) (Fig. 1). Thus, LdUPRT plays an exclusive role in pyrimidine salvage in the parasite, a function that profoundly impacts the capacity of the parasite to survive as the amastigote in a rodent model.
To characterize the biochemical and kinetic properties of LdUPRT and to evaluate the involvement of LdUPRT in the noteworthy vulnerability of three different genera of protozoan parasite to uracil- or 5-fluorouracil-mediated growth inhibition when the pyrimidine biosynthetic pathway is genetically compromised, recombinant LdUPRT was purified and characterized. Kinetic parameters to the naturally occurring substrates, as well as to several important uracil analogs, were determined, and the L. donovani enzyme, unlike its T. gondii counterpart (17), was shown to form a stable tetramer in the absence of GTP. Furthermore, profound substrate inhibition of LdUPRT to nucleobase substrates was demonstrated, providing a mechanism by which L. donovani, T. gondii, T. cruzi, or T. brucei, genetically deficient in pyrimidine biosynthesis, would exhibit a dramatic growth sensitivity to exogenous uracil. It is conjectured that this substrate inhibition of LdUPRT by uracil affords the parasite a protective mechanism to protect its nutritionally indispensable purine salvage mechanism and to maintain an equilibrium between purine and pyrimidine nucleotide pools in the parasite.
EXPERIMENTAL PROCEDURES
Chemicals and Reagents
Uracil, 5-fluorouracil, 4-thiouracil, PRPP, GTP, isopropyl β-d-1-thiogalactopyranoside (IPTG), and metal salts were purchased from Sigma-Aldrich. Ni-NTA-agarose beads were from Qiagen. Complete Mini EDTA-free protease inhibitor was bought from Roche Applied Science. Biosafe Coomassie and Bio-Rad protein dye were acquired from Bio-Rad Laboratories Life Science Research. Oligonucleotide primers were obtained from Integrated DNA Technologies, Inc. (Coralville, IA), and Phusion® High-Fidelity PCR Master Mix was from Fisher Scientific. ChampionTM pET Directional TOPO expression kit was purchased from Invitrogen. The Agilent 8453 UV-visible diode array spectrophotometer was from Agilent Technologies (Santa Clara, CA). All other chemicals and reagents were of the highest quality commercially available.
Expression and Purification of LdUPRT and T. gondii UPRT (TgUPRT) in Escherichia coli
The cloning of LdUPRT into the pET 200/D-TOPO® E. coli expression vector has been previously reported (5). The full-length TgUPRT cDNA was amplified by PCR using the forward primer 5′-GAGGCCACCTGGGCCATGGCGCAGGTCCCAGCGAG-3′ and reverse primer 5′-GAGGCCAGCCCGGCCCTACATGGTTCCAAAGTACCGGTCACCGAA-3′ (SfiI restriction sites are in bold, and unique triads are underlined) from a previously reported TgUPRT cDNA construct (18). The insert was ligated in to the pET 200/D-TOPO® vector containing an NH2-terminal His6 tag and transformed into One Shot® Top10 chemically competent E. coli. The fidelity and orientation of the recombinant plasmid was verified by DNA sequencing. The LdUPRT and TgUPRT expression constructs in the pET 200/D-TOPO® vector were transformed into BL21StarTM (DE3) One Shot® E. coli according to the ChampionTM pET Directional TOPO user manual and plated on LB plates containing 50 μg/ml kanamycin. Transformants were picked and expanded in 200 ml or 1 liter of LB medium to an A600 ∼0.6, and protein expression was induced with 1 mm IPTG for 16 h at 37 °C with constant shaking.
The E. coli from the 16-h culture was harvested by centrifugation and resuspended in a buffer containing 50 mm NaH2PO4, 300 mm NaCl, 10 mm imidazole, pH 8.0, and EDTA-free protease inhibitors. The cells were ruptured by sonication on ice with six 10-s bursts at 200–300 W with a 10-s cooling period between each burst. The lysate was centrifuged at 10,000 × g for 30 min at 4 °C to pellet the cellular debris, and the supernatant was collected. The clarified lysate was incubated with a 50% Ni-NTA slurry at 4 °C for 1 h with continuous shaking. The lysate-Ni-NTA mixture was loaded onto a column and washed twice in 50 mm NaH2PO4, 1 m NaCl, 20 mm imidazole, pH 8.0 buffer and eluted in buffer consisting of 50 mm NaH2PO4, 300 mm NaCl, and 250 mm imidazole, pH 8.0. The purified recombinant proteins were buffer-exchanged into a final storage buffer containing 50 mm KCl, 50 mm KH2PO4, 5% glycerol, pH 8.0, using 7,000 molecular weight cutoff ZebaTM spin desalting columns (ThermoFisher Scientific). Concentrated LdUPRT and TgUPRT preparations were obtained by ultrafiltration employing Amicon Ultra-10K centrifugal filter units (EMD Millipore Corp., Billerica, MA), and protein concentrations were determined using the Bio-Rad Bradford total protein assay system.
LdUPRT Kinetics
All kinetic parameters were determined using a published spectrophotometric method based on monitoring the absorbance change at a specified wavelength under steady state conditions (18, 19). Each assay mixture was prepared by adding the substrates to a buffer containing 50 mm Tris-HCl, 5 mm MgCl2, and 2 mm DTT, pH 7.5 (TMD 50), unless otherwise noted. For each substrate, the assay mixture was blanked prior to assay to remove background caused by substrates or reagents. Kinetic traces were initiated by addition of LdUPRT enzyme to the pre-equilibrated assay mixture, and data were collected for total of 120 s at a fixed wavelength. The assays were based on the small but significant differential molar absorption coefficients (Δϵ) between the substrate and product, e.g., uracil and UMP at a given wavelength. The fixed wavelengths and extinction coefficients employed varied depending upon the nucleobase substrate and are described below. All kinetic parameters were calculated by the suite of algorithms available in GraphPad Prism 4.0.
The kinetic parameters for uracil were determined using an assay mixture consisting of TMD 50 buffer, 1 mm PRPP, and various concentrations of uracil ranging from 5 μm to 1.5 mm, whereas the Km value for PRPP was ascertained in TMD 50 buffer, 250 μm uracil, and PRPP concentrations ranging from 25 μm to 2.5 mm. To evaluate the effect of GTP on PRPP kinetics, 2 mm GTP was added to the assay mixture described above. UMP formation was determined at a wavelength of 280 nm, and the rates were calculated using a differential molar extinction coefficient of Δϵ = 1419 m−1 cm−1.
The activity of LdUPRT toward the nucleobase analogs 5-fluorouracil and 4-thiouracil were determined in TMD 50 buffer, 1 mm PRPP, pH 7.5, containing either 5–150 μm 5-fluorouracil or 1–80 μm 4-thiouracil. Steady state kinetics were performed based on the expenditure of 5-fluorouracil at 303 nm and the formation of 4-thiouridine-5′-monophosphate at 320 nm, respectively. The differential extinction coefficient for 5-fluorouracil was calculated to be Δϵ = 923 m−1 cm−1 (at pH 7.5; λmax 303 nm), and the differential extinction coefficient for 4-thiouridine-5′-monophosphate was previously determined to be ϵ = 16300 m−1 cm−1 (at pH 7.5; λmax 320 nm according to the brochure from Jena Bioscience (Jena, Germany).
Effect of pH on LdUPRT Activity
The pH optimum of LdUPRT was evaluated by measuring the activity between pH 6.0 and 10.0 at 0.5 pH unit increments and also at pH 5.8 using either a 50 mm CHES, 50 mm Bis-Tris or 50 mm Tricine buffer. Concentrated NaOH and HCl were used to adjust the pH in the three buffer solutions. To measure LdUPRT activity, 250 μm of uracil, 1 mm PRPP, 2 mm MgCl2, and 2 mm DTT were added to each buffer, and the rate of UMP formation was measured spectrophotometrically as described above.
Ion Dependence of LdUPRT
The effects of an assortment of divalent cations on LdUPRT activity was verified in 50 mm Tris-HCl, 2 mm DTT, 250 μm uracil, 1 mm PRPP, pH 7.5, to which 2 mm of one of the following cations was added: MgCl2, MnCl2, BaCl2, CoCl2, CaCl2, NiCl2, and ZnCl2. Control experiments were performed both in the absence of divalent cation and in the presence of 10 mm EDTA. UMP formation was determined as described above.
Size Exclusion Chromatography
Either LdUPRT or TgUPRT at a concentration of 1.0 mg/ml was injected in a volume of 100 μl of 50 mm KCl, 50 mm KH2PO4, pH 8.0 buffer onto a Superose 12 10/300 GL column (GE Healthcare) and eluted with 1 column volume of 50 mm KCl, 50 mm KH2PO4, pH 8.0 buffer at a flow rate of 0.4 ml/min. Parallel runs were also conducted in the presence of 2 mm GTP in both the loading and elution buffers. Protein in the eluates was monitored by absorption at 280 nm. Estimated molecular weights were calibrated using a gel filtration marker kit from Sigma-Aldrich. Size exclusion chromatography of LdUPRT and TgUPRT was performed after 3-h incubations in 3 m urea, and 3 m urea was added to both the loading and elution buffers.
Parasite Cell Culture
The creation and characterization of Δuprt, Δcps, and Δumps L. donovani lines have been reported previously (3, 5). Wild type and null mutant promastigotes were continuously cultured in 26 °C in pH 7.4 DME-L medium that was supplemented with 10% Serum Plus® (SAFC Biosciences, Lenexa, KS), 1 mg/ml hemin, and 100 μm hypoxanthine as a purine source. The Δuprt, Δcps, and Δumps transgenic strains were routinely maintained in the drugs in which the homologous gene replacement events were selected.
Growth Assays
The abilities of wild type and mutant cells to grow in a range of uracil concentrations (4 μm to 4 mm) were determined by placing 5.0 × 103 promastigotes into individual wells of a 96-well cell culture plate containing 0.2 ml of growth medium. Additional uracil sensitivity experiments were performed using the same protocol but with either 250 μm cytidine, 2 mm dihydroorotate, or 2 mm orotate added to the growth medium. 5-Fluorouracil and 4-thiouracil sensitivity experiments were conducted using the same protocol, again as a function of multiple 5-fluorouracil (40 nm to 40 μm) or 4-thiouracil (1 μm to 1 mm) concentrations. 250 μm cytidine was added to the growth medium in these growth experiments with the two uracil analogs. At the end of each growth experiment, parasites were counted using the AlamarBlue® (BIOSOURCE) cell viability assay (20). Reduction of AlamarBlue was monitored at 570 and 600 nm on a Multiskan Ascent plate reader (Thermo Labsystems, Vantaa, Finland). The percentage of dye reduction was calculated according to the formula delineated in the manufacturer's pamphlet, and the largest reduction was expressed as maximum growth.
Substrate Inhibition Profiles
The ability of high concentrations of uracil, 5-fluorouracil, and 4-thiouracil to inhibit their own phosphoriboyslation by LdUPRT was gauged in TMD 50 buffer containing 1 mm PRPP and 150–1500 μm concentrations of the uracil or 5-fluorouracil or 75–1500 μm concentrations of 4-thiouracil employing the spectrophotometric methods described above. To evaluate whether uracil-mediated substrate inhibition was reversible or irreversible, 2.0 μg of purified LdUPRT was incubated in the absence or presence of either 1.0 mm PRPP, 1.5 mm uracil, or both 1.0 mm PRPP and 1.5 mm uracil for 5 min and diluted in TMD buffer just prior to assay, and LdUPRT activity assessed in the presence of 1.0 mm PRPP and either 75 μm or 1.5 mm uracil. The thermostability of LdUPRT was evaluated by incubating 2.0-μg aliquots of purified LdUPRT in the absence or presence of either 1.0 mm PRPP, 75 μm uracil, 1.5 mm uracil, both 1.0 mm PRPP and 75 μm uracil, or both 1.0 mm PRPP and 1.5 mm uracil at 62 °C for various time points up to 20 min; diluting into TMD buffer; and assaying residual LdUPRT activity in TMD buffer to which 1.0 mm PRPP and 200 μm uracil were added.
RESULTS
LdUPRT Expression and Purification
LdUPRT was robustly expressed from the pET 200/D-TOPO construct using the BL21StarTM (DE3) One Shot® E. coli expression system and IPTG induction (Fig. 2). A visible band was observed at 27 kDa, consistent with the predicted molecular mass (Fig. 2). This band was not observed in uninduced E. coli. His6-LdUPRT (henceforth just referred to as LdUPRT) was subsequently purified to virtual homogeneity by affinity chromatography (Fig. 2, lanes D–F). Overall yield was roughly 4–6 mg of purified recombinant protein per liter of bacterial culture. The purified LdUPRT was stable with no measurable loss of enzymatic activity for >1 month when maintained at 4 °C in the elution buffer to which 5% glycerol was added.
FIGURE 2.
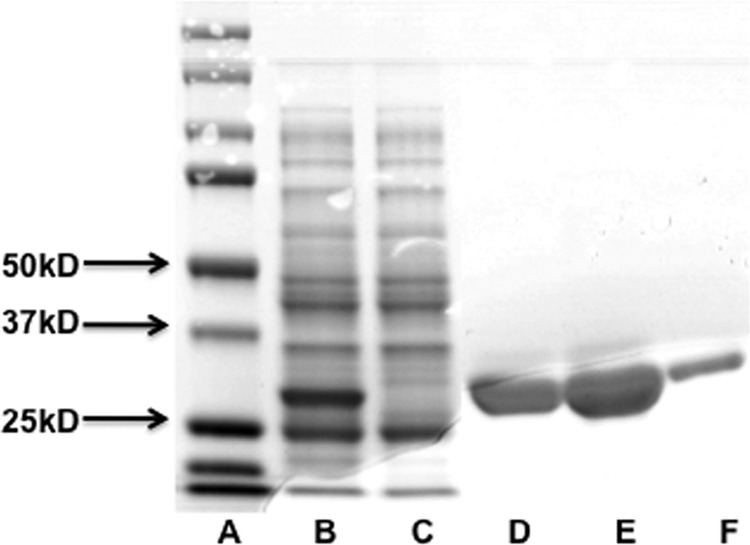
Purification of LdUPRT. LdUPRT was overexpressed in E. coli and purified to homogeneity over a Ni-NTA column. Lane A, Precision Plus Protein Standards; lane B, 10,000 × g crude cell lysates of IPTG-treated BL21StarTM (DE3) One Shot® E. coli transformed with LdUPRT; lane C, 10,000 × g cell lysates of uninduced BL21StarTM (DE3) One Shot® E. coli; lanes D–F, final elution fractions from the Ni-NTA column. Molecular weight markers are shown in lane A.
Enzyme Kinetics
Freshly purified recombinant LdUPRT was enzymatically active and catalyzed the phosphoribosylation of uracil efficiently. LdUPRT displayed a pH optimum between 7.5 and 8.5 with sharp drops in activity 0.5 pH units outside this range, as well as an absolute requirement for the presence of a divalent cation (Fig. 3). Maximum activity of LdUPRT was achieved with Mg2+, although LdUPRT was almost as efficient with Co2+ and Mn2+. Ba2+, Ca2+, Ni2+, and Zn2+ did not support LdUPRT catalysis. Michaelis-Menten plots of steady state kinetic data obtained at pH 7.5 and 5.0 mm MgCl2 revealed apparent Km values of 20.4 and 99.3 μm for uracil and PRPP, respectively (Fig. 4, A and B). The kinetic data for the bisubstrate reaction were collected at concentrations of invariant substrate that were ∼10-fold greater than the experimentally determined Km value. Because the affinity of TgUPRT, the only uracil phosphoribosyltransferase (UPRT) of parasitic origin that has been previously characterized, for PRPP was reduced by the addition of GTP (17), the Km value for PRPP was also determined in the presence of GTP. Incubation of LdUPRT with 2 mm GTP treatment did not, however, affect the Km value of LdUPRT for PRPP (Fig. 4B). A Vmax value of 13.6 ± 1.4 μmol/min/mg protein for LdUPRT was computationally derived (Fig. 4, A and B). A kcat value of 6.19 s−1 was then calculated from the kinetic data, and the catalytic efficiency (kcat/Km) was computed to be 0.303 s−1 μm−1. LdUPRT also displayed high affinities for the pyrimidine analogs 4-thiouracil and 5-fluorouracil with calculated Km values of 7.1 and 6.4 μm, respectively (Fig. 4, C and D). The calculated Vmax values of LdUPRT for the two nucleobase analogs were similar, 1.1 ± 0.18 and 1.3 ± 0.12 μmol/min/mg protein for 4-thiouracil and 5-fluorouracil, respectively.
FIGURE 3.
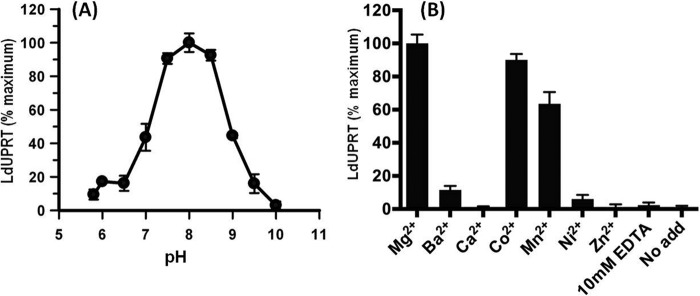
pH and divalent cation profiles of LdUPRT. Initial rates of LdUPRT activity were determined as a function of pH at 0.5 pH units from pH 6.0 to 10.0 and at pH 5.8 as described under “Experimental Procedures.” The data are presented as percentages of maximum activity (pH 8.0) as a function of pH (A). LdUPRT activity was also assessed as a function of the divalent cation in the assay mixture (B). All cations were present at a concentration of 2 mm as the chloride salt. Controls included no divalent cation and 10 mm EDTA. The data are calculated as percentages of maximum LdUPRT activity. The data points in both panels are the averages ± standard deviations obtained for three separate experiments.
FIGURE 4.

Michaelis-Menten kinetics for LdUPRT. LdUPRT activity was measured spectrophotometrically as a function of uracil concentration at a fixed 1.0 mm PRPP concentration (A) and as a function of PRPP concentration at 250 μm uracil in the absence and presence of 2 mm GTP (B). Michael-Menten kinetics were also collected as a function of 4-thiouracil (C) and 5-fluorouracil (D) concentrations in the presence of 1.0 mm PRPP. All data are the means ± standard deviations of three replicates. Kinetic parameters were calculated in GraphPad Prism 4.0.
Oligomerization State
Size exclusion chromatography indicated that LdUPRT migrated with a molecular mass just under 100 kDa, consistent with a tetrameric oligomerization state (Fig. 5). In contrast, purified recombinant TgUPRT migrated on the size exclusion column with a molecular mass consistent with a dimeric quaternary state. A dimeric structure has been previously reported for TgUPRT based on its sedimentation properties in sucrose gradients (17). The addition of 2 mm GTP to TgUPRT, known to stabilize higher order structures of the protein (17), induced an oligomeric state that was either a trimeric or quaterneric structure (Fig. 5). The quaternary structure of LdUPRT, as expected, was unaffected by 2 mm GTP (data not shown).
FIGURE 5.
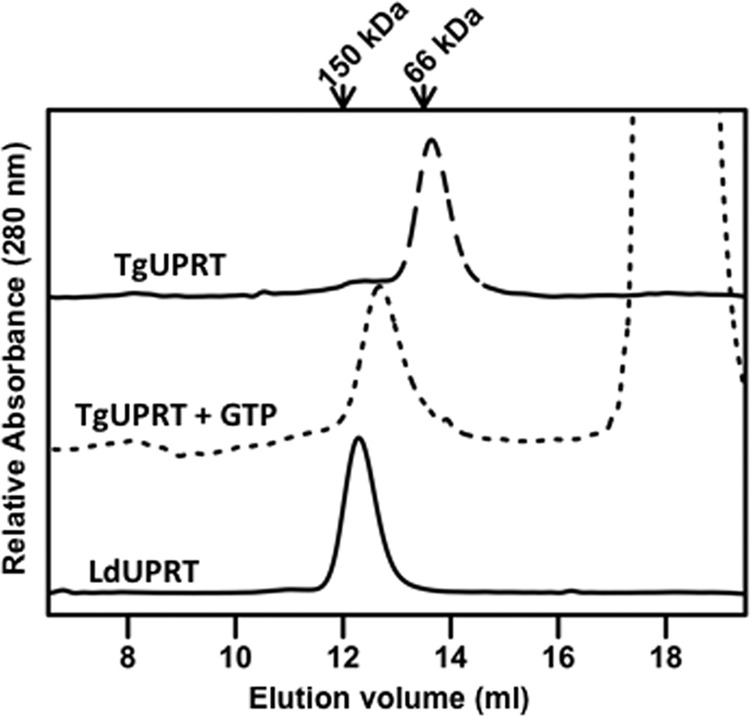
Size exclusion chromatography of purified LdUPRT and TgUPRT. 1.0 mg/ml purified TgUPRT in the absence or presence of 2 mm GTP or purified LdUPRT were chromatographed over a Superose 12 10/300 GL column, and eluted protein was monitored by absorbance at 280 nm and plotted as a function of elution volume. Molecular weight standards included carbonic anhydrase (Mr = ∼29 kDa), bovine serum albumin (Mr = ∼66 kDa), and alcohol dehydrogenase (Mr = ∼150 kDa).
Effect of Added Pyrimidines on the Sensitivity of L. donovani to Uracil
It has been previously reported that the growth of L. donovani rendered auxotrophic for pyrimidines through genetic lesions in the de novo pyrimidine biosynthesis pathway, specifically strains in which either the CPS or UMPS ORFs have been deleted, is inhibited by high concentrations of uracil in the growth medium (5). Uracil is not, however, growth inhibitory to wild type L. donovani (5). Similarly, T. gondii (11), T. cruzi (12), and T. brucei (13) with genetic defects in pyrimidine biosynthesis pathway are susceptible to uracil-mediated growth inhibition, whereas their wild type counterparts are not. The previously published uracil sensitivity experiments with L. donovani were conducted in the absence of additional pyrimidines in the culture medium (5). To determine whether this curious growth inhibitory effect of uracil on pyrimidine auxotrophic L. donovani was impacted by the presence of other pyrimidines, the uracil susceptibility of Δcps and Δumps promastigotes to uracil was evaluated in the absence or presence of pyrimidine biosynthetic or salvage intermediates (Fig. 6). Whereas both Δcps and Δumps promastigotes were sensitive to uracil in the absence or presence of 250 μm cytidine in the culture medium, the uracil supersensitivity of the Δcps line was abrogated by the addition of either 2 mm orotate or 2 mm dihydroorotate (Fig. 6). Neither orotate nor dihydroorotate, however, affected the sensitivity of the Δumps null mutant to uracil (Fig. 6, C and D).
FIGURE 6.

Inhibition of L. donovani growth and LdUPRT activity by high substrate concentrations. The abilities of wild type (■), Δcps (▴), and Δumps (●) L. donovani promastigotes to grow in various concentrations of uracil (A) or in various concentrations of uracil plus 250 μm cytidine (B), 2 mm orotate (C), or 2 mm dihydroorotate (D) were assessed as described under “Experimental Procedures.” The data are those from one of at least three independent experiments, all of which gave virtually identical results.
LdUPRT Substrate Inhibition
The ability of dihydroorotate and orotate to alleviate uracil-mediated growth inhibition of L. donovani promastigotes harboring a genetic lesion in CPS implied that uracil was triggering a pyrimidine deficiency explicitly in cells with genetic lesions in pyrimidine biosynthesis. One possible mechanism by which pyrimidine deficiency could be selectively induced in pyrimidine auxotrophic L. donovani is by uracil-mediated substrate inhibition of LdUPRT. To test this conjecture, the ability of high concentrations of uracil to inhibit nucleobase phosphoribosylation by LdUPRT was determined. As shown in Fig. 7A, concentrations of uracil 10-fold higher than the Km value dramatically diminished the capacity of LdUPRT to convert uracil to UMP. 1.5 mm uracil diminished LdUPRT activity by ∼90%. To test whether TgUPRT was also prone to substrate inhibition by uracil, TgUPRT activity was also measured as a function of uracil concentration in the assay. As shown in Fig. 7B, TgUPRT catalytic activity is also markedly inhibited by high uracil in a dose-dependent manner similar to the pattern of inhibition obtained with LdUPRT.
FIGURE 7.

Substrate inhibition of LdUPRT and TgUPRT. LdUPRT (A) and TgUPRT (B) activity was determined spectrophotometrically at a variety of high uracil concentrations in the assay mixture. LdUPRT activity was also ascertained spectrophotometrically as a function of high concentrations of 4-thiouracil (C) or 5-fluorouracil (D). The data presented are the means ± standard deviations from three distinct measurements.
To examine in more detail the mechanism by which uracil elicited inhibition of LdUPRT, the reversibility of this substrate inhibition was examined. Purified LdUPRT samples were preincubated with 1.0 mm PRPP, 1.5 mm uracil, or both 1.0 mm PRPP and 1.5 mm uracil and then examined for LdUPRT activity. None of the preincubation conditions affected LdUPRT activity, and the activity detected remained sensitive to inhibition by high uracil concentrations (Fig. 8A). The thermolability of LdUPRT was also tested using the same conditions as those employed for the reversibility assays. 1.0 mm PRPP stabilized LdUPRT to heat inactivation at 62 °C, whereas 1.5 mm uracil did not impact thermolability. Furthermore, addition of either 75 μm or 1.5 mm uracil to the enzyme in the absence or presence of 1.0 mm PRPP had no effect on the heat inactivation profile of the enzyme (Fig. 8B and data not shown). Attempts to examine changes in LdUPRT secondary structure induced by uracil by circular dichroism spectroscopy were precluded by the high level of absorbance of 1.5 mm uracil in the far ultraviolet spectral range.
FIGURE 8.
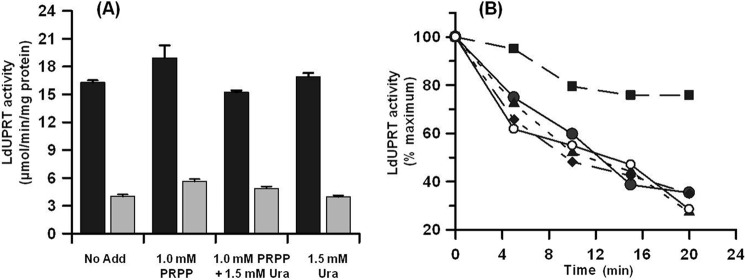
Reversibility and thermostability of uracil-induced substrate inhibition of LdUPRT. Purified LdUPRT was preincubated in the absence or presence of either 1.0 mm PRPP, both 1.0 mm PRPP and 1.5 mm uracil, or 1.5 mm uracil alone, as described under “Experimental Procedures,” then diluted into TMD buffer, and assayed for LdUPRT activity in the presence of 1.0 mm PRPP and either 75 μm (black bars) or 1.5 mm uracil (gray bars), respectively (A). The thermostability of LdUPRT was assessed at 62 °C in TMD buffer in the absence (●) or presence of either 1.0 mm PRPP (■), both 1.0 mm PRPP and 75 μm uracil (▴), both 1.0 mm PRPP and 1.5 mm uracil (♦), 75 μm uracil alone (data not shown), or 1.5 mm uracil alone (○), and residual activity was measured as described under “Experimental Procedures” (B).
Inhibition of LdUPRT Activity and L. donovani Growth by Uracil Analogs
LdUPRT was also susceptible to substrate inhibition by high concentrations of the uracil analogs 4-thiouracil and 5-fluorouracil (Fig. 7, C and D). To determine whether the substrate sensitivity of LdUPRT to 4-thiouracil and 5-fluorouracil could also affect the growth susceptibility of Δcps and Δumps L. donovani to the two uracil analogs, the growth of wild type and the two pyrimidine auxotrophs was assessed over a range of analog concentrations. These experiments were performed in medium supplemented with 250 μm cytidine, which does not affect uracil sensitivity of Δcps and Δumps L. donovani (Fig. 6B) but is required for their survival and growth. Both the Δcps and Δumps null mutants exhibited sensitivity to 4-thiouracil with EC50 values of 82.3 and 67.3 μm, respectively, whereas wild type parasites, as well as a Δuprt L. donovani strain, were refractory to 4-thiouracil concentrations in the medium as high as 1 mm (Fig. 9A). Unlike uracil, which was growth inhibitory toward the pyrimidine auxotrophs, 4-thiouracil at concentrations >500 μm killed the mutant parasites. The pyrimidine auxotrophs were also more sensitive to 5-fluorouracil than the wild type or Δuprt mutant, although the differences were not as dramatic as for uracil (Fig. 6A) and 4-thiouracil (Fig. 9A). EC50 values obtained for wild type, Δuprt, Δcps, and Δumps promastigotes were 1.65, 1.33, 0.49, and 0.24 μm, respectively (Fig. 9B). These 5-fluorouracil growth sensitivity experiments were also carried out in growth medium supplemented with 250 μm cytidine as the requisite source of preformed pyrimidine for the pyrimidine biosynthesis null mutants.
FIGURE 9.
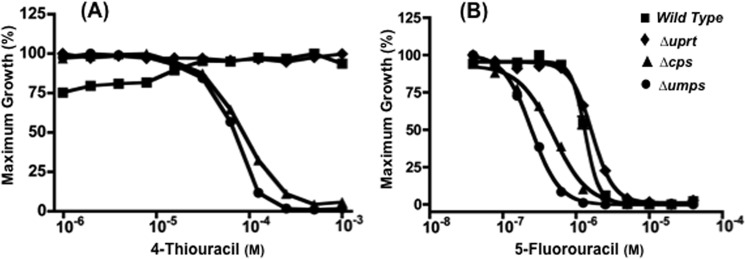
Sensitivity of wild type and null mutant L. donovani to 4-thiouracil and 5-fluorouracil. The abilities of wild type (■), Δuprt (♦), Δcps (▴), and Δumps (●) L. donovani promastigotes to grow in various concentrations of 4-thiouracil (A) or 5-fluorouracil (B) were assessed as described under “Experimental Procedures.” Growth data are presented from a single representative experiment, which has been repeated at least three times with similar results.
DISCUSSION
A genetic dissection of the pyrimidine pathway has established that LdUPRT is the sole enzyme capable of salvaging preformed pyrimidines to the nucleotide level in L. donovani and that all pyrimidine nucleosides that can satisfy the nutritional requirements of strains genetically auxotrophic for pyrimidines are ultimately converted to uracil prior to phosphoribosylation by LdUPRT (5). Introduction of a Δuprt lesion into wild type L. donovani obliterates pyrimidine salvage in both life cycle stages of the parasite and, when introduced into a Δcps line that lacks an intact biosynthetic pathway but is still capable of manifesting a robust infection, reduces parasite burdens in both liver and spleen to zero (5). To investigate this key pyrimidine salvage enzyme in more detail, recombinant LdUPRT was purified to homogeneity, and its kinetic parameters were determined (Figs. 2 and 4). LdUPRT displays a neutral pH optimum, requires a divalent cation for activity, and exhibits affinities toward its naturally occurring substrates that are equivalent to that previously reported for the T. gondii enzyme (18). LdUPRT also recognizes the uracil analogs 4-thiouracil and 5-fluorouracil with affinities similar to that of uracil (Fig. 4, C and D). Molecular sizing of LdUPRT implied that the L. donovani enzyme was a physiologically active tetramer (Fig. 5), similar to the T. gondii counterpart for which high resolution crystal structures were determined in a number of catalytic states (17, 21). In contrast to the T. gondii enzyme (17), the activity of LdUPRT was not augmented by GTP (Fig. 4B).
It has been previously observed that Δcps and Δumps L. donovani promastigotes exhibit a collateral supersensitivity to uracil, a nucleobase that is neither growth inhibitory nor cytotoxic toward wild type parasites (5). This effect of uracil on pyrimidine auxotrophic L. donovani is specific for the nucleobase and is growth inhibitory in nature rather than lethal (5). This substrate inhibition of LdUPRT by uracil was reversible and did not affect the thermostability of the enzyme (Fig. 8). The fact that two distinct genetic lesions in the pyrimidine pathway instigate this susceptibility to uracil substantiates that it is triggered by a deficit in pyrimidine biosynthesis capacity and not by some ancillary event in either of the null mutant lines. Analogous pyrimidine biosynthetic mutants of other protozoan parasites, including Δcps T. gondii (11), Δcps T. cruzi (12), and Δumps T. brucei (13), all display this predisposition to be growth inhibited by high concentrations of uracil in the culture medium. Heretofore, no mechanism has been established for this intriguing growth inhibition of the normally nondetrimental nucleobase toward pyrimidine biosynthesis mutants of protozoan parasites, although Ali et al. (13) conjectured that excessive uracil influx might bring about nucleotide pool imbalances in T. brucei.
The ability of orotate and dihydroorotate, which are both metabolic intermediates in the pyrimidine biosynthetic pathway (Fig. 1), to eliminate the uracil hypersensitivity of the Δcps line (Fig. 6, C and D) implied that the provision of an exogenous source of pyrimidine nucleotides was alleviating a pyrimidine starvation state that was being instigated by uracil. Cytidine, however, which is deaminated to uridine, cleaved to uracil, phosphoribosylated to UMP, and ultimately distributed into all other pyrimidines in the parasite, does not impact the uracil supersensitivity of the Δcps or Δumps strain (Fig. 6B). It should be noted that neither orotate nor dihydroorotate, which cannot be converted into UMP by L. donovani harboring a Δumps lesion (Fig. 1), alleviated the susceptibility of the Δumps strain to uracil (Fig. 6, C and D). Taken together, we conjectured that uracil at high concentrations was instigating an intracellular depletion of pyrimidine nucleotides by impairing salvage because the growth inhibition in the Δcps knock-out could be alleviated by de novo production of UMP but not by a precursor of UMP synthesis that is incorporated through LdUPRT. This hypothesis was tested directly by verifying that high concentrations of uracil (>10-fold Km concentrations) triggered substrate inhibition of LdUPRT that inhibited activity >90% at the highest uracil concentration tested, i.e., 1.5 mm (Fig. 7A). Purified TgUPRT was also inhibited by high uracil concentrations (Fig. 7B), intimating that substrate inhibition by uracil may be a common feature of protozoan UPRTs. Substrate inhibition of UPRT by uracil remains to be evaluated for the T. cruzi and T. brucei UPRT enzymes but is a plausible mechanism to account for the susceptibility of pyrimidine auxotrophs to uracil-instigated growth inhibition in those species. Both T. cruzi and T. brucei accommodate a UPRT gene within their respective genomes (2, 22, 23), and the UPRT enzyme has been detected in both species (24), although neither has been investigated in detail. The biological significance for LdUPRT substrate inhibition may pertain to PRPP sparing. PRPP is a critical substrate for purine salvage, an indispensable nutritional function for all protozoan parasites (1), and a process that is known to be mediated through two PRPP-dependent phosphoribosyltransferases: HGPRT and XPRT, in L. donovani (25). Thus, high concentrations of the nucleobase would prioritize PRPP for usage in purine salvage when pyrimidine pools are replete. Because L. donovani amastigotes within the phagolysosome are exposed to RNA degradation products that they can salvage, this substrate inhibition of LdUPRT by uracil can also ensure the maintenance of a balanced supply of pyrimidine and purine nucleotides for the parasite under conditions when pyrimidine pools are replete.
Similarly, 4-thiouracil, which has been used as a tag for evaluating gene expression and transcriptional profiling on a genome-wide level (26, 27), was not toxic to wild type L. donovani promastigotes, although the Δcps and Δumps null mutants were killed by high concentrations of the analog (Fig. 9A). The marked inhibition of LdUPRT activity by concentrations of 4-thiouracil that also induced parasite growth inhibition (Fig. 9A) implicates the disruption of pyrimidine salvage as the sole mechanism of growth disruption induced by 4-thiouracil treatment of the pyrimidine auxotrophs. This conclusion is supported by the observed greater sensitivity of LdUPRT to substrate inhibition by 4-thiouracil versus uracil (Fig. 7, A and C) that was reflected in the greater sensitivity of the Δcps and Δumps parasites to growth inhibition by 4-thiouracil compared with uracil (Figs. 6A and 8A).
Although LdUPRT was also predisposed to substrate inhibition by high levels of 5-fluorouracil (Fig. 7D), it is difficult to reconcile this incomplete substrate inhibition of the enzyme with the observed toxicity toward L. donovani promastigotes that was observed at concentrations of 5-fluorouracil 2–3 orders of magnitude lower than those that inhibit LdUPRT. Although the precise mechanism of 5-fluorouracil toxicity toward L. donovani promastigotes is unknown, the Δuprt cell line, which is capable of de novo pyrimidine synthesis, was just as susceptible to 5-fluorouracil as the wild type strain (Fig. 9B). Despite the demonstration herein that 5-fluorouracil is a substrate for LdUPRT, L. donovani promastigotes in which LdUPRT has been genetically deleted (Δuprt) are essentially as sensitive to 5-fluorouracil as wild type parasites (EC50 = 1.65 μm versus 1.33 μm, respectively; Fig. 9B), revealing another comparably efficient means for phosphoribosylating 5-fluorouracil. Collectively, these data intimate that 5-fluorouracil is phosphoribosylated via both LdUPRT and orotate phosphoribosyltransferase, a component of the UMPS bifunctional enzyme (Fig. 1), and biochemical evidence that purified recombinant UMPS is capable of phosphoribosylating 5-fluorouracil directly supports this contention (data not shown). Because it is not feasible to generate a conditionally lethal Δumps/Δuprt double knock-out caused by the lack of a pyrimidine salvage bypass mechanism (Fig. 1), it cannot be definitively determined using genetic approaches that UPRT and UMPS are the exclusive routes by which the fluorinated pyrimidine is salvaged. Collectively, these data intimate that the production of the 5-fluoro-UMP from 5-fluorouracil by LdUPRT and/or UMPS is sufficient to account for the observed toxicity of the fluorinated pyrimidine in pyrimidine prototrophs. The enhanced 5-fluorouracil toxicity observed in pyrimidine auxotrophs is likely more complicated but may result from a combination of factors including competition between 5-fluorouracil and uracil for phosphoribosylation by LdUPRT, competition between UMP and 5-fluoro-UMP for further metabolism by downstream enzymes, and/or more efficient incorporation of 5-fluorouracil into the nucleotide pool and RNA in the absence of de novo UMP production. This is consistent with the model proposed by Ali et al. (13) to explain the hypersensitivity of T. brucei pyrimidine auxotrophs to 5-fluorouracil.
In summary, we have performed a biochemical and kinetic characterization of LdUPRT, the sole enzyme capable of incorporating preformed host pyrimidines into the parasite nucleotide pool. The kinetic characterization of LdUPRT revealed a remarkable inhibition of the enzyme by its uracil substrate that can account for the unique susceptibility of L. donovani harboring genetic lesions in the pyrimidine biosynthetic pathway to the nucleobase. This substrate inhibition of UPRT enzymes appears to be a general mechanism by which purine auxotrophs can equilibrate purine and pyrimidine nucleotide pools and offers a means by which purine incorporation, an essential nutritional function, is ceded preference over pyrimidine salvage, a nonessential process, when the parasites have access to nucleotide precursors. Finally, because mammalian cells lack an analogous UPRT enzyme (28), the capacity of LdUPRT to recognize cytotoxic nucleobase analogs offers a potential therapeutic strategy by which cytotoxic uracil derivatives could be selectively incorporated into the parasite nucleotide pool without impacting the human host. 5-Fluorouracil and 4-thiouracil, however, are not prospective candidates as pro-drugs for which LdUPRT activation is required because 5-fluorouracil, an anti-neoplastic agent employed pervasively in the treatment of gastrointestinal cancers (29), exhibits unacceptable toxicity toward mammalian cells (30), whereas 4-thiouracil is nontoxic toward wild type L. donovani (Fig. 9A). A structure-activity study of uracil analogs has been previously carried out with a partially purified TgUPRT preparation (31), but no similar study has been performed with LdUPRT. Extrapolating the results of such a structure-activity analysis performed on LdUPRT to intact Leishmania parasites, however, is complicated somewhat by the existence of another pyrimidine phosphoribosyltransferase activity, i.e., the orotate phosphoribosyltransferase, the penultimate enzyme in de novo pyrimidine biosynthesis that can also presumably recognize pyrimidine analogs (3). Indeed, 5-fluorouracil is a known substrate for the mammalian orotate phosphoribosyltransferase enzyme (32). Regardless, the absence of a mammalian equivalent raises the possibility of exploiting LdUPRT as a selective mechanism for activating potential antileishmanial pyrimidine nucleobase analogs.
This work was supported, in whole or in part, by National Institutes of Health Grant AI023682 (to B. U.).
- PRPP
- 5-phosphoribosyl-1-pyrophosphate
- UMPS
- UMP synthase
- CPS
- carbamoyl phosphate synthetase
- LdUPRT
- L. donovani uracil phosphoribosyltransferase
- IPTG
- isopropyl β-d-1-thiogalactopyranoside
- TgUPRT
- T. gondii uracil phosphoribosyltransferase
- UPRT
- uracil phosphoribosyltransferase
- Ni-NTA
- nickel-nitrilotriacetic acid
- CHES
- 2-(cyclohexylamino)ethanesulfonic acid
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine.
REFERENCES
- 1. Carter N., Rager N., Ullman B. (2003) Purine and pyrimidine transport and metabolism, in Molecular and Medical Parasitology (Marr J. J., Komuniecki R., eds) pp. 197–223, Academic Press Limited, London [Google Scholar]
- 2. El-Sayed N. M., Myler P. J., Blandin G., Berriman M., Crabtree J., Aggarwal G., Caler E., Renauld H., Worthey E. A., Hertz-Fowler C., Ghedin E., Peacock C., Bartholomeu D. C., Haas B. J., Tran A. N., Wortman J. R., Alsmark U. C., Angiuoli S., Anupama A., Badger J., Bringaud F., Cadag E., Carlton J. M., Cerqueira G. C., Creasy T., Delcher A. L., Djikeng A., Embley T. M., Hauser C., Ivens A. C., Kummerfeld S. K., Pereira-Leal J. B., Nilsson D., Peterson J., Salzberg S. L., Shallom J., Silva J. C., Sundaram J., Westenberger S., White O., Melville S. E., Donelson J. E., Andersson B., Stuart K. D., Hall N. (2005) Comparative genomics of trypanosomatid parasitic protozoa. Science 309, 404–409 [DOI] [PubMed] [Google Scholar]
- 3. French J. B., Yates P. A., Soysa D. R., Boitz J. M., Carter N. S., Chang B., Ullman B., Ealick S. E. (2011) The Leishmania donovani UMP synthase is essential for promastigote viability and has an unusual tetrameric structure that exhibits substrate-controlled oligomerization. J. Biol. Chem. 286, 20930–20941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ivens A. C., Peacock C. S., Worthey E. A., Murphy L., Aggarwal G., Berriman M., Sisk E., Rajandream M. A., Adlem E., Aert R., Anupama A., Apostolou Z., Attipoe P., Bason N., Bauser C., Beck A., Beverley S. M., Bianchettin G., Borzym K., Bothe G., Bruschi C. V., Collins M., Cadag E., Ciarloni L., Clayton C., Coulson R. M., Cronin A., Cruz A. K., Davies R. M., De Gaudenzi J., Dobson D. E., Duesterhoeft A., Fazelina G., Fosker N., Frasch A. C., Fraser A., Fuchs M., Gabel C., Goble A., Goffeau A., Harris D., Hertz-Fowler C., Hilbert H., Horn D., Huang Y., Klages S., Knights A., Kube M., Larke N., Litvin L., Lord A., Louie T., Marra M., Masuy D., Matthews K., Michaeli S., Mottram J. C., Muller-Auer S., Munden H., Nelson S., Norbertczak H., Oliver K., O'Neil S., Pentony M., Pohl T. M., Price C., Purnelle B., Quail M. A., Rabbinowitsch E., Reinhardt R., Rieger M., Rinta J., Robben J., Robertson L., Ruiz J. C., Rutter S., Saunders D., Schafer M., Schein J., Schwartz D. C., Seeger K., Seyler A., Sharp S., Shin H., Sivam D., Squares R., Squares S., Tosato V., Vogt C., Volckaert G., Wambutt R., Warren T., Wedler H., Woodward J., Zhou S., Zimmermann W., Smith D. F., Blackwell J. M., Stuart K. D., Barrell B., Myler P. J. (2005) The genome of the kinetoplastid parasite, Leishmania major. Science 309, 436–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wilson Z. N., Gilroy C. A., Boitz J. M., Ullman B., Yates P. A. (2012) Genetic dissection of pyrimidine biosynthesis and salvage in Leishmania donovani. J. Biol. Chem. 287, 12759–12770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Stark G. R., Wahl G. M. (1984) Gene amplification. Annu. Rev. Biochem. 53, 447–491 [DOI] [PubMed] [Google Scholar]
- 7. Wahl G. M., Padgett R. A., Stark G. R. (1979) Gene amplification causes overproduction of the first three enzymes of UMP synthesis in N-(phosphonacetyl)-l-aspartate-resistant hamster cells. J. Biol. Chem. 254, 8679–8689 [PubMed] [Google Scholar]
- 8. Suttle D. P., Stark G. R. (1979) Coordinate overproduction of orotate phosphoribosyltransferase and orotidine-5′-phosphate decarboxylase in hamster cells resistant to pyrazofurin and 6-azauridine. J. Biol. Chem. 254, 4602–4607 [PubMed] [Google Scholar]
- 9. Opperdoes F. R. (1987) Compartmentation of carbohydrate metabolism in trypanosomes. Annu. Rev. Microbiol. 41, 127–151 [DOI] [PubMed] [Google Scholar]
- 10. Opperdoes F. R. (1988) Glycosomes may provide clues to the import of peroxisomal proteins. Trends. Biochem. Sci. 13, 255–260 [DOI] [PubMed] [Google Scholar]
- 11. Fox B. A., Bzik D. J. (2002) De novo pyrimidine biosynthesis is required for virulence of Toxoplasma gondii. Nature 415, 926–929 [DOI] [PubMed] [Google Scholar]
- 12. Hashimoto M., Morales J., Fukai Y., Suzuki S., Takamiya S., Tsubouchi A., Inoue S., Inoue M., Kita K., Harada S., Tanaka A., Aoki T., Nara T. (2012) Critical importance of the de novo pyrimidine biosynthesis pathway for Trypanosoma cruzi growth in the mammalian host cell cytoplasm. Biochem. Biophys. Res. Commun. 417, 1002–1006 [DOI] [PubMed] [Google Scholar]
- 13. Ali J. A., Tagoe D. N., Munday J. C., Donachie A., Morrison L. J., de Koning H. P. (2013) Pyrimidine biosynthesis is not an essential function for Trypanosoma brucei bloodstream forms. PLoS One 8, e58034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Arakaki T. L., Buckner F. S., Gillespie J. R., Malmquist N. A., Phillips M. A., Kalyuzhniy O., Luft J. R., Detitta G. T., Verlinde C. L., Van Voorhis W. C., Hol W. G., Merritt E. A. (2008) Characterization of Trypanosoma brucei dihydroorotate dehydrogenase as a possible drug target. Structural, kinetic and RNAi studies. Mol. Microbiol. 68, 37–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hassan H. F., Coombs G. H. (1986) A comparative study of the purine- and pyrimidine-metabolising enzymes of a range of trypanosomatids. Comp. Biochem. Physiol. B. 84, 219–223 [PubMed] [Google Scholar]
- 16. Shi W., Schramm V. L., Almo S. C. (1999) Nucleoside hydrolase from Leishmania major. Cloning, expression, catalytic properties, transition state inhibitors, and the 2.5-A crystal structure. J. Biol. Chem. 274, 21114–21120 [DOI] [PubMed] [Google Scholar]
- 17. Schumacher M. A., Bashor C. J., Song M. H., Otsu K., Zhu S., Parry R. J., Ullman B., Brennan R. G. (2002) The structural mechanism of GTP stabilized oligomerization and catalytic activation of the Toxoplasma gondii uracil phosphoribosyltransferase. Proc. Natl. Acad. Sci. U.S.A. 99, 78–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Carter D., Donald R. G., Roos D., Ullman B. (1997) Expression, purification, and characterization of uracil phosphoribosyltransferase from Toxoplasma gondii. Mol. Biochem. Parasitol. 87, 137–144 [DOI] [PubMed] [Google Scholar]
- 19. Natalini P., Ruggieri S., Santarelli I., Vita A., Magni G. (1979) Baker's yeast UMP:pyrophosphate phosphoribosyltransferase. Purification, enzymatic and kinetic properties. J. Biol. Chem. 254, 1558–1563 [PubMed] [Google Scholar]
- 20. Mikus J., Steverding D. (2000) A simple colorimetric method to screen drug cytotoxicity against Leishmania using the dye Alamar Blue. Parasitol. Int. 48, 265–269 [DOI] [PubMed] [Google Scholar]
- 21. Schumacher M. A., Carter D., Scott D. M., Roos D. S., Ullman B., Brennan R. G. (1998) Crystal structures of Toxoplasma gondii uracil phosphoribosyltransferase reveal the atomic basis of pyrimidine discrimination and prodrug binding. EMBO J. 17, 3219–3232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Berriman M., Ghedin E., Hertz-Fowler C., Blandin G., Renauld H., Bartholomeu D. C., Lennard N. J., Caler E., Hamlin N. E., Haas B., Böhme U., Hannick L., Aslett M. A., Shallom J., Marcello L., Hou L., Wickstead B., Alsmark U. C., Arrowsmith C., Atkin R. J., Barron A. J., Bringaud F., Brooks K., Carrington M., Cherevach I., Chillingworth T. J., Churcher C., Clark L. N., Corton C. H., Cronin A., Davies R. M., Doggett J., Djikeng A., Feldblyum T., Field M. C., Fraser A., Goodhead I., Hance Z., Harper D., Harris B. R., Hauser H., Hostetler J., Ivens A., Jagels K., Johnson D., Johnson J., Jones K., Kerhornou A. X., Koo H., Larke N., Landfear S., Larkin C., Leech V., Line A., Lord A., Macleod A., Mooney P. J., Moule S., Martin D. M., Morgan G. W., Mungall K., Norbertczak H., Ormond D., Pai G., Peacock C. S., Peterson J., Quail M. A., Rabbinowitsch E., Rajandream M. A., Reitter C., Salzberg S. L., Sanders M., Schobel S., Sharp S., Simmonds M., Simpson A. J., Tallon L., Turner C. M., Tait A., Tivey A. R., Van Aken S., Walker D., Wanless D., Wang S., White B., White O., Whitehead S., Woodward J., Wortman J., Adams M. D., Embley T. M., Gull K., Ullu E., Barry J. D., Fairlamb A. H., Opperdoes F., Barrell B. G., Donelson J. E., Hall N., Fraser C. M., Melville S. E., El-Sayed N. M. (2005) The genome of the African trypanosome Trypanosoma brucei. Science 309, 416–422 [DOI] [PubMed] [Google Scholar]
- 23. El-Sayed N. M., Myler P. J., Bartholomeu D. C., Nilsson D., Aggarwal G., Tran A. N., Ghedin E., Worthey E. A., Delcher A. L., Blandin G., Westenberger S. J., Caler E., Cerqueira G. C., Branche C., Haas B., Anupama A., Arner E., Aslund L., Attipoe P., Bontempi E., Bringaud F., Burton P., Cadag E., Campbell D. A., Carrington M., Crabtree J., Darban H., da Silveira J. F., de Jong P., Edwards K., Englund P. T., Fazelina G., Feldblyum T., Ferella M., Frasch A. C., Gull K., Horn D., Hou L., Huang Y., Kindlund E., Klingbeil M., Kluge S., Koo H., Lacerda D., Levin M. J., Lorenzi H., Louie T., Machado C. R., McCulloch R., McKenna A., Mizuno Y., Mottram J. C., Nelson S., Ochaya S., Osoegawa K., Pai G., Parsons M., Pentony M., Pettersson U., Pop M., Ramirez J. L., Rinta J., Robertson L., Salzberg S. L., Sanchez D. O., Seyler A., Sharma R., Shetty J., Simpson A. J., Sisk E., Tammi M. T., Tarleton R., Teixeira S., Van Aken S., Vogt C., Ward P. N., Wickstead B., Wortman J., White O., Fraser C. M., Stuart K. D., Andersson B. (2005) The genome sequence of Trypanosoma cruzi, etiologic agent of Chagas disease. Science 309, 409–415 [DOI] [PubMed] [Google Scholar]
- 24. Hammond D. J., Gutteridge W. E. (1982) UMP synthesis in the kinetoplastida. Biochim. Biophys. Acta 718, 1–10 [DOI] [PubMed] [Google Scholar]
- 25. Boitz J. M., Ullman B. (2006) A conditional mutant deficient in hypoxanthine-guanine phosphoribosyltransferase and xanthine phosphoribosyltransferase validates the purine salvage pathway of Leishmania donovani. J. Biol. Chem. 281, 16084–16089 [DOI] [PubMed] [Google Scholar]
- 26. Cleary M. D., Meiering C. D., Jan E., Guymon R., Boothroyd J. C. (2005) Biosynthetic labeling of RNA with uracil phosphoribosyltransferase allows cell-specific microarray analysis of mRNA synthesis and decay. Nat. Biotechnol. 23, 232–237 [DOI] [PubMed] [Google Scholar]
- 27. Zeiner G. M., Cleary M. D., Fouts A. E., Meiring C. D., Mocarski E. S., Boothroyd J. C. (2008) RNA analysis by biosynthetic tagging using 4-thiouracil and uracil phosphoribosyltransferase. Methods Mol. Biol. 419, 135–146 [DOI] [PubMed] [Google Scholar]
- 28. Pfefferkorn E. R., Pfefferkorn L. C. (1977) Specific labeling of intracellular Toxoplasma gondii with uracil. J. Protozool. 24, 449–453 [DOI] [PubMed] [Google Scholar]
- 29. Longley D. B., Harkin D. P., Johnston P. G. (2003) 5-fluorouracil: mechanisms of action and clinical strategies. Nat. Rev. Cancer 3, 330–338 [DOI] [PubMed] [Google Scholar]
- 30. Shoemaker L. K., Arora U., Rocha Lima C. M. (2004) 5-fluorouracil-induced coronary vasospasm. Cancer Control 11, 46–49 [DOI] [PubMed] [Google Scholar]
- 31. Iltzsch M. H., Tankersley K. O. (1994) Structure-activity relationship of ligands of uracil phosphoribosyltransferase from Toxoplasma gondii. Biochem. Pharmacol. 48, 781–792 [DOI] [PubMed] [Google Scholar]
- 32. Peters G. J., Laurensse E., Leyva A., Lankelma J., Pinedo H. M. (1986) Sensitivity of human, murine, and rat cells to 5-fluorouracil and 5′-deoxy-5-fluorouridine in relation to drug-metabolizing enzymes. Cancer Res. 46, 20–28 [PubMed] [Google Scholar]