

Background: SDH5 is in tumors. However, the functional role of SDH5 in lung cancer remains unknown.
Results: SDH5 expression regulates EMT.
Conclusion: SDH5 functions as a critical protein in the regulation of EMT by modulating the GSK-3β-β-catenin signaling pathway.
Significance: SDH5 may be a prognostic biomarker and potential therapeutic target for lung cancer metastasis.
Keywords: Biomarkers, E-cadherin, EMT, Lung Cancer, Metastasis, Mitochondrial Protein, SDH5
Abstract
We demonstrate that loss of succinate dehydrogenase 5 (SDH5) expression initiates epithelial-mesenchymal transition (EMT), which is visualized by the repression of E-cadherin and up-regulation of vimentin in lung cancer cell lines and clinical lung cancer specimens. In SDH5 knock-out mice, lung epithelial cells exhibited elevated mesenchymal markers, which is characteristic of EMT. Using a human lung xenograft-mouse model, we observed that knocking down endogenous SDH5 in human carcinoma cells leads to the development of multiple lymph node metastases. Moreover, our data indicate that SDH5 functions as a critical protein in regulating EMT by modulating the glycogen synthase kinase (GSK)-3β-β-catenin signaling pathway. These results reveal a critical role for SDH5 in EMT and suggest that SDH5 may be a prognostic biomarker and potential therapeutic target for lung cancer metastasis.
Introduction
Lung cancer is one of the most common cancers in the world (1, 2), and it is a leading cause of cancer death in men and women in China. Lung cancer is generally a treatable disease in the absence of metastasis (3). Thus, early diagnosis of patients who develop lung cancer metastasis could reduce the mortality and morbidity associated with this disease (3, 4). The development of metastasis depends on the invasion and migration of cancer cells from the primary tumor into the surrounding tissues. To acquire such invasive abilities, carcinoma cells may inherit unique phenotypic changes, such as epithelial-mesenchymal transition (EMT)2 (5). EMT is a process characterized by a loss of cell adhesion, repression of E-cadherin expression, and increased cell motility. EMT is a highly conserved cellular process that allows polarized, generally immotile epithelial cells to convert to motile, mesenchymal-like cells. This process was first recognized as a feature of embryogenesis, which is vital for morphogenesis during embryonic development, and more recently, it has been implicated in promoting carcinoma invasion and metastasis. During EMT, three major changes occur: (i) morphological changes from a cobblestone-like monolayer of epithelial cells to dispersed, spindle-shaped mesenchymal cells with migratory protrusions; (ii) changes in differentiation markers from cell-cell junction proteins and cytokeratin intermediate filaments to vimentin filaments and fibronectin; and (iii) acquisition of invasiveness through the extracellular matrix (6, 7).
Decreased E-cadherin expression or gain of vimentin expression is closely correlated with various indices of lung cancer progression, including the grade, local invasiveness, dissemination into blood, and tumor relapse after radiotherapy (8, 9).
Succinate dehydrogenase 5 (SDH5), originally named EMI5/YOL07, is required for the flavination of succinate dehydrogenase. SDH5 is associated with increased risk for the development of several kinds of cancer, and it is mutated in paraganglioma and gastrointestinal stromal tumors (10). SDH5 is down-regulated in various types of human cancers, and it plays an important role in tumor development (11–13).
Thus, SDH5 likely functions as a tumor suppressor in cancer development; however, its role and mechanism in lung cancer metastasis are largely unknown. Here, we demonstrate that loss of SDH5 facilitates EMT, leading to lung cancer metastasis.
EXPERIMENTAL PROCEDURES
Cell Culture and Clinical Specimens
Lung cancer cell lines, including NCI-H23, NCI-H1299, CRL-5908, NCI-H1975, CaLu-3, A549, Slu-02, PG49, and HTB-55 cells, were obtained from ATCC and maintained in Dulbecco's modified Eagle's medium (DMEM; Invitrogen) containing 10% fetal bovine serum (FBS; Invitrogen).
The Institutional Review Board of China approved the retrieval of cancer specimens and the connection with clinical data from our institute (approval ID 8435672). Cell lysates were subjected to Western blot analysis or immunohistochemical staining.
In Vitro Migration Assay
For migration assays, 5 × 104 cells were plated in the top chamber of a Transwell insert (24-well insert, pore size, 8 mm; Corning), and serum-containing medium was placed in the lower chamber. After incubation for 48 h, cells that did not migrate or invade through the pores were removed with a cotton swab. The cells on the lower surface of the membrane were stained with Cell Stain (Chemicon, Tokyo, Japan) and quantified by measuring absorbance at 560.
Analysis of the Wnt Signaling Pathway
Cells were treated with WNT- or control-conditioned medium (Wnt-CM (ATCC number CRL-2647) and L-CM, respectively) for 24 h, and Wnt signaling was monitored by various assays, including Western blotting, GSK-3β kinase assays (Boshida, Wuhan, China), luciferase reporter gene assays (Chemicon), and fluorescence confocal microscopy (Sigma).
Orthotopic Animal Model and Imaging
All experimental procedures were approved by the Institutional Animal Care and Use Committee of China. The lungs of male nude mice (6–8 weeks of age) were exposed and injected with 5 × 105 cells suspended in 20 μl of phosphate-buffered saline (PBS). One week after injection, surgical staples were removed, and the tumor growth and local metastasis were monitored by bioluminescent imaging (BLI; Xenogen).
Plasmid Constructs, Conditioned Medium, and Antibodies
Plasmids for SDH5 and PP2A were obtained from Sigma. For cDNA transfection, cells (5 × 105 cells/well) were seeded in a 6-well plate (Costar) at 70–80% confluence before transfection. Transfection was carried out using Lipofectamine PLUS (Invitrogen), according to the manufacturer's instructions. Wnt-CM and L-CM) were collected according to the directions from ATCC, and they were added to the cells for 24 h. The anti-SDH5 polyclonal antibody was obtained from Biocompare. Okadaic acid (OA), anti-GSK-3β, anti-phospho-GSK-3β (Ser-9), anti-actin, anti-E-cadherin, anti-β-catenin, anti-ZEB1, and anti-vimentin were obtained from Sigma. Anti-human-specific pan-cytokeratin was purchased from Abcam. Anti-Snail, anti-Twist, and anti-TGF were obtained from Invitrogen.
siRNA Oligonucleotides and Delivery Methods
Three pairs of siRNA oligonucleotides for human SDH5 and PP2A were obtained from Invitrogen. siRNA oligonucleotides (20 μm) were transfected into cells by using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol.
Immunoprecipitation and Western Blot Analysis
For immunoprecipitation, transfected Slu-02 cells were washed twice with cold PBS and rinsed in 1.5 ml of cold lysis buffer (50 mm Tris-HCl (pH 7.5), 150 mm NaCl, 0.1% Triton X-100, 1 mm sodium orthovanadate, 1 mm sodium fluoride, 1 mm sodium pyrophosphate, 10 mg/ml aprotinin, 10 mg/ml leupeptin, 2 mm phenylmethylsulfonyl fluoride, and 1 mm EDTA) for 20 min on ice. The immunocomplexes were subjected to Western blot analysis according to the manufacturer's protocol.
GSK-3β Kinase Assay
A fluorescence peptide substrate-based assay was used to assess GSK-3β kinase activity (Omnia Ser/Thr Recombinant kit; Invitrogen). Briefly, the GSK-3β complex was prepared from equal amounts of cell lysates by immunoprecipitation, and it was incubated with 10 μm of Ser/Thr peptide substrate in kinase reaction buffer (containing 1 mm ATP and 1 mm DTT) for 20 min at 30 °C. Fluorescence intensity was recorded by measuring the A485 in a 96-well plate. Relative GSK-3β activity was calculated using untreated cells (equal to 1).
Luciferase Reporter Gene Assay
For the reporter gene assay, cells seeded in 24-well plates were transfected with β-catenin, firefly luciferase reporter gene construct (TOP or FOP; 200 ng), and 1 ng of pRL-SV40 Renilla luciferase (as an internal control). Cell extracts were prepared 24 h after transfection, and luciferase activity was measured using the Dual-Luciferase Reporter Assay System (Promega).
Fluorescence Confocal Microscopy
NCI-H23 cells were transfected with control or SDH5 siRNA for 72 h. Then, the cells were fixed in 4% formaldehyde and subjected to indirect immunofluorescence microscopy with anti-β-catenin. The fluorescein isothiocyanate (FITC)-conjugated anti-IgG was purchased from Molecular Probes. Confocal immunofluorescence microscopy (Olympus) was performed using an Olympus confocal microscope, according to the manufacturer's protocol (×40 magnification).
BLI
BLI was performed using an IVIS Imaging System (Xenogen). Images and bioluminescent signals were acquired and analyzed using Living Image and Xenogen software. Briefly, three mice were anesthetized, injected with d-luciferin (150 mg/kg, intraperitoneally), and imaged 10 min after injection for 3 min.
Histology and Immunohistochemical Staining
Tumors were removed, weighed, fixed in 5% formalin, and prepared for histological analysis. Consecutive tumor sections were stained with H&E, SDH5, E-cadherin, vimentin, and β-catenin. Immunohistochemical staining was carried out using the ABC staining kit (Santa Cruz Biotechnology) and a secondary biotinylated antibody to mouse IgG (Invitrogen). Lung cancer patient tissues were washed with PBS, inflated, and fixed with 10% buffered formalin. The sections were paraffin-embedded, cut into 5-μm sections, and stained with routine H&E.
Targeted Inactivation of the SDH5 Gene by Homologous Recombination
We recently reported the KO strategy (9). The linearized targeting construct was electroporated into 129/C57BL/6 ES cells, and the targeted clones were selected with G418 and ganciclovir. Resistant clones were screened for homologous recombination by PCR and confirmed by Southern blot analysis. Two independent AIP1+/lox ES clones were injected into WT blastocysts. Chimeras were further bred with WT females for germ line transmission. SDH5+/lox mice were mated with β-actin Cre mice to generate SDH5+/− mice with a deletion of both the targeting region and the Neo gene. KO mice were obtained by mating the heterozygous (SDH5+/−) male and female.
Genotyping SDH5 Knock-out Mice
Genotyping was performed by PCR with SDH5WT and SDH5KO primers in the same reaction system. A 4.5- and 5.0-kb fragment were amplified separately in SDH5 (wild type) and SDH5 mice (homozygous), respectively, and both fragments were amplified in the SDH5 mice (heterozygous). Opg expression was detected via routine RT-PCR and Western blotting analyses.
Statistical Analysis
All error bars in graphical data represent the mean ± S.E. Student's two-tailed t test was used to determine the statistical relevance between groups, and p < 0.05 was considered significant.
RESULTS
SDH5 Regulates EMT in Vitro
We detected SDH5 expression in the following lung cancer cell lines: EKVX, HOP-62, and NCI-H23 cells express high levels of SDH5; Slu-01 expresses low levels of SDH5; and Slu-02, NCI-H226, NCI-H460, and NCI-H522 do not express SDH5 (Fig. 1A). We also detected E-cadherin, vimentin, and ZEB1 expression in these lung cancer cell lines: NCI-H23 and Slu-01 express high levels of vimentin and ZEB1, but low levels of E-cadherin; Slu-02 and NCI-H226 displayed the opposite expression pattern (Fig. 1B). We also demonstrated that SDH5 does not contribute to cell proliferation in vitro and that there is no statistical significance between Slu-02 and NCL-H226 versus NCL-H23 or Slu-01 cells (Fig. 1C).
FIGURE 1.

SDH5 regulates EMT in lung cancer cells. A, SDH5 expression was detected in lung cancer cell lines. EKVX, HOP-62, and NCI-H23 express high levels of SDH5; Slu-01 expresses low levels of SDH5; and Slu-02, NCI-H226, NCI-H460, and NCI-H522 do not express SDH5. B, immunofluorescence staining of E-cadherin and vimentin is shown in Slu-02, NCI-H226, NCI-H23, and Slu-01 cell lines with DAPI counterstaining (magnification, ×100). C, cells were seeded in serum-free medium for 48 h, and the relative cell number was determined by 3-(4, 5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. There is no statistical significance between Slu-02 and NCI-H226 cells versus NCI-H23 or Slu-01 cells (p > 0.05) at different time points. D, elevated SDH5 levels reverse EMT in vitro. The morphology of Slu-02 and NCI-H226 cells transfected with either the control vector or SDH5 was observed by phase-contrast microscopy (magnification, ×100). E, expression of epithelial or mesenchymal markers in Slu-02 and NCI-H226 cells transfected with either the control vector or SDH5 was analyzed by Western blotting. β-Actin was used as a loading control. F, knockdown of SDH5 initiates EMT in vitro. NCI-H23 and Slu-01 cells were infected with control lentivirus or lentivirus-expressing shRNA specific to SDH5, and the expressing cells were selected with puromycin. Morphology was revealed by phase-contrast microscopy (magnification, ×100). G, the expression of mesenchymal and epithelial markers in SDH5-knockdown cells was analyzed by Western blotting (p < 0.01). H, effect of SDH5 on cell migration in vitro is shown. Slu-02 and NCI-H226 transfected with either the control vector or SDH5 and NCI-H23 and Slu-01 infected with control lentivirus or lentivirus-expressing shRNA specific to SDH5 were plated in Transwell chambers for 48 h, and quantitative measurements of migrating cells were determined. Data are presented as the mean ± S.E. (error bars; p < 0.01). Each experiment was performed twice, with triplicates for each assay.
In addition, we established that SDH5 could regulate EMT. As was reported previously, cells undergoing an EMT or mesenchymal-epithelial transition (MET) display transient morphologic and biologic changes that modify cell polarity, contact with neighboring cells, and cell motility. These phenotypic changes are reminiscent of those observed in Slu-02 and NCI-H226 cells, which, when transfected with SDH5, displayed a clear morphological transition from spindle-like fibroblastic (control) to cobblestone-like cells (transfected with SDH5) with well organized cell contact and polarity (Fig. 1D). The effect of SDH5 expression on increasing E-cadherin and reducing vimentin expression was observed in Slu-02 and NCI-H226 (Fig. 1E). In contrast, when endogenous SDH5 expression was knocked down in two different human lung cancer cells (NCI-H23 and Slu-01), EMT clearly occurred, based on changes in cell morphology and biomarker expression (Fig. 1, F and G). Moreover, the expression of SDH5 significantly impacted cell motility in vitro (Fig. 1H). Taken together, these data indicate that SDH5 is a potent inhibitor of EMT.
SDH5 Prevents β-Catenin Nuclear Translocation, and β-Catenin Overexpression Reverses SDH5-mediated MET
To understand the possible mechanism by which SDH5 inhibits EMT, we examined the effect of SDH5 on the GSK-3β-β-catenin signaling pathway. In the canonical Wnt pathway, GSK-3β-mediated β-catenin degradation is inhibited, leading to the accumulation of β-catenin in the nucleus and the transactivation of β-catenin/T cell factor (TCF) target genes (14). Thus, the hallmark of β-catenin signaling in both normal and neoplastic tissues is nuclear translocation. By knocking down endogenous SDH5 with siRNA, we observed accumulation of cytoplasmic β-catenin, nuclear translocation of β-catenin, and reduced membrane-associated β-catenin (Fig. 2A).
FIGURE 2.

SDH5 inactivates β-catenin, and β-catenin overexpression reverses SDH5-mediated MET. A, SDH5 prevents β-catenin nuclear translocation. NCI-H23 cells were transfected with control or SDH5-siRNA. The subcellular localization of β-catenin was visualized by confocal microscopy (magnification, ×500) and Western blotting. B, total β-catenin mRNA was measured by RT-PCR. C, SDH5 inhibits β-catenin/TCF transcriptional activity. NCI-H23 cells treated with control- or SDH5-siRNA, and Slu-02 and NCI-H226 cells treated with control and SDH5 were transfected with the TCF-responsive promoter reporter (TOP-flash) or nonresponsive control reporter (FOP-flash); then, luciferase activity was measured as the ratio of TOP and FOP. Relative luciferase activity is presented as the mean ± S.E. (error bars) from each sample after normalizing to the control (= 1). The asterisk indicates statistical significance (p < 0.01). D and E, β-catenin overexpression reverses SDH5-mediated MET. Increasing amounts of β-catenin were transfected in Slu-02 (D) or NCI-H23 (E) cells for 24 h. Total cell lysates were probed with antibodies against E-cadherin, vimentin, and β-catenin. β-Catenin/TCF transcription activity was measured as described previously. F and G, the morphology of Slu-02 (F) and NCI-H23 (G) cells was observed by phase-contrast microscopy (magnification, ×100).
Total β-catenin mRNA did not change after siRNA knockdown of SDH5 in NCI-H23 cells (Fig. 2B), but β-catenin/TCF transcriptional activity (TOP/FOP) increased (Fig. 2C). Consistently, overexpression of SDH5 in Slu-02 and NCI-H226 cell lines decreased β-catenin/TCF transcriptional activity (TOP/FOP) (Fig. 2C).
Because SDH5 can activate GSK-3β, which decreases cytosolic β-catenin protein levels and nuclear β-catenin transcriptional activity (Fig. 2, A and C), we examined whether the inhibitory effect of SDH5 could be reversed by overexpressing β-catenin. In SDH5-transfected cells, increasing amounts of β-catenin cDNA restored EMT, as determined by marker expression, and increased β-catenin transcriptional activity and morphology (Fig. 2, D and F). Similarly, elevated expression of β-catenin protein levels and nuclear β-catenin transcriptional activity in NCI-H23-KD cells induced EMT in a dose-dependent manner (Fig. 2E). The morphology of these cells also changed (Fig. 2G).
SDH5 Regulates PP2A to Active GSK-3β
After Slu-02 cells were transfected with SDH5, GSK-3β appeared to directly associate with SDH5 based on immunoprecipitation experiments (Fig. 3A). Moreover, GSK-3β activity was significantly elevated, as determined by Ser-9 (negative regulatory site) phosphorylation levels and a decrease in β-catenin/TCF transcriptional activity assays (TOP/FOP) (Fig. 3, B and C). Because SDH5 is not a phosphatase, the mechanism of GSK-3β activation by SDH5 may be mediated by a separate phosphatase associated with this complex. PP2A is a heterotrimeric complex containing a catalytic subunit, a structural subunit, and a variable regulatory subunit. PP2A has been shown to regulate GSK-3β phosphorylation. In our study, co-immunoprecipitation data (Fig. 3D) indicated that SDH5 could form a complex with GSK-3β and PP2A.
FIGURE 3.

SDH5 is associated with PP2A and modulates the GSK-3β-β-catenin signaling pathway. A, Slu-02 cells were transfected with the control vector (VC) or SDH5. GSK-3β was immunoprecipitated from cell lysates, and the membranes were probed with antibodies against SDH5 or GSK-3β. B, SDH5 inhibits GSK-3β phosphorylation at Ser-9. Slu-02 cells were treated with the control and SDH5, and cell lysates were blotted with p-GSK-3β (Ser-9) and GSK-3β antibodies. β-Actin was used as a loading control. C, SDH5 activates GSK-3β kinase activity. Kinase activities were determined as described under “Experimental Procedures.” Relative GSK-3β kinase activities are shown as the mean ± S.E. (error bars) of each sample after normalizing to untreated Slu-02 cells. D, PP2A is critical for SDH5-mediated regulation of GSK-3β-β-catenin signaling and EMT. SDH5 complexes are associated with both GSK3β and PP2A. Slu-02 cells were transfected with the control vector or SDH5. Cell lysates were immunoprecipitated (IP) with GSK-3β, PP2A, and SDH5 antibodies and probed with FLAG and PP2A or GSK3β antibodies, respectively. E, NCI-H23 cells were co-transfected with PP2A-siRNA or treated with OA, and GSK-3β activity was determined. Both OA and PP2A-siRNA inactivate GSK-3β kinase activity (p < 0.01). F, PP2A plays a role in SDH5-mediated regulation of EMT and Ser-9 phosphorylation of GSK-3β. Slu-02 cells transfected with either VC or SDH5 were treated with OA (25 nm, 24 h) or co-transfected with PP2A-specific siRNA (100 pmol, 24 h). Cell lysates were subjected to Western blotting.
To further assess the direct effect of PP2A on GSK-3β-β-catenin activity, we examined the role of endogenous PP2A in SDH5-mediated modulation of GSK-3β-β-catenin signaling. SDH5-expressing cells were treated with a PP2A inhibitor, OA, or PP2A-siRNA. Both OA and PP2A-siRNA treatment abolished SDH5-mediated dephosphorylation of GSK-3β on Ser-9, EMT (Fig. 3F), and GSK-3β kinase activity (Fig. 3E). These data indicate that PP2A is critical for SDH5-mediated GSK-3β activation and MET responses. Thus, SDH5 inhibits GSK-3β activity through PP2A.
SDH5 Activates GSK-3β and Antagonizes Wnt-mediated EMT
Wnt signaling is a key inducer of EMT during embryonic development and cancer progression. We tested whether manipulating SDH5 levels in various cell lines could modulate Wnt-induced EMT. Whereas Wnt only slightly elicited EMT (Fig. 4A, left two lanes) in NCI-H23, it significantly increased EMT when endogenous SDH5 was knocked down by SDH5-siRNA (Fig. 4A, right two lanes). Consistently, β-catenin/TCF transcriptional activity was increased upon knockdown of SDH5 (Fig. 4B).
FIGURE 4.

SDH5 antagonizes Wnt-mediated EMT. Knockdown of SDH5 inactivates GSK-3β, promotes β-catenin activity, and enhances Wnt-induced EMT in NCI-H23 cells. Cells were co-transfected with the control or SDH5-siRNA and TOP or FOP. Then, cells were treated with L- or Wnt-CM. A, cell lysates were subjected to Western blotting. B, relative luciferase expression was measured as described above. C and D, in contrast, restoring SDH5 expression in Slu-02 cells (SDH5-negative) prevented Wnt-induced EMT. Asterisks indicate statistically significant differences in cells transfected with the control vector versus SDH5. Error bars, S.E.
In contrast, restoring SDH5 expression in Slu-02 (SDH5-negative cell) prevented Wnt-induced EMT and β-catenin/TCF transcriptional activity (Fig. 4, C and D), strongly suggesting that SDH5 is an antagonist of Wnt-mediated EMT. However, overexpression of SDH5 did not affect the expression of Frizzled-1 (Wnt receptor) in NCI-H226 cell lines (supplemental Fig. S3).
Down-regulation of SDH5 Promotes Tumor Metastasis in Vivo
Because NCI-H23 cells have low metastatic potential, decreased SDH5 expression, and can initiate EMT (Fig. 1), we examined the metastatic potential of KD (SDH5 knockdown) versus Con-expressing NCI-H23 cells using an orthotopic mouse model. Stable luciferase activity was confirmed in each subline to ensure equal levels before injection. BLI was used to monitor tumor growth and the onset of metastases. One week after injection, BLI (Fig. 5A) detected multiple metastatic lesions in various sites in animals injected with NCI-H23-KD cells. In contrast, control mice exhibited only small primary tumors 5 weeks after injection, and none of these animals showed any signs of metastases (Fig. 5A). All mice bearing KD (SDH5 knockdown) cells developed metastasis (Fig. 5A), and H&E staining showed that all of these mice developed lung cancer, with or without metastasis (Fig. 6). Immunohistochemistry (IHC) showed that the majority of tumor cells strongly expressed vimentin (Fig. 6), whereas they exhibited weak staining of E-cadherin and cytokeratin (Fig. 6). Moreover, in the localization experiment, SDH5 was expressed in lung epithelial cells, and in the co-localization experiment, we SDH5 was expressed in lung epithelial cells but not in bronchial cells (supplemental Fig. S2).
FIGURE 5.
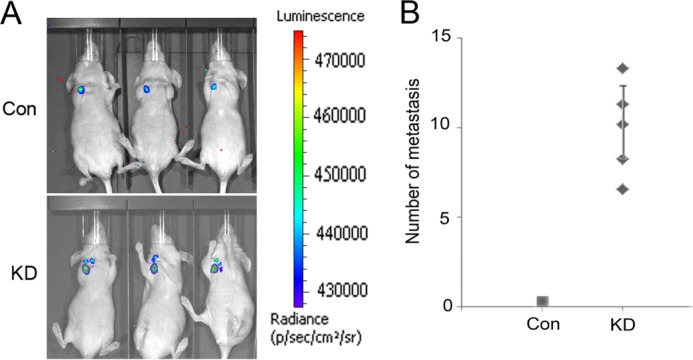
SDH5 down-regulation promotes tumor metastasis in vivo. A, representative BLI images of mice bearing NCI-H23-KD tumors with metastatic lesions. Mice (n = 3) were imaged 10 days later to determine local tumor growth and metastasis. B, number of metastatic nodules in individual mice bearing Con or KD tumors at the time of sacrifice.
FIGURE 6.
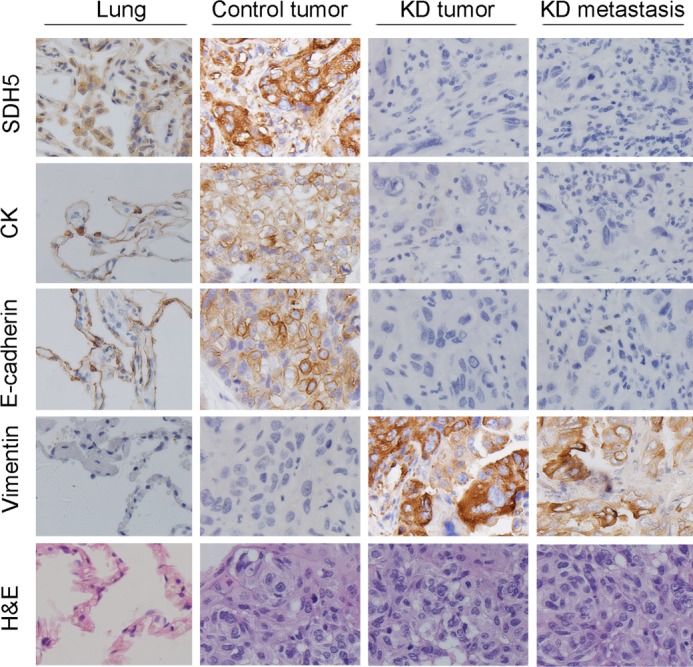
H&E and IHC staining for control mice and mice bearing KD tumors. H&E staining showed primary tumors without detectable metastasis in control mice and the lymph node metastases in mice bearing KD tumors 2 weeks after injection (magnification, ×100). IHC showed that a majority of the KD tumors had strong vimentin staining and weak E-cadherin, cytokeratin, and SDH5 staining (magnification, ×100).
SDH5 Knock-out Mice Exhibit Mesenchymal Characteristics in Lung Epithelial Cells
To further determine the impact of SDH5 on phenotypic changes in normal lung tissue, SDH5 knock-out (KO; SDH5−/−) mice were employed (15). The strategy for targeted deletion of the mouse SDH5 gene is outlined in our previous study (15). This strategy effectively disrupts the coding regions of SDH5 that are required for its activity. Moreover, after homologous recombination, it can introduce a translation stop codon in-frame, preventing translation downstream of the SDH5 sequence. Following heterozygous mating, homozygotes were identified and distinguished form heterozygotes and wild-type mice by PCR (Fig. 7B). RT-PCR and Western blot analyses failed to detect SDH5 expression in SDH knock-out mice (Fig. 7, C and D). In SDH5WT (SDH5+/+) mice, SDH5 expression was associated with lung epithelial cells (Fig. 7E). Consistent with in vitro data (Figs. 3 and 4), Ser-9 phosphorylation of GSK-3β was higher in SDH5−/− mice than that in the SDH5+/+ mice (Fig. 7F), suggesting that GSK-3β activity was suppressed in SDH5−/− mice. Additionally, lung epithelial cells in SDH5−/− mice exhibit decreased E-cadherin and elevated vimentin expression (Fig. 7E).
FIGURE 7.
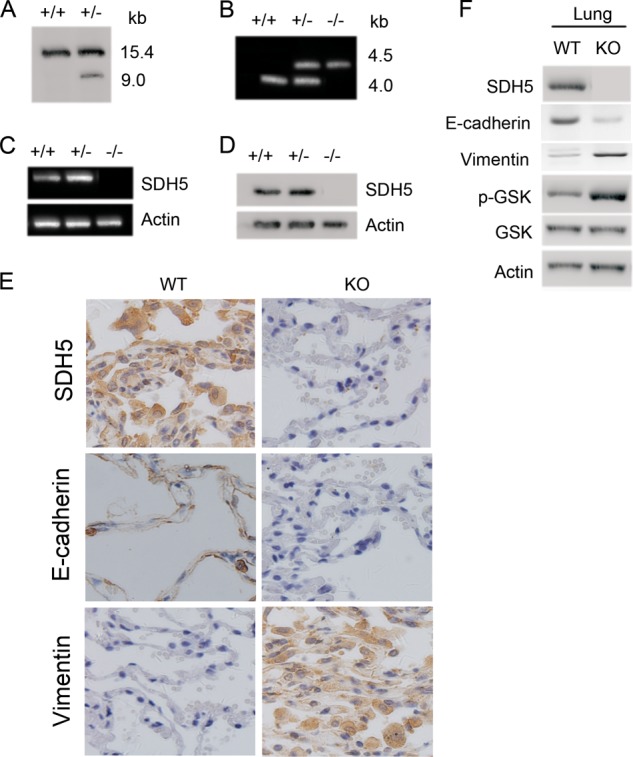
Genotyping of SDH5 knock-out mice that exhibit mesenchymal characteristics in lung epithelial cells. A, Southern blot analysis by using ES cells. Genomic DNA was digested with EcoRV: +/+, wide ES clone; +/−, positive targeted clone. B, genotyping of SDH5 knock-out mice by PCR. 4.5- and 4.0-kb fragments could be separately amplified in SDH5 (wild type and homozygous mice); both fragments were amplified in SDH5 heterozygous mice. C and D, RT-PCR (C) and Western blot analysis (D) of RNA and protein, respectively, from the liver of wild type (+/+), heterozygous (+/−), and homozygous (−/−) SDH5 mice. The absence of SHD5 expression in SDH5 mice was confirmed at both the mRNA and protein levels. E, elevated mesenchymal markers in lung cells of SHD5−/− mice. Expression of SHD5, E-cadherin, and vimentin in paraffin-embedded sections of lung tissue was determined by IHC (magnification, ×100). F, reduced GSK-3β activity and vimentin in SHD5−/− mice and increased E-cadherin in SHD5−/− mice. Lung homogenates from WT and KO mice were subjected to Western blotting.
SDH5 Expression Regulates EMT Markers in Lung Cancer Patients
Next, we examined the relationship between SDH5 expression and EMT markers in lung cancer patients. Specimens from different stages of human lung cancer patients with or without lymph nodes metastasis were chosen by positron emission tomography/computed tomography (Fig. 8A). Based on the TNM Staging System for Lung Cancer, we selected patients from stage I and stage III. Detailed information for the chosen patients is listed in Table 1. H&E staining verified lung cancer patients with or without lymph node metastasis (Fig. 8B). Loss of SDH5 and E-cadherin, as well as increased vimentin, p-GSK-3β, and β-catenin levels were clearly detected in tissues from lung cancer patients with lymph node metastasis (Fig. 8C). There was a significant correlation between the levels of SDH5 and E-cadherin and an inverse correlation between the levels of SDH5 and vimentin in all of the samples tested. Taken together, our human in vivo data are consistent with our in vitro data from various cancer cell lines.
FIGURE 8.

SDH5 in primary cancer lung cancer is correlated with risk of lymph node metastasis. A, SDH5 regulates EMT in lung cancer with different stages of disease, including lung cancer patients with or without metastasis, as determined by positron emission tomography/computed tomography. B, H&E staining was used to identify lung cancer patients with or without lymph nodes metastasis (magnification, ×100). C, loss of SDH5 expression correlates with changes in EMT marker in clinical specimens from lung cancer patients. Expression levels of SDH5, E-cadherin, vimentin, β-catenin, and p-GSK-3β (Ser-9) in normal (n = 3) and lung cancer (n = 3) tissues were determined by Western blotting. Densitometry was used to determine relative protein levels, and all proteins were normalized to the levels of β-actin.
TABLE 1.
Detailed patient information
“Primary” refers to patients without lymph node metastasis, and “metastasis” refers to patients with lymph node metastasis.
| Tumor | Sex | Age | TNM | Stage |
|---|---|---|---|---|
| Primary | Male | 44 | T1N0M0 | I |
| Primary | Male | 56 | T2N0M0 | I |
| Primary | Female | 45 | T1N0M0 | I |
| Primary | Male | 76 | T2N0M0 | I |
| Primary | Female | 56 | T1N0M0 | I |
| Metastasis | Female | 66 | T2N2M0 | III |
| Metastasis | Female | 42 | T1N2M0 | III |
| Metastasis | Female | 68 | T2N2M0 | III |
| Metastasis | Male | 67 | T2N2M0 | III |
| Metastasis | Male | 54 | T1N2M0 | III |
DISCUSSION
SDH5 is mitochondrial protein. In a recent study, Rutter and co-workers (10) showed that SDH5 is required for the flavination of SDHA, which is necessary for SDH assembly and function. Moreover, SDH5 is recognized as a novel endocrine tumor suppressor gene, and it is associated with high risk for the development of several tumor types (10–12). However, the role of SDH5 in lung cancer remains unknown. In this study, we show that SDH5 functions as a critical protein that modulates GSK-3β-β-catenin signaling and EMT in human lung cancer. The physical interaction between SDH5 and GSK-3β facilitates GSK-3β activation through Ser-9 dephosphorylation, which decreases nuclear β-catenin accumulation and transcriptional activity. These results reveal the potent inhibitory function of SDH5 on Wnt-β-catenin signaling. Within the SDH5-GSK-3β complex, SDH5 functions as a negative regulator of Wnt-β-catenin signaling. The role of Ser-9 phosphorylation of GSK-3β in Wnt/β-catenin signaling is still controversial (16–19). For example, Ser-9 phosphorylation of GSK-3β is not correlated with Wnt-mediated GSK-3β activity in certain cell types (20, 21). However, other studies demonstrated that many growth factors, such as insulin growth factor, transforming growth factor-β, and epidermal growth factor, could increase β-catenin accumulation through Ser-9 phosphorylation of GSK-3β (21, 22). Inactivation of GSK-3β through Ser-9 phosphorylation is involved in hepatitis B virus-x protein-mediated β-catenin stabilization in hepatocellular carcinoma cells (23). Our analysis of several lung cancer cell lines revealed that Ser-9 phosphorylation of GSK-3β is involved in SDH5-mediated β-catenin stability and transcriptional activity, suggesting that the effect of Ser-9 phosphorylation on β-catenin signaling is cell type-dependent.
We further examined the relationship between SDH5 expression and EMT markers in lung cancer patients. Loss of SDH5 and E-cadherin, as well as increased vimentin and p-GSK-3β, were clearly detected in tissues from lung cancer patients with lymph node metastasis (Fig. 8). Moreover, SDH5 did not affect EMT regulators, including Snail, Slug, Twist, and members of the TGF signaling pathway (supplemental Fig. S1), but it affect the expression of ZEB1 (Fig. 1).
In general, β-catenin has a dual role in EMT: it not only enhances cell-cell adhesion by associating with cadherin complexes in adherens junctions of cell membrane, but it also functions as a transcriptional co-activator by interacting with TCF transcription factor complexes in the nucleus. The induction of EMT by nuclear β-catenin has been explored during development in both cell lines and tumors. Several studies suggest that β-catenin-mediated transcription induces Slug or Twist1 gene expression, which further represses E-cadherin and contributes to EMT (24, 25, 26). Our data show that loss of SDH5 in cells can lead to the accumulation of nuclear β-catenin (Fig. 2A). Thus, we believe that SDH5 can modulate the dynamic switching between membrane- and nuclear-associated β-catenin (Fig. 2), which is a critical determinant of EMT. Taken together, these data indicate that SDH5 is a key inhibitor of EMT.
The majority of human carcinomas exhibit an epithelial phenotype. To break away from neighboring cells and invade adjacent tissue layers or peripheral lymph nodes, carcinoma cells often lose cell-cell adhesion and acquire motility. In general, 60% of newly diagnosed lung cancer patients have local invasive cancer, and almost all of these patients eventually develop metastatic disease, accounting for most cancer deaths (27, 28). Thus, detection of metastatic potential at an early stage could lead to an increase in disease-free survival rates. In regard to the clinical outcome of lung cancer progression, the presence of lymph node invasion is associated with the lowest 10-year progression-free survival rate. In our orthotopic lung cancer animal model (Figs. 5 and 6), mice bearing SDH5-knockdown cells displayed a dramatic increase in the incidence of lymph node metastases and the number of metastatic sites where tissues clearly exhibit mesenchymal characteristics. Wnt signaling has been identified as a determinant of lung cancer metastasis to the brain and bone. Similarly, our data indicate that down-regulation of SDH5 increases the propensity of lung cancer cells to metastasize to lymph nodes (Fig. 5). Consistently, SDH5−/− mice exhibit alterations in EMT markers in the lung tissue (Fig. 7).
In summary, this study delineates the functional role of SDH5 in EMT, which explains how loss of SDH5 in lung cancer underlies the onset of aggressive metastatic lung cancer. We believe that assessing SDH5 expression in lung cancer specimens could be a valuable prognostic biomarker for the risk of lung cancer metastasis, and the delineation of SDH5 function could provide a potential intervention strategy for lung cancer metastasis.
This work was supported by National Natural Science Foundation of China Grant 81101776 and China Postdoctoral Science Foundation Grant 2013T60256.

This article contains supplemental Figs. S1–S3.
- EMT
- epithelial-mesenchymal transition
- BLI
- bioluminescent imaging
- CM
- conditioned medium
- IHC
- immunohistochemistry
- MET
- mesenchymal-epithelial transition
- OA
- okadaic acid
- SDH5
- succinate dehydrogenase 5
- TCF
- T cell factor
- PP2A
- protein phosphatase A
- TOP
- TCF-responsive promoter reporter
- FOP
- nonresponsive control reporter.
REFERENCES
- 1. Jakobsen J. N., Sørensen J. B. (2013) Clinical impact of Ki-67 labeling index in non-small cell lung cancer. Lung Cancer 79, 1–7 [DOI] [PubMed] [Google Scholar]
- 2. Kaag D. (2013) Carboplatin dose calculation in lung cancer patients with low serum creatinine concentrations using CKD-EPI and Cockcroft-Gault with different weight descriptors. Lung Cancer 79, 54–58 [DOI] [PubMed] [Google Scholar]
- 3. Chen Z., Cheng K., Walton Z., Wang Y., Ebi H., Shimamura T., Liu Y., Tupper T., Ouyang J., Li J., Gao P., Woo M. S., Xu C., Yanagita M., Altabef A., Wang S., Lee C., Nakada Y., Peña C. G., Sun Y., Franchetti Y., Yao C., Saur A., Cameron M. D., Nishino M., Hayes D. N., Wilkerson M. D., Roberts P. J., Lee C. B., Bardeesy N., Butaney M., Chirieac L. R., Costa D. B., Jackman D., Sharpless N. E., Castrillon D. H., Demetri G. D., Jänne P. A., Pandolfi P. P., Cantley L. C., Kung A. L., Engelman J. A., Wong K. K. (2012) A murine lung cancer co-clinical trial identifies genetic modifiers of therapeutic response. Nature 483, 613–617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Couzin-Frankel J. (2012) Clinical trials: experimental cancer therapies move to the front line. Science 335, 282–283 [DOI] [PubMed] [Google Scholar]
- 5. Vuoriluoto K., Haugen H., Kiviluoto S., Mpindi J. P., Nevo J., Gjerdrum C., Tiron C., Lorens J. B., Ivaska J. (2011) Vimentin regulates EMT induction by Slug and oncogenic H-Ras and migration by governing Axl expression in breast cancer. Oncogene 30, 1436–1448 [DOI] [PubMed] [Google Scholar]
- 6. Ponnusamy M. P., Lakshmanan I., Jain M., Das S., Chakraborty S., Dey P., Batra S. K. (2010) MUC4 mucin-induced epithelial to mesenchymal transition: a novel mechanism for metastasis of human ovarian cancer cells. Oncogene 29, 5741–5754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Roy L. D., Sahraei M., Subramani D. B., Besmer D., Nath S., Tinder T. L., Bajaj E., Shanmugam K., Lee Y. Y., Hwang S. I., Gendler S. J., Mukherjee P. (2011) MUC1 enhances invasiveness of pancreatic cancer cells by inducing epithelial to mesenchymal transition. Oncogene 30, 1449–1459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bokobza S. M., Ye L., Kynaston H., Jiang W. G. (2011) Growth and differentiation factor 9 (GDF-9) induces epithelial-mesenchymal transition in prostate cancer cells. Mol. Cell. Biochem. 349, 33–40 [DOI] [PubMed] [Google Scholar]
- 9. Cao Y., Zhang L., Kamimura Y., Ritprajak P., Hashiguchi M., Hirose S., Azuma M. (2011) B7-H1 overexpression regulates epithelial-mesenchymal transition and accelerates carcinogenesis in skin. Cancer Res. 71, 1235–1243 [DOI] [PubMed] [Google Scholar]
- 10. Hao H. X., Khalimonchuk O., Schraders M., Dephoure N., Bayley J. P., Kunst H., Devilee P., Cremers C. W., Schiffman J. D., Bentz B. G., Gygi S. P., Winge D. R., Kremer H., Rutter J. (2009) SDH5, a gene required for flavination of succinate dehydrogenase, is mutated in paraganglioma. Science 325, 1139–1142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kaelin W. G., Jr. (2009) SDH5 mutations and familial paraganglioma: somewhere Warburg is smiling. Cancer Cell 16, 180–182 [DOI] [PubMed] [Google Scholar]
- 12. Kunst H. P., Rutten M. H., de Mönnink J. P., Hoefsloot L. H., Timmers H. J., Marres H. A., Jansen J. C., Kremer H., Bayley J. P., Cremers C. W. (2011) SDHAF2 (PGL2-SDH5) and hereditary head and neck paraganglioma. Clin. Cancer Res. 17, 247–254 [DOI] [PubMed] [Google Scholar]
- 13. Starker L. F., Delgado-Verdugo A., Udelsman R., Björklund P., Carling T. (2010) Expression and somatic mutations of SDHAF2 (SDH5), a novel endocrine tumor suppressor gene in parathyroid tumors of primary hyperparathyroidism. Endocrine 38, 397–401 [DOI] [PubMed] [Google Scholar]
- 14. Li C., Zhou C., Wang S., Feng Y., Lin W., Lin S., Wang Y., Huang H., Liu P., Mu Y. G., Shen X. (2011) Sensitization of glioma cells to tamoxifen-induced apoptosis by PI3-kinase inhibitor through the GSK-3β/β-catenin signaling pathway. PLoS One 6, e27053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liu J., Zhu J., Wang C. (2013) Generation of SDH5 gene knock-out and neo gene knock-in in mouse model. J. Clin. Oncol. Cancer Res. 40, 1789–1792 [Google Scholar]
- 16. Toth M. J., Ward K., van der Velden J., Miller M. S., Vanburen P., Lewinter M. M., Ades P. A. (2011) Chronic heart failure reduces Akt phosphorylation in human skeletal muscle: relationship to muscle size and function. J. Appl. Physiol. 110, 892–900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhang Y., Shu Y. M., Wang S. F., Da B. H., Wang Z. H., Li H. B. (2010) Stabilization of mismatch repair gene PMS2 by glycogen synthase kinase 3β is implicated in the treatment of cervical carcinoma. BMC Cancer 10, 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Matsuda T., Zhai P., Maejima Y., Hong C., Gao S., Tian B., Goto K., Takagi H., Tamamori-Adachi M., Kitajima S., Sadoshima J. (2008) Distinct roles of GSK-3α and GSK-3β phosphorylation in the heart under pressure overload. Proc. Natl. Acad. Sci. U.S.A. 105, 20900–20905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Brabant G., Müller G., Horn R., Anderwald C., Roden M., Nave H. (2005) Hepatic leptin signaling in obesity. FASEB J. 19, 1048–1050 [DOI] [PubMed] [Google Scholar]
- 20. Ramachandran R., Zhao X. F., Goldman D. (2011) Ascl1a/Dkk/β-catenin signaling pathway is necessary and glycogen synthase kinase-3β inhibition is sufficient for zebrafish retina regeneration. Proc. Natl. Acad. Sci. U.S.A. 108, 15858–15863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chen H. Z., Liu Q. F., Li L., Wang W. J., Yao L. M., Yang M., Liu B., Chen W., Zhan Y. Y., Zhang M. Q., Cai J. C., Zheng Z. H., Lin S. C., Li B. A., Wu Q. (2012) The orphan receptor TR3 suppresses intestinal tumorigenesis in mice by down-regulating Wnt signalling. Gut 61, 714–724 [DOI] [PubMed] [Google Scholar]
- 22. Kitazawa M., Cheng D., Tsukamoto M. R., Koike M. A., Wes P. D., Vasilevko V., Cribbs D. H., LaFerla F. M. (2011) Blocking IL-1 signaling rescues cognition, attenuates tau pathology, and restores neuronal β-catenin pathway function in an Alzheimer's disease model. J. Immunol. 187, 6539–6549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yang W. J., Chang C. J., Yeh S. H., Lin W. H., Wang S. H., Tsai T. F., Chen D. S., Chen P. J. (2009) Hepatitis B virus X protein enhances the transcriptional activity of the androgen receptor through c-Src and glycogen synthase kinase-3β kinase pathways. Hepatology 49, 1515–1524 [DOI] [PubMed] [Google Scholar]
- 24. Yang M. H., Chen C. L., Chau G. Y., Chiou S. H., Su C. W., Chou T. Y., Peng W. L., Wu J. C. (2009) Comprehensive analysis of the independent effect of Twist and Snail in promoting metastasis of hepatocellular carcinoma. Hepatology 50, 1464–1474 [DOI] [PubMed] [Google Scholar]
- 25. Zhang Y., Yan W., Chen X. (2011) Mutant p53 disrupts MCF-10A cell polarity in three-dimensional culture via epithelial-to-mesenchymal transitions. J. Biol. Chem. 286, 16218–16228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang Y., Yan W., Jung Y. S., Chen X. (2012) Mammary epithelial cell polarity is regulated differentially by p73 isoforms via epithelial-to-mesenchymal transition. J. Biol. Chem. 287, 17746–17753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Janku F., Berry D. A., Gong J., Parsons H. A., Stewart D. J., Kurzrock R. (2012) Outcomes of phase II clinical trials with single-agent therapies in advanced/metastatic non-small cell lung cancer published between 2000 and 2009. Clin Cancer Res. 18, 6356–6363 [DOI] [PubMed] [Google Scholar]
- 28. Shinagare A. B., Okajima Y., Oxnard G. R., Dipiro P. J., Johnson B. E., Hatabu H., Nishino M. (2012) Unsuspected pulmonary embolism in lung cancer patients: comparison of clinical characteristics and outcome with suspected pulmonary embolism. Lung Cancer 78, 161–166 [DOI] [PMC free article] [PubMed] [Google Scholar]