

Summary
Following adherence of neutrophils to the endothelium, neutrophils undergo a major morphological change that is a necessary prelude to their extravasation. We show here that this shape change is triggered by an elevation of cytosolic inositol (1,4,5)-trisphosphate (IP3), to provoke physiological Ca2+ influx through a store-operated mechanism. This transition from a spherical to ‘flattened’ neutrophil morphology is rapid (∼100 seconds) and is accompanied by an apparent rapid expansion of the area of the plasma membrane. However, no new membrane is added into the plasma membrane. Pharmacological inhibition of calpain-activation, which is triggered by Ca2+ influx during neutrophil spreading, prevents normal cell flattening. In calpain-suppressed cells, an aberrant form of cell spreading can occur where an uncoordinated and localised expansion of the plasma membrane is evident. These data show that rapid neutrophil spreading is triggered by Ca2+ influx, which causes activation of calpain and release of furled plasma membrane to allow its apparent ‘expansion’.
Key words: Ca2+ signalling; Cell spreading; Neutrophils; Phagocytosis; Inositol (1,4,5)-trisphosphate; IP3
Introduction
An essential feature of the way in which neutrophils behave in the body is their capacity to change morphology and undertake significant and rapid changes in cell shape. During chemotaxis, neutrophils develop a leading front edge and a trailing uropod, and during phagocytosis, they form localised pseudopodia and phagosomes. Not surprisingly, the mechanism and intracellular signalling events controlling chemotaxis (Kay et al., 2008) and phagocytosis (Swanson, 2008) continue to be widely studied. However, the mechanism by which neutrophils change shape rapidly has been less well studied, even though the qualitatively different and slower cell spreading behaviour in non-immune cells is increasingly understood (Zhang et al., 2008; Huang et al., 2009; Cuvelier et al., 2007). Despite this lack of attention, neutrophil spreading is the key event in determining the rate of neutrophil extravasation both physiologically and pathologically. After rolling along the endothelium, the neutrophil flattens onto the endothelial surface (‘spreading’) at sites of inflammation in response to cues from the endothelial surface. This dramatic cell shape change minimises the shear force of blood flow on the neutrophil that would otherwise tend to cause cell rolling. The flattening morphology transition is also a necessary prelude to neutrophil extravasation, either between the endothelial cells or through an individual cell, and subsequent chemotaxis in the extravascular space (Ley et al., 2007).
Pioneering work by Maxfield showed that the spreading of neutrophils and macrophages onto surfaces is preceded by large global cytosolic free Ca2+ signals (Kruskal et al., 1986: Kruskal and Maxfield, 1987). These cytosolic free Ca2+ signals were caused by adhesive signals to the cell from the substrata, and it has subsequently been shown that immobilisation of β2 integrin by surfaces (Jaconi et al., 1991; Pettit and Hallett, 1996) or cross-linking by anti-integrin antibodies (Ng-Sikorski et al., 1991: Petersen et al., 1993) triggers similar Ca2+ signals. There is, however, a major problem in designing experiments to investigate the causal link between the Ca2+ signal and the cell spreading event. The stimulus for the Ca2+ signal (i.e. a surface) cannot be studied in isolation from the spreading response as both the delivery of the signal and the ability to respond depend on the same event, namely contact with the same surface. In our studies, we have overcome this problem by studying neutrophils in conditions that permit contact with a surface in the absence of signalling and flattening, and subsequently imposing changes in their cytosolic chemistry using (non-contact) photolytic manipulation. Using this approach, we have found that the elevation of cytosolic free Ca2+ triggered by photolytic generation of cytosolic inositol (1,4,5)-trisphosphate (IP3) is sufficient to induce a rapid cell shape change. The membrane expansion accompanying this cell shape change occurred without the insertion of new membrane, and thus probably results from unfurling of wrinkled plasma membrane (Dewitt and Hallett, 2007; Hallett and Dewitt, 2007). This is also suggested from biophysical measurement (Herant et al., 2005) and the manipulation of neutrophil membrane tension (Houk et al., 2012). The cell spreading and membrane expansion depended on activation of the Ca2+-activated protease, calpain. Calpain activation, triggered by Ca2+ influx, was therefore the key step for the rapid neutrophil flattening morphology transition.
Results
Morphology changes during neutrophil flattening
Rapid spreading onto glass can be triggered in adherent neutrophils by elevating cytosolic free Ca2+ using a chemotactic stimulus (Fig. 1). The cytosolic free Ca2+ concentration and cell morphology of the non-stimulated neutrophils is stable for many minutes before stimulation but after the peptide-induced cytosolic free Ca2+ signal, the cells flatten onto the surface over the subsequent 80 seconds. Although there is no obvious increase in cell–substrate contact area immediately on addition of the peptide or at the peak of the cytosolic free Ca2+ signal (Fig. 1A), a dramatic increase in the area in contact with the glass substrate is evident immediately afterwards (Fig. 1). Upon reconstructing the cell surface of the cells from confocal optical sections, it can be seen that both the cell adhesion footprint increases and cell thickness decreases to a point limited by the thickness of the nuclear lobes within the cell. The individual lobes of the multi-lobed nucleus within the non-spread cell are held with the longer axis upright and are restrained from sedimenting by the resistance of cell periphery (Fig. 1B,C). However, after the Ca2+ signal, the restraint to sedimentation of the nuclear lobes is relaxed and the lobes lie on their long axis and adopt the characteristic polymorphonuclear display (Fig. 1B,C). A striking change during flattening is in that the apparent cell surface area increased by up to 200% in these experiments. As a spherical surface is the minimum that encloses a given volume, it is not surprising that the transition to a non-spherical cell morphology is accompanied by an increase in the apparent cell surface area.
Fig. 1.
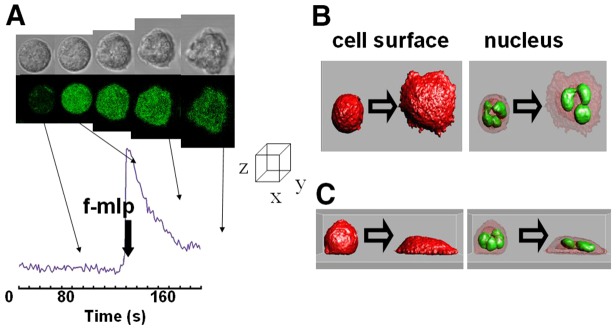
Rapid cell spreading in neutrophils is induced by a Ca2+ signal. (A) The upper series of images show the phase contrast images and the lower series the fluo4 intensity (as a readout of Ca2+ concentration) of an individual human neutrophil undergoing cell spreading in response to f-mlp (1 µM). The graph shows the time course of cytosolic free Ca2+ concentration with times at which the images were taken indicated and the time of addition of f-mlp shown. (B,C) The cell surface (stained with DiI) and the nucleus (stained with acridine orange) have been reconstructed from confocal optical slices of an adherent but not ‘spread’ and a spread cell and are shown in orthogonal planes (B,C) as isosurfaces (Imaris). Each of the examples shown are representative of at least 20 experiments using neutrophils taken from at least four different donors.
Uncaging IP3 triggers neutrophil flattening
Studies using soluble stimuli (such as those shown in Fig. 1) are of limited value for studying the mechanism of the morphology change. First, only cells which are adherent to some extent can be studied (otherwise the addition of stimulus would simply wash away the cell). Second, chemotactic stimuli do not provide a simple cytosolic signal. Occupancy of chemotactic receptors generates a number of chemical changes within the cytosol and the cell membrane, each of which could contribute to the effect. Third, soluble stimuli do not reflect the physiological situation, where ICAM-1 molecules on the endothelial cells engage β2 integrin on the neutrophil to trigger the morphology change. For these reasons, studies of neutrophils spreading onto stimulating surfaces are often performed. However, there is also an important logical problem in designing experiments to investigate the link between the Ca2+ signal and the cell spreading. As the surface onto which the neutrophil sediments both delivers the stimulus Jaconi et al., 1991; Pettit and Hallett, 1996) and is required for the spreading response to be evident, ‘stimulus’ and ‘response’ become logically entangled. In this study, we have overcome these problems by imposing a defined chemical change within the cytosol on neutrophils that are in contact with an underlying (non-stimulatory) surface by using photolytic uncaging of Ca2+-elevating agents.
Uncaging cytosolic caged IP3 will generate biologically active IP3 within the cell on command. On exposure to the uncaging illumination, the characteristics of the Ca2+ signal were variable between individual neutrophils, but three phases could often be distinguished (Fig. 2B), an initial lag (0–5 seconds), followed by a small rise or shoulder (100–200 nM), before a single large and transient Ca2+ signal (peaking at 800 nM). As uncaging fluorescein by this method occurred without delay (Fig. 2A), the lag period between UV illumination and the onset of the Ca2+ signal was attributed to the time required to generate sufficient IP3 to breach the threshold cytosolic concentration of ∼20 µM that is required to release of Ca2+ from intracellular storage sites (Davies-Cox et al., 2001) and trigger Ca2+ influx across the plasma membrane. For neutrophils on glass, this IP3 uncaging strategy induced a rapid transformation to the flattened ‘spread’ morphology in all cells studied in which a Ca2+ influx signal was triggered (n>50, Fig. 2C; supplementary material Movie 1). Exposure of non-loaded neutrophils to uncaging wavelengths or exposure of caged-IP3-loaded cells to non-uncaging wavelengths failed to increase the spreading rate. Furthermore, the effect cannot be attributed to the photolytic products other than IP3 because uncaging other caged compounds (including nitr5, see later) failed to trigger cell spreading. IP3-induced spreading was also triggered in neutrophils on an integrin-engaging surface (fibronectin-coated glass), showing that the flattening response was not unique to the glass surface (Fig. 3B). However, fibronectin was not routinely used as it was necessary to select cells in contact with the surface before as the surface itself had triggered the Ca2+ signals (Jaconi et al., 1991; Pettit and Hallett, 1996). The Ca2+ signal triggered by IP3 uncaging in neutrophils on this surface was also more complex than on glass, with multiple peaks, reminiscent of the Ca2+ signals, which occur during integrin-mediate phagocytosis (Dewitt and Hallett, 2002; Dewitt et al., 2003). The complexity of the Ca2+ signal has been attributed to cycles of Ca2+-induced integrin untethering and mobilisation followed by further integrin-induced Ca2+ signalling (Dewitt and Hallett, 2002).
Fig. 2.
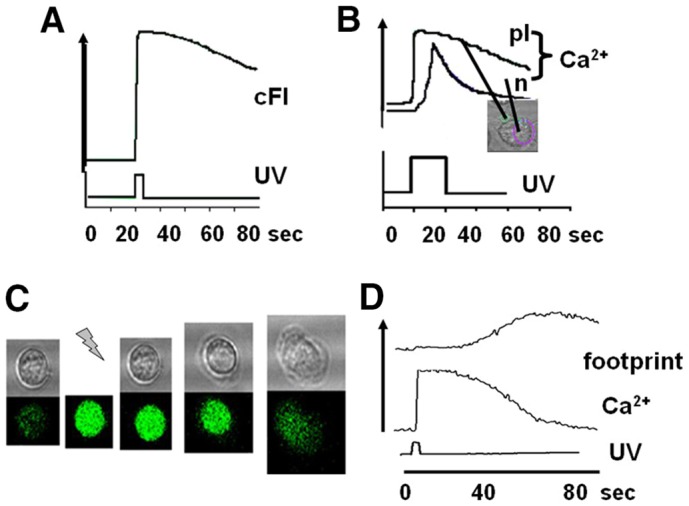
Uncaging cytosolic IP3. (A) The effect of uncaging caged fluorescein is shown. There is no delay between the onset of UV illumination and the increase of fluorescent signal. (B) The effect of uncaging caged cytosolic IP3 on cytosolic free Ca2+ is shown for a platelet (pl) and neutrophil (n) in the same microscopic field. The upper traces show the intensity of fluo4 and the lower trace the uncaging illumination (at 360 nm). (C) The upper series of images shows the phase contrast images and the lower series the fluo4 intensity (Ca2+) of an individual human neutrophil undergoing cell spreading in response to uncaging of caged IP3. In the series of images the time at which uncaging occurred is shown by the ‘lightning bolt’. (D) Graph of the cell–substrate contact area (footprint, upper line); cytosolic free Ca2+ change (middle line) and uncaging illumination (UV, lower line). Each of the examples shown in this figure are representative of at least 50 experiments using neutrophils taken from at least four different donors. Examples of cell spreading induced by uncaging IP3 are also shown in supplementary material Movie 1.
Fig. 3.

Integrin engagement and inhibition of Ca2+ influx affects the IP3-induced Ca2+ signal. The rate of cell spreading and cytosolic free Ca2+ signalling induced by uncaging IP3 in neutrophils on (A) glass, (B) fibronectin-coated glass substrates and (C) in the presence of Ni2+ (1 mM) which blocks Ca2+ influx are shown as a series of phase contrast images on which the fluo4 intensity has been superimposed (top section) and quantified in the graphs which show the cytosolic free Ca2+ signal (top line), the UV illumination intensity (middle line), and the cell footprint (bottom line) as described in Fig. 1.
The crucial role of Ca2+ influx across the plasma membrane
In all cases in which IP3 was uncaged (n>50), the onset of cell spreading occurred after the Ca2+ influx signal and never before (i.e. during the IP3-induced Ca2+ release event). However, given that there was a delay in the spreading response, this data did not distinguish whether the IP3-induced store Ca2+ release (i.e. the shoulder level of Ca2+) or the larger Ca2+ influx signal was responsible for the morphology change. In order to investigate this, cytosolic free Ca2+ was elevated to the same level achieved by IP3-induced store release (∼100 nM above resting cytosolic free Ca2+) by uncaging ‘caged Ca2+’ (the nitrobenzhydrol tetracarboxylate Ca2+ chelator nitr5). Uncaging caged Ca2+ elevated the cytosolic free Ca2+ concentration without stimulating Ca2+ influx. Although the concentration of cytosolic free Ca2+ in this experiment approximately matched that of the IP3-induced Ca2+ release, neutrophil flattening was not triggered (Fig. 4A–C). We found that nitr5 itself or its photolytic release products did not prevent neutrophils from spreading because f-mlp (formyl-methionyl-leucyl-phenylalanine) was able to induce normal cell spreading in nitr5-loaded cells after photolysis of nitr5 (Fig. 4A–C; supplementary material Movie 2). This confirmed (1) that photolysis itself was not a stimulus for neutrophil spreading, and (2) that the Ca2+-release signal induced by IP3 was below the critical concentration or in the wrong cellular location to trigger spreading. This result is consistent with an earlier study in which it was necessary to elevate global cytosolic free Ca2+ to an unphysiologically high level (∼30 µM) to induce neutrophil flattening (Pettit and Hallett, 1998). That the crucial Ca2+ component was Ca2+ influx was confirmed by using Ni2+ ions to block Ca2+ influx without inhibiting IP3-induced Ca2+ store release (see Hillson and Hallett, 2007); this inhibited IP3-induced neutrophil spreading (Fig. 3C). We therefore concluded that IP3 itself was not the crucial component for cell spreading, but rather that it was physiological Ca2+ influx through Ca2+ channels opened on the plasma membrane as a result of IP3-mediated Ca2+ store release.
Fig. 4.

Factors affecting neutrophil spreading. The effect of uncaging Ca2+ (nitr5) to a level of the initial (release) phase of the caged IP3-induced Ca2+ signal. (A) The matching of initial (release) phase of the caged IP3-induced Ca2+ signal (upper graph ‘cIP3’) and nitr5 uncaging followed by addition of f-mlp (lower graph). (B) Graphs are showing the effect of f-mlp addition from A in more detail, with the accompanying effect on the cell ‘footprint’ and (C) images of the cell and its cytosolic free Ca2+ from which the data was taken. (D–F) The effect of the pre-treatment of neutrophils with cytochalasin B (5 µg/ml) before uncaging IP3: (D) a series of phase contrast and fluo4 images, (E) quantification of cytosolic free Ca2+, footprint and cell roundness [taken as (cell perimeter/π.max cell dimension)], and (F) changes in the reconstructed isosurface of the cell and the position of the nucleus, with the arrowheads indicating local sites of cell deformation. An example of the ‘wriggling response’ to IP3 uncaging in cytochalasin-B-treated neutrophils is shown in supplementary material Movie 2.
Actin-polymerisation-independent surface area changes
Although a rise in cytosolic free Ca2+ is not required for actin polymerisation in neutrophils (al-Mohanna and Hallett, 1990), actin polymerisation clearly drives cell spreading because the IP3-induced Ca2+ influx signal failed to trigger spreading in neutrophils treated with the actin polymerisation inhibitor cytochalasin B (Fig. 4D,E). However, under these conditions, the IP3-induced Ca2+ signal triggered a noticeable periodic loss of sphericity of the cells as they appeared to ‘wriggle’ (Fig. 4D,E; supplementary material Movie 2). This loss of sphericity could not be attributed to the nuclear lobes ‘pushing against’ the plasma membrane as 3D imaging of the cells showed that the lobes of the multi-lobular nucleus remained in a tight formation and that bulges in the cell surface did not correlate with places that could be directly ‘pushed’ by nuclear lobes (Fig. 4F). As the spherical shape is the minimum surface area for a given volume, loss of sphericity must be accompanied by an increase in surface area. Cell wriggling therefore was a manifestation of an actin-polymerisation-independent cell surface area increase.
Cell surface area expansion without new membrane insertion
The plasma membrane has no facility to stretch beyond ∼5% of its length as lateral forces pull apart the phospholipids in the bilayer. The mechanism by which the surface area of the neutrophil apparently expands must therefore involve either the addition of new membrane, possibly by fusion of vesicles from within the cell, or the unfurling of its wrinkled membrane (Dewitt and Hallett, 2007; Hallett and Dewitt, 2007). The latter possibility was proposed over 30 years ago (Erickson and Trinkaus, 1976), but, until now, it has proved difficult to provide evidence for the mechanism of membrane expansion. The two possibilities can be distinguished by the use of FM1-43, which binds to membrane and increases the quantum yield of its fluorescence. The addition of new internal membranes to the plasma membrane as the surface area expands would thus give an increase in the total cellular FM1-43 fluorescence with the intensity of FM1-43 in the ‘expanding’ plasma membrane remaining constant. In contrast, the unfurling of wrinkled membrane would result in the total cellular FM1-43 fluorescence remaining constant, while FM1-43 intensity in the ‘expanding’ plasma membrane would decrease. The latter was found to be the case, with total cell FM1-43 fluorescence remaining constant during cell spreading and the fluorescence intensity of plasma membrane decreasing (Fig. 5A,B). This is despite the abundance of intracellular membranes within the cell that are capable of being staining by FM1-43, which are revealed by permeabilising the plasma membrane (Fig. 5B). In cells with asymmetric spreading, the FM1-43 signal intensity in the sub-region of the membrane, which did not expand, remained constant (Fig. 5B,C). Although these measurements were performed with the confocal pinhole opened wide to increase the thickness of optical sections, and so minimise any artefactual decrease in fluorescence signal owing to the movement of the spreading cell below the confocal plane, this possibility was excluded by the use of a second membrane marker, DiI. As the DiI intensity would be subject to the same optical or cell movement artefacts as FM1-43, the ratio of FM1-43 intensity to DiI intensity would be insensitive to these potential artefacts. However, unlike FM1-43, DiI was loaded into the membrane before observation and was absent from the extracellular medium during the experiment. Hence, DiI would decrease in intensity as a result of either the addition of new (unstained) intracellular membrane or unfurling of wrinkled membrane. The ratio of FM1-43:DiI would therefore have greater sensitivity than the FM1-43 signal alone. This ratio (FM1-43:DiI) failed to increase on cell spreading, and, in fact, often decreased slightly (Fig. 5C), and was the same in regions of the cells that had flattened and those that had not. Although this approach might not detect a small amount of membrane insertion (given fluorescent signal increases <10%), such an insertion would not contribute significantly to the increase in surface area. These data were therefore consistent with the unwrinkling mechanism for membrane expansion being dominant rather than the insertion of new membrane.
Fig. 5.

The mechanism of membrane expansion during cell spreading. Time series of images of neutrophils in the presence of FM1-43 with the accompanying phase contrast images for (A) an unstimulated neutrophil, which remains approximately spherical and has a constant FM1-43 signal (quantified in the graph as ‘footprint’ and FM intensity respectively), (B) a neutrophil undergoing rapid spreading, with the final fluorescent image resulting from the permeabilisation of the plasma membrane with digitonin (DGT; 150 µM), and (C) a rapidly spreading neutrophil pre-stained with DiI (red) in the presence of FM1-43 (green) as in the previous two panels. In C, the ratio of the FM1-43 and DiI signals is shown together with the change in cell footprint.
The crucial role of calpain activation
It has been proposed that plasma membrane wrinkles are held in place by calpain-sensitive cytoskeletal elements (Hallett and Dewitt, 2007; Dewitt and Hallett, 2007), and that localised high Ca2+ restricted to wrinkled membrane (Brasen et al., 2010) might activate sub-plasma membrane calpain-1, whose Kd is ∼30 µM (Goll et al., 2003). Calpain activity, monitored using a ratiometric fluorescence approach, to avoid the effect of optical path-length changes during cell spreading on the measurement (Dewitt et al., 2009; see Materials and Methods), increased during cell spreading (Fig. 6A). However, measuring activity using this approach gives limited information because (1) it is not possible to synchronously monitor cytosolic free Ca2+ (fura2 or fluo4 requires excitation in the 340–390 nm or 470–490 nm regions thus preventing simultaneous calpain measurement), and (2) it is not possible to time the onset of calpain activation and so correlate this to cell spreading (the signal observed was the result of the accumulation of the product of calpain activation, which accompanied neutrophil spreading). However, the method showed that calpain was activated and permitted effect of the use of pharmacological inhibitors of calpain to be established. Two classes of calpain inhibitors were used, mono-halide mercaptoacrylates, such as PD 150606 (Adams et al., 2012), which acts on a regulatory site required for Ca2+ triggering of the proteolytic activity, and calpeptin, which inhibits the proteolytic domain of the enzyme. The spreading morphological changes were reduced by these inhibitors at concentrations which inhibited the calpain activity increase associated with cell spreading. Electric cell-substrate impedance sensing (ECIS) showed that calpain inhibition significantly reduced the rate of neutrophils spreading on either the electrode itself or fibronectin-coated electrodes (Fig. 6B). The inhibitors also had a dramatic and clear inhibitory effect on IP3-induced neutrophil spreading: on IP3 uncaging, the evoked Ca2+ signal remained at levels that would stimulate rapid cell spreading (Fig. 6C), but the majority of cells failed to spread on either glass or integrin-engaging fibronectin (Fig. 6C,D). However, on glass, a small number of calpain-suppressed cells exhibited a slow and aberrant type of cell spreading. Unlike in cells with active calpain, where cell spreading was synchronised and uniform over the majority of the cell periphery (Figs 1,3), these calpain-suppressed cells flattened by producing numerous small bleb-like structures, which subsequently coalesced into a larger area and gave rise to an asymmetrical spreading response (Fig. 7A; supplementary material Movie 3). The FM1-43 signal in these mini-blebs decreased (Fig. 7B), which is consistent with them resulting from localised plasma membrane unwrinkling (Fig. 7D). We propose that, in the absence of full calpain activity, the release of membrane wrinkles is slowed sufficiently for the cellular events that normally accompany rapid neutrophil spreading to be temporally resolved into two clear stages of membrane expansion: first, the release of surface wrinkles, which is, second, permissive for the subsequent actin-dependent morphology change.
Fig. 6.

The role of calpain activity in neutrophil spreading. (A) Calpain activity monitored by the ratio of the signals from fluorogenic substrate Suc-LY-AMC and fluorescein in two neutrophils undergoing spreading is shown as a series of ratio images from which the graph was constructed. (B) The effect of a calpain inhibitor on the impedance, quantified by ECIS as a measure of spreading of neutrophils. The graphs shown are the means (black lines) and ±s.d. (grey lines) for five electrodes (the total cell number was ∼125). (C) Sample data comparing the effect of two calpain inhibitors (calpeptin, CPTN) and PD151746 (PDa) with a solvent control (DMSO). In all sections, uncaging of IP3 was at 30 seconds, and the fluo4 (Ca2+) and phase contrast images are shown together with the quantification of cytosolic free Ca2+ and the contact area of the cell (footprint). (D) The percentage of neutrophils that did not spread but were adherent (grey area of bars) and those that spread (black area of bars) is shown for three calpain inhibitors [PD150606 (PD1), PD151746(PD2) and calpeptin (CPT)] after IP3 uncaging. Cells were cultured on glass or fibronectin (indicated by ‘F’).
Fig. 7.

Aberrant spreading mechanism in calpain-inhibited cells. (A) A series of phase contrast images that are typical of neutrophils that spread after calpain inhibition. Unlike uninhibited cells, clear ‘bleb-like’ structures form before spreading. The complete data set for this is shown in supplementary material Movie 3. (B) A close up view of a ‘bleb-like’ structure in a neutrophil pre-stained with DiI (red) in the presence of FM1-43 (green) showing that FM1-43 intensity decreases, consistent with unfurling of existing wrinkled membrane, rather than the addition of new membrane. (C) The kinetics of cell spreading under calpain inhibition, where ‘steps’ in the increase in cell area are observed that correlate with the formation of ‘bleb-like’ structures (indicated by the asterisks). (D,E) Our hypothesis for the underlying mechanism at a macroscale (D), where the wrinkled membrane locally and patchily unfurls; and at a micro-scale (E), where the molecules holding the wrinkles in place are cleaved by activated calpain.
Discussion
In this study, we have shown that elevated cytosolic IP3 is an initiator of the Ca2+ influx that stimulates the morphological flattening of neutrophils. The Ca2+ influx is sufficient to activate cytosolic calpain, an event that is crucial for permitting spreading to proceed and for the apparent expansion in plasma membrane area. Because the apparent expansion is not accompanied by the addition of new membrane, we propose that the wrinkles on the surface of the neutrophils represent a ‘membrane reservoir’ and that calpain mediates the unfurling of these wrinkles.
Activation of phospholipase C (PLC), which generates IP3, probably plays a crucial role in physiological neutrophil spreading. Inhibition of PLC activity by compound U73122 (Bleasdale et al., 1990) has a profound inhibitory effect on neutrophil spreading (Smith et al., 1996). Similarly, in lymphocytes, the IP3-generating enzyme (PLCγ2) was found to be crucial for cell spreading (Weber et al., 2008). However, the authors of this latter study concluded that, in these lymphocytes, IP3 itself was not involved because cell spreading occurred in mutant cells lacking the three types of IP3 receptor (Weber et al., 2008). Clearly, our study shows that photolytically generated IP3, produced to a level sufficient to trigger a Ca2+ signal, induces cell spreading in human neutrophils. However, we must be careful not to extrapolate from this that IP3 is involved in neutrophil spreading physiologically. Indeed, at the integrin-engaging surface in myeloid cells, there is a strong and localised production of phosphotidylinositol (3,4,5)-trisphosphate (PIP3) (Dewitt et al., 2006), which might be involved in Ca2+ influx by a non-IP3 route. The data in this present paper shows that the Ca2+ influx itself, however it is generated physiologically, is a signal for rapid neutrophil spreading.
In other cell types, there is growing evidence for a role of calpain-2 in focal adhesion complexes and slow motility (Franco et al., 2004; Franco and Huttenlocher 2005). Neutrophils, however, mainly express calpain-1 and do not form focal adhesion complexes, and there are two reports that calpain inhibitors increase neutrophil motility (Lokuta et al., 2003; Katsube et al., 2008). Although it is not clear whether this latter effect was a result of decreased cell adhesion, there is a significant body of evidence showing that calpain inhibitors are effective in preventing neutrophil extravasation during experimental inflammation in animal models (Noble et al., 1998; Ikeda et al., 2002; Tissier et al., 2004; Cuzzocrea et al., 2000; Marzocco et al., 2004; Yoshifuji et al., 2005) and that calpain inhibition prevents full extension of pseudopodia during phagocytosis by neutrophils (Dewitt and Hallett, 2002) and spreading by lymphocytes (Stewart et al., 1998) and platelets (Croce et al., 1999). In our studies, calpain-inhibited cells were essentially quiescent, or sluggish even after IP3 uncaging (e.g. see Fig. 6). The ‘blebbing prelude’ to slow cell spreading, which we observed in calpain-suppressed neutrophils (Fig. 7), might occur physiologically in some cells (Fackler and Grosse, 2008; Charras and Paluch, 2008), especially in Dictyostelium, where recent reports have shown that blebbing even accompanies chemotaxis (Yoshida and Soldati, 2006; Langridge and Kay, 2006).
Our study therefore demonstrates that rapid cell spreading is triggered by the influx of Ca2+ through physiologically opened Ca2+ channels. This depends on calpain activation to release tethered undulations (wrinkles) in the plasma membrane, which, in turn, permits the apparent plasma membrane expansion. It is possible that the activation of calpain in neutrophils leads to the cleavage of an important set of cytoskeletal plasma membrane linkage molecules, including talin (Sampath et al., 1998) and ezrin (Potter et al., 1998), which are responsible for maintaining the membrane its is wrinkled state and thus acts as the permissive step in rapid neutrophil spreading (Dewitt and Hallett, 2007; Hallett and Dewitt, 2007).
Methods and Materials
Cell preparation and probe loading
Human neutrophils were isolated from the blood of healthy volunteers as described previously (Pettit and Hallett, 1995) as approved by SMREC ethics board (Cardiff University School of Medicine Research Ethics Committee, Cardiff, UK) with the informed consent of participants. The neutrophils were then suspended in Krebs medium (120 mM NaCl, 4.9 mM KCl 1.2 mM KH2PO4, 1.2 mM MgSO4, 1.3 mM CaCl2, 25 mM HEPES and 0.1% BSA, adjusted to pH 7.4 with NaOH). The neutrophils were loaded with the fluorescent Ca2+ probe fluo4 acetoxylmethyl ester (AM) (Molecular Probes; 1–5 µM) as described in detail elsewhere (Hillson et al., 2007). This produced cytosolic concentrations of the probes of 50–100 µM. When loading neutrophils with both Ca2+ probe and caged-IP3 or caged Ca2+ (nitr5), the incubation with the caged IP3 propionyloxymethyl ester (PM) (Alexis Biochem) or nitr5-AM (Biochem) was started before adding the fluo4-AM compound. Loading with caged compounds was performed in the dark to prevent uncontrolled uncaging by ambient light. The times for loading of the probes were: fluo4-AM, 40 minutes, IP3 (caged)-PM, 1 hour; and nitr5-AM (1 hour). Extreme care was taken during the isolation and loading procedures to avoid mechanical activation of the cells, which can cause them to adhere rapidly and spontaneously to uncoated glass. Under the isolation conditions used here, neutrophils adhere to glass, but remain in the apparently ‘spherical’ (i.e. non-spread) morphology.
Ca2+ measurement and manipulation
Cytosolic free Ca2+ was measured as described in detail elsewhere (Hillson et al., 2007) using the resonant scanning head of the Leica RS confocal microscope, which had a frequency of ∼40 MHz. Neutrophils were allowed to adhere to the glass coverslip mounted onto a thermostatically controlled stage (37±0.1°C) in Krebs medium and soluble stimuli such as f-mlp (Sigma) were added to them during a data acquisition run. During image acquisition, uncaging was achieved by positioning of an additional dichroic mirror and band-pass filter to illuminate the microscopic field with UV (330–300 nm) light (430 DCLPO2 dichroic; Omega Optics) from a 100 W mercury arc lamp. It was shown, using caged fluorescein as a surrogate read-out, that this procedure generated photolytic product linearly with no initial time delay. After loading with caged IP3 from its permeant ester, human neutrophils retain the ability to undergo adhesion and spontaneous spreading. In addition, other neutrophil functions, such as chemotaxis and phagocytosis were unimpaired. However, cell–cell adhesion (aggregation) in suspension was often observed to increase. This effect was attributed to the ester loading method, presumably esterase-cleavage products, rather than caged IP3, as loading with other esters (e.g. those of fluo4, fluo3, fura2) can also induce this. The Ca2+ and morphological responses reported here were also mimicked in individual neutrophils that had been microinjected with non-esterified forms of fluo4 and caged IP3 (Dewitt et al., 2003). Given that no esterase products are generated under these latter conditions, none of the phenomena reported can be attributed to these side products. From knowledge of the relative uncaging indices for caged IP3 and nitr5 (εφ) (Ellis-Davies, 2007), photolysis was estimated to released IP3 at a rate of 4 µM/second.
Quantification of Ca2+ and morphological changes
Adjustments to gain and offset were applied to whole time-series data sets to allow genuine comparisons within a time sequence. Ratio images (F/Fo) were taken to negate differences in Ca2+ probe loading, but this never revealed Ca2+ signals that were not evident in the raw fluorescent intensity data set. Cytosolic free Ca2+ was estimated using the following equation, which the Kd for fluo4 taken as 516 nM (Hillson and Hallett, 2007), Ca2+ = Kd[F−Fmin]/[Fmax−F]. Fmin and Fmax were found for Ca2+-free and Ca2+-saturating probes as described (Hillson et al., 2007). The cell morphology change during neutrophil spreading was dramatic and easily quantified in populations by counting individual cells. For time relationships, the cell contact area (footprint) was measured frame by frame using Image Master (PTI) or ImageJ software. The kinetics of cell spreading was also quantified by ‘electric cell-substrate impedance sensing’ (ECIS, Applied Biophysics), which is a non-invasive means of monitoring the kinetics of spreading of very small populations of neutrophils (about five cells per electrode) onto a 250-µm-diameter gold electrode coated with fibronectin. As neutrophils spread, they reduce the effective area of the electrode and the impedance signal rises (Wegener et al., 2000). All measurements were performed at 37°C and the final outcome was confirmed by microscopic inspection of the electrodes. For 3D reconstruction, optical xy planes were taken at z planes through cells whose plasma membrane had been stained with DiI (Invitrogen) or PKH (Sigma), and with either the nuclear membrane stained with PKH or DNA content stained with Acridine Orange (Sigma). Optical sectioning was performed in real time as the cell morphology changed, using a fast resonant scanning system (Leica RS 2) and the data later reconstructed using Imaris software (Bitplane). All data shown are representative of a number of different experimental runs from at least three different blood donors as the source of neutrophils.
Calpain measurement and manipulation
Calpain activity was assessed in neutrophils by dual labelling with fluorescein from its diacetate ester (fluorescein diacetate, Sigma) and the cell-permeant fluorogenic calpain substrate, Suc-Leu-Tyr-7-amido-4-methyl coumarin (Suc-LY-AMC). The calpain-derived product, AMC was measured by excitation at 360 nm and fluorescein by excitation at 470 nm using a rapid changing monochromator (DeltaRAM, PTI, UK) and emission >510 nm was detected using an intensified CCD camera (PTI, UK). Ratio images (360:470 nm) were acquired in real time using ImageMaster software (PTI, UK). Calpain activity was inhibited by pre-incubation (15 minutes) with either calpeptin, which inhibits the proteolytic domain, or PD150606 or PD 151746 (50 µM), which inhibit the Ca2+ activation domain.
Measurement of membrane expansion
The membrane was stained by the addition of FM1-43 (Molecular Probes) to neutrophils in suspension (8 µM) and imaged confocally (Leica RS SP2; excitation 488 nm, emission 530–560 nm) with the pinhole set open (2 AU). The dye was present throughout the experiment. In experiments with DiI, the cell membranes were pre-stained (as described above) before the addition of FM1-43.
Imaging and quantification
Confocal imaging was achieved using a resonant scanning head for the Leica RS confocal laser microscope with a 63× oil immersion objective (NA 1.32) (HCX- PL- APO). The confocal pinhole was set to 1 AU for all experiments except where FM1-43 was used, where it was adjusted to 2 AU in order to increase the thickness of the confocal slice sufficiently to monitor total cell fluorescence. Neutrophils were allowed to adhere onto the glass coverslip mounted on a thermostatically controlled stage (37±0.1°C) in Krebs medium and soluble stimuli (f-mlp) were delivered or uncaging achieved during image acquisition. Adjustments to gain and offset were applied to whole time-series data sets to allow genuine comparisons within a time sequence. ‘Raw fluorescent’ images with no post acquisition contrast enhancement or other manipulation were used for quantification. The intensity and area occupied by the spreading cell was measured using either Leica analysis software, ImageMaster analysis software (PTI) or Image J (NIH). The number of individual neutrophils analysed exceeded 50 in all experiments. All data shown were representative of at least three different experimental runs from different blood donors as the source of neutrophils.
Supplementary Material
Acknowledgments
We are grateful to members of the Neutrophil Signalling Group for their donations of blood.
Footnotes
Author contributions
S.D. performed the majority of experiments presented; R.J.F. performed the 3D reconstructions; M.B.H. and S.D. supervised the work; and M.B.H. wrote the paper.
Funding
This work was supported by the Wellcome Trust (UK) [grant number 077962/Z/06/Z to M.B.H. and S.D.]. R.J.F. was in receipt of the Cecil Prosser studentship from University of Wales. Deposited in PMC for release after 6 months.
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.124917/-/DC1
References
- Adams S. E., Parr C., Miller D. J., Allemann R. K., Hallett M. B. (2012). Potent inhibition of Ca2+-dependent activation of calpain-1 by novel ercaptoacrylates. Medchemcomm 3, 566–570 10.1039/c2md00280a [DOI] [Google Scholar]
- al-Mohanna F. A., Hallett M. B. (1990). Actin polymerization in neutrophils is triggered without a requirement for a rise in cytoplasmic Ca2+. Biochem. J. 266, 669–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleasdale J. E., Thakur N. R., Grembans R. S., Bundy G. L., FitzPatrick F. A., Smith R. J., Bunting S. (1990). Selective inhibition of receptor-coupled phospholipase C-dependent processes in human platelets and polymorphonuclear neutrophils. J. Pharmacol. Exp. Ther. 255, 756–768 [PubMed] [Google Scholar]
- Brasen J. C., Olsen L. F., Hallett M. B. (2010). Cell surface topology creates high Ca2+ signalling microdomains. Cell Calcium 47, 339–349 10.1016/j.ceca.2010.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charras G., Paluch E. (2008). Blebs lead the way: how to migrate without lamellipodia. Nat. Rev. Mol. Cell Biol. 9, 730–736 10.1038/nrm2453 [DOI] [PubMed] [Google Scholar]
- Croce K., Flaumenhaft R., Rivers M., Furie B., Furie B. C., Herman I. M., Potter D. A. (1999). Inhibition of calpain blocks platelet secretion, aggregation, and spreading. J. Biol. Chem. 274, 36321–36327 10.1074/jbc.274.51.36321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuvelier D., Théry M., Chu Y. S., Dufour S., Thiéry J. P., Bornens M., Nassoy P., Mahadevan L. (2007). The universal dynamics of cell spreading. Curr. Biol. 17, 694–699 10.1016/j.cub.2007.02.058 [DOI] [PubMed] [Google Scholar]
- Cuzzocrea S., McDonald M. C., Mazzon E., Siriwardena D., Serraino I., Dugo L., Britti D., Mazzullo G., Caputi A. P., Thiemermann C. (2000). Calpain inhibitor I reduces the development of acute and chronic inflammation. Am. J. Pathol. 157, 2065–2079 10.1016/S0002-9440(10)64845-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies-Cox E. V., Laffafian I., Hallett M. B. (2001). Control of Ca2+ influx in human neutrophils by inositol 1,4,5-trisphosphate (IP3) binding: differential effects of micro-injected IP3 receptor antagonists. Biochem. J. 355, 139–143 10.1042/0264-6021:3550139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewitt S., Hallett M. B. (2002). Cytosolic free Ca2+ changes and calpain activation are required for β integrin-accelerated phagocytosis by human neutrophils. J. Cell Biol. 159, 181–189 10.1083/jcb.200206089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewitt S., Hallett M. B. (2007). Leukocyte membrane “expansion”: a central mechanism for leukocyte extravasation. J. Leukoc. Biol. 81, 1160–1164 10.1189/jlb.1106710 [DOI] [PubMed] [Google Scholar]
- Dewitt S., Laffafian I., Hallett M. B. (2003). Phagosomal oxidative activity during β2 integrin (CR3)-mediated phagocytosis by neutrophils is triggered by a non-restricted Ca2+ signal: Ca2+ controls time not space. J. Cell Sci. 116, 2857–2865 10.1242/jcs.00499 [DOI] [PubMed] [Google Scholar]
- Dewitt S., Tian W., Hallett M. B. (2006). Localised PtdIns(3,4,5)P3 or PtdIns(3,4)P2 at the phagocytic cup is required for both phagosome closure and Ca2+ signalling in HL60 neutrophils. J. Cell Sci. 119, 443–451 10.1242/jcs.02756 [DOI] [PubMed] [Google Scholar]
- Dewitt S., Darley R. L., Hallett M. B. (2009). Translocation or just location? Pseudopodia affect fluorescent signals. J. Cell Biol. 184, 197–203 10.1083/jcb.200806047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis-Davies G. C. R. (2007). Caged compounds: photorelease technology for control of cellular chemistry and physiology. Nat. Methods 4, 619–628 10.1038/nmeth1072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson C. A., Trinkaus J. P. (1976). Microvilli and blebs as sources of reserve surface membrane during cell spreading. Exp. Cell Res. 99, 375–384 10.1016/0014-4827(76)90595-4 [DOI] [PubMed] [Google Scholar]
- Fackler O. T., Grosse R. (2008). Cell motility through plasma membrane blebbing. J. Cell Biol. 181, 879–884 10.1083/jcb.200802081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco S. J., Huttenlocher A. (2005). Regulating cell migration: calpains make the cut. J. Cell Sci. 118, 3829–3838 10.1242/jcs.02562 [DOI] [PubMed] [Google Scholar]
- Franco S. J., Rodgers M. A., Perrin B. J., Han J., Bennin D. A., Critchley D. R., Huttenlocher A. (2004). Calpain-mediated proteolysis of talin regulates adhesion dynamics. Nat. Cell Biol. 6, 977–983 10.1038/ncb1175 [DOI] [PubMed] [Google Scholar]
- Goll D. E., Thompson V. F., Li H. Q., Wei W., Cong J. Y. (2003). The calpain system. Physiol. Rev. 83, 731–801 [DOI] [PubMed] [Google Scholar]
- Hallett M. B., Dewitt S. (2007). Ironing out the wrinkles of neutrophil phagocytosis. Trends Cell Biol. 17, 209–214 10.1016/j.tcb.2007.03.002 [DOI] [PubMed] [Google Scholar]
- Herant M., Heinrich V., Dembo M. (2005). Mechanics of neutrophil phagocytosis: behavior of the cortical tension. J. Cell Sci. 118, 1789–1797 10.1242/jcs.02275 [DOI] [PubMed] [Google Scholar]
- Hillson E. J., Hallett M. B. (2007). Localised and rapid Ca2+ micro-events in human neutrophils: conventional Ca2+ puffs and global waves without peripheral-restriction or wave cycling. Cell Calcium 41, 525–536 10.1016/j.ceca.2006.10.010 [DOI] [PubMed] [Google Scholar]
- Hillson E. J., Dewitt S., Hallett M. B. (2007). Optical methods for the measurement and Manipulation of cytosolic free Ca2+ in neutrophils. Methods Mol. Biol. 412, 125–137 [DOI] [PubMed] [Google Scholar]
- Houk A. R., Jilkine A., Mejean C. O., Boltyanskiy R., Dufresne E. R., Angenent S. B., Altschuler S. J., Wu L. F., Weiner O. D. (2012). Membrane tension maintains cell polarity by confining signals to the leading edge during neutrophil migration. Cell 148, 175–188 10.1016/j.cell.2011.10.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y., Arora P., McCulloch C. A., Vogel W. F. (2009). The collagen receptor DDR1 regulates cell spreading and motility by associating with myosin IIA. J. Cell Sci. 122, 1637–1646 10.1242/jcs.046219 [DOI] [PubMed] [Google Scholar]
- Ikeda Y., Young L. H., Lefer A. M. (2002). Attenuation of neutrophil-mediated myocardial ischemia-reperfusion injury by a calpain inhibitor. Am. J. Physiol. 282, H1421–H1426 [DOI] [PubMed] [Google Scholar]
- Jaconi M. E. E., Theler J. M., Schlegel W., Appel R. D., Wright S. D., Lew P. D. (1991). Multiple elevations of cytosolic-free Ca2+ in human neutrophils: initiation by adherence receptors of the integrin family. J. Cell Biol. 112, 1249–1257 10.1083/jcb.112.6.1249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsube M., Kato T., Kitagawa M., Noma H., Fujita H., Kitagawa S. (2008). Calpain-mediated regulation of the distinct signaling pathways and cell migration in human neutrophils. J. Leukoc. Biol. 84, 255–263 10.1189/jlb.0907664 [DOI] [PubMed] [Google Scholar]
- Kay R. R., Langridge P., Traynor D., Hoeller O. (2008). Changing directions in the study of chemotaxis. Nat. Rev. Mol. Cell Biol. 9, 455–463 [DOI] [PubMed] [Google Scholar]
- Kruskal B. A., Maxfield F. R. (1987). Cytosolic free calcium increases before and oscillates during frustrated phagocytosis in macrophages. J. Cell Biol. 105, 2685–2693 10.1083/jcb.105.6.2685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruskal B. A., Shak S., Maxfield F. R. (1986). Spreading of human neutrophils is immediately preceded by a large increase in cytosolic free calcium. Proc. Natl. Acad. Sci. USA 83, 2919–2923 10.1073/pnas.83.9.2919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langridge P. D., Kay R. R. (2006). Blebbing of Dictyostelium cells in response to chemoattractant. Exp. Cell Res. 312, 2009–2017 10.1016/j.yexcr.2006.03.007 [DOI] [PubMed] [Google Scholar]
- Ley K., Laudanna C., Cybulsky M. I., Nourshargh S. (2007). Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat. Rev. Immunol. 7, 678–689 10.1038/nri2156 [DOI] [PubMed] [Google Scholar]
- Lokuta M. A., Nuzzi P. A., Huttenlocher A. (2003). Calpain regulates neutrophil chemotaxis. Proc. Natl. Acad. Sci. USA 100, 4006–4011 10.1073/pnas.0636533100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzocco S., Di Paola R., Autore G., Mazzon E., Pinto A., Caputi A. P., Thiemermann C., Cuzzocrea S. (2004). Calpain inhibitor I reduces intestinal ischemia-reperfusion injury in the rat. Shock 21, 38–44 10.1097/01.shk.0000095056.62263.b2 [DOI] [PubMed] [Google Scholar]
- Ng-Sikorski J., Andersson R., Patarroyo M., Andersson T. (1991). Calcium signaling capacity of the CD11b/CD18 integrin on human neutrophils. Exp. Cell Res. 195, 504–508 10.1016/0014-4827(91)90402-G [DOI] [PubMed] [Google Scholar]
- Noble K. E., Yong K., Khwaja A. (1998). Neutrophil transendothelial migration is regulated by the calcium dependent protease calpain. Blood 92, Suppl.12198 [Google Scholar]
- Petersen M., Williams J. D., Hallett M. B. (1993). Cross-linking of CD11b or CD18 signals release of localized Ca2+ from intracellular stores in neutrophils. Immunology 80, 157–159 [PMC free article] [PubMed] [Google Scholar]
- Pettit E. J., Hallett M. B. (1995). Early Ca2+ signalling events in neutrophils detected by rapid confocal laser scanning. Biochem. J. 310, 445–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettit E. J., Hallett M. B. (1996). Localised and global cytosolic Ca2+ changes in neutrophils during engagement of Cd11b/CD18 integrin visualised using confocal laser scanning reconstruction. J. Cell Sci. 109, 1689–1694 [DOI] [PubMed] [Google Scholar]
- Pettit E. J., Hallett M. B. (1998). Release of ‘caged’ cytosolic Ca2+ triggers rapid spreading of human neutrophils adherent via integrin engagement. J. Cell Sci. 111, 2209–2215 [DOI] [PubMed] [Google Scholar]
- Potter D. A., Tirnauer J. S., Janssen R., Croall D. E., Hughes C. N., Fiacco K. A., Mier J. W., Maki M., Herman I. M. (1998). Calpain regulates actin remodeling during cell spreading. J. Cell Biol. 141, 647–662 10.1083/jcb.141.3.647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampath R., Gallagher P. J., Pavalko F. M. (1998). Cytoskeletal interactions with the leukocyte integrin beta2 cytoplasmic tail. Activation-dependent regulation of associations with talin and alpha-actinin. J. Biol. Chem. 273, 33588–33594 10.1074/jbc.273.50.33588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith R. J., Justen J. M., McNab A. R., Rosenbloom C. L., Steele A. N., Detmers P. A., Anderson D. C., Manning A. M. (1996). U-73122: a potent inhibitor of human polymorphonuclear neutrophil adhesion on biological surfaces and adhesion-related effector functions. J. Pharmacol. Exp. Ther. 278, 320–329 [PubMed] [Google Scholar]
- Stewart M. P., McDowall A., Hogg N. (1998). LFA-1-mediated adhesion is regulated by cytoskeletal restraint and by a Ca2+-dependent protease, calpain. J. Cell Biol. 140, 699–707 10.1083/jcb.140.3.699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson J. A. (2008). Shaping cups into phagosomes and macropinosomes. Nat. Rev. Mol. Cell Biol. 9, 639–649 10.1038/nrm2447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tissier S., Lancel S., Marechal X., Mordon S., Depontieu F., Scherpereel A., Chopin C., Neviere R. (2004). Calpain inhibitors improve myocardial dysfunction and inflammation induced by endotoxin in rats. Shock 21, 352–357 10.1097/00024382-200404000-00010 [DOI] [PubMed] [Google Scholar]
- Weber M., Treanor B., Depoil D., Shinohara H., Harwood N. E., Hikida M., Kurosaki T., Batista F. D. (2008). Phospholipase C-γ2 and Vav cooperate within signaling microclusters to propagate B cell spreading in response to membrane-bound antigen. J. Exp. Med. 205, 853–868 10.1084/jem.20072619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegener J., Keese C. R., Giaever I. (2000). Electric cell-substrate impedance sensing (ECIS) as a noninvasive means to monitor the kinetics of cell spreading to artificial surfaces. Exp. Cell Res. 259, 158–166 10.1006/excr.2000.4919 [DOI] [PubMed] [Google Scholar]
- Yoshida K., Soldati T. (2006). Dissection of amoeboid movement into two mechanically distinct modes. J. Cell Sci. 119, 3833–3844 10.1242/jcs.03152 [DOI] [PubMed] [Google Scholar]
- Yoshifuji H., Umehara H., Maruyama H., Itoh M., Tanaka M., Kawabata D., Fujii T., Mimori T. (2005). Amelioration of experimental arthritis by a calpain-inhibitory compound: regulation of cytokine production by E-64-d in vivo and in vitro. Int. Immunol. 17, 1327–1336 10.1093/intimm/dxh311 [DOI] [PubMed] [Google Scholar]
- Zhang X., Jiang G., Cai Y., Monkley S. J., Critchley D. R., Sheetz M. P. (2008). Talin depletion reveals independence of initial cell spreading from integrin activation and traction. Nat. Cell Biol. 10, 1062–1068 10.1038/ncb1765 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.