

Abstract
Functional elucidation of causal genetic variants and elements requires precise genome editing technologies. The type II prokaryotic CRISPR (clustered regularly interspaced short palindromic repeats)/Cas adaptive immune system has been shown to facilitate RNA-guided site-specific DNA cleavage. We engineered two different type II CRISPR/Cas systems and demonstrate that Cas9 nucleases can be directed by short RNAs to induce precise cleavage at endogenous genomic loci in human and mouse cells. Cas9 can also be converted into a nicking enzyme to facilitate homology-directed repair with minimal mutagenic activity. Lastly, multiple guide sequences can be encoded into a single CRISPR array to enable simultaneous editing of several sites within the mammalian genome, demonstrating easy programmability and wide applicability of the RNA-guided nuclease technology.
Precise and efficient genome-targeting technologies are needed to enable systematic reverse engineering of causal genetic variations by allowing selective perturbation of individual genetic elements. Although genome-editing technologies such as designer zinc fingers (ZFs) (1–4), transcription activator–like effectors (TALEs) (4–10), and homing meganucleases (11) have begun to enable targeted genome modifications, there remains a need for new technologies that are scalable, affordable, and easy to engineer. Here, we report the development of a class of precision genome-engineering tools based on the RNA-guided Cas9 nuclease (12–14) from the type II prokaryotic clustered regularly interspaced short palindromic repeats (CRISPR) adaptive immune system (15–18).
The Streptococcus pyogenes SF370 type II CRISPR locus consists of four genes, including the Cas9 nuclease, as well as two noncoding CRISPR RNAs (crRNAs): trans-activating crRNA (tracrRNA) and a precursor crRNA (pre-crRNA) array containing nuclease guide sequences (spacers) interspaced by identical direct repeats (DRs) (fig. S1) (19). We sought to harness this prokaryotic RNA-programmable nuclease system to introduce targeted double-stranded breaks (DSBs) in mammalian chromosomes through heterologous expression of the key components. It has been previously shown that expression of tracrRNA, pre-crRNA, host factor ribonuclease (RNase) III, and Cas9 nuclease is necessary and sufficient for cleavage of DNA in vitro (12, 13) and in prokaryotic cells (20, 21). We codon-optimized the S. pyogenes Cas9 (SpCas9) and RNase III (SpRNase III) genes and attached nuclear localization signals (NLSs) to ensure nuclear compartmentalization in mammalian cells. Expression of these constructs in human 293FT cells revealed that two NLSs are most efficient at targeting SpCas9 to the nucleus (Fig. 1A). To reconstitute the non-coding RNA components of the S. pyogenes type II CRISPR/Cas system, we expressed an 89-nucleotide (nt) tracrRNA (fig. S2) under the RNA polymerase III U6 promoter (Fig. 1B). Similarly, we used the U6 promoter to drive the expression of a pre-crRNA array comprising a single guide spacer flanked by DRs (Fig. 1B). We designed our initial spacer to target a 30–base pair (bp) site (protospacer) in the human EMX1 locus that precedes an NGG trinucleotide, the requisite protospacer-adjacent motif (PAM) (Fig. 1C and fig. S1) (22, 23).
Fig. 1.
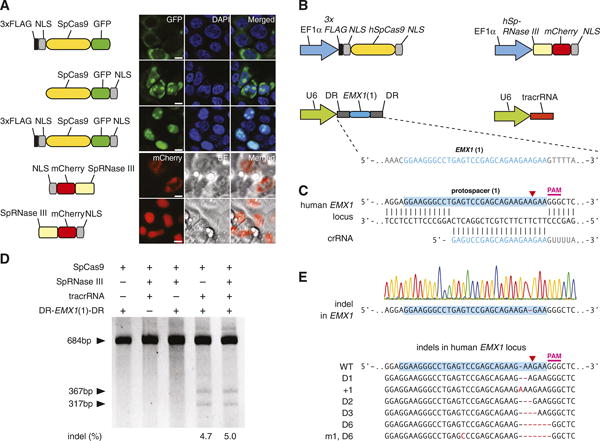
The type II CRISPR locus from S. pyogenes SF370 can be reconstituted in mammalian cells to facilitate targeted DSBs of DNA. (A) Engineering of SpCas9 and SpRNase III with NLSs enables import into the mammalian nucleus. GFP indicates green fluorescent protein; scale bars, 10 μm. (B) Mammalian expression of human codon–optimized SpCas9 (hSpCas9) and SpRNase III (hSpRNase III) genes were driven by the elongation factor 1α (EF1 α) promoter, whereas tracrRNA and pre-crRNA array (DR-Spacer-DR) were driven by the U6 promoter. A protospacer (blue highlight) from the human EMX1 locus with PAM was used as template for the spacer in the pre-crRNA array. (C) Schematic representation of base pairing between target locus and EMX1-targeting crRNA. Red arrow indicates putative cleavage site. (D) SURVEYOR assay for SpCas9-mediated indels. (E) An example chromatogram showing a microdeletion, as well as representative sequences of mutated alleles identified from 187 clonal amplicons. Red dashes, deleted bases; red bases, insertions or mutations.
To test whether heterologous expression of the CRISPR system (SpCas9, SpRNase III, tracrRNA, and pre-crRNA) can achieve targeted cleavage of mammalian chromosomes, we transfected 293FT cells with different combinations of CRISPR/Cas components. Because DSBs in mammalian DNA are partially repaired by the indel-forming non-homologous end joining (NHEJ) pathway, we used the SURVEYOR assay (fig. S3) to detect endogenous target cleavage (Fig. 1D and fig. S2B). Cotransfection of all four required CRISPR components resulted in efficient cleavage of the protospacer (Fig. 1D and fig. S2B), which was subsequently verified by Sanger sequencing (Fig. 1E). SpRNase III was not necessary for cleavage of the protospacer (Fig. 1D), and the 89-nt tracrRNA is processed in its absence (fig. S2C). Similarly, maturation of pre-crRNA does not require RNase III (Fig. 1D and fig. S4), suggesting that there may be endogenous mammalian RNases that assist in pre-crRNA maturation (24–26). Removing any of the remaining RNA or Cas9 components abolished the genome cleavage activity of the CRISPR/Cas system (Fig. 1D). These results define a minimal three-component system for efficient RNA-guided genome modification in mammalian cells.
Next, we explored the generalizability of RNA-guided genome editing in eukaryotic cells by targeting additional protospacers within the EMX1 locus (Fig. 2A). To improve codelivery, we designed an expression vector to drive both pre-crRNA and SpCas9 (fig. S5). In parallel, we adapted a chimeric crRNA-tracrRNA hybrid (Fig. 2B, top) design recently validated in vitro (12), where a mature crRNA is fused to a partial tracrRNA via a synthetic stem loop to mimic the natural crRNA:tracrRNA duplex (Fig. 2B, bottom). We observed cleavage of all protospacer targets when SpCas9 is coexpressed with pre-crRNA (DR-spacer-DR) and tracrRNA. However, not all chimeric RNA designs could facilitate cleavage of their genomic targets (Fig. 2C and table S1). We then tested targeting of additional genomic loci in both human and mouse cells by designing pre-crRNAs and chimeric RNAs targeting the human PVALB and the mouse Th loci (fig. S6). We achieved efficient modification at all three mouse Th and one PVALB targets by using the crRNA:tracrRNA duplex, thus demonstrating the broad applicability of the CRISPR/Cas system in modifying different loci across multiple organisms (table S1). For the same protospacer targets, cleavage efficiencies of chimeric RNAs were either lower than those of crRNA:tracrRNA duplexes or undetectable. This may be due to differences in the expression and stability of RNAs, degradation by endogenous RNA interference machinery, or secondary structures leading to inefficient Cas9 loading or target recognition.
Fig. 2.
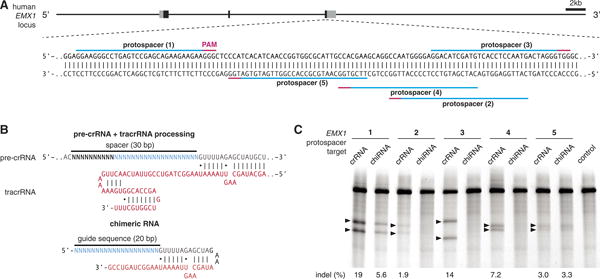
SpCas9 can be reprogrammed to target multiple genomic loci in mammalian cells. (A) Schematic of the human EMX1 locus showing the location of five protospacers indicated by blue lines with corresponding PAM in magenta. (B) Schematic of the pre-crRNA:tracrRNA complex (top) showing hybridization between the direct repeat (gray) region of the pre-crRNA and tracrRNA. Schematic of a chimeric RNA design (12) (bottom). tracrRNA sequence is shown in red and the 20-bp spacer sequence in blue. (C) SURVEYOR assay comparing the efficacy of Cas9-mediated cleavage at five protospacers in the human EMX1 locus. Each protospacer was targeted by using either processed pre-crRNA:tracrRNA complex (crRNA) or chimeric RNA (chiRNA). Arrowheads indicate cleavage products for each protospacer target.
Effective genome editing requires that nucleases target specific genomic loci with both high precision and efficiency. To investigate the specificity of RNA-guided genome modification, we analyzed single-nucleotide mismatches between the spacer and its mammalian protospacer target (Fig. 3A). We observed that single-base mismatch up to 11 bp 5′ of the PAM completely abolished genomic cleavage by SpCas9, whereas spacers with mutations farther upstream retained activity against the protospacer target (Fig. 3B). This is consistent with previous bacterial and in vitro studies of Cas9 specificity (12, 20). Furthermore, SpCas9 is able to mediate genomic cleavage as efficiently as a pair of TALE nucleases (TALENs) targeting the same EMX1 protospacer (Fig. 3, C and D).
Fig. 3.
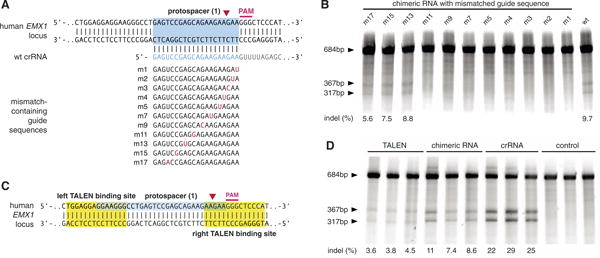
Evaluation of the SpCas9 specificity and comparison of efficiency with TALENs. (A) EMX1-targeting chimeric crRNAs with single point mutations were generated to evaluate the effects of spacer-protospacer mismatches. (B) SURVEYOR assay comparing the cleavage efficiency of different mutant chimeric RNAs. (C) Schematic showing the design of TALENs that target EMX1. (D) SURVEYOR gel comparing the efficiency of TALEN and SpCas9 (N = 3).
Targeted modification of genomes ideally avoids mutations arising from the error-prone NHEJ mechanism. The wild-type SpCas9 is able to mediate site-specific DSBs, which can be repaired through either NHEJ or homology-directed repair (HDR). We engineered an aspartate-to-alanine substitution (D10A) in the RuvC I domain of SpCas9 to convert the nuclease into a DNA nickase (SpCas9n, Fig. 4A) (12, 13, 20), because nicked genomic DNA is typically repaired either seamlessly or through high-fidelity HDR. SURVEYOR (Fig. 4B) and sequencing of 327 amplicons did not detect any indels induced by SpCas9n. However, nicked DNA can in rare cases be processed via a DSB intermediate and result in a NHEJ event (27). We then tested Cas9-mediated HDR at the same EMX1 locus with a homology repair template to introduce a pair of restriction sites near the protospacer (Fig. 4C). SpCas9 and SpCas9n catalyzed integration of the repair template into EMX1 locus at similar levels (Fig. 4D), which we further verified via Sanger sequencing (Fig. 4E). These results demonstrate the utility of CRISPR for facilitating targeted genomic insertions. Given the 14-bp (12 bp from the seed sequence and 2 bp from PAM) target specificity (Fig. 3B) of the wild-type SpCas9, the use of a nickase may reduce off-target mutations.
Fig. 4.
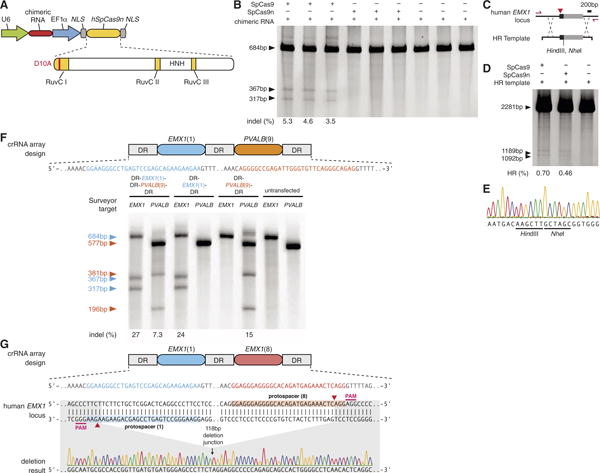
Applications of Cas9 for homologous recombination and multiplex genome engineering. (A) Mutation of the RuvC I domain converts Cas9 into a nicking enzyme (SpCas9n). HNH, histidine-asparagine-histidine endonuclease domain. (B) Coexpression of EMX1-targeting chimeric RNA with SpCas9 leads to indels, whereas SpCas9n does not (N = 3). (C) Schematic representation of the recombination strategy. A homology repair (HR) template is designed to insert restriction sites into EMX1 locus. Primers used to amplify the modified region are shown as red arrows. (D) Restriction fragment length polymorphism gel analysis. Arrows indicate fragments generated by HindIII digestion. (E) Example chromatogram showing successful recombination. (F) SpCas9 can facilitate multiplex genome modification by using a crRNA array that contains two spacers targeting EMX1 and PVALB. Schematic showing the design of the crRNA array (top). Both spacers mediate efficient protospacer cleavage (bottom). (G) SpCas9 can be used to achieve precise genomic deletion. Two spacers targeting EMX1 (top) mediated a 118-bp genomic deletion (bottom).
Lastly, the natural architecture of CRISPR loci with arrayed spacers (fig. S1) suggests the possibility of multiplexed genome engineering. By using a single CRISPR array encoding a pair of EMX1- and PVALB-targeting spacers, we detected efficient cleavage at both loci (Fig. 4F). We further tested targeted deletion of larger genomic regions through concurrent DSBs by using spacers against two targets within EMX1 spaced by 119 bp and observed a 1.6% deletion efficacy (3 out of 182 amplicons, Fig. 4G), thus demonstrating the CRISPR/Cas system can mediate multiplexed editing within a single genome.
The ability to use RNA to program sequence-specific DNA cleavage defines a new class of genome engineering tools. Here, we have shown that the S. pyogenes CRISPR system can be heterologously reconstituted in mammalian cells to facilitate efficient genome editing; an accompanying study has independently confirmed high-efficiency RNA-guided genome targeting in several human cell lines (28). However, several aspects of the CRISPR/Cas system can be further improved to increase its efficiency and versatility. The requirement for an NGG PAM restricts the target space of SpCas9 to every 8 bp on average in the human genome (fig. S7), not accounting for potential constraints posed by crRNA secondary structure or genomic accessibility resulting from chromatin and DNA methylation states. Some of these restrictions may be overcome by exploiting the family of Cas9 enzymes and its differing PAM requirements (22, 23) across the microbial diversity (17). Indeed, other CRISPR loci are likely to be transplantable into mammalian cells; for example, the Streptococcus thermophilus LMD-9 CRISPR1 system can also mediate mammalian genome cleavage (fig. S8). Lastly, the ability to carry out multiplex genome editing in mammalian cells enables powerful applications across basic science, biotechnology, and medicine (29).
Supplementary Material
Acknowledgments
We thank the entire Zhang lab for their support and advice; P. A. Sharp for generous help with Northern blot analysis; C. Jennings, R. Desimone, and M. Kowalczyk for helpful comments; and X. Ye for help with confocal imaging. L.C. and X.W. are Howard Hughes Medical Institute International Student Research Fellows. D.C. is supported by the Medical Scientist Training Program. P.D.H. is a James Mills Pierce Fellow. X.W. is supported by NIH grants R01-GM34277 and R01-CA133404 to P. A. Sharp, X.W.'s thesis adviser. L.A.M. is supported by Searle Scholars, R. Allen, an Irma T. Hirschl Award, and a NIH Director's New Innovator Award (DP2AI104556). F.Z. is supported by a NIH Director's Pioneer Award (DP1MH100706); the Keck, McKnight, Gates, Damon Runyon, Searle Scholars, Klingenstein, and Simons foundations; R. Metcalfe; M. Boylan; and J. Pauley. The authors have no conflicting financial interests. A patent application has been filed relating to this work, and the authors plan on making the reagents widely available to the academic community through Addgene and to provide software tools via the Zhang lab Web site (www.genome-engineering.org).
Footnotes
References and Notes
- 1.Porteus MH, Baltimore D. Science. 2003;300:763. doi: 10.1126/science.1078395. [DOI] [PubMed] [Google Scholar]
- 2.Miller JC, et al. Nat Biotechnol. 2007;25:778. doi: 10.1038/nbt1319. [DOI] [PubMed] [Google Scholar]
- 3.Sander JD, et al. Nat Methods. 2011;8:67. doi: 10.1038/nmeth.1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wood AJ, et al. Science. 2011;333:307. doi: 10.1126/science.1207773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Christian M, et al. Genetics. 2010;186:757. doi: 10.1534/genetics.110.120717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang F, et al. Nat Biotechnol. 2011;29:149. doi: 10.1038/nbt.1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miller JC, et al. Nat Biotechnol. 2011;29:143. doi: 10.1038/nbt.1755. [DOI] [PubMed] [Google Scholar]
- 8.Reyon D, et al. Nat Biotechnol. 2012;30:460. doi: 10.1038/nbt.2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boch J, et al. Science. 2009;326:1509. doi: 10.1126/science.1178811. [DOI] [PubMed] [Google Scholar]
- 10.Moscou MJ, Bogdanove AJ. Science. 2009;326:1501. doi: 10.1126/science.1178817. [DOI] [PubMed] [Google Scholar]
- 11.Stoddard BL. Q Rev Biophys. 2005;38:49. doi: 10.1017/S0033583505004063. [DOI] [PubMed] [Google Scholar]
- 12.Jinek M, et al. Science. 2012;337:816. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gasiunas G, Barrangou R, Horvath P, Siksnys V. Proc Natl Acad Sci USA. 2012;109:E2579. doi: 10.1073/pnas.1208507109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Garneau JE, et al. Nature. 2010;468:67. doi: 10.1038/nature09523. [DOI] [PubMed] [Google Scholar]
- 15.Deveau H, Garneau JE, Moineau S. Annu Rev Microbiol. 2010;64:475. doi: 10.1146/annurev.micro.112408.134123. [DOI] [PubMed] [Google Scholar]
- 16.Horvath P, Barrangou R. Science. 2010;327:167. doi: 10.1126/science.1179555. [DOI] [PubMed] [Google Scholar]
- 17.Makarova KS, et al. Nat Rev Microbiol. 2011;9:467. doi: 10.1038/nrmicro2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bhaya D, Davison M, Barrangou R. Annu Rev Genet. 2011;45:273. doi: 10.1146/annurev-genet-110410-132430. [DOI] [PubMed] [Google Scholar]
- 19.Deltcheva E, et al. Nature. 2011;471:602. doi: 10.1038/nature09886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sapranauskas R, et al. Nucleic Acids Res. 2011;39:9275. doi: 10.1093/nar/gkr606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Magadán AH, Dupuis ME, Villion M, Moineau S. PLoS ONE. 2012;7:e40913. doi: 10.1371/journal.pone.0040913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Deveau H, et al. J Bacteriol. 2008;190:1390. doi: 10.1128/JB.01412-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mojica FJ, Díez-Villaseñor C, García-Martínez J, Almendros C. Microbiology. 2009;155:733. doi: 10.1099/mic.0.023960-0. [DOI] [PubMed] [Google Scholar]
- 24.Jinek M, Doudna JA. Nature. 2009;457:405. doi: 10.1038/nature07755. [DOI] [PubMed] [Google Scholar]
- 25.Malone CD, Hannon GJ. Cell. 2009;136:656. doi: 10.1016/j.cell.2009.01.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Meister G, Tuschl T. Nature. 2004;431:343. doi: 10.1038/nature02873. [DOI] [PubMed] [Google Scholar]
- 27.Certo MT, et al. Nat Methods. 2011;8:671. doi: 10.1038/nmeth.1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mali P, et al. Science. 2013;339:823. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carr PA, Church GM. Nat Biotechnol. 2009;27:1151. doi: 10.1038/nbt.1590. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.