

Abstract
BACKGROUND
Current immunoassays for the chemokine RANTES (regulated on activation, normal T-cell expressed and secreted) are not tailored for specific isoforms that exist endogenously, despite the fact that variants with modified activity are known to exist. This is surprising in view of this protein’s ubiquitous increased presence in many diseases and that the 2 established isoforms are truncated by enzymes also correlated to disease. An in-depth population survey of RANTES heterogeneity in the context of multiple diseases via a mass spectrometric immunoassay (MSIA) may resolve this issue.
METHODS
We developed an MSIA for RANTES and endogenous variants apparent in human plasma. Samples from multiple cohorts of individuals (type 2 diabetes, congestive heart failure, history of myocardial infarction, and cancer patients) were run in parallel with samples from healthy individuals (239 people total). We used 230 μL of plasma per individual and tabulated relative percent abundance (RPA) values for identified isoforms.
RESULTS
We detected at least 19 variants, including the dipeptidyl peptidase IV (DPP-IV)-truncated variant. The majority of variants were unreported in the literature. Identifiable modifications included N- and/or C-terminal truncations, oxidation, glycation, and glycosylation. We observed statistically significant differences in RPA values for multiple variants between disease cohorts and recognized prospective disease-specific protein profiles for RANTES.
CONCLUSIONS
Because of widespread interest in the clinical value of RANTES, the protein diversity established here may aid in the design of future, fully quantitative assays. Equally important, an inclusive qualitative understanding of RANTES heterogeneity may present new insights into the relationship between RANTES and disease.
Chemokines are essential to the initiation and maintenance of inflammation. They perform their duty via the mechanisms of chemotaxis, whereby leukocytes are attracted to higher gradients of chemokines. More than 40 chemokines and 20 chemokine receptors have been identified. The chemokine superfamily is made up of 4 main groups (C, CC, CXC, and CX3C), based on the position of the first 2 cysteine residues in a conserved 4-cysteine motif found pervasively among chemokines (RANTES is a CC chemokine, hence its alternative name: CCL5) (1). The receptors are G-protein coupled, 7-transmembrane receptors named accordingly to the class of chemokine they bind (e.g., RANTES/CCL5 binds to the CCRX receptor subset) (2). Proinflammatory chemokine receptors tend to have less discriminative ligand-binding specificities, whereas receptors involved in normal leukocyte trafficking have higher specificities (2–4). RANTES2 (which stands for regulated on activation, normal T-cell expressed and secreted) binds to CCR1, CCR3, CCR5, and DARC receptors and correspondingly has an integral role in inflammation, cell recruitment, and T-cell activation. RANTES can be released from multiple sources, including activated T lymphocytes and monocytes/macrophages, epithelial cells, bronchial epithelium, and dermal fibroblast and renal tubular epithelium (5). The native wild type is composed of 68 amino acids and has a molecular weight of 7847 Da with both disulfide bonds intact (notably, the signal-containing protein is 91 aa in size). To date, there are 2 well-established RANTES variants: truncated versions made up of residues [3–68] and [4–68]. These variants are the products of 2 separate regulatory enzymes [dipeptidyl peptidase IV (DPP-IV) and cathepsin G, respectively] and have modified chemotactic and antiviral functionality compared with that of intact RANTES (6, 7). Furthermore, there is evidence that an O-linked glycosylated RANTES variant may exist (8). Consequently, research concerned with RANTES, which was discovered in 1988 via a cDNA study of T cells (9), has in a short amount of time begun to unravel a molecule of depth that appears to be connected to an array of pathophysiological conditions that arise in humans.
The clinical significance of RANTES is considerable, as it has been associated with many diseases including kidney-related complications (e.g., renal failure and renal cancer), autoimmune diseases (e.g., arthritis, diabetes, and glomerulonephritis), sepsis-induced disseminated intravascular coagulation in infants, and several forms of carcinoma including breast and cervical cancer (5, 10–12). Indeed, plasma RANTES concentrations have been found to be increased in order of cancer stage (I, II, III, or IV) (5). Furthermore, a large population study found higher plasma concentrations of RANTES in subjects with type 2 diabetes (T2D) and impaired glucose tolerance (IGT) relative to healthy controls (13). Concerning its role in viral disease, RANTES was discovered to be an effective antiviral agent that restricts the entry of CCR5-tropic HIV-1 strains by means of the CCR5 receptor (6). The [3–68] variant form of RANTES was subsequently found to also inhibit HIV to the same degree but to lack the same binding affinity to the CCR1 receptor. Thus, the impact of RANTES microheterogeneity in biological systems is as important as its quantitative fluctuation.
Commercially available RANTES assays exist widely as ELISAs and appear to be the primary tool for non-variant-specific quantification of plasma RANTES in disease-related studies (5, 14, 15). For reference, 1 such assay has a sensitivity of 2 ng/L, a measuring range of 31.2-2000 ng/L, and intraassay and interassay CVs of 3.6% and 10.3%, respectively (15). Healthy circulating plasma concentrations appear to be around 3 μg/L (5). The ELISA approach lacks the intrinsic ability to differentiate between wild-type and variant forms of RANTES. The consideration of microheterogeneity, however, may help to explain potential measurement discrepancies between assays. This additional element of multiplexed variant detectability enables a view of the genetic and posttranslational variants of proteins, which may have significant diagnostic value in personalized medicine. Indeed, as McIntosh wrote, “Researchers should be encouraged to evaluate and report as a secondary or even primary analysis the natural variation of the proteome in the clinical bio-samples they analyze” (16).
Thus, future quantitative studies of RANTES stand to benefit from a more complete survey of qualitatively different variants of RANTES as they exist in healthy and diseased populations. Toward this end, this report focuses on mapping the structural differences of RANTES found in a variety of diseases. As a result, we provide an alternative qualitative and semiquantitative method for high-throughput population-based analysis of RANTES that may assist in expanding our understanding of this molecule’s complex nature in biological systems.
Materials and Methods
STUDY SUBJECTS AND SAMPLE COLLECTION AND PREPARATION
We prepared and derivatized 1,1′-carbonyldiimidazole (CDI)-activated affinity pipette tips with mouse monoclonal antihuman RANTES antibody (R&D Systems), as described for other antibodies (17). For the development of the assay, we used bulk human plasma from a healthy female donor. We acquired 239 additional human samples under institutional review board approval and grouped them into nonoverlapping cohorts as follows: samples from 37 healthy individuals (plasma); 29 healthy individuals (serum); 50 T2D patients (plasma); 25 individuals with congestive heart failure (CHF) and T2D (plasma); 17 individuals with CHF, a history of myocardial infarction (hMI), and T2D (plasma); 25 individuals with CHF and hMI (plasma); 29 individuals with CHF only (plasma); and 27 individuals with cancer (prostate, breast, and colon cancer) (plasma). In all plasma samples, EDTA was used as the anticoagulant. Cohorts were age and sex matched. We pretreated 230 μL human plasma with 115 μL of a detergent solution containing 4.5% Tween 20, 150 mmol/L octyl-β-glucopyranoside, 1.5 mol/L ammonium acetate, and concentrated PBS (0.67 mol/L sodium phosphate, 1 mol/L sodium chloride), for a total analytical volume of 345 μL. We immobilized negative control mass spectrometric immunoassay (MSIA) tips with monoclonal antihuman C-peptide and insulin antibody (AbD Serotec).
PLASMA MASS SPECTROMETRIC IMMUNOASSAYS
RANTES was extracted with the aid of a Beckman Multimek 96 pipetting robot by repeatedly (500 repetitions) drawing and expelling (back into the analytical volume) 125 μL aliquots of the analytical volume through an anti-RANTES affinity pipette. After extraction, we rinsed the pipettes with HBS-P (0.01 mol/L HEPES, 0.15 mol/L NaCl, 0.05% vol/vol surfactant P20, pH 7.4), H2O, 100 mmol/L Tris hydrochloride (pH 4.6), and H2O (in this order, each rinse consisting of 10 repetitions of 150 μL), after which RANTES was eluted and prepared for MALDI-TOF MS by drawing 4 μL MALDI matrix solution [saturated aqueous solution of sinapic acid, 33% (vol/vol) acetonitrile, 0.45% (vol/vol) trifluoroacetic acid (TFA)] into the pipette and depositing onto a MALDI target.
MASS SPECTROMETRY
We performed MALDI-TOF mass spectrometry (MS) by use of a Bruker Ultraflex III MALDI-TOF instrument operating in the positive-ion, delayed-extraction mode; linear mode with ion source 1 at 25.00 kV, ion source 2 at 23.50 kV, lens at 6.00 kV, 50 ns delayed extraction, deflection signal suppression up to m/z 3000, and 1 GS/s sample rate. Single measurements were acquired, per individual, using 20 000 laser shots signal-averaged to ensure good ion-counting statistics. We externally calibrated the spectra with a mixture of 4 proteins supplied by Bruker (cat. no. 208241), ranging from m/z 5734.52 (insulin [M + H]+) to m/z 12360.97 (cytochrome C [M+H]+).
DATA ANALYSIS
Individual mass spectra were baseline subtracted (Tophat algorithm) and smoothed (SavitzkyGolay algorithm; width = 0.2 m/z; cycles = 1) before peak integration by use of Bruker Daltonics flexAnalysis 3.0. Peaks representing intact RANTES and RANTES variants were integrated (by use of Intrinsic Bioprobes Inc. Zebra 1.0) and tabulated in a spreadsheet for determination of relative percent abundances.
DUAL EXTRACTION
We extracted RANTES as described above followed by elution into a new sample reservoir. We then extracted this new reservoir as if it were a plasma sample itself, generating a high-purity sample with minimal nonspecific protein binding and an exceptionally clean MALDI-TOF MS spectrum of RANTES. We performed the dual extraction by extracting 20 tips using the normal sample preparation (with the exception of using 1 mL plasma and 500 μL detergent solution per sample) followed by MSIA tip elution with 7 μL of 33% (vol/vol) acetonitrile and 0.45% (vol/vol) TFA into an Eppendorf tube containing 1 mL HBS-N (0.01 mol/L HEPES and 0.15 mol/L NaCl) and 10 μL of 3.9 g/L prealbumin antibody (Dako). The antibody was used as a carrier protein. This enriched sample was subsequently extracted using a new anti-RANTES affinity pipette and eluted as described above. We repeated the process exactly using antihuman insulin antibody-immobilized tips to serve as a negative control.
Results and Discussion
Fig. 1 shows an MSIA spectrum that is qualitatively representative of those obtained for the individuals investigated in this study (the spectrum is a resized version of that seen in Supplemental Fig. 1B, which accompanies the online version of this article at http://www.clinchem.org/content/vol56/issue9). Along with intact RANTES, the following variants were identified: 2 N-terminally truncated forms (maroon; II and III), 1 C-terminally truncated form (pink; V), 7 C- and N-terminally truncated forms (blue; IV, VI, VIII, and X-XIII), glycated forms representing intact and [3–68] (purple; XIVI and XIVII), oxidized forms (+16 m/z, unlabeled), and 4 glycosylated forms (green; XVI–XIX) (Fig. 1 and Table 1), for a total of at least 19 variants. After fine-tuning to ensure that all MS signals were specific to RANTES via a dual extraction (described below), we tentatively mass-mapped the ions represented in Fig. 1. Most of these peaks were found in the majority of the samples. With regard to specificity of peak assignments, this type of technique maybe viewed as analogous to an ultra-high-resolution Western blot that is performed hundreds of times with consistent results. Notably, peaks I–XIX were not observed in control experiments using blank tips (no antibody immobilized), antihuman C-peptide MSIA tips (single extraction), or antihuman insulin MSIA tips (single and dual extraction). Additional data on antibody specificity is included in the online Supplemental Information.
Fig. 1. Mass spectrum resulting from the targeted top-down analysis of RANTES (from online Supplemental Fig. 1B).
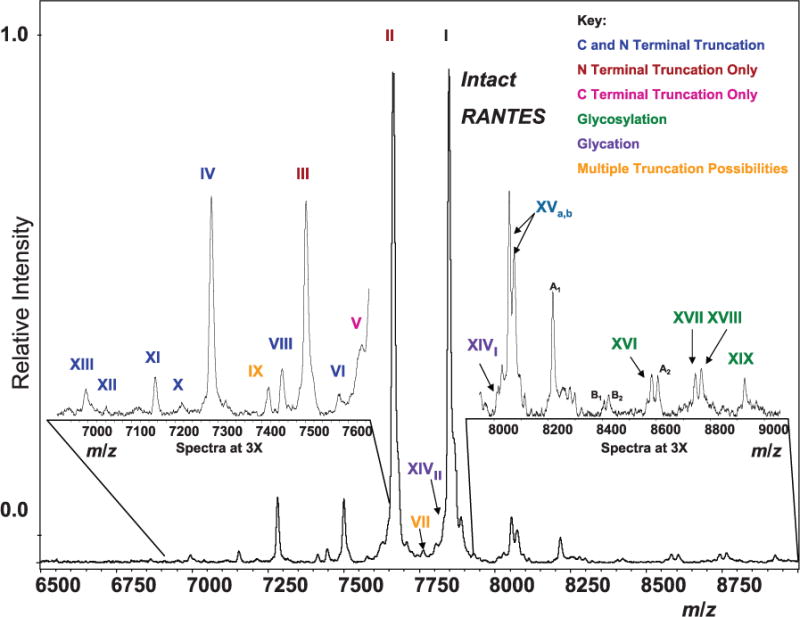
Indicated are signals from the ~19 major protein isoforms. Isoforms fall into 6 categories: C- and N-terminal truncation, N-terminal truncation only, C-terminal truncation only, multiple truncation possibilities (owing to identical masses), glycosylation, and glycation. Table 1 lists the identities of each variant. A1 and A2, signals that appeared in negative control experiments and therefore may not be related to RANTES (8215.4 and 8603.5 m/z); B1 and B2, signals that may be related to RANTES but are unknown (8404.28 and 8421.33 m/z).
Table 1.
RANTES variants evident in human plasma.a
| Isoforms | Observed [M+H]+ | Theoretical [M+H]+ | Identity |
|---|---|---|---|
| Intact | |||
| I | 7848.0 | 7848.0 | [1–68] |
| C- and N-terminal truncation | |||
| VI | 7576.3 | 7576.7 | [3–67] |
| VIII | 7445.1 | 7445.5 | [3–66] |
| IV | 7282.2 | 7282.3 | [4–66] |
| X | 7213.4 | 7213.3 | [2–63] |
| XI | 7153.0 | 7153.2 | [4–65] |
| XII | 7039.9 | 7040.1 | [4–64] |
| XIII | 6992.9 | 6993.1 | [7–66] |
| N-terminal truncation only | |||
| II | 7663.8 | 7663.8 | [3–68] |
| III | 7500.6 | 7500.6 | [4–68] |
| C-terminal truncation only | |||
| V | 7629.4 | 7629.7 | [1–66] |
| Multiple truncation possibilities | |||
| IX | 7413.3 | 7413.5 | [2–65], [4–67], or [5–68] |
| VII | 7761.6 | 7760.9 | [1–67] or [2–68] |
| Glycation | |||
| XIVII | 7826.4 | 7825.9 | [3–68] + Hex |
| XIVI | 8010.6 | 8010.1 | [1–68] + Hex |
| Glycosylation | |||
| XVI | 8580.6 | 8581.7 | [3–68] + Hex1 HexNAc3Deoxyhex1 |
| XVII | 8741.8 | 8743.8 | [3–68] + Hex2HexNAc3Deoxyhex1 |
| XVIII | 8764.5 | 8765.9 | [1–68] + Hex1 HexNAc3Deoxyhex1 |
| XIX | 8925.2 | 8928.0 | [1–68] + Hex2HexNAc3Deoxyhex1 |
| Oxidation | |||
| Unlabeled | 7863.8 | 7864.0 | [3–68] + oxidation |
| Unlabeled | 7679.4 | 7679.8 | [1–68] + oxidation |
| Matrix adducts | |||
| XVa | 8054.1 | 8054.2 | [1–68] + sinapic acid |
| XVb | 8071.9 | 8072.2 | [1–68] + sinapic acid |
Roman numerals correspond to labeled peaks in Fig. 1.
To ensure accurate mass-mapping assignments, we designed the development of a secondary extraction process (dual extraction) to support the removal of nonspecifically bound proteins and create an exemplar full-scan spectral view of endogenous RANTES microheterogeneity in humans. Typically, 1 MSIA affinity pipette is used to extract a protein from a single plasma sample, which is then rinsed and immediately eluted. Here, we eluted many affinity pipettes from pooled human plasma into a single buffered solution to create a sample with much lower nonspecific protein concentrations and significantly higher RANTES concentration. This consequently created a vastly cleaner MSIA MALDI-TOF MS spectrum with amplified signals corresponding to the weakest endogenous RANTES species present within humans (see online Supplemental Information).
Relative percent abundance (RPA) values of isoforms can be obtained after integrating all mass spectral peak areas (previously determined by a verified protein variant map, e.g., Fig. 1), followed by dividing the peak area of each isoform by the summed areas of all isoforms and finally multiplying by 100. To test the reproducibility of this form of semiquantification, we ran a single sample 96 times (i.e., 96 MSIA tips extracted RANTES from 96 samples from the same individual). In this test, RANTES variants [1–68], [3–68], [4–68], and [4–66] had RPA values (SDs) of 77.82% (0.53%), 20.3% (0.49%), 1.26% (0.07%), and 0.62% (0.05%). The ranges for these variants were observed to be 77.08%–79.60%, 18.67%–20.97%, 1.08%–1.43%, and 0.51%–0.76%, respectively. After establishing a map of RANTES heterogeneity, we scrutinized individual classes of posttranslational modifications in more detail as follows.
TRUNCATION
RANTES was found consistently in a multitude of N-and C-terminally truncated forms (10 individual truncated isoforms were identified). The 2 previously known truncated variants, [3–68] (II) and [4–68] (III), are generated by 2 separate regulatory enzymes: DPP-IV and cathepsin G (6, 7).RANTES, as with most chemokines, contains its receptor binding motif at the N-terminus; thusiftruncation proceeds, modified chemotactic activity ensues. Therefore, the information content is 2-fold within such a multiplexed assay: indirectly, the enzymatic activity/expression, and directly, the relative abundance of inactive and active protein variants. In this context, this information is in the form of DPP-IV and cathepsin G activity from the abundance of [3–68] and [4–68] and each individual’s relative ability to induce and/or block signal transduction on target receptors (e.g., full-length RANTES vs DPP-IV-cleaved RANTES). As for the other 8 alternate truncated forms of RANTES found here, the enzymatic activity they indirectly represent remains unknown. More than a decade ago, Proost et al. (18) reported on 2 related C-C chemokines, monocyte chemotactic proteins 1 and 2 (MCP-1 and –2), and the discovery that they exist in several N- and/or C-terminally truncated forms with corresponding modified activity. Notably, they observed that N-truncated forms were almost completely devoid of activity but C-terminus-only truncated forms retained full activity. Furthermore, they observed a particular N-truncated form of MCP-2 to act as a natural inhibitor for MCP-2, MCP-1, and RANTES in regard to chemotactic activity. Proost et al. also found an analogous case with RANTES [3–68]—they observed the variant to also act as a natural inhibitor against full-length RANTES and as an effective antagonist of HIV entry into mononuclear cells (19).
We expect these N-terminal–truncated variants to be devoid of activity with increasing lack of receptor specificity the further the variant is truncated inward. Conversely, we expect the C-terminal truncations to have little effect on biological activity. This distinction may be superfluous in this case, however, because most of the C-truncated variants are also N-truncated. A case-in-point example regarding the imperative of acknowledging all truncated isoforms of a protein is the calcium regulatory protein parathyroid hormone (PTH). PTH exists in several embodiments including N and/or C truncations. PTH bindsto2 separate receptors: the classic receptor specific for the amino terminus and an as-of-yet uncloned receptor on the carboxyl terminus, the 2 exerting opposite biological effects in modulating calcium concentrations (20, 21). Interestingly, PTH microheterogeneity has been studied for almost half a century, but the possibility of a C-terminal receptor binding domain was assumed to not exist until very recently. We recently produced a multiplexed assay that can simultaneously distinguish between these active forms (22). Thus, it seems logical that additional assays stand to profit from the ability to unambiguously identify, in a multiplexed manner, an entire full-length protein form. Conversely, as with most “biased” quantification proteomic approaches whereby only 1 or 2 surrogate peptides (resulting from a tryptic digest) are monitored to represent an entire set of protein variants as they exist endogenously, the degree of protein heterogeneity presented here would have been disguised and unacknowledged. This would have been true for all truncated forms if the surrogate peptide were chosen from a midregion. Even if a surrogate peptide were chosen from a terminal region (e.g., to represent [3–68]), the assay would still be blind to C-terminal truncation and the possibility of discovering more variants. The MSIA analytical approach allows for the continual analysis of all of these variants.
GLYCOSYLATION AND GLYCATION
We mass-mapped the presence of at least 4 putative O-linked glycosylated variants that appear to exist on both intact and truncated forms of RANTES (observable only on [1–68] and [3–68]; more were not detected corresponding to the other variants likely owing to their already low relative abundance). A ladderlike structure was observed reaching its largest potential at a mass of 8926.9 Da, representing intact RANTES with Deoxyhex1HexNAc3Hex2 (see Fig. 1 and Table 1). Importantly, the ratio of O-glycosylated (Deoxyhex1HexNAc3Hex1) intact (XVIII) to O-glycosylated (Deoxyhex1HexNAc3Hex1) [3–68] (XVI) was found to be conserved relative to the ratio of intact (I) and [3–68] (II) among essentially all samples in the population study, providing further verification of the mass-mapping assignments (Fig. 2). About 2 decades ago, Kameyoshi et al. (8) discovered a putative RANTES variant that possessed the same N-terminal sequence as intact RANTES yet had an increase in mass of approximately 500 mass units [full-length intact protein was detected at 8355 (10) m/z]. It was assumed to be an O-glycosylated variant due to the absence of Edman degradation sequence information at a hypothesized dual-serine O-glycosylation site (at residues 4 and 5). Interestingly, they observed that this variant retained eosinophil chemotactic activity similar to the native form (8). This putative variant was not detected in our study. A few years later, Proost et al. (in the same study mentioned previously (18)) found O-linked glycosylated variants of MCP-1 that had a 2- to 3-fold decrease in chemotactic function on monocytes and human acute monocytic leukemia cell lines (THP1) relative to intact MCP-1. Because of the structural relatedness between RANTES and MCP-1, it remains quite possible for RANTES O-linked glycosylation variants to have modified activity as well. Regardless, it is essential tobe aware of RANTES in all of its structural embodiments.
Fig. 2. Mass spectra from a patient with T2D and one without.

Table 1 lists the identities of the individual variants. Note the correlation between the ratio of [1–68] (I) and [3–68] (II) to the ratio of XXIII to XIXII (both glycosylated with Deoxyhex1HexNAc3Hex1). Also note the increase in glycation observed in the T2D sample.
The glycated signatures of [1–68] and [3–68] were detected in the majority of samples. Glycation is common among plasma proteins. To date, however, there do not appear to be any previous reports of RANTES glycation. As we previously demonstrated with vitamin D–binding protein (DBP) (23), significant population differences in DBP glycation levels between healthy patients and those with T2D, cardiovascular disease, and cancer were found to be common. Predictably, there is a markedly high increase in glycation in the diabetic and cancer samples studied here. Fig. 2 displays the RANTES spectrum of a non-T2D (cardiovascular disease) patient compared with that of a T2D patient. A glycation peak is observed in the T2D spectrum labeled XIVI, but this peak is absent in the non-T2D spectrum. Several population trends such as this were encountered (Figs. 3–5).
Fig. 3. Stacked bar plot population data for mixed cohort of about 250 individuals: [1–68] and [3–68].

Healthy, T2D, CHF (with or without T2D and/or MI), and cancer [1–68] averages were 7.54%, 48.31%, 44.99%, and 43.12%. T2D, CHF (with or without T2D and/or MI), and cancer [1–68] were significantly different from the healthy cohort at P < 0.001. Healthy, T2D, CHF (with or without T2D and/or hMI), and cancer [3–68] averages were 78.15%, 33.66%, 44.99%, and 43.12%. Healthy [3–68] was significantly different from the T2D, CHF (with or without T2D and/or hMI), and cancer cohorts at P < 0.001.
Fig. 5. Population data for mixed cohort of 239 individuals: glycated RANTES.
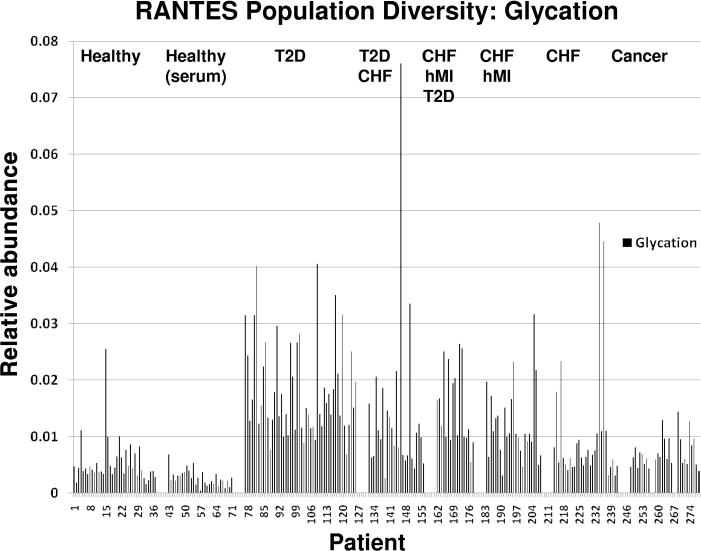
Averages of glycated RANTES in healthy, T2D, and CHF (with or without T2D and MI) were 0.55%, 1.84%, and 1.36%. Healthy glycated RANTES was significantly different from the T2D and CHF (with or without T2D and MI) cohort at P < 0.001.
POPULATION STUDY
Figs. 3–5 represent a population of 239 individuals in different states of health and the diversity of RANTES found therein. The relative abundance for several distinct categories of RANTES heterogeneity is presented. We observed significant RPA differences between all disease cohorts. The subsequent figures compare the relative abundance of [1–68] and [3–68] (Fig. 3), oxidation (Fig. 4), glycation (Fig. 5), and [4–66], [3–66], [3–67], and [1–66] (see online Supplemental Information) across all of the disease states. Obvious variation in RANTES profiles begin to emerge in this larger context. For example, in Fig. 3, a large increase in the RPA of [1–68] and a large decrease in the RPA of [3–68] is observed in the majority of the diseased individuals relative to healthy controls. In Fig. 4, glycation increases significantly in the diseased cohorts, with the T2D individuals unsurprisingly exhibiting the highest relative abundance, having an average value and range of 1.84% (0.84%) and 0.68%–4.06% compared to the healthy cohort value of 0.43% (0.35%) and range of 0.05%–2.55% (the glycation calculation comes from adding together the glycated [1–68] and [3–68]). In Fig. 5, a markedly higher RPA of oxidized RANTES is observed in the diseased individuals, with the highest increase observed in those with a history of myocardial infarction [average value and range of 9.65% (7.16%) and 0.27%–30.86% compared with the healthy cohort value of 1.42% (0.69%) and 0.17%–3.63%]. Finally, the differences in the relative percent abundances of the lowest-level truncated variants is presented in online Supplemental Fig. 2, with microdifferences observed between most disease states including the abnormal higher relative abundance of [1–66] in the CHF and hMI individuals, with an average value and range of 3.05% (7.54%) and 0.02%–36.67% compared with the healthy cohort value of 1.07% (0.58%) and 0.10%–4.26%. Recognizing heterogeneity in this context of multiple diseases presents a unique prospect for diagnosing more than 1 disease, using independent variant markers, from a single protein analysis. It is only after looking at protein phenotypes (e.g., concentration and posttranslational modifications) in the context of populations that deviations in protein multiplicity—including changes in population frequency and relative abundance—become clear and thus biomedically applicable (23, 24).
Fig. 4. Population data for mixed cohort of 239 individuals: oxidized RANTES.
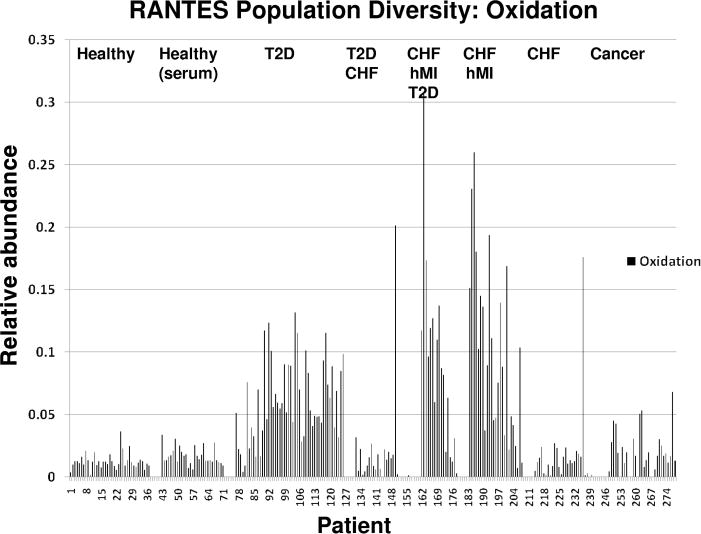
Averages of oxidized RANTES in healthy, T2D, CHF and MI, and cancer were 1.23%, 6.12%, 9.65%, and 2.24%. Healthy oxidized RANTES was significantly different from the T2D and hMI cohorts at P < 0.001 and significantly different from the cancer cohort at P = 0.0013.
Notably, owing to the ex vivo activity of enzymes in plasma (confirmed by our laboratory by observing the intact form to be processed to [3–68]), future population studies may be designed with the use of collection tubes containing enzyme inhibitors. This will guarantee an accurate readout of bioactive (and inactive) RANTES isoforms at the time of collection, which will also be beneficial to establishing interlaboratory standardizations. Regardless, the results illustrate the necessity to acknowledge RANTES diversity and optimistically suggest that it may hold multipurpose clinical utility.
In the span of 2 decades, studies have quickly taken the comprehension of RANTES from a single gene product found in T cells to a molecule that appears to be related with several diseases, expressed in a range of tissues, and acutely regulated via truncation by specific enzymes. In this report, we introduce 16 new forms of the molecule apparent in human plasma in the context of disease populations. The structural variety RANTES exhibits in different disease states should influence the design of future quantitative clinical assays (choice of variant to monitor, internal standard, etc.). The population diversity explored here strongly suggests that RANTES variants should be monitored, independent from one another, to assess RANTES chemokine function and regulation accurately in the context of disease.
Supplementary Material
Acknowledgments
Research Funding: R.W. Nelson, NIH grant R01 DK082542; C.R. Borges, NIH grant R01 DK082542.
Role of Sponsor: The funding organizations played no role in the design of study, choice of enrolled patients, review and interpretation of data, or preparation or approval of manuscript.
Footnotes
Nonstandard abbreviations: RANTES, regulated on activation, normal T-cell expressed and secreted; DPP-IV, dipeptidyl peptidase IV; T2D, type 2 diabetes; IGT, impaired glucose tolerance; CDI, 1,1′-carbonyldiimidazole; CHF, congestive heart failure; hMI, history of myocardial infarction; HBS-P, 0.01 mol/L HEPES, 0.15 mol/L NaCl, and 0.05% vol/vol surfactant P20, pH 7.4; MSIA, mass spectrometric immunoassay; TFA, trifluoroacetic acid; MS, mass spectrometry; HBS-N, 0.01 mol/L HEPES and 0.15 mol/L NaCl; RPA, relative percent abundance; MCP, monocyte chemotactic protein; PTH, parathyroid hormone; DBP, vitamin D–binding protein.
Author Contributions: All authors confirmed they have contributed to the intellectual content of this paper and have met the following 3 requirements: (a) significant contributions to the conception and design, acquisition of data, or analysis and interpretation of data; (b) drafting or revising the article for intellectual content; and (c) final approval of the published article.
Authors’ Disclosures of Potential Conflicts of Interest: Upon manuscript submission, all authors completed the Disclosures of Potential Conflict of Interest form. Potential conflicts of interest:
Employment or Leadership: R.W. Nelson, Intrinsic Bioprobes, Inc.
Consultant or Advisory Role: R.W. Nelson, Intrinsic Bioprobes, Inc.
Stock Ownership: R.W. Nelson, Intrinsic Bioprobes, Inc.
Honoraria: None declared.
Expert Testimony: None declared.
References
- 1.Nelson PJ, Krensky AM. Chemokines, chemokine receptors, and allograft rejection. Immunity. 2001;14:377–86. doi: 10.1016/s1074-7613(01)00118-2. [DOI] [PubMed] [Google Scholar]
- 2.Rossi D, Zlotnik A. The biology of chemokines and their receptors. Annu Rev Immunol. 2000;18:217–42. doi: 10.1146/annurev.immunol.18.1.217. [DOI] [PubMed] [Google Scholar]
- 3.Murphy PM, Baggiolini M, Charo IF, Hebert CA, Horuk R, Matsushima K, et al. International union of pharmacology. XXII. Nomenclature for chemokine receptors. Pharmacol Rev. 2000;52:145–76. [PubMed] [Google Scholar]
- 4.Segerer S, Nelson PJ, Schlondorff D. Chemokines, chemokine receptors, and renal disease: from basic science to pathophysiologic and therapeutic studies. J Am Soc Nephrol. 2000;11:152–76. doi: 10.1681/ASN.V111152. [DOI] [PubMed] [Google Scholar]
- 5.Niwa Y, Akamatsu H, Niwa H, Sumi H, Ozaki Y, Abe A. Correlation of tissue and plasma RANTES levels with disease course in patients with breast or cervical cancer. Clin Cancer Res. 2001;7:285–9. [PubMed] [Google Scholar]
- 6.Lim JK, Lu W, Hartley O, DeVico AL. N-terminal proteolytic processing by cathepsin G converts RANTES/CCL5 and related analogs into a truncated 4–68 variant. J Leukoc Biol. 2006;80:1395–404. doi: 10.1189/jlb.0406290. [DOI] [PubMed] [Google Scholar]
- 7.Oravecz T, Pall M, Roderiguez G, Gorrell MD, Ditto M, Nguyen NY, et al. Regulation of the receptor specificity and function of the chemokine RANTES (regulated on activation, normal T cell expressed and secreted) by dipeptidyl peptidase IV (CD26)-mediated cleavage. J Exp Med. 1997;186:1865–72. doi: 10.1084/jem.186.11.1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kameyoshi Y, Dorschner A, Mallet AI, Christophers E, Schroder JM. Cytokine RANTES released by thrombin-stimulated platelets is a potent attractant for human eosinophils. J Exp Med. 1992;176:587–92. doi: 10.1084/jem.176.2.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schall TJ, Jongstra J, Dyer BJ, Jorgensen J, Clayberger C, Davis MM, Krensky AM. A human T cell-specific molecule is a member of a new gene family. J Immunol. 1988;141:1018–25. [PubMed] [Google Scholar]
- 10.Krensky AM, Ahn YT. Mechanisms of disease: regulation of RANTES (CCL5) in renal disease. Nat Clin Pract Nephrol. 2007;3:164–70. doi: 10.1038/ncpneph0418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Azenshtein E, Luboshits G, Shina S, Neumark E, Shahbazian D, Weil M, et al. The CC chemokine RANTES in breast carcinoma progression: regulation of expression and potential mechanisms of promalignant activity. Cancer Res. 2002;62:1093–102. [PubMed] [Google Scholar]
- 12.Ng PC, Li K, Leung TF, Wong RP, Li G, Chui KM, et al. Early prediction of sepsis-induced disseminated intravascular coagulation with interleukin-10, interleukin-6, and RANTES in preterm infants. Clin Chem. 2006;52:1181–9. doi: 10.1373/clinchem.2005.062075. [DOI] [PubMed] [Google Scholar]
- 13.Herder C, Haastert B, Muller-Scholze S, Koenig W, Thorand B, Holle R, et al. Association of systemic chemokine concentrations with impaired glucose tolerance and type 2 diabetes: results from the Cooperative Health Research in the Region of Augsburg Survey S4 (KORA S4) Diabetes. 2005;54(Suppl 2):S11–7. doi: 10.2337/diabetes.54.suppl_2.s11. [DOI] [PubMed] [Google Scholar]
- 14.Kaburagi Y, Shimada Y, Nagaoka T, Hasegawa M, Takehara K, Sato S. Enhanced production of CC-chemokines (RANTES, MCP-1, MIP-1alpha, MIP-1beta, and eotaxin) in patients with atopic dermatitis. Arch Dermatol Res. 2001;293:350–5. doi: 10.1007/s004030100230. [DOI] [PubMed] [Google Scholar]
- 15.Christodoulakos GE, Lambrinoudaki IV, Economou EV, Papadias C, Vitoratos N, Panoulis CP, et al. Circulating chemoattractants RANTES, negatively related to endogenous androgens, and MCP-1 are differentially suppressed by hormone therapy and raloxifene. Atherosclerosis. 2007;193:142–50. doi: 10.1016/j.atherosclerosis.2006.05.045. [DOI] [PubMed] [Google Scholar]
- 16.McIntosh M. The need to characterize and report the normal heterogeneity of proteins in clinical biological samples. J Proteome Res. 2007;6:2913. [Google Scholar]
- 17.Niederkofler EE, Tubbs KA, Kiernan UA, Nedelkov D, Nelson RW. Novel mass spectrometric immunoassays for the rapid structural characterization of plasma apolipoproteins. J Lipid Res. 2003;44:630–9. doi: 10.1194/jlr.D200034-JLR200. [DOI] [PubMed] [Google Scholar]
- 18.Proost P, Struyf S, Couvreur M, Lenaerts JP, Conings R, Menten P, et al. Posttranslational modifications affect the activity of the human monocyte chemotactic proteins MCP-1 and MCP-2: identification of MCP-2(6–76) as a natural chemokine inhibitor. J Immunol. 1998;160:4034–41. [PubMed] [Google Scholar]
- 19.Proost P, De Meester I, Schols D, Struyf S, Lambeir AM, Wuyts A, et al. Amino-terminal truncation of chemokines by CD26/dipeptidyl-peptidase IV: conversion of RANTES into a potent inhibitor of monocyte chemotaxis and HIV-1-infection. J Biol Chem. 1998;273:7222–7. doi: 10.1074/jbc.273.13.7222. [DOI] [PubMed] [Google Scholar]
- 20.D’Amour P, Brossard JH, Rousseau L, Nguyen-Yamamoto L, Nassif E, Lazure C, et al. Structure of non-(1–84) PTH fragments secreted by parathyroid glands in primary and secondary hyperparathyroidism. Kidney Int. 2005;68:998–1007. doi: 10.1111/j.1523-1755.2005.00493.x. [DOI] [PubMed] [Google Scholar]
- 21.D’Amour P. Circulating PTH molecular forms: what we know and what we don’t. Kidney Int Suppl. 2006;S29:33. doi: 10.1038/sj.ki.5001599. [DOI] [PubMed] [Google Scholar]
- 22.Lopez MF, Rezai T, Sarracino DA, Prakash A, Krastins B, Athanas M, et al. Selected reaction monitoring-mass spectrometric immunoassay responsive to parathyroid hormone and related variants. Clin Chem. 2010;56:281–90. doi: 10.1373/clinchem.2009.137323. [DOI] [PubMed] [Google Scholar]
- 23.Borges CR, Rehder DS, Jarvis JW, Schaab MR, Oran PE, Nelson RW. Full-length characterization of proteins in human populations. Clin Chem. 2010;56:202–11. doi: 10.1373/clinchem.2009.134858. [DOI] [PubMed] [Google Scholar]
- 24.Nedelkov D, Phillips DA, Tubbs KA, Nelson RW. Investigation of human protein variants and their frequency in the general population. Mol Cell Proteomics. 2007;6:1183–7. doi: 10.1074/mcp.M700023-MCP200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.