

Abstract
Cardiac oxidative stress has been implicated in the pathogenesis of hypertrophy, cardiomyopathy and heart failure. Systemic deletion of the gene encoding adipose triglyceride lipase (ATGL), the enzyme that catalyzes the rate-limiting step of triglyceride lipolysis, results in a phenotype characterized by severe steatotic cardiac dysfunction. The objective of the present study was to investigate a potential role of oxidative stress in cardiac ATGL deficiency. Hearts of mice with global ATGL knockout were compared to those of mice with cardiomyocyte-restricted overexpression of ATGL and to those of wildtype littermates. Our results demonstrate that oxidative stress, measured as lucigenin chemiluminescence, was increased ~ 6-fold in ATGL-deficient hearts. In parallel, cytosolic NADPH oxidase subunits p67phox and p47phox were upregulated 4–5-fold at the protein level. Moreover, a prominent upregulation of different inflammatory markers (tumor necrosis factor α, monocyte chemotactant protein-1, interleukin 6, and galectin-3) was observed in those hearts. Both the oxidative and inflammatory responses were abolished upon cardiomyocyte-restricted overexpression of ATGL. Investigating the effect of oxidative and inflammatory stress on nitric oxide/cGMP signal transduction we observed a ~ 2.5-fold upregulation of soluble guanylate cyclase activity and a ~ 2-fold increase in cardiac tetrahydrobiopterin levels. Systemic treatment of ATGL-deficient mice with the superoxide dismutase mimetic Mn(III)tetrakis (4-benzoic acid) porphyrin did not ameliorate but rather aggravated cardiac oxidative stress. Our data suggest that oxidative and inflammatory stress seems involved in lipotoxic heart disease. Upregulation of soluble guanylate cyclase and cardiac tetrahydrobiopterin might be regarded as counterregulatory mechanisms in cardiac ATGL deficiency.
Abbreviations: ATGL, adipose triglyceride lipase; ATGL(−/−), adipose triglyceride lipase knockout; BH2, dihydrobiopterin, [2-amino-6-(1,2-dihydroxypropyl)-7,8-dihydro-1H-pteridin-4-one]; BH4, tetrahydrobiopterin, [(6R)-2-amino-6-[(1R,2S)-1,2-dihydroxypropyl]-5,6,7,8-tetrahydropteridin-4(1H)-one]; DEA/NO, 2,2-diethyl-1-nitroso-oxyhydrazine; DAG, diacylglycerol; eNOS, endothelial nitric oxide synthase; FFA, free fatty acid; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; (s)GC, (soluble) guanylate cyclase; IL-6, interleukin 6; Mac-2, galectin-3; MCP-1, monocyte chemotactic protein-1; MnTBAP, Mn(III)tetrakis (4-benzoic acid) porphyrin chloride; NADPH, nicotinamide adenine dinucleotide phosphate; iNOS, inducible nitric oxide synthase; nNOS, neuronal nitric oxide synthase; NO, nitric oxide; NOX, NADPH oxidase; ONOO−, peroxynitrite; PBS, phosphate-buffered saline; PKC, protein kinase C; PPARα, peroxisome proliferator receptor α; SOD, superoxide dismutase; TG, triacylglycerol; TNFα, tumor necrosis factor α; VASP, vasodilator-stimulated phosphoprotein; pVASP, phosphorylated vasodilator-stimulated phosphoprotein
Keywords: Adipose triglyceride lipase, Cardiac hypertrophy, Oxidative stress, Inflammation, NADPH oxidase
Graphical abstract
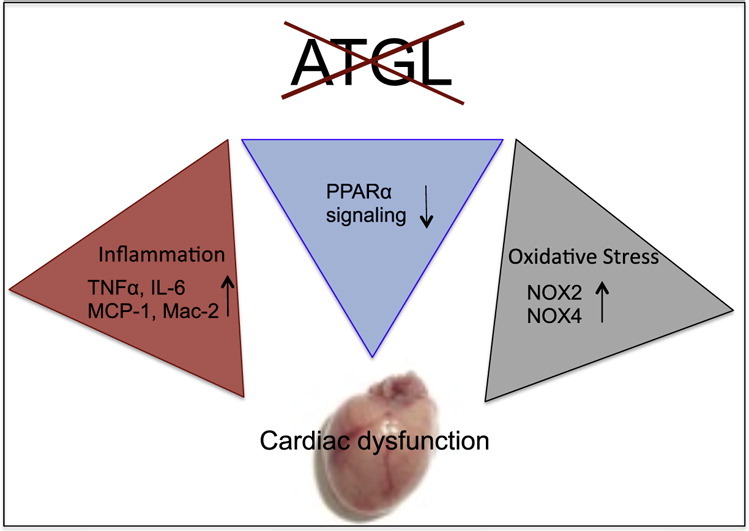
Highlights
-
•
ATGL(−/−) mice suffer from severe cardiac oxidative stress originating from upregulation of NOX2-dependent NADPH oxidase.
-
•
Inflammation markers TNFα, MCP-1, IL-6, and Mac-2 are increased in cardiac ATGL deficiency.
-
•
Activity of sGC and cardiac BH4 levels are elevated in ATGL(−/−) hearts.
-
•
Systemic treatment of ATGL(−/−) mice with the SOD mimetic MnTBAP did not ameliorate oxidative stress.
1. Introduction
Cardiac lipotoxicity is regarded as sum of various cell-damaging processes: Besides disruption of organelle membranes and activation of the apoptotic machinery, ER stress and oxidative stress have been identified as crucial components of the lipotoxic response [1]. Different enzymatic sources of oxidative stress including NADPH oxidase (for review, see [2]), uncoupled nitric oxide synthase (NOS; [3]), and xanthine oxidase [4], have been implicated in the development of cardiac dysfunction. Recent evidence points to an essential role of NADPH oxidases (NAD(P)H:oxygen oxidoreductases; EC 1.6.3.1) as mediators of lipid-driven oxidative stress. Thus, diet-induced obesity in rats resulted in ectopic fat deposition and concomitant upregulation of the NADPH oxidase 2 (NOX2) system in liver and heart [5]. Similarly, a previous study on Lep/Lep mice, a genetic model of obesity, suggested that cardiac contractile dysfunction is related to enhanced NOX2-dependent oxidative stress [6]. Besides NOX2, NOX4 has been described as the second important NADPH oxidase isoform in the heart. However, its distinct role in cardiac pathophysiology is unclear yet, since both beneficial and harmful effects have been demonstrated in different animal models of NOX4 overexpression or knockout [7,8].
With the recent discovery of adipose triglyceride lipase (ATGL) a key enzyme of triglyceride catabolism was identified [9]. ATGL catalyzes the initial step of the triglyceride lipolysis cascade i.e. the hydrolytic cleavage of triacylglycerols (TGs) into free fatty acids (FFAs) and diacylglycerols (DAGs). The enzyme is predominantly expressed in adipose tissue, but it is also present to a lesser extent in cardiac muscle, skeletal muscle, and other tissues. Systemic ablation of ATGL in mice (ATGL(−/−)) yielded a phenotype with increased whole body fat mass and neutral lipids accumulating in adipose and non-adipose tissues [10]. In cardiac muscle, ATGL deficiency caused an age-dependent increase of myocyte lipid droplets in number and size. In parallel, echocardiographic analysis demonstrated the progressive development of ventricular hypertrophy with abnormal myocardial texture and systolic dysfunction [10]. Using Langendorff perfusion of isolated hearts we have recently demonstrated that the contractile and microvascular response to ß-adrenergic stimulation was drastically reduced in ATGL deficiency [11]. Contrariwise, transgenic mice with supraphysiological protein expression of ATGL in cardiomyocytes (WT/MHC-A35) showed reduced triglyceride levels in the heart as well as improved basal pump function and protection against experimentally induced pathophysiological stress [12].
Concerning the prevalence of obesity- and diabetes-related human cardiomyopathy that is characterized by myocardial fat deposition and compromised contractile function, ATGL-deficient and overexpressing mice are invaluable tools to study lipotoxic heart disease. Using these animal models we aimed to investigate the potential role of oxidative stress in the development and progression of lipid-driven cardiomyopathy with special emphasis on the specific role of cardiac NADPH oxidases.
2. Theory
ATGL(−/−) mice suffer from severe cardiac function due to abnormal accumulation of triglycerides within cardiomyocytes. Since oxidative stress has been identified as crucial component of cardiac lipotoxicity in experimental animals [5,6] we hypothesized that a similar mechanism might be operative in ATGL-deficient hearts. The theory will be tested on the one hand by identifying potential sources of oxidative stress and on the other hand by chronic treatment of animals with a superoxide dismutase mimetic.
3. Materials and methods
3.1. Materials
2,2-Diethyl-1-nitroso-oxyhydrazine (DEA/NO), 1-benzyl-3-(5-hydroxymethylfur-2-yl) indazole (YC-1) and Mn(III)tetrakis (4-benzoic acid) porphyrin chloride (MnTBAP) (Enzo Life Sciences AG, Lausen, Switzerland) were purchased through Eubio (Vienna, Austria). Antibodies were purchased as indicated: NOX2 and p67phox (BD Transduction Laboratories, Heidelberg, Germany); p47phox (Novus Biologicals, Cambridge, UK); VASP (Cell Signaling through New England Biolabs, Frankfurt, Germany); NOX4 (Abcam, Cambridge, UK); pVASP (Calbiochem through VWR, Vienna, Austria); Mac-2 (galectin-3; Cedarlane through Szabo-Scandic, Vienna, Austria); phosphorylated protein kinase C (PKC; pan; Thr 410 of human PKCζ; Cell Signaling); GAPDH (Sigma, Vienna, Austria). [3H]l-Arginine (40 Ci/mmol) and [α-32P]GTP (400 Ci/mmol) were obtained from PerkinElmer (Vienna, Austria); gp91ds-tat was synthesized by piChem (Graz, Austria). ChemiGlow™ was obtained from Biozym (Vienna, Austria). Protease Inhibitor Cocktail tablets (Complete™) were from Roche (Vienna, Austria). Materials for real-time PCR experiments were from Applied Biosystems (Vienna, Austria). GenElute™ Mammalian Total RNA Miniprep Kit, RIPA buffer, and all other chemicals were from Sigma (Vienna, Austria).
3.2. Animals
Homozygous ATGL(−/−) mice [10] and corresponding wild-type (WT) littermates were used for this study. In addition, WT and ATGL(−/−) mice with cardiomyocyte-specific overexpression of ATGL termed WT/MHC-A35 mice and ATGL(−/−)/MHC-A35 mice, respectively [12,13], were used for most experiments. After weaning, animals received standard laboratory mouse chow and water ad libitum. Mice were housed in approved cages and kept on a regular 12 h dark/light cycle. Animals were sacrificed at 9 weeks of age. Hearts were dissected, cleaned, and frozen for further biochemical analysis. All animals received care in accordance with the Austrian law on experimentation with laboratory animals (last amendment, 2012), which is based on the US National Institutes of Health guidelines.
3.3. MnTBAP treatment
WT and ATGL(−/−) mice (aged 6 weeks) were treated with MnTBAP (5 mg × kg− 1 × d− 1; i.p.) dissolved in phosphate-buffered saline (PBS) as vehicle once per day for 3 weeks. Application of vehicle to WT and ATGL(−/−) mice had no effect. After sacrifice of the animals, hearts were excised and plasma was collected and frozen for biochemical analysis. Cardiac and plasma MnTBAP levels were measured by HPLC as described [14]. The protocol was approved by the Austrian Federal Ministry for Science and Research (BMWF-66.007/0002-II/3b/2012).
3.4. NADPH oxidase activity
Hearts were homogenized in 10 volumes of PBS containing Complete™ using a glass potter Elvehjem homogenizer. Total homogenates (50–100 μg of protein) were incubated in PBS containing diethylenetriamine pentaacetic acid (100 μM) at 37 °C for 30 min in the absence or presence of the NADPH oxidase inhibitor gp91ds-tat (50 μM) [15], the SOD mimetic MnTBAP (10 μM) or Cu, Zn SOD (500 U/ml). Thereafter, NADPH (300 μM) was added to activate NADPH oxidases, followed by addition of lucigenin at a non-redox cycling concentration of 5 μM [16]. Lucigenin-derived chemiluminescence was measured every 10 s for 3 min using a TriCarb® 2100TR Liquid Scintillation Counter (PerkinElmer, Vienna, Austria). Results were corrected for protein-deficient blanks and expressed as cpm per μg protein.
3.5. Determination of NOS activity
Hearts were homogenized in 10 volumes of a 50 mM triethanolamine/HCl buffer (pH 7.4) containing 1% (v/v) ß-mercaptoethanol, 10 mM 3-[(3-cholamidopropyl)dimethyl-ammonio]-1-propanesulfonate, 0.5 mM EDTA and Complete™ with a glass potter Elvehjem homogenizer. Homogenates were centrifuged at 10,000 g for 10 min at 4 °C. Supernatants were assayed for NOS activity as conversion of [3H]l-arginine to [3H]l-citrulline as previously described [17]. Results were corrected for enzyme-deficient blanks and recovery of l-citrulline.
3.6. Real-time PCR
Total RNA was isolated from homogenized hearts using the GenElute™ Mammalian Total RNA Miniprep Kit including DNAse treatment of samples. RNA was transcribed to cDNA using the High Capacity cDNA Reverse Transcription Kit. Real-time PCR analysis was performed with ~ 25 ng cDNA using TaqMan® Universal PCR Master Mix and pre-designed TaqMan® Gene Expression Assays for NOX2 (Mm 00432775_m1), NOX4 (Mm 00479246_m1), tumor necrosis factor α (TNFα; Mm 00443258_m1), monocyte chemotactic protein-1 (MCP-1; Mm 00441243_g1), and interleukin 6 (IL-6; Mm 00446190_m1). Reactions were carried out on a 7300 Real-Time PCR System (Applied Biosystems, Vienna, Austria). Cycling conditions were as follows: 2 min at 50 °C, 10 min at 95 °C, and 40 cycles of 15 s at 95 °C and 1 min at 60 °C. Data were analyzed according to the 2 −∆∆Ct method using cyclophilin (Mm 00835365_g1) as the reference gene. Lack of amplification was verified in no-template controls.
3.7. Western blot analysis
Denatured samples containing 20 μg of protein were separated by SDS-PAGE (8% gel) and transferred onto nitrocellulose membranes. After blocking, membranes were probed with antibodies raised against NOX2 (1:5000), NOX4 (1:1000), p67phox (1:5000), p47phox (1:750), pPKC (pan; 1:1000), VASP (1:1000), pVASP (1:1000), Mac-2 (1:5000), or GAPDH (1:50,000), followed by treatment with horseradish peroxidase-conjugated anti-rabbit IgG for NOX2, NOX4, p47phox, pPKC, and VASP (1:5000), anti-mouse IgG for p67phox, pVASP, and GAPDH (1:5000) or anti-rat (1:5000) for Mac-2. Immunoreactive bands were visualized by chemiluminescence using ChemiGlow™ and quantified densitometrically using the E.A.S.Y. Win 32 software (Herolab, Vienna, Austria).
3.8. Determination of sGC activity
Hearts were homogenized in 10 volumes of a 50 mM triethanolamine/HCl buffer (pH 7.4), supplemented with 2 mM dithiothreitol, 2 mM EDTA and Complete™, using a glass potter Elvehjem homogenizer. After centrifugation at 10,000 g for 15 min (4 °C), supernatants were removed and centrifuged at 100,000 g for 1 h (4 °C). For determination of sGC activity, cytosols (20–60 μg) were incubated for 20 min at 37 °C in a total volume of 0.1 ml of a 50 mM triethanolamine/HCl buffer (pH 7.4), containing 0.1 mM [α-32P]GTP (~ 300,000 cpm), 5 mM MgCl2, 1 mM cGMP, 1 mM isobutylmethylxanthine, 1 mM EGTA, 2 mM dithiothreitol, 5 mM creatine phosphate, and 15 mU of creatine phosphokinase. Reactions were started with 0.1 mM DEA/NO or a combination of 0.1 mM DEA/NO and 0.1 mM YC-1. Basal activity was assayed in the presence of 5 mM MnCl2. Reactions were terminated by ZnCO3 precipitation, and [32P]cGMP was isolated by column chromatography as described previously [18]. Results were corrected for enzyme-deficient blanks and recovery of cGMP.
3.9. Determination of cardiac cGMP levels
Frozen hearts (30–50 mg) were pulverized using a dismembrator and extracted in 5% trichloroacetic acid. Samples were centrifuged at 4000 g for 15 min at 4 °C. Supernatants were collected and washed three times for 5 min with water-saturated ether. Cardiac cGMP was quantified by radioimmunoassay as described [19].
3.10. Determination of cardiac BH4 and BH2
Frozen hearts (40–50 mg) were homogenized with 8 volumes of a chilled mixture containing 100 mM HClO4 and 85 mM H3PO4 using a glass potter Elvehjem homogenizer. Homogenates were centrifuged for 10 min at 20,000 g (4 °C). Supernatants (50 μl) were oxidized under acidic and alkaline conditions with 10 μl of a mixture of 0.1 M I2 and 0.5 M KI at ambient temperature for 60 min in the dark. Subsequently, excess I2 was eliminated with 10 μl of 0.2 M ascorbic acid and samples were neutralized with either 1 M NaOH or 1.7 M H3PO4. After centrifugation at 20,000 g for 5 min (4 °C), samples were loaded onto a LiChrospher 100 RP 18 HPLC column, 5 μm particle size (Merck, Darmstadt, Germany), using a mixture of 95% NaH2PO4 (20 mM, pH 3.0) and 5% methanol as mobile phase (flow rate of 1 ml/min). Pteridines were quantified with a fluorescence detector at excitation and emission wavelengths of 350 and 440 nm, respectively, by interpolation using a calibration curve that was measured daily with authentic BH4 (5–300 nM). Data were expressed as pmol per g wet weight.
3.11. Staining of cardiac lipids
Heart specimens were fixed with 2.5% glutaraldehyde in 0.01 M cacodylate buffer pH 7.3. After dehydration, specimens were embedded in Agar 100 resin (Plano, Wetzlar, Germany). Semi-thin sections (1 μm) were stained with azur-methylene blue according to standard methods.
3.12. Protein quantification
Protein concentrations were either measured by the method of Bradford [20] or using the Thermo Scientific Pierce BCA™ Protein Assay Kit (Fisher Scientific Austria GmbH, Vienna, Austria).
3.13. Statistics
Data are presented as mean values ± S.E.M. Student's t-test (unpaired) was applied for single comparisons. One-way ANOVA followed by Student–Newman–Keuls post-hoc test was applied for multiple comparisons. Differences were assumed to be significant for p < 0.05.
4. Results
4.1. NADPH oxidase-derived oxidative stress in ATGL deficiency
The involvement of NADPH oxidase-derived oxidative stress in ATGL deficiency was probed by comparing gp91ds-tat-sensitive chemiluminescence in cardiac homogenates from WT, ATGL(−/−), WT/MHC-A35 and ATGL(−/−)/MHC-A35 mice. As shown in Fig. 1A, knockout of ATGL resulted in more than 6-fold higher chemiluminescence indicating significantly increased NADPH oxidase activity. Cardiomyocyte-specific overexpression of ATGL completely reversed the effect of ATGL deficiency. Presence of the selective NADPH oxidase inhibitor gp91ds-tat (50 μM) diminished chemiluminescence by 73.1 ± 10.0 and 84.3 ± 4.5% in homogenates of WT and ATGL(−/−) hearts, respectively (Fig. 1A; inset). A comparable extent of inhibition was observed with the SOD mimetic MnTBAP (10 μM) whereas Cu, Zn SOD (500 U/ml) was less efficient (Fig. 1A, inset). Increased NADPH oxidase activity was accompanied by upregulated mRNA expression of the catalytic NOX2 subunit in ATGL-deficient hearts (Fig. 1B; inset). At the protein level, NOX2 expression tended to increase in ATGL deficiency but the observed difference did not reach statistical significance (p = 0.071) (Fig. 1B). Protein expression of the cytosolic activator subunits p47phox (Fig. 1C) and p67phox (Fig. 1D), which initiate enzyme activation by the stoichiometric assembly with the plasma membrane-attached p22phox/NOX2 heterodimer, was significantly increased in ATGL deficiency, with 5.4 ± 0.4 and 4.4 ± 0.3-fold higher protein levels of p47phox (Fig. 1C) and p67phox (Fig. 1D), respectively. The effect of ATGL deficiency on NADPH oxidase subunit expression was reversed upon overexpression of ATGL in cardiomyocytes (Fig. 1B, C, D). Fig. 2 shows the effect of ATGL deficiency on NOX4 mRNA and protein expression. NOX4 mRNA was raised ~ 3-fold in ATGL(−/−) hearts (Fig. 2A), and a significant increase of 50% was observed at the protein level (Fig. 2B).
Fig. 1.

NOX2-dependent oxidative stress in cardiac ATGL deficiency. (A) gp91ds-tat-sensitive lucigenin chemiluminescence was measured in cardiac homogenates prepared from WT (open bars), ATGL(−/−) (solid bars), WT/MHC-A35 (striped bars), and ATGL(−/−)/MHC-A35 mice (gray bars) as described in Materials and methods. Data represent mean values ± S.E.M. of 4 individual experiments. Inset: Inhibition of NADPH-induced chemiluminescence by gp91ds-tat (50 μM), Cu, Zn SOD (500 U/ml) or MnTBAP (10 μM) in hearts of WT (open bars) and ATGL(−/−) mice (solid bars). (B) Cardiac NOX2 mRNA and protein expression were measured by qPCR and Western blot, respectively. Data are expressed as folds of WT controls (WT = 1) and represent mean values ± S.E.M. of 6 experiments. qPCR measurements were performed in triplicates. (C, D) Protein expression of p47phox and p67phox subunits, respectively. Data are expressed as folds of WT controls (WT = 1) and represent mean values ± S.E.M. of 6 individual experiments. (E) Representative Western blots of NOX2, p47phox, p67phox, and GAPDH; *p < 0.05 ATGL(−/−) vs WT.
Fig. 2.

Cardiac NOX4 is upregulated in ATGL deficiency. (A) NOX4 mRNA was measured in hearts of WT (open bars), ATGL(−/−) (solid bars), WT/MHC-A35 (striped bars), and ATGL(−/−)/MHC-A35 mice (gray bars) by qPCR as described. Data represent mean values ± S.E.M. of 3–5 experiments performed in triplicate. (B) Protein expression of NOX4 was measured by Western blot analysis. Data are expressed as folds of WT controls (WT = 1) and represent mean values ± S.E.M. of 6 individual experiments. A representative Western blot is shown as inset; *p < 0.05 ATGL(−/−) vs WT.
4.2. Inflammatory markers are upregulated in ATGL deficiency
To test whether proinflammatory processes are involved in cardiac ATGL deficiency we measured mRNA levels of the well-established markers TNFα, MCP-1, and IL-6. As illustrated in Fig. 3A, ATGL deficiency led to a ~ 3-fold increase in TNFα mRNA expression. For the chemotactant protein MCP-1, an even more pronounced effect was observed as mRNA levels were elevated ~ 17-fold as compared to WT controls (Fig. 3B). Likewise, mRNA expression of the proinflammatory cytokine IL-6 was raised nearly 4-fold in ATGL(−/−) hearts (Fig. 3C). The proinflammatory response to ATGL deficiency was abolished by cardiomyocyte-specific overexpression of ATGL. As shown in Fig. 3D and F, protein expression of Mac-2, a lectin that has been implicated in the development of cardiac fibrosis and ventricular dysfunction in experimental animals [21,22] and human surveys [21] was elevated ~ 11-fold in hearts of ATGL(−/−) mice compared to WT controls. The lectin was not detectable in hearts with cardiomyocyte-restricted overexpression of ATGL. To test if PKC is involved in the inflammatory response, enzyme activation was measured as phosphorylation of a crucial threonine located in the activation loop. Threonine phosphorylation was increased more than 2-fold in cardiac homogenates of ATGL(−/−) mice (Fig. 3E and F). The effect of ATGL deficiency on PKC phosphorylation was reversed by cardiomyocyte-restricted overexpression of ATGL.
Fig. 3.

Upregulation of inflammatory markers in ATGL deficiency. (A) TNFα mRNA expression in cardiac homogenates of WT (open bars), ATGL(−/−) (solid bars), WT/MHC-A35 (striped bars), and ATGL(−/−)/MHC-A35 mice (gray bars) was measured by qPCR using TaqMan probes. (B, C) MCP-1 and IL-6 mRNA expression, respectively. Data represent mean values ± S.E.M. of 4–6 experiments performed in triplicate; *p < 0.05 ATGL(−/−) and ATGL(−/−)/MHC-A35 vs WT. (D) Protein expression of Mac-2 was measured by Western blot analysis. Data are expressed as folds of WT controls (WT = 1) and represent mean values ± S.E.M. of 9 individual experiments. (E) Activation of cardiac PKCs was measured by Western blot as phosphorylation of a crucial threonine. Data are expressed as folds of WT controls (WT = 1) and represent mean values ± S.E.M. of 9 individual experiments. *p < 0.05 ATGL(−/−) vs WT. (F) Representative Western blots of Mac-2, pPKC (pan), and GAPDH.
4.3. Cardiac NO/cGMP signaling
The potential consequences of oxidative stress for cardiac NO/cGMP signaling in ATGL deficiency were studied by measuring various parameters characteristic for different steps of the signaling cascade. As shown in Fig. 4A, cardiac NOS activity determined as conversion of l-arginine to l-citrulline was not affected by any genetic manipulation. Ca2 +-independent activity was not detectable (data not shown), arguing against significant iNOS expression in any experimental group. Protein expression of eNOS and nNOS was not affected (data not shown). However, activity of sGC, the major downstream target of NOS-derived NO, was significantly increased in ATGL(−/−) hearts (Fig. 4B). Basal cGMP formation measured in the presence of Mn2 + was increased from 0.023 ± 0.002 to 0.077 ± 0.011 nmol × mg− 1 × min− 1 (Fig. 4B; inset). Upon stimulation with the NO donor DEA/NO or co-stimulation with the NO-sensitizing drug YC-1 [23], the rates of cGMP formation were enhanced ~ 2.6- and ~ 2.4-fold, respectively. Thus, sGC activity was significantly increased at different activation states of the enzyme. Cardiac-specific overexpression of ATGL resulted in sGC activities identical to that of WT animals (Fig. 4B). Quantification of cardiac cGMP by radioimmunoassay showed that, despite the significantly higher sGC activity in ATGL(−/−) hearts, cGMP levels were virtually identical in all experimental groups (Fig. 4C). Similarly, cGMP-dependent phosphorylation of vasodilator-stimulated phosphoprotein (VASP) at serine 239, which represents a reliable biochemical marker of the NO/cGMP pathway [24], was not affected (Fig. 4D).
Fig. 4.

Effects on NO/cGMP signaling. (A) 10,000 g supernatants of hearts from WT (open bars), ATGL(−/−) (solid bars), WT/MHC-A35 (striped bars), and ATGL(−/−)/MHC-A35 mice (gray bars) were prepared and assayed for Ca2 +-dependent [3H]l-citrulline formation. Data are mean values ± S.E.M. of 4 experiments performed in duplicate. (B) Cardiac sGC activity was measured in cytosols stimulated with 0.1 mM DEA/NO with and without YC-1 (0.1 mM). The inset depicts basal enzyme activity assayed in the presence of MnCl2. Data are mean values ± S.E.M. of 5 experiments performed in duplicate. *p < 0.05 ATGL(−/−) vs WT. (C) cGMP levels were determined by radioimmunoassay. Data represent mean values ± S.E.M. of 5–7 experiments performed in duplicate. (D) Phosphorylation of VASP at serine 239 was quantified by Western blot. Data are presented as the ratio of phosphorylated to total VASP protein and represent mean values ± S.E.M. of 6 individual experiments.
4.4. Cardiac BH4 levels are increased in ATGL deficiency
Oxidative stress generated in the course of different vascular and metabolic disorders may cause depletion of the essential NOS cofactor BH4 [25–27]. Reduced intracellular availability of BH4 leads to uncoupling of NOS i.e. the production of superoxide instead of NO, thereby aggravating the oxidative burden. To investigate whether this occurs in ATGL deficiency, we measured cardiac biopterin levels. Paradoxically, BH4 levels were actually increased ~ 2-fold in ATGL deficiency (Fig. 5A). In hearts, exclusively overexpressing ATGL in cardiomyocytes, BH4 values were comparable to those of WT animals. By contrast, cardiac BH2 was not significantly affected by any genetic manipulation (Fig. 5B). Calculations of the BH4/BH2 ratio showed a tendency towards increased values in ATGL deficiency; however, the difference did not reach statistical significance (Fig. 5C).
Fig. 5.

Cardiac BH4 is increased in ATGL deficiency. (A, B) BH4 and BH2 were quantified in hearts of WT (open bars), ATGL(−/−) (solid bars), WT/MHC-A35 (striped bars), and ATGL(−/−)/MHC-A35 mice (gray bars) by HPLC analysis. (C) BH4/BH2 ratio. Data represent mean values ± S.E.M. of 8 individual experiments. *p < 0.05 ATGL(−/−) vs WT.
4.5. MnTBAP did not reverse the effects of ATGL deficiency
As shown previously [14], systemic application of the SOD mimetic MnTBAP [28,29] to WT and ATGL(−/−) mice resulted in pharmacologically effective tissue concentrations of the drug (data not shown). However, MnTBAP treatment did not reverse but rather exacerbated ATGL deficiency-mediated oxidative stress (Fig. 6A). With respect to cardiac biopterins, a similarly adverse effect of the compound was observed: MnTBAP-treatment of ATGL(−/−) mice resulted in a reduction of BH4 levels similar to those measured in WT hearts (Fig. 6B). Likewise, MnTBAP treatment of ATGL(−/−) mice resulted in a decline of the cardiac BH4/BH2 ratio to values observed in WT animals (Fig. 6C).
Fig. 6.

Systemic application of MnTBAP (5 mg × kg− 1 × day− 1) aggravates oxidative stress in ATGL deficiency. (A) gp91ds-tat-sensitive lucigenin chemiluminescence was measured in cardiac homogenates prepared from MnTBAP-treated WT (open bars) and ATGL(−/−) mice (solid bars) and compared to that of non-treated animals. Data represent mean values ± S.E.M. of 4 individual experiments. *p < 0.05 non-treated and MnTBAP-treated ATGL(−/−) vs non-treated WT; §p < 0.05 non-treated ATGL(−/−) vs MnTBAP-treated ATGL(−/−). (B, C) Cardiac BH4 and BH2 were determined by HPLC. Data represent mean values ± S.E.M. of 4 individual experiments. *p < 0.05 ATGL(−/−) vs WT.
5. Discussion
The present study was designed to investigate the role of oxidative stress in cardiac dysfunction of ATGL(−/−) mice. Our data suggest considerable upregulation of NOX2/p47phox/p67phox subunit expression in those hearts, which was associated with increased superoxide production evident as chemiluminescence sensitive to the selective peptide inhibitor gp91ds-tat. Upregulation of NOX2 mRNA expression suggests the involvement of a transcriptional mechanism. Cardiomyocyte-specific overexpression of ATGL resulted in complete reversal of NADPH oxidase upregulation, confirming that the observed effect was causally related to ATGL deficiency. Concerning the cardiac phenotype of the animal model [10], these results agree well with previous reports showing that activation of NOX2-dependent NADPH oxidase contributes to the progression of cardiac hypertrophy in heart failure [30,31].
Upregulation of NOX4 expression in ATGL-deficient hearts is difficult to interpret since the role of this isoform in cardiac function is discussed ambiguously [7,8]. Following the concept of Zhang and colleagues [8], increased expression of NOX4 is protective against chronic overload stress with the specific function to preserve myocardial capillary blood flow by inducing angiogenesis. On the other hand, exacerbation of cardiac dysfunction, fibrosis, and apoptosis in response to pressure overload has been observed in experimental animals with cardiomyocyte-specific overexpression of NOX4 [7]. Therefore, additional work is necessary to elucidate the specific role of NOX4 in cardiac ATGL deficiency.
Cardiac oxidative stress was accompanied by increased expression of TNFα, MCP-1, and IL-6 pointing to inflammatory processes involved in ATGL(−/−) hearts. In addition, prominent upregulation of cardiac Mac-2, a lectin that is highly expressed and secreted by macrophages and monocytes [32] and induces chemotactic, phagocytic, and lipid scavenging processes [33–35] points to severe chronic cardiac inflammation.
Concerning the important role of NO/cGMP signaling in cardiac function and assuming that enhanced superoxide production might compromise this pathway we studied whether this signaling route is affected by ATGL deficiency. Two major effects were observed: ATGL deficiency resulted in significant upregulation of sGC activity and increased levels of BH4. Latter finding was quite unexpected as it disagrees with the concept that oxidative stress is invariably accompanied by depletion of BH4. However, chronic administration of BH4 to mice suffering from experimentally induced heart disease was reported to result in marked suppression of cardiac remodeling and hypertrophy as well as in reduced oxidative stress [36]. Moreover, enhanced BH4 biosynthesis in cardiomyocytes was described as adaptive response to proinflammatory stimuli [37]. Therefore, in ATGL deficiency, increased cardiac BH4 biosynthesis and/or recycling may serve as an endogenous protective mechanism against tissue injury caused by oxidative and inflammatory stress with the overall aim to maintain the cellular redox equilibrium. The molecular mechanisms that finally result in increased BH4 levels in ATGL deficiency might comprise enhanced de novo synthesis via the GTP cyclohydrolase pathway and/or more efficient BH4 recycling by dihydrofolate reductase. However, elucidation of these mechanisms was beyond the scope of this study.
Different explanations are conceivable to link ATGL deficiency to oxidative inflammatory stress and increased sGC activity (Scheme 1). Conceivably, severely impaired peroxisome proliferator receptor α (PPARα) signaling in ATGL deficiency [38] may initiate a sequelae of events that eventually lead to the observed effects. Thus, downregulation of PPARα signaling has been demonstrated to favor the development of cardiomyocyte hypertrophy ([39], path 1). Interestingly, mice lacking PPARα have been described to develop cardiac dysfunction due to oxidative stress ([40], path 2). Moreover, the PPARα-deficient cardiac phenotype is characterized by enhanced expression of proinflammatory and fibrotic markers [39] not unlike our observations with ATGL(−/−) mice (path 3). Elevation of cardiac TNFα levels has been associated with enhanced expression/activation of NADPH oxidases in the course of chronic ventricular remodeling ([41], path 4).
Scheme 1.

Signaling pathways that link cardiac ATGL deficiency to oxidative inflammatory stress and increased sGC activity. Impaired PPARα signaling in ATGL deficiency induces cardiac hypertrophy (path 1) that is compensated by increased sGC activity (path 6). Impaired PPARα signaling leads to increased superoxide production by NADPH oxidases (path 2) and augmented expression of inflammatory markers (path 3). Elevated TNFα levels induce expression of cardiac NADPH oxidases via activation of PKC (path 4). Lipid droplet surface-binding proteins directly initiate oxidative and inflammatory processes (path 5). NADPH oxidase-generated superoxide scavenges NO to form ONOO− (path 7). Reduced NO availability induces activation of cardiac sGC. NADPH oxidase-derived superoxide production enhances cardiac hypertrophy and contractile dysfunction (path 8).
Protein kinase C (PKC) has been described as important mediator of TNFα-induced NADPH oxidase activation [42]. In ATGL-deficient hearts, activation of PKCs measured as phosphorylation of a crucial threonine was increased more than 2-fold, indeed suggesting that PKCs link upregulation of TNFα to NADPH oxidase activation. Further studies are necessary to characterize the type of PKC involved, its activation pattern as well as to clarify if systemic inhibition of PKCs (e.g. by ruboxistaurin) ameliorates cardiac oxidative inflammatory stress in ATGL(−/−) mice.
Concerning the strong evidence that cGMP is antihypertrophic (for review, see [43]), increased sGC activity might serve to counteract the hypertrophic effects of ATGL deficiency (path 6). Alternatively, sGC might be activated as a stress response to enhanced superoxide formation by cardiac NADPH oxidases. Superoxide will scavenge NO to form peroxynitrite ([44], path 7), a reaction that efficiently outcompetes formation of ferrous-nitrosyl-sGC and consequent enzyme activation [45]. In return, cardiac sGC could be activated to maintain cGMP signaling despite reduced NO bioavailability.
Albeit our data suggest that absence of distinct FFA-derived PPARα ligands accounts for oxidative and inflammatory processes in ATGL deficiency, pronounced accumulation of lipids in cardiac sections of ATGL(−/−) mice (Fig. S1; supplement) implies an alternative signaling route: It is feasible that lipid droplets per se contribute to the induction of the oxidative inflammatory response. Lipid droplets contain cell-type specific surface-binding proteins with distinct functions that have not fully been characterized thus far. Recently, perilipin 5, a lipid droplet coating protein, that is highly expressed in the heart has been reported to suppress the oxidative burden in this tissue [46]. Thus, distinct alterations in lipid droplet proteome might serve as signal to induce oxidative and inflammatory mechanisms.
For unknown reasons and contrary to expectations, systemic treatment of ATGL(−/−) mice with the SOD mimetic MnTBAP did not ameliorate but rather aggravated cardiac oxidative stress as determined in vitro. The drug was chosen on the basis of results from in vitro experiments demonstrating effective inhibition of superoxide production in WT and ATGL(−/−) hearts (Fig. 1A, inset). Moreover, a recent study on PPARγ(−/−) mice convincingly demonstrated prevention of superoxide-mediated development of lethal cardiomyopathy by MnTBAP [47]. The inability of the compound to exert its protective effects in the ATGL-deficient animal model may originate from insufficient reactivity or steric hindrance in a lipid-overloaded cellular environment. Alternatively, there have been reports questioning the quality, composition and reactivity of commercially available MnTBAP [48,49]. Follow-up studies with NADPH oxidase inhibitors such as gp91ds-tat [15] or with antioxidants such as or apocynin [50] or TEMPOL [51] are necessary to clarify the role of oxidative stress in lipotoxic heart disease.
In summary, our data demonstrate that ATGL deficiency (i) upregulates NADPH oxidase expression and superoxide formation, (ii) increases expression of inflammatory markers, (iii) increases BH4 levels and the BH4/BH2 ratio, and (iv) upregulates sGC without affecting cardiac cGMP levels and VASP phosphorylation. These results suggest that the adverse effects of ATGL deficiency on cardiac performance involve oxidative and inflammatory stress.
The following are the supplementary data related to this article.
Staining of lipid droplets in sections of hearts from WT and ATGL(−/−) mice. Semithin cross sections of cardiac muscle from control (A) and ATGL(−/−) (B) animals stained with azur-methylene blue. Numerous lipid droplets up to 30 μm in diameter covering more than 50% of the total are visible in cardiac sections from ATGL(−/−) mice.
Acknowledgement
We thank Dr. A.C.F. Gorren for critical reading of the manuscript and helpful discussions. We thank Silvia Schauer and Gerald Höfler (Institute of Pathology, Medical University of Graz) for performing and interpreting the histological experiments. The excellent technical assistance of Ines Neubacher is gratefully acknowledged. This work was supported by the Austrian Science Fund (Grants F3003, P24005, and W901 DK Molecular Enzymology to B.M.).
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
Contributor Information
Astrid Schrammel, Email: astrid.schrammel-gorren@uni-graz.at.
Marion Mussbacher, Email: marion.mussbacher@uni-graz.at.
Sarah Winkler, Email: sarah.winkler@uni-graz.at.
Guenter Haemmerle, Email: guenter.haemmerle@uni-graz.at.
Heike Stessel, Email: heike.stessel@uni-graz.at.
Gerald Wölkart, Email: gerald.woelkart@uni-graz.at.
Rudolf Zechner, Email: rudolf.zechner@uni-graz.at.
Bernd Mayer, Email: mayer@uni-graz.at.
References
- 1.Brookheart R.T., Michel C.I., Schaffer J.E. As a matter of fat. Cell Metab. 2009;10:9–12. doi: 10.1016/j.cmet.2009.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Murdoch C.E., Zhang M., Cave A.C., Shah A.M. NADPH oxidase-dependent redox signalling in cardiac hypertrophy, remodelling and failure. Cardiovasc. Res. 2006;71:208–215. doi: 10.1016/j.cardiores.2006.03.016. [DOI] [PubMed] [Google Scholar]
- 3.Takimoto E., Champion H.C., Li M., Ren S., Rodriguez E.R., Tavazzi B., Lazzarino G., Paolocci N., Gabrielson K.L., Wang Y., Kass D.A. Oxidant stress from nitric oxide synthase-3 uncoupling stimulates cardiac pathologic remodeling from chronic pressure load. J. Clin. Invest. 2005;115:1221–1231. doi: 10.1172/JCI21968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Engberding N., Spiekermann S., Schaefer A., Heineke A., Wiencke A., Muller M., Fuchs M., Hilfiker-Kleiner D., Hornig B., Drexler H., Landmesser U. Allopurinol attenuates left ventricular remodeling and dysfunction after experimental myocardial infarction: a new action for an old drug? Circulation. 2004;110:2175–2179. doi: 10.1161/01.CIR.0000144303.24894.1C. [DOI] [PubMed] [Google Scholar]
- 5.Feillet-Coudray C., Sutra T., Fouret G., Ramos J., Wrutniak-Cabello C., Cabello G., Cristol J.P., Coudray C. Oxidative stress in rats fed a high-fat high-sucrose diet and preventive effect of polyphenols: involvement of mitochondrial and NAD(P)H oxidase systems. Free Radic. Biol. Med. 2009;46:624–632. doi: 10.1016/j.freeradbiomed.2008.11.020. [DOI] [PubMed] [Google Scholar]
- 6.Li S.Y., Yang X., Ceylan-Isik A.F., Du M., Sreejayan N., Ren J. Cardiac contractile dysfunction in Lep/Lep obesity is accompanied by NADPH oxidase activation, oxidative modification of sarco(endo)plasmic reticulum Ca2 +-ATPase and myosin heavy chain isozyme switch. Diabetologia. 2006;49:1434–1446. doi: 10.1007/s00125-006-0229-0. [DOI] [PubMed] [Google Scholar]
- 7.Kuroda J., Ago T., Matsushima S., Zhai P., Schneider M.D., Sadoshima J. NADPH oxidase 4 (Nox4) is a major source of oxidative stress in the failing heart. Proc. Natl. Acad. Sci. U. S. A. 2010;107:15565–15570. doi: 10.1073/pnas.1002178107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang M., Brewer A.C., Schroder K., Santos C.X., Grieve D.J., Wang M., Anilkumar N., Yu B., Dong X., Walker S.J., Brandes R.P., Shah A.M. NADPH oxidase-4 mediates protection against chronic load-induced stress in mouse hearts by enhancing angiogenesis. Proc. Natl. Acad. Sci. U. S. A. 2010;107:18121–18126. doi: 10.1073/pnas.1009700107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zimmermann R., Strauss J.G., Haemmerle G., Schoiswohl G., Birner-Gruenberger R., Riederer M., Lass A., Neuberger G., Eisenhaber F., Hermetter A., Zechner R. Fat mobilization in adipose tissue is promoted by adipose triglyceride lipase. Science. 2004;306:1383–1386. doi: 10.1126/science.1100747. [DOI] [PubMed] [Google Scholar]
- 10.Haemmerle G., Lass A., Zimmermann R., Gorkiewicz G., Meyer C., Rozman J., Heldmaier G., Maier R., Theussl C., Eder S., Kratky D., Wagner E.F., Klingenspor M., Hoefler G., Zechner R. Defective lipolysis and altered energy metabolism in mice lacking adipose triglyceride lipase. Science. 2006;312:734–737. doi: 10.1126/science.1123965. [DOI] [PubMed] [Google Scholar]
- 11.Wölkart G., Schrammel A., Dörffel K., Haemmerle G., Zechner R., Mayer B. Cardiac dysfunction in adipose triglyceride lipase deficiency: treatment with a PPARalpha agonist. Br. J. Pharmacol. 2012;165:380–389. doi: 10.1111/j.1476-5381.2011.01490.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kienesberger P.C., Pulinilkunnil T., Sung M.M., Nagendran J., Haemmerle G., Kershaw E.E., Young M.E., Light P.E., Oudit G.Y., Zechner R., Dyck J.R. Myocardial ATGL overexpression decreases the reliance on fatty acid oxidation and protects against pressure overload-induced cardiac dysfunction. Mol. Cell. Biol. 2012;32:740–750. doi: 10.1128/MCB.06470-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schoiswohl G., Schweiger M., Schreiber R., Gorkiewicz G., Preiss-Landl K., Taschler U., Zierler K.A., Radner F.P., Eichmann T.O., Kienesberger P.C., Eder S., Lass A., Haemmerle G., Alsted T.J., Kiens B., Hoefler G., Zechner R., Zimmermann R. Adipose triglyceride lipase plays a key role in the supply of the working muscle with fatty acids. J. Lipid Res. 2010;51:490–499. doi: 10.1194/jlr.M001073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Oury T.D., Thakker K., Menache M., Chang L.Y., Crapo J.D., Day B.J. Attenuation of bleomycin-induced pulmonary fibrosis by a catalytic antioxidant metalloporphyrin. Am. J. Respir. Cell Mol. Biol. 2001;25:164–169. doi: 10.1165/ajrcmb.25.2.4235. [DOI] [PubMed] [Google Scholar]
- 15.Rey F.E., Cifuentes M.E., Kiarash A., Quinn M.T., Pagano P.J. Novel competitive inhibitor of NAD(P)H oxidase assembly attenuates vascular O(2)(−) and systolic blood pressure in mice. Circ. Res. 2001;89:408–414. doi: 10.1161/hh1701.096037. [DOI] [PubMed] [Google Scholar]
- 16.Li Y., Zhu H., Kuppusamy P., Roubaud V., Zweier J.L., Trush M.A. Validation of lucigenin (bis-N-methylacridinium) as a chemilumigenic probe for detecting superoxide anion radical production by enzymatic and cellular systems. J. Biol. Chem. 1998;273:2015–2023. doi: 10.1074/jbc.273.4.2015. [DOI] [PubMed] [Google Scholar]
- 17.Mayer B., Klatt P., Werner E.R., Schmidt K. Molecular mechanisms of inhibition of porcine brain nitric oxide synthase by the antinociceptive drug 7-nitro-indazole. Neuropharmacology. 1994;33:1253–1259. doi: 10.1016/0028-3908(94)90024-8. [DOI] [PubMed] [Google Scholar]
- 18.Schultz G., Böhme E. Guanylate cyclase. GTP pyrophosphate-lyase (cyclizing), EC 4.6.1.2. In: Bergmeyer H.U., Bergmeyer J., Graßl M., editors. Meth Enzym Anal. Verlag Chemie; Weinheim, Germany: 1984. pp. 379–389. [Google Scholar]
- 19.Schmidt K., Mayer B., Kukovetz W.R. Effect of calcium on endothelium-derived relaxing factor formation and cGMP levels in endothelial cells. Eur. J. Pharmacol. 1989;170:157–166. doi: 10.1016/0014-2999(89)90536-0. [DOI] [PubMed] [Google Scholar]
- 20.Bradford M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein–dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 21.Sharma U.C., Pokharel S., van Brakel T.J., van Berlo J.H., Cleutjens J.P., Schroen B., Andre S., Crijns H.J., Gabius H.J., Maessen J., Pinto Y.M. Galectin-3 marks activated macrophages in failure-prone hypertrophied hearts and contributes to cardiac dysfunction. Circulation. 2004;110:3121–3128. doi: 10.1161/01.CIR.0000147181.65298.4D. [DOI] [PubMed] [Google Scholar]
- 22.Yu L., Ruifrok W.P., Meissner M., Bos E.M., van Goor H., Sanjabi B., van der Harst P., Pitt B., Goldstein I.J., Koerts J.A., van Veldhuisen D.J., Bank R.A., van Gilst W.H., Sillje H.H., de Boer R.A. Genetic and pharmacological inhibition of galectin-3 prevents cardiac remodeling by interfering with myocardial fibrogenesis. Circ. Heart Fail. 2013;6:107–117. doi: 10.1161/CIRCHEARTFAILURE.112.971168. [DOI] [PubMed] [Google Scholar]
- 23.Friebe A., Schultz G., Koesling D. Sensitizing soluble guanylyl cyclase to become a highly CO-sensitive enzyme. EMBO J. 1996;15:6863–6868. [PMC free article] [PubMed] [Google Scholar]
- 24.Oelze M., Mollnau H., Hoffmann N., Warnholtz A., Bodenschatz M., Smolenski A., Walter U., Skatchkov M., Meinertz T., Munzel T. Vasodilator-stimulated phosphoprotein serine 239 phosphorylation as a sensitive monitor of defective nitric oxide/cGMP signaling and endothelial dysfunction. Circ. Res. 2000;87:999–1005. doi: 10.1161/01.res.87.11.999. [DOI] [PubMed] [Google Scholar]
- 25.Bitar M.S., Wahid S., Mustafa S., Al-Saleh E., Dhaunsi G.S., Al-Mulla F. Nitric oxide dynamics and endothelial dysfunction in type II model of genetic diabetes. Eur. J. Pharmacol. 2005;511:53–64. doi: 10.1016/j.ejphar.2005.01.014. [DOI] [PubMed] [Google Scholar]
- 26.Landmesser U., Dikalov S., Price S.R., McCann L., Fukai T., Holland S.M., Mitch W.E., Harrison D.G. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J. Clin. Invest. 2003;111:1201–1209. doi: 10.1172/JCI14172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sasaki N., Yamashita T., Takaya T., Shinohara M., Shiraki R., Takeda M., Emoto N., Fukatsu A., Hayashi T., Ikemoto K., Nomura T., Yokoyama M., Hirata K., Kawashima S. Augmentation of vascular remodeling by uncoupled endothelial nitric oxide synthase in a mouse model of diabetes mellitus. Arterioscler. Thromb. Vasc. Biol. 2008;28:1068–1076. doi: 10.1161/ATVBAHA.107.160754. [DOI] [PubMed] [Google Scholar]
- 28.Faulkner K.M., Liochev S.I., Fridovich I. Stable Mn(III) porphyrins mimic superoxide dismutase in vitro and substitute for it in vivo. J. Biol. Chem. 1994;269:23471–23476. [PubMed] [Google Scholar]
- 29.Szabo C., Day B.J., Salzman A.L. Evaluation of the relative contribution of nitric oxide and peroxynitrite to the suppression of mitochondrial respiration in immunostimulated macrophages using a manganese mesoporphyrin superoxide dismutase mimetic and peroxynitrite scavenger. FEBS Lett. 1996;381:82–86. doi: 10.1016/0014-5793(96)00087-7. [DOI] [PubMed] [Google Scholar]
- 30.Heymes C., Bendall J.K., Ratajczak P., Cave A.C., Samuel J.L., Hasenfuss G., Shah A.M. Increased myocardial NADPH oxidase activity in human heart failure. J. Am. Coll. Cardiol. 2003;41:2164–2171. doi: 10.1016/s0735-1097(03)00471-6. [DOI] [PubMed] [Google Scholar]
- 31.Li J.M., Gall N.P., Grieve D.J., Chen M., Shah A.M. Activation of NADPH oxidase during progression of cardiac hypertrophy to failure. Hypertension. 2002;40:477–484. doi: 10.1161/01.hyp.0000032031.30374.32. [DOI] [PubMed] [Google Scholar]
- 32.Sato S., Hughes R.C. Regulation of secretion and surface expression of Mac-2, a galactoside-binding protein of macrophages. J. Biol. Chem. 1994;269:4424–4430. [PubMed] [Google Scholar]
- 33.Cinti S., Mitchell G., Barbatelli G., Murano I., Ceresi E., Faloia E., Wang S., Fortier M., Greenberg A.S., Obin M.S. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. J. Lipid Res. 2005;46:2347–2355. doi: 10.1194/jlr.M500294-JLR200. [DOI] [PubMed] [Google Scholar]
- 34.Sano H., Hsu D.K., Apgar J.R., Yu L., Sharma B.B., Kuwabara I., Izui S., Liu F.T. Critical role of galectin-3 in phagocytosis by macrophages. J. Clin. Invest. 2003;112:389–397. doi: 10.1172/JCI17592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sano H., Hsu D.K., Yu L., Apgar J.R., Kuwabara I., Yamanaka T., Hirashima M., Liu F.T. Human galectin-3 is a novel chemoattractant for monocytes and macrophages. J. Immunol. 2000;165:2156–2164. doi: 10.4049/jimmunol.165.4.2156. [DOI] [PubMed] [Google Scholar]
- 36.Moens A.L., Ketner E.A., Takimoto E., Schmidt T.S., O'Neill C.A., Wolin M.S., Alp N.J., Channon K.M., Kass D.A. Bi-modal dose-dependent cardiac response to tetrahydrobiopterin in pressure-overload induced hypertrophy and heart failure. J. Mol. Cell. Cardiol. 2011;51:564–569. doi: 10.1016/j.yjmcc.2011.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kalivendi S., Hatakeyama K., Whitsett J., Konorev E., Kalyanaraman B., Vasquez-Vivar J. Changes in tetrahydrobiopterin levels in endothelial cells and adult cardiomyocytes induced by LPS and hydrogen peroxide—a role for GFRP? Free Radic. Biol. Med. 2005;38:481–491. doi: 10.1016/j.freeradbiomed.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 38.Haemmerle G., Moustafa T., Woelkart G., Buttner S., Schmidt A., van de Weijer T., Hesselink M., Jaeger D., Kienesberger P.C., Zierler K., Schreiber R., Eichmann T., Kolb D., Kotzbeck P., Schweiger M., Kumari M., Eder S., Schoiswohl G., Wongsiriroj N., Pollak N.M., Radner F.P., Preiss-Landl K., Kolbe T., Rulicke T., Pieske B., Trauner M., Lass A., Zimmermann R., Hoefler G., Cinti S., Kershaw E.E., Schrauwen P., Madeo F., Mayer B., Zechner R. ATGL-mediated fat catabolism regulates cardiac mitochondrial function via PPAR-alpha and PGC-1. Nat. Med. 2011;17:1076–1085. doi: 10.1038/nm.2439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Smeets P.J., Teunissen B.E., Willemsen P.H., van Nieuwenhoven F.A., Brouns A.E., Janssen B.J., Cleutjens J.P., Staels B., van der Vusse G.J., van Bilsen M. Cardiac hypertrophy is enhanced in PPAR alpha−/− mice in response to chronic pressure overload. Cardiovasc. Res. 2008;78:79–89. doi: 10.1093/cvr/cvn001. [DOI] [PubMed] [Google Scholar]
- 40.Guellich A., Damy T., Lecarpentier Y., Conti M., Claes V., Samuel J.L., Quillard J., Hebert J.L., Pineau T., Coirault C. Role of oxidative stress in cardiac dysfunction of PPARalpha−/− mice. Am. J. Physiol. 2007;293:H93–H102. doi: 10.1152/ajpheart.00037.2007. [DOI] [PubMed] [Google Scholar]
- 41.Moe K.T., Aulia S., Jiang F., Chua Y.L., Koh T.H., Wong M.C., Dusting G.J. Differential upregulation of Nox homologues of NADPH oxidase by tumor necrosis factor-alpha in human aortic smooth muscle and embryonic kidney cells. J. Cell. Mol. Med. 2006;10:231–239. doi: 10.1111/j.1582-4934.2006.tb00304.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kilpatrick L.E., Sun S., Li H., Vary T.C., Korchak H.M. Regulation of TNF-induced oxygen radical production in human neutrophils: role of delta-PKC. J. Leukoc. Biol. 2010;87:153–164. doi: 10.1189/jlb.0408230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ritchie R.H., Irvine J.C., Rosenkranz A.C., Patel R., Wendt I.R., Horowitz J.D., Kemp-Harper B.K. Exploiting cGMP-based therapies for the prevention of left ventricular hypertrophy: NO* and beyond. Pharmacol. Ther. 2009;124:279–300. doi: 10.1016/j.pharmthera.2009.08.001. [DOI] [PubMed] [Google Scholar]
- 44.Huie R.E., Padmaja S. The reaction of no with superoxide. Free Radic. Biol. Med. 1993;18:195–199. doi: 10.3109/10715769309145868. [DOI] [PubMed] [Google Scholar]
- 45.Stone J.R., Marletta M.A. Soluble guanylate cyclase from bovine lung: activation with nitric oxide and carbon monoxide and spectral characterization of the ferrous and ferric states. Biochemistry. 1994;33:5636–5640. doi: 10.1021/bi00184a036. [DOI] [PubMed] [Google Scholar]
- 46.Kuramoto K., Okamura T., Yamaguchi T., Nakamura T.Y., Wakabayashi S., Morinaga H., Nomura M., Yanase T., Otsu K., Usuda N., Matsumura S., Inoue K., Fushiki T., Kojima Y., Hashimoto T., Sakai F., Hirose F., Osumi T. Perilipin 5, a lipid droplet-binding protein, protects heart from oxidative burden by sequestering fatty acid from excessive oxidation. J. Biol. Chem. 2012;287:23852–23863. doi: 10.1074/jbc.M111.328708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ding G., Fu M., Qin Q., Lewis W., Kim H.W., Fukai T., Bacanamwo M., Chen Y.E., Schneider M.D., Mangelsdorf D.J., Evans R.M., Yang Q. Cardiac peroxisome proliferator-activated receptor gamma is essential in protecting cardiomyocytes from oxidative damage. Cardiovasc. Res. 2007;76:269–279. doi: 10.1016/j.cardiores.2007.06.027. [DOI] [PubMed] [Google Scholar]
- 48.Batinic-Haberle I., Cuzzocrea S., Reboucas J.S., Ferrer-Sueta G., Mazzon E., Di Paola R., Radi R., Spasojevic I., Benov L., Salvemini D. Pure MnTBAP selectively scavenges peroxynitrite over superoxide: comparison of pure and commercial MnTBAP samples to MnTE-2-PyP in two models of oxidative stress injury, an SOD-specific Escherichia coli model and carrageenan-induced pleurisy. Free Radic. Biol. Med. 2009;46:192–201. doi: 10.1016/j.freeradbiomed.2008.09.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Reboucas J.S., Spasojevic I., Batinic-Haberle I. Quality of potent Mn porphyrin-based SOD mimics and peroxynitrite scavengers for pre-clinical mechanistic/therapeutic purposes. J. Pharm. Biomed. Anal. 2008;48:1046–1049. doi: 10.1016/j.jpba.2008.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu J., Zhou J., An W., Lin Y., Yang Y., Zang W. Apocynin attenuates pressure overload-induced cardiac hypertrophy in rats by reducing levels of reactive oxygen species. Can. J. Physiol. Pharmacol. 2010;88:745–752. doi: 10.1139/y10-063. [DOI] [PubMed] [Google Scholar]
- 51.Chess D.J., Xu W., Khairallah R., O'Shea K.M., Kop W.J., Azimzadeh A.M., Stanley W.C. The antioxidant tempol attenuates pressure overload-induced cardiac hypertrophy and contractile dysfunction in mice fed a high-fructose diet. Am. J. Physiol. 2008;295:H2223–H2230. doi: 10.1152/ajpheart.00563.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Staining of lipid droplets in sections of hearts from WT and ATGL(−/−) mice. Semithin cross sections of cardiac muscle from control (A) and ATGL(−/−) (B) animals stained with azur-methylene blue. Numerous lipid droplets up to 30 μm in diameter covering more than 50% of the total are visible in cardiac sections from ATGL(−/−) mice.