

Abstract
Histone deacetylases (HDACs), together with histone acetyltransferases (HATs), regulate gene expression by modulating the acetylation level of chromatin. HDAC3 is implicated in many important cellular processes, particularly in cancer cell proliferation and metastasis, making inhibition of HDAC3 a promising epigenetic treatment for certain cancers. HDAC3 is activated upon complex formation with both inositol tetraphosphate (IP4) and the deacetylase-activating domain (DAD) of multi-protein nuclear receptor corepressor complexes. In previous studies, we have shown that binding of DAD and IP4 to HDAC3 significantly restricts its conformational space towards its stable ternary complex conformation, and suggest this to be the active conformation. Here, we report a single mutation of HDAC3 that is capable of mimicking the stabilizing effects of DAD and IP4, without the presence of either. This mutation, however, results in a total loss of deacetylase activity, prompting a closer evaluation of our understanding of the activation of HDAC3.
Keywords: HDAC3, R265P, conformational selection, allostery, molecular recognition
Introduction
Histone deacetylases (HDACs) were originally discovered and named for their role as keepers of the histone code.1 Through deacetylation of acetyllysines located on the n-termini, or “tails,” of histone proteins, HDACs repress transcription of certain genes.2 It is now known, however, that HDACs also modulate the activity of many other nonhistone proteins, like p53 and NFκB.1 HDAC3 is a deacetylase that has quickly become a popular drug target as it not only promotes cell proliferation and metastasis in cancer cells, but is also implicated in several neurodegenerative diseases as well as in HIV-1 latency.3–6
Like nearly all other HDACs, HDAC3 is only active upon recruitment to a multiprotein nuclear receptor corepressor complex. Specifically, HDAC3 must be minimally bound to the deacetylase-activating domain (DAD) of either the silencing mediator for retinoid and thyroid hormone receptors (SMRT) or the homologous nuclear receptor co-repressor 1 (NCoR) complex.7–9
Upon resolving the crystal structure of HDAC3, bound to DAD, Watson et al. also discovered that a molecule of inositol tetraphosphate (IP4) rested at the protein-protein interface (1).10 Although it was established that IP4 is necessary for both formation of the HDAC3:DAD complex and for deacetylase activation, the underlying structural mechanism was not clear. Mutagenesis data has suggested that a delicate interdependence between IP4 and DAD is important in the activation of HDAC3.10,11
Recently, using molecular dynamics (MD) simulations, we showed that binding of either DAD or IP4 to HDAC3 results in a restriction of its conformational space, which is further exemplified once both are present in the active ternary (HDAC3:IP4:DAD) complex.12 We proposed that this rigid conformation is important for the activation of HDAC3, likely by priming the deacetylase for substrate recognition.
Here, we report an HDAC3 mutant that mimics the rigid ternary complex deacetylase conformation, without the need for DAD or IP4. A single arginine-to-proline substitution at position 265 (R265P) is capable of biasing the HDAC3 conformational ensemble towards its active state, identifying the 265 position as a critical hot spot in the allosteric activation of HDAC3. Intriguingly, despite its structural dynamics that suggest it to be constitutively active, the HDAC3R265P mutant was actually shown to be inactive.10 We discuss the implications of these results on our understanding of the general activation mechanism of HDAC3 by DAD and IP4.
Results
In our previous work, we showed that the conformational ensemble of apo HDAC3WT is restricted upon binding to DAD and IP4.12 Here, we use several structural metrics, outlined below, to compare the conformational flexibility of the inactive HDAC3R265P mutant to that of the free and complexed HDAC3WT states, based on triplicate 100-ns molecular dynamics (MD) simulations for each state.
Global stability of apo HDAC3R265P resembles that of activated HDAC3WT
Using principal component (PC) analysis, we characterized how the HDAC3 dynamics change according to its two most dominant low-frequency modes. By projecting the HDAC3:IP4, HDAC3:DAD, and HDAC3:IP4:DAD trajectories onto a PC space built from the apo HDAC3WT simulations, we previously showed a constriction of the deacetylase ensemble upon binding to DAD and IP4 (2, panels A-D).12 We note that binding of either DAD or IP4 results in comparable restriction of dynamics along these modes and the deacetylase backbone is further restricted in the active ternary complex.
The projection of the mutant HDAC3R265P trajectories onto the WT apo PC space demonstrates a bias in its conformational ensemble toward the active state (2, panel E). We also note that the mutant deacetylase, in its apo form, maintained a surprisingly low RMSD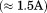 to the crystal structure of the WT deacetylase in the HDAC3:IP4:DAD complex.
to the crystal structure of the WT deacetylase in the HDAC3:IP4:DAD complex.
PC1 and PC2 (movies available in Ref.12) together describe a contractile motion around the active site. PC2 is dominated by the dynamics of Loop 1, whereas PC1 primarily results from concerted motions of Helix 6, the C-terminal helix, and Loop 6 (1). The mutated residue, R265, resides on Loop 6, where it interacts with either the solvent or IP4, if present. Nearby K25 on Loop 1 also interacts with IP4, and causes a charge-charge repulsion with R265 in the absence of IP4. The proline at this position could trivially be expected to stabilize Loop 6, both by removing one of the positive charges from this basic region, and by the rigid nature of the proline residue itself. The dramatic constriction of the conformational ensemble observed for apo HDAC3R265P, however, involves other regions quite far from the mutation site, such as the C-terminal helix and Helix 6, indicating several wide-spread allosteric networks in HDAC3 that can be triggered at position 265.
To further dissect the effect of the inactivating R265P mutation on the deacetylase conformational plasticity, we also analyzed the change in the per-residue root mean squared fluctuations (ΔRMSF) for the mutant and the WT complexed states, relative to the apo HDAC3WT fluctuations. In this way, negative ΔRMSF values indicate residues that are stabilized in the mutant or complexed states, relative to HDAC3WT in its apo form.
First, we note that the R265P mutation results in the stabilization of the same WT deacetylase regions that are stabilized upon complex formation with either DAD, IP4, or both (3). The random coil region at positions 340 to 348 (1) is the only exception to this trend, as it is only destabilized in the ternary complex, but not by DAD, IP4 or the R265P mutation alone. We also identified a striking similarity between the HDAC3R265P and the HDAC3:IP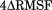 plots (3). For both the mutant and the IP4-bound, but not DAD-bound deacetylase, we observed negative peaks at the C-terminal helix, which is believed to include the nuclear localization signal (NLS) of HDAC3.13 Other motifs that are stabilized to a greater extent by DAD, for example, Loop 1, were affected to a lesser extent by either the binding of IP4 or by the mutation. The replacement of just one of the three key IP4-interacting residues appears to result in the same curious ΔRMSF signature as seen with IP4 binding.
plots (3). For both the mutant and the IP4-bound, but not DAD-bound deacetylase, we observed negative peaks at the C-terminal helix, which is believed to include the nuclear localization signal (NLS) of HDAC3.13 Other motifs that are stabilized to a greater extent by DAD, for example, Loop 1, were affected to a lesser extent by either the binding of IP4 or by the mutation. The replacement of just one of the three key IP4-interacting residues appears to result in the same curious ΔRMSF signature as seen with IP4 binding.
Geometry of apo HDAC3R265P active site channel resembles that of activated HDAC3WT
Most of the key stabilized regions in HDAC3WT form a channel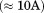 to the active site. By analyzing the radii along the length of the channel during the MD simulations, we observed that the channel in the active ternary complex maintains its geometry, whereas the apo HDAC3WT channel has large fluctuations in its geometry (4). The apo HDAC3R265P data also indicate a very restrained active site channel geometry, although not identical to that of the ternary complex deacetylase.
to the active site. By analyzing the radii along the length of the channel during the MD simulations, we observed that the channel in the active ternary complex maintains its geometry, whereas the apo HDAC3WT channel has large fluctuations in its geometry (4). The apo HDAC3R265P data also indicate a very restrained active site channel geometry, although not identical to that of the ternary complex deacetylase.
The form of this active site channel is likely an important part of the substrate recognition mechanism of HDAC3. An analysis of the key channel-lining residues highlights a cluster of hydrophobic residues at the opening of the channel, followed by more polar residues closer to the active site. This distribution of channel-lining residues is indicative of the current pharmacophore of HDAC inhibitors, as well as of the acetyllysine binding mode.14
One of the key features of the active site channel is the conserved residue Tyr298, which serves to anchor the acetyllysine substrate (5). From the multiple MD simulations, we found that Tyr298 samples two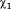 dihedral states, only one of which permits the catalytic interaction with the carbonyl oxygen of the acetyl group (5). For HDAC3WT, the binding of both IP4 and DAD shifts the rotameric population exclusively toward the catalytically relevant conformation, inward, whereas the apo HDAC3WT ensemble reflects nearly equal probability of inward and outward Tyr298 conformations. The R265P mutation, appears to also bias the Tyr298 conformation toward the catalytically relevant state, without the presence of DAD or IP4.
dihedral states, only one of which permits the catalytic interaction with the carbonyl oxygen of the acetyl group (5). For HDAC3WT, the binding of both IP4 and DAD shifts the rotameric population exclusively toward the catalytically relevant conformation, inward, whereas the apo HDAC3WT ensemble reflects nearly equal probability of inward and outward Tyr298 conformations. The R265P mutation, appears to also bias the Tyr298 conformation toward the catalytically relevant state, without the presence of DAD or IP4.
Discussion
We identified a mutation, R265P, on one of the active site loops of HDAC3, which is capable of reproducing many of the long-range stabilizing effects of IP4 or DAD binding. These results confirm the presence of an allosteric network in HDAC3 that can be triggered at position 265, either by small molecule binding, or as shown here through the R265P replacement. Fluctuations in Loop 6 (1) are directly linked to large fluctuations in Helix 6 and the C-terminal helix, the latter of which contains the NLS of HDAC3.13 These fluctuations appear to be transmitted through the protein backbone via the random coil region between residues 340 and 348, as well as the other nearby active site loops, resulting in an overall stabilized backbone upon stabilization of a single amino acid side chain. Even though its conformational ensemble suggests the HDAC3R265P mutant may be constitutively active, however, it was instead shown to result in a loss of all WT deacetylase activity.10
Activation of HDAC3 may require further stabilization
Although the structural dynamics of the HDAC3R265P mutant are biased towards the deacetylase conformation in the active ternary complex, our results do show that the mutant ensemble more closely resembles either the HDAC3:DAD or HDAC3:IP4 intermediate complexes, neither of which has been shown to be active, to our knowledge. It is possible, then that the deacetylase activity requires additional “fine tuning” of the conformational ensemble. In each of our structural analyses, we observed a synergistic restricting effect on the deacetylase dynamics when both IP4 and DAD were bound, which was not observed in the simulations of the apo HDAC3R265P mutant. The R265P mutation has been shown to preclude complex formation with DAD,10 which may be required to further refine the conformational ensemble toward the active state. We should note, however, that we have also performed simulations of the mutant bound to both IP4 and DAD (not shown here), but did not observe any further stabilization of the deacetylase backbone than was observed in the apo HDAC3R265P case. The HDAC3R265P ensemble, although biased toward the active conformation, may still be lacking necessary, yet subtle, structural rearrangements to be activated.
It is also possible that the physical presence of IP4 and DAD themselves contributes to substrate recognition or stabilization, in addition to their synergistic stabilizing effects of the deacetylase backbone. In this case, the loss of affinity for DAD and IP4 would be enough to result in a loss of deacetylase activity associated with the R265P mutation. We think this is unlikely, however, because even though DAD is necessary for histone recognition of HDAC3,15 the HDAC activity assay used to study the mutant utilizes a peptide substrate that can be deacetylated by a number of HDACs, which do not specifically require DAD.10
R265P mutation may alter substrate specificity
It seems, then, that due to rather subtle details in its dynamics, the HDAC3R265P mutant is unable to deacetylate the same substrate that the WT can deacetylate when bound to DAD and IP4. Because of its stability and close resemblance to the WT deacetylase in the ternary complex, however, it is plausible that this mutant may still be active toward some other acetyllysine substrates. In the future, alternative assays with multiple peptide substrates may be used to detect possible changes in deacetylase target specificity as a result of this mutation.
Implications on HDAC3 biology
A shifted substrate specificity for the HDAC3R265P mutant would be of particular biological relevance as this mutation can occur as a result of a known single nucleotide polymorphism (rs467744), not yet associated with any known phenotypes, to our knowledge. Moreover, if this intermediately restrained conformational ensemble of HDAC3 is associated with an altered substrate specificity, this would have profound implications on the role of IP4 in modulating HDAC3WT activity, since the HDAC3:IP4 complex has a comparably restrained conformational ensemble. Because HDAC3 is known to localize to both the nucleus and cytoplasm, and has been shown to modulate cellular processes even when unable to bind to DAD,15 it is possible that IP4 may serve to activate cytoplasmic HDAC3 toward other nonhistone targets, and even promote nuclear import of HDAC3 through stabilization of its NLS. The HDAC3R265P mutant, on the other hand, may be unaffected by the inositol signaling pathway and have aberrant subcellular localization.
Evolutionary origins of a constitutively active HDAC
The R265 residue is conserved in nearly all of the class I HDACs, suggesting perhaps the same allosteric network is conserved within the catalytic HDAC core.16,17 The exception, however, is HDAC8, which happens to also be the only deaetylase in its class that is constitutively active.18 Intriguingly, in HDAC8, the conserved arginine at position 265 is instead a proline. Our results on the R265P mutant provide insight into the molecular mechanism through which this constitutive activity may be conferred to HDAC8. Future assays of the HDAC3R265P mutant specificity, among that of other HDAC8-like mutants, would be necessary in order to address this exciting idea.
Computational Methods
System setup and equilibration
The systems were built using the AMBER ff99SB force field, with the ildn modification for ILE, LEU, ASP, and ASN residues.19 Parameters for both IP4 and the active site Zn center, as well as protonation states were taken from previous work.12 Each system was solvated in a 10.0 Å TIP3P octahedron, and potassium ions were added to neutralize the charge in each case: HDAC (9), HDAC:IP4 (17), HDAC:IP4:DAD (13).20 Additional potassium and chloride ions were added to simulate physiological salt concentration (0.15M). The updated Lennard Jones parameters for ions were used.21 Systems were heated to 300K at constant volume (NVT) with restraints on the protein that were gradually reduced from 200 to 0 kcal/mol/Å2 over a period of 150 ps. The Langevin thermostat was used with a collision frequency of 1.0 ps−1. The SHAKE algorithm was used to constrain bonds to nonpolar hydrogens, and a 1.0-fs time-step was used during dynamics.22 An 8.0 Å cutoff was used for nonbonded interactions. Equilibration runs (100 ps) were done in the NPT ensemble, using isotropic pressure scaling and a pressure relaxation time of 2.0 ps.
Production MD simulations
For each system, three independent simulations were started from different snapshots from the equilibration simulations (taken at arbitrary intervals after density of water box was equilibrated) with randomized velocities. All production runs were done in the same NVT conditions described above, with the exception of a 2.0-fs time-step. All simulations were done using the PMEMD module within the Amber11 simulation package.23 Production runs were performed on GPUs using Amber11 pmemd.CUDA, each for 100 ns.
Trajectory analysis
We used the ptraj module from the AmberTools package to perform RMSD, RMSF, and PC analyses. In all analyses, the protein backbone atoms were aligned to the coordinates in the crystal structure (PDB ID 4A69). For the PC analysis, we build the PC1:PC2 space using the first two principal components (over 50% of the total variance) of the three 100-ns apo HDAC3WT trajectories, using atoms only.
atoms only. atoms from all other trajectories were first aligned to the crystal structure before projecting into the PC space. For the analysis of the active site channel geometry, we used 100 snapshots taken from equally spaced intervals from the three independent 100-ns simulations of each state. We used the Caver algorithm to find tunnels in the protein, using starting points in the active site (residues 132, 255, and 145), a probe radius of 0.9 Å, and a shell radius of 6.0 Å.24 Clustering of tunnels was done using the average linkage algorithm, with a threshold of 4.
atoms from all other trajectories were first aligned to the crystal structure before projecting into the PC space. For the analysis of the active site channel geometry, we used 100 snapshots taken from equally spaced intervals from the three independent 100-ns simulations of each state. We used the Caver algorithm to find tunnels in the protein, using starting points in the active site (residues 132, 255, and 145), a probe radius of 0.9 Å, and a shell radius of 6.0 Å.24 Clustering of tunnels was done using the average linkage algorithm, with a threshold of 4.
Figure 1.
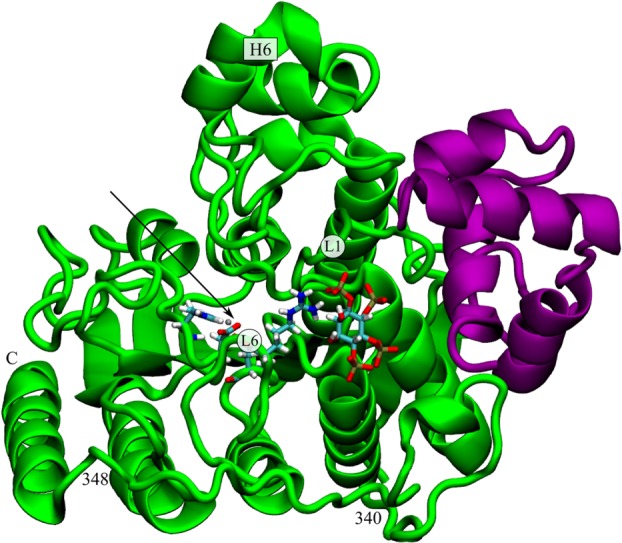
Cartoon representation of HDAC3 (green ribbon) bound to DAD (purple ribbon) and IP4 (sticks), from Protein Data Bank (PDB) identification code: 4A69.10 Arrow indicates the location of the active site. For reference, some loops and helices are identified, as well as the location of the R265 residue.
Figure 2.

Principal component analysis of HDAC3. (A) apo HDAC3, (B) IP4-bound HDAC3, (C) DAD-bound HDAC3, (D) IP4- and DAD-bound HDAC3, and (E) apo HDAC3R265P mutant dynamics are each projected onto PC1:PC2 space of the apo HDAC3WT12. A white cross in each panel indicates the projection of the HDAC3:IP4:DAD crystal structure (PDB ID: 4A6910).
Figure 3.
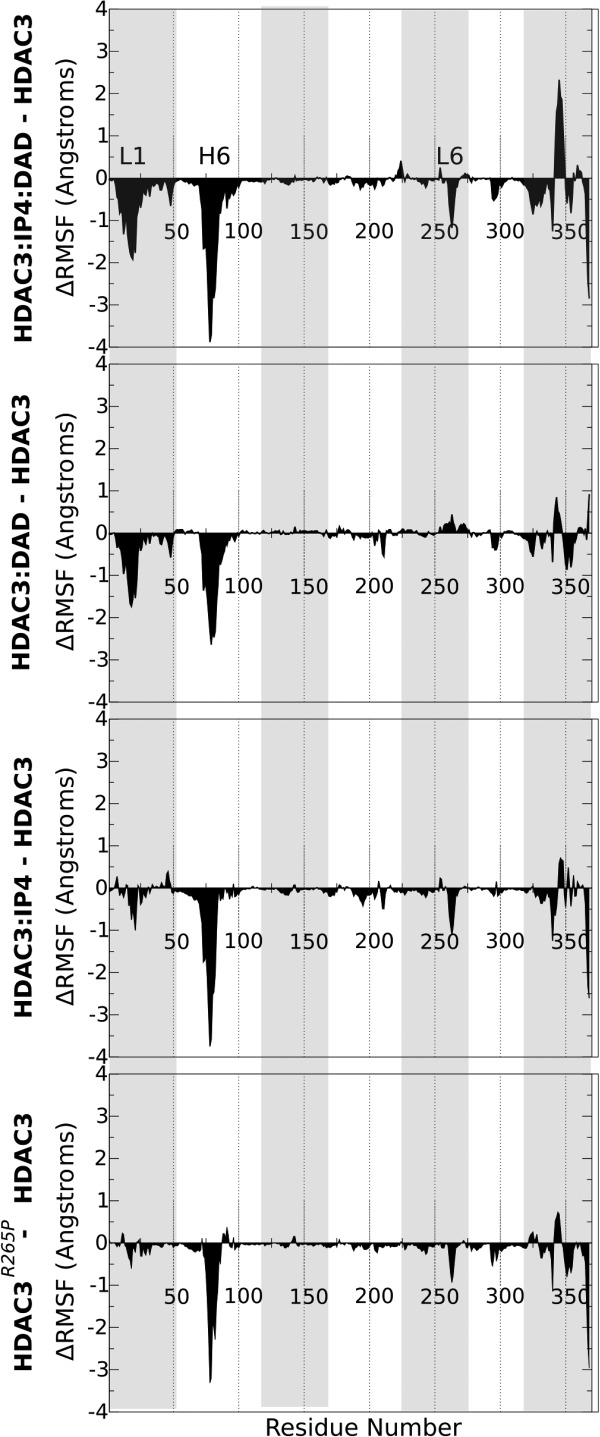
 Analysis of mutant and complexed HDAC3. Differences between per-residue RMSF values (Å) are shown for (from top to bottom) HDAC3:IP4:DAD, HDAC3:DAD, HDAC3:IP4,12 and HDAC3R265P, with reference to the apo HDAC3WT RMSF values. Negative
Analysis of mutant and complexed HDAC3. Differences between per-residue RMSF values (Å) are shown for (from top to bottom) HDAC3:IP4:DAD, HDAC3:DAD, HDAC3:IP4,12 and HDAC3R265P, with reference to the apo HDAC3WT RMSF values. Negative values correspond to stabilized regions.
values correspond to stabilized regions.
Figure 4.
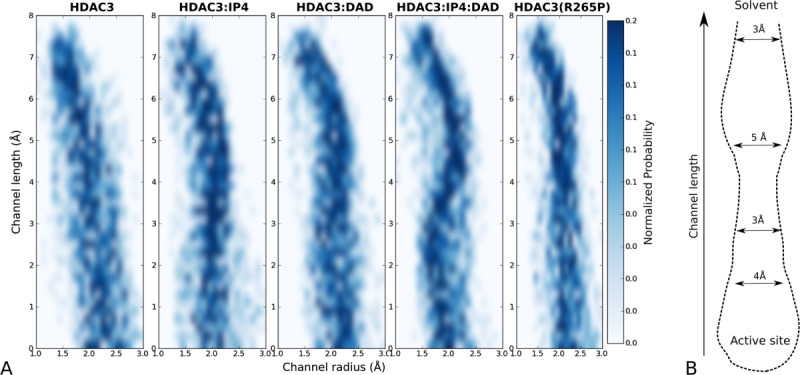
Geometry of active site channel. (A) Two-dimensional histograms showing the distribution of radii along the channel length for the apo, IP4-bound, DAD-bound, and ternary WT12 simulations, as well as for the apo R265P mutant. (B) The stable channel leading to the active site, from simulations of the HDAC3:IP4:DAD complex is shown schematically, with average diameters indicated along the channel length.
Figure 5.
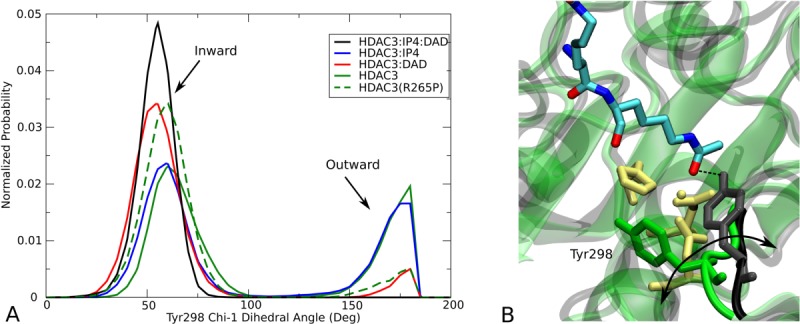
Population analysis of Tyr298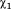 dihedral angle. (A) Normalized probability of the
dihedral angle. (A) Normalized probability of the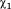 side chain dihedral angle shows two states (inward and outward) sampled in the HDAC312 simulations. (B) Cartoon representation of HDAC3 from the HDAC3:IP4:DAD (black) and apo (green) simulations are superimposed to highlight the two Tyr298 conformations. An arbitrary acetyllysine peptide substrate was docked into the active site to show the functional relevance of the inward Tyr298 conformation (hydrogen atoms are not shown).
side chain dihedral angle shows two states (inward and outward) sampled in the HDAC312 simulations. (B) Cartoon representation of HDAC3 from the HDAC3:IP4:DAD (black) and apo (green) simulations are superimposed to highlight the two Tyr298 conformations. An arbitrary acetyllysine peptide substrate was docked into the active site to show the functional relevance of the inward Tyr298 conformation (hydrogen atoms are not shown).
References
- 1.Minucci S, Pelicci PG. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat Rev Cancer. 2006;6:38–51. doi: 10.1038/nrc1779. [DOI] [PubMed] [Google Scholar]
- 2.de Ruijter AJM, van Gennip AH, Caron HN, Kemp S, van Kuilenburg ABP. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J. 2003;370:737–749. doi: 10.1042/BJ20021321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Witt O, Deubzer HE, Milde T, Oehme I. HDAC family: what are the cancer relevant targets? Cancer Lett. 2009;277:8–21. doi: 10.1016/j.canlet.2008.08.016. [DOI] [PubMed] [Google Scholar]
- 4.Hagelkruys A, Sawicka A, Rennmayr M, Seiser C. The biology of HDAC in cancer: the nuclear and epigenetic components. In: Yao TP, Seto E, editors. Histone deacetylases: the biology and clinical implication. Spinger-Verlag Berlin: Heidelberg; 2011. pp. 13–37. [Google Scholar]
- 5.Gottesfeld JM, Pandolfo M. Development of histone deacetylase inhibitors as therapeutics for neurological disease. Future Neurol. 2009;4:775–784. doi: 10.2217/fnl.09.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huber K, Doyon G, Plaks J, Fyne E, Mellors JW, Sluis-Cremer N. Inhibitors of histone deacetylases: correlation between isoform specificity and reactivation of HIV type 1 (HIV-1) from latently infected cells. J Biol Chem. 2011;286:22211–22218. doi: 10.1074/jbc.M110.180224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oberoi J, Fairall L, Watson PJ, Yang JC, Czimmerer Z, Kampmann T, Goult BT, Greenwood JA, Gooch JT, Kallenberger BC, Nagy L, Neuhaus D, Schwabe JWR. Structural basis for the assembly of the SMRT/NCoR core transcriptional repression machinery. Nat Struct Mol Biol. 2011;18:177–184. doi: 10.1038/nsmb.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li J, Wang J, Wang J, Nawaz Z, Liu JM, Qin J, Wong J. Both corepressor proteins SMRT and N-CoR exist in large protein complexes containing HDAC3. EMBO J. 2000;19:4342–4350. doi: 10.1093/emboj/19.16.4342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guenther MG, Barak O, Lazar MA. The SMRT and N-CoR corepressors are activating cofactors for histone deacetylase 3. Mol Cell Biol. 2001;21:6091–6101. doi: 10.1128/MCB.21.18.6091-6101.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Watson PJ, Fairall L, Santos GM, Schwabe JWR. Structure of HDAC3 bound to co-repressor and inositol tetraphosphate. Nature. 2012;481:335–340. doi: 10.1038/nature10728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Codina A, Love JD, Li Y, Lazar MA, Neuhaus D, Schwabe JWR. Structural insights into the interaction and activation of histone deacetylase 3 by nuclear receptor corepressors. Proc Natl Acad Sci USA. 2005;102:6009–6014. doi: 10.1073/pnas.0500299102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arrar M, Turnham R, Pierce L, de Oliveira CAF, McCammon JA. Structural insight into the separate roles of inositol tetraphosphate and deacetylase-activating domain in activation of histone deacetylase 3. Protein Sci. 2013;22:83–92. doi: 10.1002/pro.2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang WM, Tsai SC, Wen YD, Fejer G, Seto E. Functional domains of histone deacetylase-3. J Biol Chem. 2002;277:9447–9454. doi: 10.1074/jbc.M105993200. [DOI] [PubMed] [Google Scholar]
- 14.Marks P. Histone deacetylases. Curr Opin Chem Biol. 2003;3:344–351. doi: 10.1016/s1471-4892(03)00084-5. [DOI] [PubMed] [Google Scholar]
- 15.You SH, Lim HW, Sun Z, Broache M, Won KJ, Lazar MA. Nuclear receptor co-repressors are required for the histone-deacetylase activity of HDAC3 in vivo. Nat Struct Mol Biol. 2013;20:182–187. doi: 10.1038/nsmb.2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gregoretti I, Lee YM, Goodson HV. Molecular evolution of the histone deacetylase family: functional implications of phylogenetic analysis. J Mol Biol. 2004;338:17–31. doi: 10.1016/j.jmb.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 17.Dowling DP, Costanzo L, Gennadios HA, Christianson DW. Evolution of the arginase fold and functional diversity. Cell Mol Life Sci. 2008;65:2039–2055. doi: 10.1007/s00018-008-7554-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Van den Wyngaert I, de Vries W, Kremer A, Neefs J, Verhasselt P, Luyten WH, Kass SU. Cloning and characterization of human histone deacetylase 8. FEBS Lett. 2000;478:77–83. doi: 10.1016/s0014-5793(00)01813-5. [DOI] [PubMed] [Google Scholar]
- 19.Lindorff-Larsen K, Piana S, Palmo K, Maragakis P, Klepeis JL, Dror RO, Shaw DE. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins. 2010;78:1950–1958. doi: 10.1002/prot.22711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 21.Joung IS, Cheatham TE. Determination of alkali and halide monovalent ion parameters for use in explicitly solvated biomolecular simulations. J Phys Chem B. 2008;112:9020–9041. doi: 10.1021/jp8001614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ryckaert JP, Ciccotti G, Berendsen HJC. Numerical integration of the Cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J Comput Phys. 1977;23:327–341. [Google Scholar]
- 23.Case TA, Darden DA, Cheatham III TE, Simmerling CL, Wang J, Duke RE, Luo R, Walker RC, Zhang W, Merz KM, Roberts B, Hayik S, Roitberg A, Seabra G, Swails J, Goetz AW, Kolossváry I, Wong KF, Paesani F, Vanicek J, Wolf RM, Liu J, Wu X, Brozell SR, Steinbrecher T, Gohlke H, Cai Q, Ye X, Wang J, Hsieh M-J, Cui G, Roe DR, Mathews DH, Seetin MG, Salomon-Ferrer R, Sagui C, Babin V, Luchko T, Gusarov S, Kovalenko A, Kollman PA. Amber 11. San Francisco: University of California; 2010. [Google Scholar]
- 24.Chovancova E, Pavelka A, Benes P, Strnad O, Brezovsky J, Kozlikova B, Gora A, Sustr V, Klvana M, Medek P, Biedermannova L, Sochor J, Damborsky J. CAVER 3.0: A Tool for the analysis of transport pathways in dynamic protein structures. PLoS Comput Biol. 2012;8:e1002708. doi: 10.1371/journal.pcbi.1002708. [DOI] [PMC free article] [PubMed] [Google Scholar]