

Abstract
To investigate early intermediates of β2-microglobulin (β2m) amyloidogenesis, we solved the structure of β2m containing the amyloidogenic Pro32Gly mutation by X-ray crystallography. One nanobody (Nb24) that efficiently blocks fibril elongation was used as a chaperone to co-crystallize the Pro32Gly β2m monomer under physiological conditions. The complex of P32G β2m with Nb24 reveals a trans peptide bond at position 32 of this amyloidogenic variant, whereas Pro32 adopts the cis conformation in the wild-type monomer, indicating that the cis to trans isomerization at Pro32 plays a critical role in the early onset of β2m amyloid formation.
Keywords: β2-microglobulin, dialysis-related amyloidosis, nanobodies, X-ray crystallography, proline isomerization, protein conformation
Introduction
For many proteins, abnormal structure or metabolism results in self-aggregation, causing the formation of fibrillar structures that give rise to amyloid fibrils.1,2 The full elucidation of the mechanism of this aggregation process requires the identification of all the transitional conformational states and oligomeric structures adopted by the polypeptide chain. The identification and characterization of oligomers preceding the formation of fibrils is of particular interest because of an increasing awareness that these species are likely to play a critical role in the pathogenesis of protein deposition diseases.3,4 However, the characterization of early aggregation-prone monomeric species also remains a challenge.5
Over the years, human β2-microglobulin (β2m) has been used extensively as a model system to elucidate the molecular mechanism of amyloidosis. β2m is a small protein (99 amino acids) that adopts the typical seven-stranded β-sandwich immunoglobulin fold formed by two antiparallel β-sheets (strands ABED and strands GFC) that are connected via a single disulphide bond (Cys25–Cys80).6,7 β2m is part of the major histocompatibility complex I (MHC I), a protein complex that is involved in the presentation of antigenic peptides.6,8 In healthy individuals, the catabolism of β2m involves the shedding of the complex from the cell membrane, the dissociation of β2m from the complex and the excretion of the freed β2m by the kidney. However, patients suffering from renal failure cannot clear the free β2m from the serum causing the β2m concentration to rise 5–60 times above the normal level of 0.1 µM. In persistent dialysis patients, excess β2m undergoes self-association to form amyloid fibrils that deposit in the musculo-skeletal system of the patient, causing dialysis related amyloidosis (DRA).9 Although an elevated concentration is not sufficient for aggregation, it is probably a decisive factor in causing the disease.1,2,7
It is well known that the cis–trans isomerization rates of peptidyl–prolyl amide bond are slow, even in random coil polypeptide chains10 and that cis–trans isomerization is the rate limiting step in many structural transitions.11–13 By studying the folding kinetics at pH 7 and 37°C, Radford and coworkers demonstrated that β2m folds by two parallel routes involving two native-like intermediates. One folding intermediate contains the peptidyl-Pro32 amide bond in the cis conformation while the other has this bond in a trans conformation, with a rate limiting isomerization from trans to cis causing a minor fraction to fold slower. The folding intermediate containing this non-native trans-proline isomer was identified as a direct precursor of dimeric species and oligomers that accumulate before the development of amyloid fibrils.14,15 Miranker's team independently confirmed the importance of this backbone isomerization in β2m fibrillogenesis, using divalent copper as oligomerization trigger. Addition of Cu2+ initiates the isomerization of a conserved cis proline at position 32, thereby facilitating the formation of amyloid fibrils.16,17 Consistent with these models, Pro32 also adopts the trans conformation in the X-ray structure of a domain-swapped dimer of ΔN6 β2m—another amyloidogenic β2-microglobulin variant—confirming that the cis to trans isomerization at Pro32 plays a critical role in the onset of β2m amyloid formation.5
The impact of several Pro32 mutations on the amyloidogenic properties of β2m has been analyzed to link peptidyl–prolyl cis–trans isomerization to fibril formation. In the presence of Cu2+, Pro32Ala β2m oligomers are formed within one minute, while the oligomerization of wild type β2m shows a kinetic profile of 1 h.16 Similarly, the Pro32Gly mutation causes a dramatic enhancement in the rate of amyloid fibril elongation.14 Remarkably, this mutant folds considerably faster than the wild-type β2m, involving only one native-like intermediate. It was suggested that Gly32 adopts a trans conformation in the folded protein as in the folding intermediate that was identified as a direct precursor of amyloidogenesis, explaining faster fibril elongation of the mutant because this trans-peptide species generates the amyloidogenic properties of β2m.14
Aiming to further characterize the aggregation-prone nature of the folding intermediate that serves as the precursor for amyloidosis, we used nanobody-assisted X-ray crystallography to solve the structure of Pro32Gly β2m (P32G β2m). One nanobody (Nb24) that was previously identified to block the aggregation of the ΔN6 amyloidogenic variant, was used in this study to co-crystallize the amyloidogenic P32G β2m variant as a monomer.
Results
The P32G mutation promotes fibril elongation of β2m under physiological conditions
The ability of recombinant wild-type β2m and two amyloidogenic β2-microglobulin variants, P32G β2m and ΔN6 β2m, to elongate β2m seeds under physiological conditions was monitored by measuring the fluorescence increase of the amyloid-specific dye Thioflavin T (ThT) or by visualizing the amyloids using negative stain electron microscopy (EM). Amyloid fibrils from P32G β2m and ΔN6 β2m could be detected after 14 days while wild-type β2m fibrils could only be observed after 30 days (Fig. 1). Although wild-type β2m was able to elongate β2m seeds into amyloid fibrils under physiological conditions, the fibrillogenesis process was completed significantly faster with the P32G β2m and ΔN6 β2m variants, confirming their amyloidogenic properties.14,15,18
Figure 1.
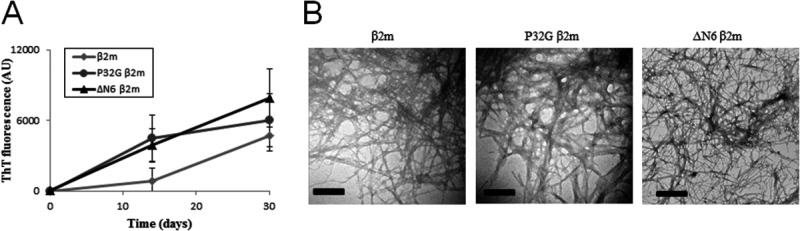
Seeded fibrillogenesis of β2m, P32G β2m, and ΔN6 β2m. (A) ThT fluorescence increase was used to monitor the progress of fibril elongation of β2m (light gray diamonds), P32G β2m (dark gray circles), and ΔN6 β2m (black triangles). (B) EM images confirmed the presence of P32G β2m fibrils and ΔN6 β2m fibrils after 14 days incubation whereas β2m fibrils only appeared after 30 days incubation. The scale bar represents 200 nm.
Nanobodies efficiently block P32G β2m fibrillogenesis under physiological conditions
Nanobodies are single domain antibodies harboring the full antigen-binding capacity of naturally occurring heavy chain antibodies,19,20 and have been used successfully as crystallization chaperones.5,21,22 Several nanobodies with nM to µM range dissociation constants for β2m were tested as fibrillogenesis inhibitors by incubating wild type β2m, P32G β2m, and ΔN6 β2m with β2m fibril seeds in the presence or absence of each nanobody. A nanobody raised against an irrelevant antigen (Nb108) was included in these seeding experiments as a negative control.
Using ThT fluorescence increase to follow fibrillogenesis, several nanobodies (Nb22, Nb23, Nb24, Nb29, and Nb30) were identified to inhibit fibril elongation of β2m, P32G β2m, and ΔN6 β2m (data not shown). According to the relative ThT fluorescence increase, virtually no fibrils or other aggregates are formed from the P32G β2m•Nb24 complex indicating that this nanobody (50 µM) fully protects the amyloidogenic variant against aggregation (Fig. 2). These results were confirmed by negative stain EM. The fact that we were able to purify the P32G β2m•Nb24 complex as a stable heterodimer by size exclusion chromatography prompted us to test Nb24 as a cocrystallization chaperone for monomeric P32G β2m, aiming at characterizing its aggregation-prone properties.
Figure 2.

The effect of an inhibitory nanobody (Nb24) and an unrelated nanobody (Nb108) on the seeded fibrillogenesis of β2m (after 30 days incubation), P32G β2m (after 14 days incubation), and ΔN6 β2m (after 14 days incubation), monitored by ThT fluorescence and EM imaging. The scale bar represents 200 nm.
Nanobody-assisted crystallization of the P32G amyloidogenic variant of β2m
Atomic-level structural investigation of the key conformational intermediates of amyloidosis remains a challenge due to the dynamic equilibrium between diverse structural species. Fibril formation of β2m in vivo usually takes several years and intermediate species are short lived and highly unstable. The use of specific antibodies offers promising strategies for probing the process of fibril formation by biophysical methods including X-ray crystallography.5,23,24
Aiming to characterize the aggregation-prone nature of the folding intermediate that serves as the precursor for amyloidosis, we used nanobody-assisted X-ray crystallography to solve the structure of Pro32Gly β2m (P32G β2m). P32G β2m was mixed in a 1:1 molar ratio with Nb24 in 20 mM Tris, 150 mM NaCl at pH 7.5. After separating the complex by size exclusion chromatography, the purified complex was easily concentrated (8 mg/mL) as a soluble entity and subjected to different crystallization screens. Using the hanging drop vapor diffusion method, diffracting crystals were formed in 0.1M MES (pH 6.5) using 1.6M MgSO4 as the precipitant.
X-ray diffraction data of the P32G β2m•Nb24 complex were processed until 2.6 Å resulting in a CC1/2 of 62.3% and an I/sigI of 1 was reached at 3.2 Å (Table I). The P32G β2m•Nb24 complex was crystallized in space group P42212 and the asymmetric unit contains two molecules P32G β2m each bound to one Nb24 (RMSD between the two P32G β2m•Nb24 complexes = 0.42 Å calculated over 884 main chain atoms) [Fig. 3(A)]. Nb24 binds the last residues of β-strand C and the loops linking β-strands C to D and β-strands E to F [Fig. 3(C)], very similar to the interaction observed in the ΔN6 β2m•Nb24 complex.5 When compared to β2m in complex in the MHC I (PDB entry 1DUZ),25 the binding of Nb24 has little effect on the main-chain conformation of its binding epitope. The loops that are part of the Nb24 epitope (C to D and E to F) have an RMSD of 0.33 Å calculated over 60 main chain atoms [Fig. 3(C)]. The higher overall RMSD of 1.7 Å calculated over the 380 main chain atoms is mainly due to structural differences concentrated in two regions. The first area is located at the loop connecting β-strands B to C and the second region comprises the β-strand D up to the loop linking β-strands D to E. These two segments of the protein do not interact with the nanobody. When we exclude these two regions, an RMSD value of 0.61 Å (calculated over 300 main chain atoms) could be calculated and the overall structure adopts native-like conformation.
Table I.
Data-Collection, Refinement and Validation Statistics of the Structure of the P32G β2m•Nb24 Complex
| Data Set | P32G β2m•Nb24 |
|---|---|
| Data-collection | |
| X-ray source | Diamond IO3 |
| X-ray wavelength (Å) | 0.97950 |
| Temperature (K) | 100 |
| Space group | P 42212 |
| Unit-cell parameters | |
| a, b, c (Å) | 96.8, 96.8, 167.8 |
| α, β, γ(°) | 90.0, 90.0, 90.0 |
| Resolution range (Å) | 48.43–2.6 (2.67–2.60) |
| Total/Unique reflections | 362392/25281 |
| Rmerge (%)a | 35.7 (392) |
| Rmeas (%)b | 37.0 (406) |
| Data completeness (%) | 99.9 (99.8) |
| Average I/σ | 8.5 (1.0) |
| Redundancy | 14.3 (14.7) |
| Wilson B factor (Å2) | 56.1 |
| CC(1/2) | 99.7 (62.3) |
| Refinement | |
| Correlation coefficients | |
| Correlation coefficient Fo − Fc | 0.948 |
| Correlation coefficient Fo − Fc Free | 0.914 |
| Rwork/Rfreec | 22.84/27.69 |
| Total number | |
| Amino acid residues | 445 |
| Water molecules | 50 |
| Ligand atoms | 8 |
| rmsd | |
| Bond length (Å) | 0.0150 |
| Bond angles (°) | 1.5567 |
| Average atomic B-factor (Å2) | |
| Protein atoms | 58.67 |
| Solvent atoms | 35.0 |
| Ramachandran plot (%) | |
| Favored regions | 97.70 |
| Allowed regions | 2.30 |
| Disallowed regions | 0 |
| PDB entry | 4KDT |
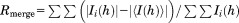
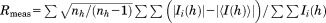
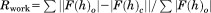 , F(h)o and F(h)c are observed and calculated structure factor amplitudes, respectively. A random subset of data (5%) was used for the Rfree calculation.
, F(h)o and F(h)c are observed and calculated structure factor amplitudes, respectively. A random subset of data (5%) was used for the Rfree calculation.
Figure 3.
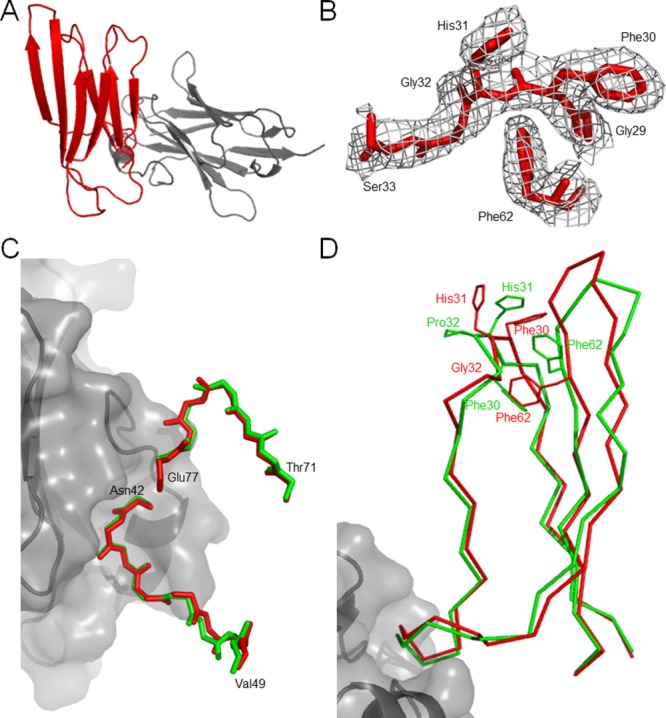
Crystal structure of P32G β2m•Nb24 complex. (A) The P32G β2m•Nb24 complex as a stable heterodimer with P32G β2m in red and Nb24 in gray. (B) The 2Fo – Fc omit density map is shown for residues Gly29, Phe30, His31, Ser33, and Phe62. The map is contoured at 1 σ within 1.6 Å of the residues. (C) Comparison between the P32G β2m•Nb24 structure (red) and the β2m structure in the MHC I complex (PDB entry 1DUZ25;green) focusing on the high similarity of the main-chain conformation in the binding region of P32G β2m to Nb24 (in gray surface representation). (D) Comparison between P32G β2m•Nb24 structure (red) and the β2m structure (PDB entry 1DUZ25; green) focusing on the key structural consequences of the cis–trans isomerization at position 32.
Whereas Pro32 consistently adopts the cis conformation in wild-type β2m (PBD entries: 1DUZ, 1JNJ, 1LDS)25–27 Gly32 adopts the trans conformation in the P32G β2m monomer (current structure) [Fig. 3(B)]. This isomerization causes Phe30 to move out of the hydrophobic core to adopt a solvent exposed configuration. The freed space is filled by Phe62, causing compensating structural rearrangements of β-strand D up to the loop linking β-strands D to E (Glu50 to Phe62) [Fig. 3(D)].
Discussion
Amyloid fibril formation generally occurs via a nucleation-dependent oligomerization process characterized by a lag phase.4,28,29 During this rate-limited phase, amyloidogenic intermediates are formed while the conversion into amyloid fibrils only occurs in the following elongation phase. The formation of these amyloidogenic intermediates involves the disruption of the native structure to a greater or lesser extent, in order to allow self-association and the formation of a cross-β-sheet structure that is the hallmark of amyloid fibrils. The lag phase can be shortened or ultimately abolished in vitro by adding fibrillar seeds, by changing the experimental conditions or by using fibrillogenic mutants.4,24,28–32
Local fluctuations of one or more regions of the β2m monomer causing the generation of precursors that are prone to spontaneous self-assembly are also at the origin of DRA.7 It has been shown that a β2m conformer with a non-native peptidyl-Pro32 trans peptide bond serves as a direct precursor of dimeric species and oligomers that accumulate before the development of amyloid fibrils, linking cis–trans isomerization of the Pro32 imidic peptide bond to β2m fibrillogenesis.14,15 From our structure, it appears that the trans conformer at position 32 is the most stable folded conformation of P32G β2m, explaining the intrinsic amyloidogenic nature of this mutant. Consistent with this notion, the trans conformation at position 32 has already been observed in other amyloidogenic variants of β2m including the domain-swapped dimer of ΔN6 β2m5 and Pro32Ala β2m.16 The trans peptide bond at position 32 as observed in Pro32Gly β2m causes several structural rearrangements that may increase to susceptibility of β2m to aggregate and form cross-β-sheet structures.
Regular β-sheets are inherently aggregation-prone because the motive for β H-bonding with any other β-strand is available.33 Natural β-sheet proteins are designed to avoid this interaction and make use of different blocking features to prevent edge-to-edge β aggregation by the formation of intersubunit β H-bonds.33 For β-sandwich proteins, a very common strategy to avoid aggregation is the presence of a charged side chain of lysine, arginine, glutamic acid, aspartic acid, or histidine. In monomeric β-sandwich proteins, such charged residues are capable to simultaneously form hydrophobic sheet-packing interactions while exposing their charged side chains to the solvent, preventing edge-to-edge association through electrostatic repulsion between these exposed charges.34–36
β2m (a typical β-sandwich protein) contains four edge strands: strands A and G form one pair at one side of the protein and strands C and D form a second pair at the opposite side (Fig. 4). Focusing on these vulnerable strands, the wild-type protein contains several gatekeepers including His51 to protect strand D and Lys91 to protect strand G, thus avoiding edge-to-edge aggregation.27 In our structure of P32G β2m, Lys91 is also exposed and remains the gatekeeper for the first edge pair formed by strands A and G (Fig. 4). The trans peptide bond caused by the Pro32Gly mutation is linked to significant structural changes in edge strand D of the ABED antiparallel β-sheet and in the D to E loop. However, His51 points towards the hydrophobic core of P32G β2m, as it does in wild type β2m, where it still acts as a gatekeeper to avoid edge-to-edge aggregation of strands C and D (Fig. 4).
Figure 4.
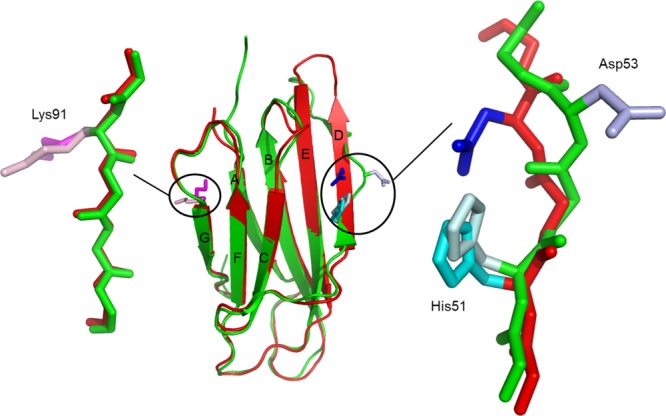
Structural consequences of the cis–trans isomerization at position 32 on three gatekeepers for P32G β2m (red) in comparison with β2m (PDB entry 1DUZ25 green). The gatekeeper for β-strands A and G is Lys91 (magenta for P32G β2m and light pink for β2m) and remains solvent exposed. For the second edge pair (β-strands C and D) there are two gatekeepers, but only His51 (cyan for P32G β2m and light cyan for β2m) remains in its original orientation while Asp53 (blue for P32G β2m and light blue for β2m) rotates and the β bulge gets lost for P32G β2m, forming a more aggregation-competent surface.
The presence of β bulges to kink β-strands is a second approach to avoid edge-to-edge aggregation by distorting the geometry and accentuate the twist of the associating strands.35 Remarkably, Asp53—a key residue that is usually forming a β bulge by pointing outwards and distorting the geometry of the D-strand26,27—is rotated inward in P32G β2m and the β bulge is lost, resulting in a more continuous D-strand that is probably more prone to intermolecular pairing (Fig. 4).
The trans peptide bond at position 32 also causes Phe30 to become solvent exposed and Phe62 to repack within the hydrophobic core to fill the freed space. This structural rearrangement of these large hydrophobic residues causes a significant increase in the surface hydrophobicity of the protein, apparently reducing its intrinsic solubility. Concomitantly, the inward rotation of the Asp53 side chain also contributes to the increase of the surface hydrophobicity (Fig. 5). Increased surface hydrophobicity has been linked to aggregation and fibril formation.37,38
Figure 5.
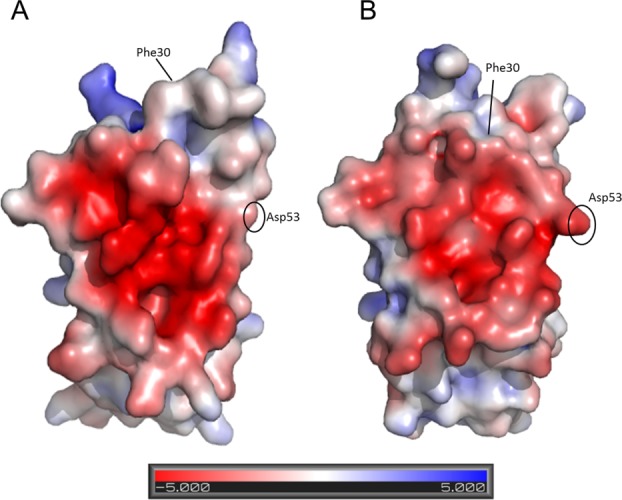
Consequences of the cis–trans isomerization at position 32 on the surface electrostatic potential. (A) P32G β2m colored by electrostatic surface potential on the solvent accessible surface. (B) β2m (PDB entry 1DUZ25) colored by electrostatic surface potential on the solvent accessible surface. Visualization is done by setting the values at which the surface colors are clamped at red (−5 kT/e) for negatively charged regions and blue (+5 kT/e) for positively charged regions.
In conclusion, we were able to trap the P32G amyloidogenic variant of β2m as a monomer and solve its structure by nanobody-assisted X-ray crystallography. The structure of monomeric P32G β2m in complex with Nb24 reveals a trans peptide bond at position 32, whereas Pro32 adopts the cis conformation in the wild-type monomer. Consistent with earlier work on the native protein14 and on the amyloidogenic ΔN6 5,39 and Pro32Ala16 variants, this study identifies the cis-to-trans isomerization at position 32 in the β2m monomer as an early event in amyloidogenesis that makes the protein surface more hydrophobic and probably renders it more susceptible to edge-to-edge β aggregation by the formation of intersubunit β H-bonds and confirms the key role of Pro32 to safeguard the aggregation prone β-sheets of β2m against edge-to-edge β association.
Material and Methods
Expression and purification of the proteins
β2m, ΔN6 β2m, and P32G β2m were expressed in Escherichia coli and purified out of inclusion bodies as described.40,41 All nanobodies were expressed in E. coli and purified from the periplasm as described.5
Fibrillogenesis under physiological conditions
Fibrils of β2m, P32G β2m, or ΔN6 β2m were grown at 37°C in the presence of β2m seeds with agitation in microtiter plates essentially as described by Radford and coworkers.18 Lyophilized protein was dissolved in 25 mM sodium phosphate, 25 mM sodium acetate buffer containing 0.5% sodium azide at pH 7.0 at a concentration of 0.5 mg/mL to which 10% of heparin-stabilized β2m seeds were added. Samples of 200 µL were agitated at 250 rmp at 37°C. Fibril formation was followed in time by measuring the ThT fluorescence increase (excitation 440 nm, emission 480 nm). In a typical experiment, the average of 10 replicates was normalized to the signal from buffer only containing β2m seeds. The presence of long straight fibrils was confirmed by negative-stain electron microscopy. For microscopy, protein samples (5 µL) were applied dropwise on carbon-coated grids (Formvar/carbon on 400 Mesh Copper). The grids were washed with water, stained with 1% uranyl acetate and the samples were analyzed on a Jeol JEM-1400 electron microscope at 100 kV.
The same experimental conditions (37°C, 250 rpm) were used to measure the inhibitory effect of nanobodies on the seeded fibrillogenesis of β2m, P32G β2m, and ΔN6 β2m. Lyophilized nanobodies (0.8 mg/mL) were dissolved in buffer (25 mM sodium phosphate, 25 mM sodium acetate buffer containing 0.5% sodium azide at pH 7.0) containing 10% β2m heparin-stabilized seeds before β2m, P32G β2m, or ΔN6 β2m was added to the reaction mixture (0.5 mg/mL). ThT fluorescence measurements were done as described above. The inhibitory effect of a nanobody (in %) was estimated from the ratio of the ThT fluorescence of samples containing a particular nanobody and samples without nanobody, incubated under the same conditions in the same microtiter plate.
Crystallization, data-collection, and structure determination of the P32G β2m•Nb24 complex
P32G β2m was mixed with equimolar amounts of Nb24 in 20 mM Tris, 150 mM NaCl, pH 7.5. After 2 h of incubation at 4°C, a size exclusion chromatography was performed in the same buffer to purify the complex. Fractions containing the complex were collected and concentrated to 8 mg/mL by ultrafiltration. Crystals were grown at 20°C using the hanging drop vapor diffusion method by mixing equal volumes of the complex with a reservoir solution containing 0.1M MES (pH 6.5) and 1.6M MgSO4. Before data collection, crystals were flash frozen in liquid nitrogen using 15% glycerol as the cryo-protectant. Diffraction data were collected at the IO3 beamline (Diamond, UK) at 100 K. Data indexing, integration and scaling were done using the XDS suite.42 Data were processed to 2.6 Å, resulting in a CC1/2 of 62.3% as cut-off and an I/sigma I of 1 was reached at 3.2 Å resolution.43 The crystal structure of the P32G β2m•Nb24 complex was solved by molecular replacement with PhaserMR44 using the separate coordinates of the ΔN6 β2m monomer and Nb24 (both from PDB entry 2X89)5 as the models. Initial model building was done automatically using ARP/wARP45,46 and the model was refined manually using COOT.47 Structure refinement was carried out using Refmac5.48 TLS refinement was implemented in the refinement protocol using four individual TLS groups determined by TLSMD.49,50 The MolProbity server was used to stereochemically validate the model.51 Data collection and refinement statistics are summarized in Table I. Electrostatic surface calculations were prepared with PDB2PQR52 using the PARSE force field and APBS.53 Figures were prepared using Yasara54 or Pymol.55
References
- 1.Bellotti V, Mangione P, Stoppini M. Biological activity and pathological implications of misfolded proteins. Cell Mol Life Sci. 1999;55:977–991. doi: 10.1007/s000180050348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dobson CM. An accidental breach of a protein's natural defenses. Nat Struct Mol Biol. 2006;13:295–297. doi: 10.1038/nsmb0406-295. [DOI] [PubMed] [Google Scholar]
- 3.Stefani M, Dobson CM. Protein aggregation and aggregate toxicity: new insights into protein folding, misfolding diseases and biological evolution. J Mol Med. 2003;81:678–699. doi: 10.1007/s00109-003-0464-5. [DOI] [PubMed] [Google Scholar]
- 4.Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- 5.Domanska K, Vanderhaegen S, Srinivasan V, Pardon E, Dupeux F, Marquez JA, Giorgetti S, Stoppini M, Wyns L, Bellotti V, Steyaert J. Atomic structure of a nanobody-trapped domain-swapped dimer of an amyloidogenic β2-microglobulin variant. Proc Natl Acad Sci USA. 2011;108:1314–1319. doi: 10.1073/pnas.1008560108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bjorkman PJ, Saper MA, Samraoui B, Bennett WS, Strominger JL, Wiley DC. Structure of the human class I histocompatibility antigen, HLA-A2. Nature. 1987;329:506–512. doi: 10.1038/329506a0. [DOI] [PubMed] [Google Scholar]
- 7.Radford SE, Gosal WS, Platt GW. Towards an understanding of the structural molecular mechanism of β2-microglobulin amyloid formation in vitro. Biochim Biophys Acta. 2005;1753:51–63. doi: 10.1016/j.bbapap.2005.07.006. [DOI] [PubMed] [Google Scholar]
- 8.Kozlowski S, Takeshita T, Boehncke W, Takahashi H, Boyd LF, Germain RN, Berzofsky JA, Margulies DH. Excess β2-microglobulin promoting functional peptide association with purified soluble class I MHC molecules. Nature. 1991;349:74–77. doi: 10.1038/349074a0. [DOI] [PubMed] [Google Scholar]
- 9.Gejyo F, Odani S, Yamada T, Honma N, Saito H, Suzuki Y, Nakagawa Y, Kobayashi H, Maruyama Y, Hirasawa Y, Suzuki M, Arakawa M. β2-microglobulin: a new form of amyloid protein associated with chronic hemodialysis. Kidney Int. 1986;30:385–390. doi: 10.1038/ki.1986.196. [DOI] [PubMed] [Google Scholar]
- 10.Brandts JF, Halvorson HR, Brennan M. Consideration of the possibility that the slow step in protein denaturation reactions is due to cistrans isomerism of proline residues. Biochemistry. 1975;14:4953–4963. doi: 10.1021/bi00693a026. [DOI] [PubMed] [Google Scholar]
- 11.Andreotti AH. Native state proline isomerization: an intrinsic molecular switch. Biochemistry. 2003;42:9515–9524. doi: 10.1021/bi0350710. [DOI] [PubMed] [Google Scholar]
- 12.Lu KP, Finn G, Lee TH, Nicholson LK. Prolyl cistrans isomerization as a molecular timer. Nat Chem Biol. 2007;3:619–629. doi: 10.1038/nchembio.2007.35. [DOI] [PubMed] [Google Scholar]
- 13.Joseph AP, Srinivasan N, de Brevern AG. Cistrans peptide variations in structurally similar proteins. Amino Acids. 2012;43:1369–1381. doi: 10.1007/s00726-011-1211-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jahn TR, Parker MJ, Homans SW, Radford SE. Amyloid formation under physiological conditions proceeds via a native-like folding intermediate. Nat Struct Mol Biol. 2006;13:195–201. doi: 10.1038/nsmb1058. [DOI] [PubMed] [Google Scholar]
- 15.Eichner T, Radford SE. A generic mechanism of β2-microglobulin amyloid assembly at neutral pH involving a specific proline switch. J Mol Biol. 2009;386:1312–1326. doi: 10.1016/j.jmb.2009.01.013. [DOI] [PubMed] [Google Scholar]
- 16.Eakin CM, Berman AJ, Miranker AD. A native to amyloidogenic transition regulated by a backbone trigger. Nat Struct Mol Biol. 2006;13:202–208. doi: 10.1038/nsmb1068. [DOI] [PubMed] [Google Scholar]
- 17.Blaho DV, Miranker AD. Delineating the conformational elements responsible for Cu2+-induced oligomerization of β2-microglobulin. Biochemistry. 2009;48:6610–6617. doi: 10.1021/bi900540j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Myers SL, Jones S, Jahn TR, Morten IJ, Tennent GA, Hewitt AW, Radford SE. A systematic study of the effect of physiological factors on β2-microglobulin amyloid formation at neutral pH. Biochemistry. 2006;45:2311–2321. doi: 10.1021/bi052434i. [DOI] [PubMed] [Google Scholar]
- 19.Hamers-Casterman C, Atarhouch T, Muyldermans S, Robinson G, Hamers C, Bajyana Songa E, Bendahman N, Hamers R. Naturally occurring antibodies devoid of light chains. Nature. 1993;363:446–448. doi: 10.1038/363446a0. [DOI] [PubMed] [Google Scholar]
- 20.Muyldermans S, Cambillau C, Wyns L. Recognition of antigens by single-domain antibody fragments: the superfluous luxury of paired domains. Trends Biochem Sci. 2001;26:230–235. doi: 10.1016/s0968-0004(01)01790-x. [DOI] [PubMed] [Google Scholar]
- 21.Steyaert J, Kobilka BK. Nanobody stabilization of G protein-coupled receptor conformational states. Curr Opin Struct Biol. 2011;21:567–572. doi: 10.1016/j.sbi.2011.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baranova E, Fronzes R, Garcia-Pino A, Van Gerven N, Papaostolou D, Péhau-Arnaudet G, Pardon E, Steyaert J, Howorka S, Remaut H. SbsB structure and lattice reconstruction unveil Ca2+ triggered S-layer assembly. Nature. 2012;487:119–122. doi: 10.1038/nature11155. [DOI] [PubMed] [Google Scholar]
- 23.Glabe CG. Conformation-dependent antibodies target diseases of protein misfolding. Trends Biochem Sci. 2004;29:542–547. doi: 10.1016/j.tibs.2004.08.009. [DOI] [PubMed] [Google Scholar]
- 24.Dumoulin M, Dobson CM. Probing the origins, diagnosis and treatment of amyloid diseases using antibodies. Biochimie. 2004;86:589–600. doi: 10.1016/j.biochi.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 25.Khan AR, Baker BM, Ghosh P, Bissison WE, Wiley DC. The structure and stability of an HLA-A*0201/octameric tax peptide complex with an empty conserved peptide-N-terminal binding site. J Immunol. 2000;164:6398–6405. doi: 10.4049/jimmunol.164.12.6398. [DOI] [PubMed] [Google Scholar]
- 26.Verdone G, Corazza A, Viglino P, Pettirossi F, Giorgetti S, Mangione P, Andreola A, Stoppini M, Bellotti V, Esposito G. The solution structure of human β2-microglobulin reveals the prodromes of its amyloid transition. Protein Sci. 2002;11:487–499. doi: 10.1110/ps.29002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Trinh CH, Smith DP, Kalverda AP, Phillips SEV, Radford SE. Crystal structure of monomeric human β2-microglobulin reveals clues to its amyloidogenic properties. Proc Natl Acad Sci USA. 2002;99:9971–9976. doi: 10.1073/pnas.152337399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Naiki H, Hashimoto N, Suzuki S, Kimura H, Nakakuki K, Gejyo F. Establishment of a kinetic model of dialysis-related amyloid fibril extension in vitro. Amyloid. 1997;4:223–232. [Google Scholar]
- 29.Xue W, Homans SW, Radford SE. Systematic analysis of nucleation-dependent polymerization reveals new insights into the mechanism of amyloid self-assembly. Proc Natl Acad Sci USA. 2008;105:8926–8931. doi: 10.1073/pnas.0711664105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chi EY, Krishnan S, Kendrick BS, Chang BS, Carpenter JF, Randolph TW. Roles of conformational stability and colloidal stability in the aggregation of recombinant human granulocyte colony-stimulating factor. Protein Sci. 2003;12:903–13. doi: 10.1110/ps.0235703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Smith DP, Jones S, Serpell LC, Sunde M, Radford SE. A systematic investigation into the effect of protein destabilisation on β2-microglobulin amyloid formation. J Mol Biol. 2003;330:943–954. doi: 10.1016/s0022-2836(03)00687-9. [DOI] [PubMed] [Google Scholar]
- 32.Uversky VN, Fink AL. Conformational constraints for amyloid fibrillation: the importance of being unfolded. Biochim Biophys Acta. 2004;1698:131–153. doi: 10.1016/j.bbapap.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 33.Richardson JS, Richardson DC, Tweedy NB, Gernert KM, Quinn TP, Hecht MH, Erickson BW, Yan Y, McClain RD, Donlan ME, Surles MC. Looking at proteins: representations, folding, packing, and design. Biophys J. 1992;63:1186–1209. [PMC free article] [PubMed] [Google Scholar]
- 34.Otzen DE, Kristensen O, Oliveberg M. Designed protein tetramer zipped together with a hydrophobic Alzheimer homology: a structural clue to amyloid assembly. Proc Natl Acad Sci USA. 2000;97:9907–9912. doi: 10.1073/pnas.160086297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Richardson JS, Richardson DC. Natural β-sheet proteins use negative design to avoid edge-to-edge aggregation. Proc Natl Acad Sci USA. 2002;99:2754–2759. doi: 10.1073/pnas.052706099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thirumalai D, Klimov DK, Dima RI. Emerging ideas on the molecular basis of protein and peptide aggregation. Curr Opin Struct Biol. 2003;13:146–159. doi: 10.1016/s0959-440x(03)00032-0. [DOI] [PubMed] [Google Scholar]
- 37.Esposito G, Corazza A, Viglino P, Verdone G, Pettirossi F, Fogolari F, Makek A, Giorgetti S, Mangione P, Stoppini M, Bellotti V. Solution structure of β2-microglobulin and insights into fibrillogenesis. Biochim Biophys Acta. 2005;1753:76–84. doi: 10.1016/j.bbapap.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 38.Valleix S, Gillmore J, Bridoux F, Mangione PP, Dogan A, Nedelec B, Boimard M, Touchard G, Goujon J, Lacombe C, Lozeron P, Adams D, Lacroix C, Maisonobe T, Planté-Bordeneuve V, Vrana JA, Theis JD, Giorgetti S, Porcari R, Ricagno S, Bolognesi M, Stoppini M, Delpech M, Pepys MB, Hawkins PN, Bellotti V. Hereditary systemic amyloidosis due to Asp76Asn variant of β2-microglobulin. N Engl J Med. 2012;366:2276–2283. doi: 10.1056/NEJMoa1201356. [DOI] [PubMed] [Google Scholar]
- 39.Eichner T, Kalverda AP, Thompson GS, Homans SW, Radford SE. Conformational conversion during amyloid formation at atomic resolution. Mol Cell. 2011;41:161–172. doi: 10.1016/j.molcel.2010.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Esposito G, Michelutti R, Verdone G, Viglino P, Hernández H, Robinson CV, Amoresano A, Dal Piaz F, Monti M, Pucci P, Mangione P, Stoppini M, Merlini G, Ferri G, Bellotti V. Removal of the N-terminal hexapeptide from human β2-microglobulin facilitates protein aggregation and fibril formation. Protein Sci. 2000;9:831–845. doi: 10.1110/ps.9.5.831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McParland VJ, Kad NM, Kalverda AP, Brown A, Kirwin-Jones P, Hunter MG, Sunde M, Radford SE. Partially unfolded states of β2-microglobulin and amyloid formation in vitro. Biochemistry. 2000;39:8735–8746. doi: 10.1021/bi000276j. [DOI] [PubMed] [Google Scholar]
- 42.Kabsch W. XDS. Acta Crystallogr D Biol Crystallogr 66. 2010:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Karplus PA, Diederichs K. Linking crystallographic model and data quality. Science. 2012;25:1030–1033. doi: 10.1126/science.1218231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Perrakis A, Morris R, Lamzin VS. Automated protein model building combined with iterative structure refinement. Nat Struct Biol. 1999;6:458–463. doi: 10.1038/8263. [DOI] [PubMed] [Google Scholar]
- 46.Morris RJ, Zwart PH, Cohen S, Fernandez FJ, Kakaris M, Kirillova O, Vonrhein C, Perrakis A, Lamzin VS. Breaking good resolutions with ARP/wARP. J Synchrotron Radiat. 2004;11:56–59. doi: 10.1107/s090904950302394x. [DOI] [PubMed] [Google Scholar]
- 47.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystollogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structure by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 49.Winn MD, Isupov MA, Murshudov GN. Use of the TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr D Biol Crystallogr. 2001;57:122–133. doi: 10.1107/s0907444900014736. [DOI] [PubMed] [Google Scholar]
- 50.Painter J, Merritt EA. Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr D Biol Crystallogr. 2006;62:439–450. doi: 10.1107/S0907444906005270. [DOI] [PubMed] [Google Scholar]
- 51.Chen VB, Arendall WBIII, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystollogr D Biol Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dolinsky TJ, Nielsen JE, McCammon JA, Baker NA. PDB2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004;32:W665–W667. doi: 10.1093/nar/gkh381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci USA. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Krieger E, Koraimann G, Vriend G. Increasing the precision of comparative models with YASARA NOVA: a self-parameterizing force field. Proteins. 2002;47:393–402. doi: 10.1002/prot.10104. [DOI] [PubMed] [Google Scholar]
- 55.Delano W. The PyMOL molecular graphics system. San Carlos, CA: DeLano Scientific; 2002. [Google Scholar]