

Abstract
Many proteins function by interacting with other small molecules (ligands). Identification of ligand-binding sites (LBS) in proteins can therefore help to infer their molecular functions. A comprehensive comparison among local structures of LBSs was previously performed, in order to understand their relationships and to classify their structural motifs. However, similar exhaustive comparison among local surfaces of LBSs (patches) has never been performed, due to computational complexity. To enhance our understanding of LBSs, it is worth performing such comparisons among patches and classifying them based on similarities of their surface configurations and electrostatic potentials. In this study, we first developed a rapid method to compare two patches. We then clustered patches corresponding to the same PDB chemical component identifier for a ligand, and selected a representative patch from each cluster. We subsequently exhaustively as compared the representative patches and clustered them using similarity score, PatSim. Finally, the resultant PatSim scores were compared with similarities of atomic structures of the LBSs and those of the ligand-binding protein sequences and functions. Consequently, we classified the patches into ∼2000 well-characterized clusters. We found that about 63% of these clusters are used in identical protein folds, although about 25% of the clusters are conserved in distantly related proteins and even in proteins with cross-fold similarity. Furthermore, we showed that patches with higher PatSim score have potential to be involved in similar biological processes.
Keywords: protein-ligand interactions, ligand-binding site, exhaustive comparison, molecular surfaces, electrostatics potentials
Introduction
Despite the rapid growth of genetic information and the number of protein structures deposited in the Protein Data Bank (PDB),1 functions of many proteins are not clearly understood. The functions of such proteins have often been assigned based on the analogy to their homologs with known functions, because proteins with highly similar sequences and structures tend to be evolutionarily related and have similar functions.2–7 However, homologs proteins are not always available, and some proteins with similar sequences but dissimilar structures exist due to protein conformational plasticity, ligand-binding, solvent effects, or mutations, such as EF-hand calcium-binding proteins.8,9 Thus, identifications of the functions for those proteins based on their homologs are not always accurate.
To compensate for these weak points of homology-based function identification, various methods to identify binding sites of small chemical compounds (ligands) in proteins have been developed.10–13 Since many proteins accomplish their characteristic biochemical functions by interacting with ligands, such as substrates, cofactors and inhibitors, identification of ligand-binding sites (LBSs) in proteins can help to infer their functions. One of the most successful methods is based on a similarity search in databases for proteins in which local structures or surfaces of LBSs are similar to those of a query protein with unknown functions.14–27 Among these methods, the surface-based methods, such as eF-seek23 and Pocket-Surfer,25 are reportedly able to detect similar LBSs independent of sequence and fold similarity.14,22,23,25,28 In addition, a heterogeneous method more targeted to druggable LBSs has been recently developed.26
Local atomic structures and physicochemical characteristics of LBSs of many proteins have been compared, in order to understand their relationships and to classify structural motifs that are useful for inference of LBSs and functions in proteins. These comparisons have revealed LBSs shared by proteins bearing different folds.16,21,22,29–33 Kinjo and Nakamura recently conducted an all-against-all comparison of 186,485 LBSs, a much larger number of LBSs as compared to previous analyses,16,21,22,29–32 using a relational database search method and alignment refinement.33 They analyzed the similarity networks of LBSs and discovered a large number of similar structural motifs of LBSs with cross-fold similarity.
Exhaustive comparisons of LBSs at atomic structure level have been performed. However, to our knowledge, such comparisons at molecular surface level have never been attempted, due to huge computational complexity required for comparisons among many local surfaces of LBSs (hereafter, referred to as a “patch”). This is because the number of vertices representing a patch is much larger than that of atoms in LBS. For example, the average number of atoms involved in LBS of adenosine-5′-triphosphate (ATP) is 74, while its average patch is constructed of 821 vertices (these numbers were obtained from the patch data used in eF-seek23. Despite this daunting task, it is worthwhile to perform an exhaustive comparison among patches and then to classify them based on similarities of their surface configurations and electrostatic potentials (EPot), for a better understanding of LBSs at surface level.
In this study, we developed a rapid similarity search method for comparing a huge number of patches, by applying a hash table. Patches, except for DNA, RNA, monoatomic ions, and unknown ligands, were extracted from molecular surface data stored at the eF-site.34 We prepared representative patches by performing hierarchical clustering for patches with the same three letter code of the PDB chemical component identifier (Ligand ID), using patch similarity based on their surface configurations and their EPots (hereafter, the score for patch similarity is referred as “PatSim”). Subsequently, to obtain the canonical surface patterns of patches, we classified them into clusters based on PatSim scores through an exhaustive comparison of the representative patches. Furthermore, we compared PatSim scores with similarities of atomic structures of LBSs, and with similarities of sequences and functions of proteins to which ligands bind, in order to understand the characteristics of LBSs from various viewpoints.
Results
Hierarchical clustering of patches in each chemical identifier
The atomic coordinates of 148,199 ligands for 8283 ligand IDs (4794 out of these had more than two atomic coordinates) were obtained from 33,053 PDB entries (PDB ID) with molecular surfaces available at eF-site.34 The patches for these ligands were extracted from surfaces of their binding proteins, according to the patch definition (see Materials and Methods section). Consequently, 141,194 patches, each constructed of >20 vertices, for 7851 ligand IDs were obtained from 31,950 PDB IDs (4538 out of these had more than two patches) (Supporting Information Table SII).
To select representative patches, we performed an all-against-all comparison among patches with identical ligand IDs, using our similarity search method (see Materials and Methods section), and performed a hierarchical clustering of patches in each ligand ID. (In total, 477,712,729 patch pairs were compared). The clustering threshold was determined as a threshold that can make the largest number of clusters (see more details in Supporting Information Table SIII). The computation of the comparisons took 64 days on a cluster machine consisting of 5 nodes, each with an 8-core processor (Intel Xeon X5460 3.2 GHz). If the similarity search for a patch pair took 10–30 seconds as a previously reported surface-based method takes,35 the total computation would take 1382–4149 days on the same system. In this sense, our method is considered to be faster than the reported method. Consequently, 26,059 representative patches made for 7851 ligand IDs were selected from 17,135 PDB entries. The number of representative patches for the 20 most abundant ligand IDs is shown in Supporting Information Table SIV.
Exhaustive comparison of the representative patches
An exhaustive comparison among the 26,059 patches was performed. Namely, a total of 339,522,711 patch pairs were compared, and then hierarchically clustered using PatSim scores. The entire computation was performed in 29 days on a cluster machine consisting of 3 nodes, each with an 8-core processor (Intel Xeon X5460 3.2 GHz).
To understand the relationship between PatSim score and structural similarity of proteins from which patches are extracted, protein fold similarities in each cluster were investigated at different clustering thresholds. We assigned SCOP codes (SCOP concise classification strings; SCCS)36 to each patch, and then quantitatively analyzed SCOP hierarchical classification (family, superfamily, fold and class) to find the level where all of patches in each cluster share common SCCS. In fact, only about half of the representatives, 13,431 patches, had SCCS assigned; however, we assigned SCCS to 4428 unassigned patches based on sequence similarity to proteins of the assigned patches (see more details in Fig. S2); that is in total, 17,804 patches (68% of the representative patches) for 6835 ligand IDs. The number of clusters (≥2 patches) classified in different SCOP classifications at different clustering thresholds is shown in Supporting Information Table SV. Two patch area sizes (psize) >0 Å2 and ≥200 Å2 were considered, because smaller patches are likely to have greater chances of matching other patches, and so they might generate background noise in this analysis. Consequently, we found that the essential features with psize >0 Å2 are the same as those with ≥200 Å2, and so we only mention hereafter for the cases with ≥200 Å2. Note that although some unassigned patches in a cluster were simply ignored, this limitation would not seriously affect a tendency in this analysis. In addition, clusters of which patches did not share any common SCCS even in class level were classified them into “others” level.
At the threshold of 0.1, which could make the largest number of clusters of which patches shared common folds at family or superfamily level (see more details in Supporting Information Table SV), and for patches with psize ≥200 Å2, there were 264 different SCCSs in SCOP fold level shared in 1852 clusters, with an average of 7 clusters per fold [Fig. 1A)]. These clusters may be regarded as well-characterized patterns of patches. Among them, there were 57-folds shared in ≥10 clusters, and 27-folds shared in ≥20 clusters. The 20 most abundant SCOP folds are shown in Figure 1(B,C). Protein kinase-like (d.144) and TIM beta/alpha-barrel (c.1) were most often shared in 148 and 136 clusters, respectively. Both protein folds are quite common in living cells. Proteins with protein kinase-like folds regulate most biological processes, such as signaling and regulatory processes, in a living organism by chemically adding phosphate groups to other proteins.37 Fifteen distinct enzyme families use TIM beta/alpha-barrel fold to catalyze different reactions.38 The diversity of these folds and other folds, such as phosphorylase/hydrolase-like (c.56), alpha/beta-hydrolases (c.69), Rossmann-fold (c.2), and immunoglobulin-like (b.1), was previously reported.33,39–41 In addition, immunoglobulin-like fold (b.1) is remarkably used in 141 clusters of the others level, indicating that this fold has structural diversity and tends to make heterogenetic patches resulting in diversity of patch.
Figure 1.
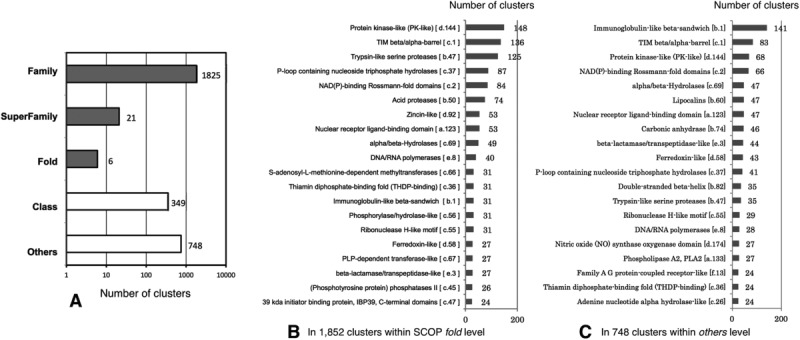
Diversity of patch clusters at the ligand-binding site in terms of protein folds at a clustering threshold of 0.1 (psize of ≥200 Å2): (A) The number of patch clusters to which the given SCOP levels are assigned for total 2949 clusters. The patch clusters indicated by dark bars are within SCOP fold. (B) The 20 most abundant SCOP folds used in 1852 patch clusters within SCOP fold, and (C) those used in 748 patch clusters within others levels.
The number of clusters containing patches for an identical ligand ID was investigated, and the top 20 most abundant ligand IDs are shown in Table I. In 2949 clusters, patches for an adenosine-5′-diphosphate (ADP) binding site were classified into the largest number of different clusters, 91 clusters, where 32 different folds (2.8 clusters per fold) were shared. Furthermore, other patches for a protoporphyrin IX containing FE (HEM) and a flavin-adenine dinucleotide (FAD) were classified into 62 and 21 clusters, respectively, where 16 (3.9 clusters per fold) and 11 (1.9 clusters per a fold) different folds, respectively, were shared. The diversity of structural characteristics in HEM binding sites has been demonstrated.42 In addition, 13% of ligand IDs (658/5228) shared a single fold with more than two clusters. This observation indicated that these ligands, in which their proteins share a limited number of folds, have diverse patches with distinct surface configurations and different EPots, and suggested that the diversity of these patches enables their proteins to perform a variety of functions.
Table I.
The Number of Clusters Containing Patches (psize ≥200 Å2) and of Nonclustered Patches to Which SCOP Folds or CATH Topologies are Assigned, for the Top 20 Most Abundant Ligand IDs.
| Ligand name | # clusters (≥ 2 patches) to which SCOP are assigned | # patches to which SCOP is assigned, but not clustered | # SCOP fold assigned to the clusters | # clusters (≥ 2 patches) to which CATH are assigned | # patches to which CATH is assigned, but not clustered | # CATH topology assigned to the clusters | |
|---|---|---|---|---|---|---|---|
| ADP | Adenosine-5′-diphosphate | 91 (160) | 89 (171) | 32 | 102 (149) | 93 (167) | 44 |
| HEM | Protoporphyrin IX containing FE | 62 (86) | 140 (222) | 16 | 73 (75) | 166 (186) | 26 |
| FAD | Flavin-adenine dinucleotide | 21 (32) | 119 (238) | 11 | 22 (31) | 145 (212) | 18 |
| PLP | Pyridoxal-5′-phosphate | 49 (63) | 120 (187) | 7 | 48 (62) | 137 (170) | 10 |
| NAD | Nicotinamide-adenine-dinucleotide | 54 (78) | 98 (167) | 16 | 63 (69) | 130 (135) | 19 |
| ATP | Adenosine-5′-triphosphate | 49 (87) | 73 (134) | 29 | 53 (83) | 86 (121) | 31 |
| HEC | HEME C | 22 (36) | 105 (127) | 9 | 35 (23) | 111 (121) | 14 |
| FMN | Flavin Mononucleotide | 22 (41) | 58 (119) | 4 | 28 (35) | 81 (96) | 6 |
| GOL | Glycerol | 52 (119) | 15 (37) | 7 | 72 (99) | 26 (26) | 12 |
| NAP | NADP Nicotinamide-adenine-dinucleotide phosphate | 24 (45) | 60 (109) | 13 | 40 (29) | 85 (84) | 18 |
| ANP | Phosphoaminophosphonic acid-adenylate ester | 35 (66) | 46 (76) | 18 | 45 (56) | 45 (77) | 26 |
| AMP | Adenosine monophosphate | 36 (66) | 35 (58) | 13 | 48 (54) | 40 (53) | 15 |
| CL1 | Alpha chlorophyll A | 39 (45) | 70 (77) | 4 | 40 (44) | 70 (77) | 6 |
| GDP | Guanosine-5′-diphosphate | 31 (44) | 60 (74) | 11 | 33 (42) | 61 (74) | 15 |
| COA | Coenzyme A | 17 (28) | 58 (88) | 6 | 17 (28) | 55 (91) | 7 |
| NDP | NADPH Dihydro-nicotinamide-adenine-dinucleotide phosphate | 22 (30) | 54 (77) | 14 | 27 (25) | 59 (72) | 12 |
| SAH | S-Adenosyl-L-homocysteine | 29 (52) | 21 (48) | 5 | 32 (49) | 31 (38) | 11 |
| SAM | S-Adenosylmethionine | 23 (40) | 19 (44) | 5 | 21 (42) | 20 (43) | 7 |
| EPE | 4-(2-Hydroxyethyl)-1-piperazine ethanesulfonic acid | 35 (57) | 8 (21) | 2 | 41 (51) | 13 (16) | 4 |
| CIT | Citric acid | 20 (56) | 11 (18) | 5 | 37 (39) | 14 (15) | 6 |
The numbers in parentheses show the numbers of all clusters, including patches without SCCS.The symbol # indicates “the number of” CATH IDs were assigned to 20,250 patches for 7155 ligand IDs in the same way of SCOP (see details in Supporting Information 4). The high correlation, 0.96, between the number of SCOP fold assigned to clusters and that of CATH topology assigned to clusters was observed.
Relationship between similarities of protein sequences and patches
Investigating the relationship between PatSim score and sequence similarity of proteins (SeqSim) is valuable for understanding the possibility of inference of ligand species and their LBSs in a protein from their homologs proteins with known LBSs. The high-correlation between them would ensure such inference with high accuracy. Therefore, an exhaustive comparison among the protein sequences for the representative patches was performed using BLAST,43,44 and the correlation between PatSim and SeqSim scores was investigated. Note that some LBSs are composed of more than two protein chains. To deal with such LBSs, we selected only one sequence in which the protein has the largest interface with a ligand. In addition, to investigate the effect of EPot on the protein-ligand interactions, PatSim score based on only surface configurations; that is PatSim without EPot, was also compared with SeqSim score.
A large number of sequence pairs, 895,354, lacked BLAST search hits, even though some patch pairs of such sequence pairs had PatSim scores larger than 0.9. Most of the pairs were for small patches with flat surface configurations, such as that for SO4 (sulfate ion) GOL (glycerol), EDO (1,2-ethanediol), PO4 (phosphate ion), and ACT (acetate ion), which are often used as additives to stabilize protein structures. About 73% of all the sequence pairs had SeqSim between 20% and 40% in both PatSim with and without EPot. The contour plot between PatSim and SeqSim scores is shown in Figure 2-I. The range of SeqSim between 20% and 35% is considered as a problematic twilight zone for sequence alignments, where the alignment method often fails to correctly align protein pairs.45 The Pearson's correlation coefficients (PCCs) were computed for different SeqSim scores and psizes, and they are shown in Table II-A.
Figure 2.
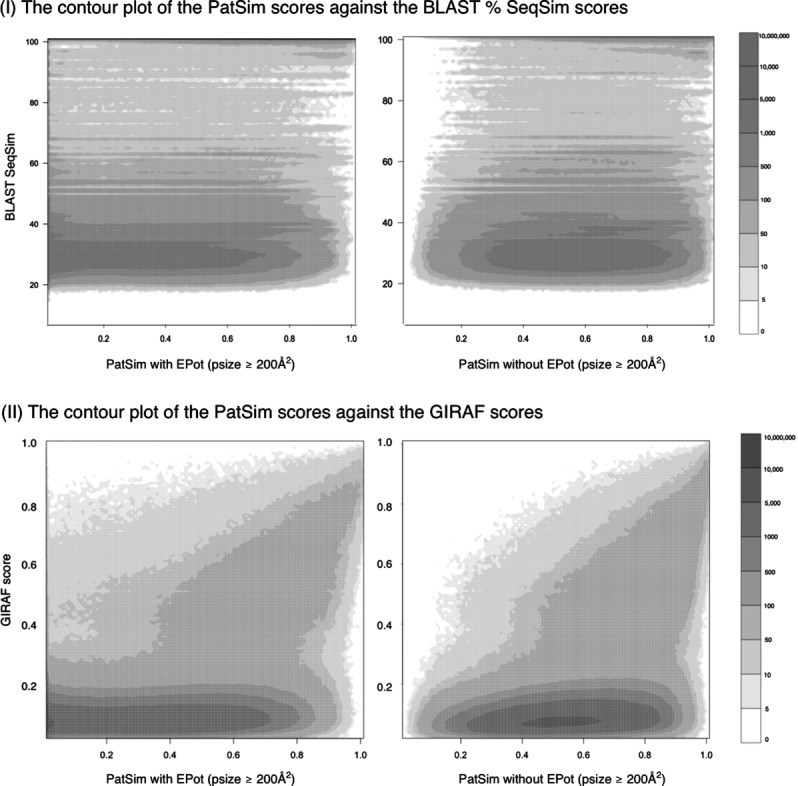
The counter plots of PatSim score with or without EPot against: (I) SeqSim score and (II) GIRAF score, for patches with psizes of ≥200 Å2.
Table II.
The Correlations of PatSim Scores With or Without EPot Against (I) BLAST % SeqSim Scores and (II) GIRAF Scores, for Patches With psize ≥ 200 Å2
| Psize (Å2) | BLAST % SeqSim | |||||
|---|---|---|---|---|---|---|
| PatSim score with EPot | PatSim score without EPot | |||||
| ≥0% | ≥40% | ≥80% | ≥0% | ≥40% | ≥80% | |
| (A) The correlations between PatSim scores and BLAST % SeqSim scores | ||||||
| > 0Å2 | 0.204 | 0.335 | 0.230 | 0.165 | 0.286 | 0.200 |
| ≥ 200Å2 | 0.255 | 0.392 | 0.263 | 0.217 | 0.347 | 0.217 |
| ≥ 500Å2 | 0.392 | 0.501 | 0.120 | 0.346 | 0.469 | 0.070 |
| GIRAF score | ||||
|---|---|---|---|---|
| PatSim score with EPot | PatSim score without EPot | |||
| Psize (Å2) | ≥0 | ≥0.2 | ≥0 | ≥0.2 |
| (B) The correlations between PatSim scores and GIRAF scores | ||||
| >0 Å2 | 0.387 | 0.437 | 0.405 | 0.442 |
| ≥200 Å2 | 0.390 | 0.437 | 0.402 | 0.444 |
| ≥500 Å2 | 0.361 | 0.492 | 0.363 | 0.503 |
Weak and modest positive correlations were observed for SeqSim score ≥0% and ≥40%, respectively. However, at SeqSim of ≥80%, PCCs largely decreased. The main reason is that there were a number of proteins accommodating different ligands on their protein surfaces, and the patches for binding different ligands to the same protein generally have distinct configurations and different EPots. Furthermore, as shown in Table II-A, PCCs for PatSim with EPot were slightly better than those for PatSim without EPot by, on average, 0.041%, 0.049%, and 0.042%, for psizes of >0 Å2, ≥200 Å2, and ≥500 Å2, respectively. As a result, PatSim and SeqSim scores were weakly correlated, suggesting that patches at LBSs can be conserved among distantly related protein sequences. On the contrary, the poor correlation also suggests that patches have unique characteristics independent of sequence homology.
Relationship between similarities of atomic structures in LBSs and patches
The next issue to address is whether any relationship exists between similarities of local atomic structures in LBSs and those of patches. To investigate this, we performed an exhaustive comparison among atomic structures in the LBSs for the representative patches using GIRAF,24 and then PCC between local structural similarity in GIRAF and PatSim score was investigated.
GIRAF scores were obtained for only 2.84% of LBS pairs (9,758,557 out of 339,522,711 pairs of 26,059 LBSs), and the other pairs were regarded as unlikely matches. In GIRAF, no matching reference sets for such pairs were retrieved by a relation algebraic procedure, in order to facilitate the computation. There was modest positive PCC between PatSim and GIRAF scores. PCCs were about 0.4 (GIRAF score >0) for both PatSim scores with and without EPot (Table II-B), but PCC for PatSim without EPot was slightly higher than that with EPot. This is because GIRAF does not consider EPot, and only considers atomic structures in LBSs. The contour plots of PatSim scores versus GIRAF scores (psize ≥200 Å2) are shown in Figure 2-II.
An ambiguous zone was found below GIRAF score of 0.2, where GIRAF sometimes failed to correctly detect pairwise similarity between LBSs (Fig. 2-II). This may be due to strict constraints for matching reference sets or the heuristic alignment method used in GIRAF. In the ambiguous zone, we found many patch pairs with high PatSim scores, as shown in Figure 3-I. This is because the differences in protein folds did not affect the surface-based search and PatSim score, but it was likely to affect the atom position-based search and its score. Above the ambiguous zone, PCCs increased to 0.437 (psize ≥200 Å2) and 0.492 (psize ≥500 Å2).
Figure 3.
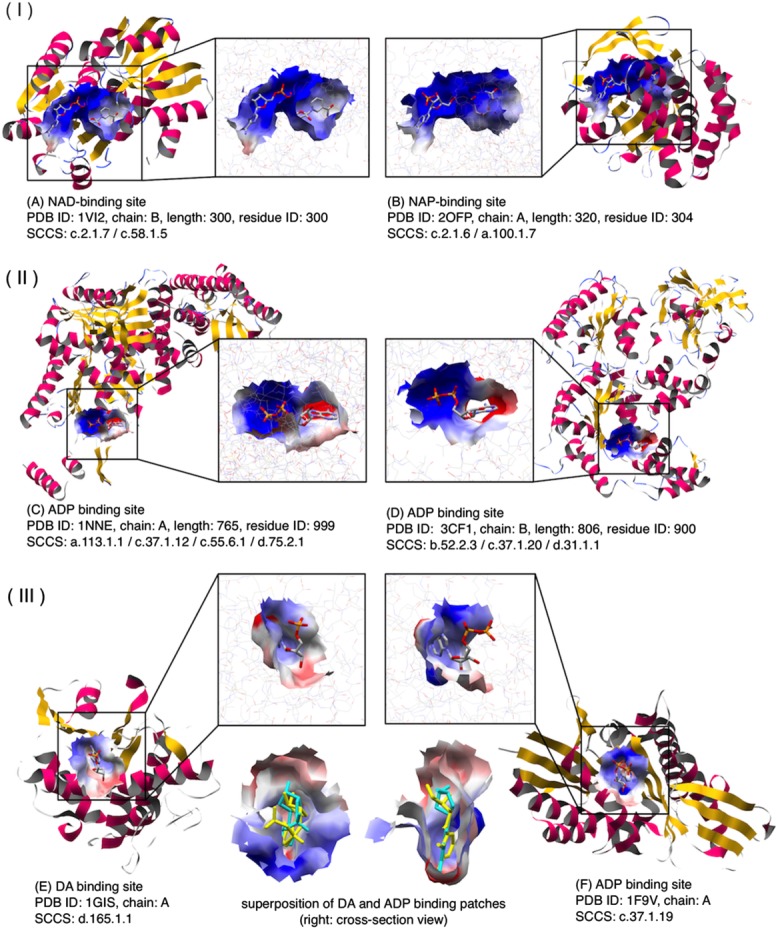
Examples of similar patches shared by protein with low atomic structural similarities: (I) (A) NAD (nicotinamide adenine dinucleotide) binding patch of shikimate 5-dehydrogenase 2 (PDB ID: 1VI2). (B) NAP (NADP nicotinamide adenine dinucleotide phosphate) binding patch of ketopantoate reductase (PDB ID: 2OFP). GIRAF score = 0.078, PatSim score = 0.945, SeqSim score = 56% (5/9; e-value = 0.96, minimum coverage = 2.9%) (II). (C) ADP-binding (adenosine-5′-diphosphate) patch of PDB ID: 1NNE. (D) ADP-binding patch of PDB ID: 3CF1. PatSim score = 0.877, No GIRAF hit, SeqSim score = 63% (7/18; e-value = 3.2). (III) (E) DA (2′-deoxyadenosine-5′-monophosphate) binding patch for the ribosome-inactivating protein alpha trichosanthin (PDB ID: 1GIS). (F) ADP binding patch for the kinesin-like protein KAR3 (PDB ID: 1F9V). Superposition of DA (cyan) and ADP (yellow) binding patches is also shown. PatSim score = 0.958, No GIRAF hit.
As a consequence, even though modest and meaningful correlation existed between PatSim score and similarity of atomic structures in LBSs, there were many similar patch pairs with PatSim scores ≥0.8 detected in the ambiguous zone. This suggested that the current surface-based method has potential to detect similar LBSs, independent of atomic structure of LBS and fold similarity.
Relationship between the similarities of functional keywords for proteins and patches
To understand the relationship between PatSim score and similarity of protein function, we assigned UniProt functional keywords (UFK)46 to each of the representative patches and then investigated functional relationships among them (see Materials and Methods section). A total of 5,947 unique UniProt accession numbers (UniProt AC) were obtained and assigned to 18,471 proteins with 22,851 patches. In addition, we assigned UniProt AC to 668 unassigned proteins with 816 patches in the same procedure of the SCCS assignment (Fig. S2); that is, in total, 19,139 proteins with 23,677 patches. The number of keywords in each category is shown in Supporting Information Table SVI. The ratio of the percent of patch pairs that possess at least one identical UFK in each range of 0.05% PatSim change was investigated in each category. Note that patches did not always have UFK in each of the categories, and so such patches were simply ignored.
As a result, the ratios for PatSim with EPot were always slightly higher than those for PatSim without EPot, in the range from 0.9 to 1.0 (Fig. 4) In particular, in the category of Biological process, which is a series of chemical reactions or other events regulated by interactions of proteins with other proteins or ligands, the ratio for PatSim with EPot was largely increased from 21.3% to 49.6%, in that range. On the contrary, the ratio for PatSim without EPot was increased from 10.9% to 29.2% in the same range. Thus, these observations indicated that there is better correlation with protein functions, especially with Biological process, when PatSim with EPot is used for similarity search of LBSs, as compared to PatSim without EPot.
Figure 4.
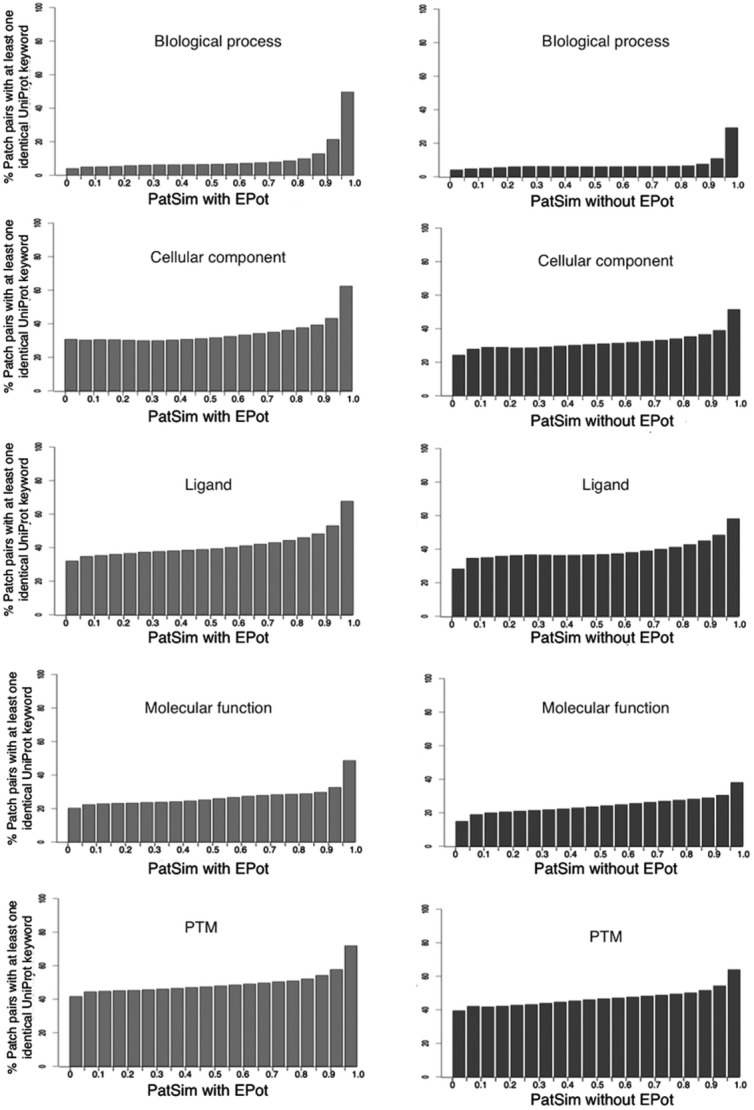
The ratio in percent of patch pairs with at least one identical UFK. In each UFK category, the number of patch pairs in which the proteins have at least one identical UFK is counted in each range of 0.05% PatSim change, and then it is divided by the total number of all patch pairs contained in the same range.
Discussion
We prepared 26,059 representative patches by selecting a patch from each cluster in the hierarchical clustering, based on local surface similarity of surface configuration and EPot. It may be possible to select representatives based on local structural similarity, as previously conducted to define structural motifs of LBSs.33 However, even if local structures of two LBSs are similar, their PatSim score is sometimes very low due to their different EPots. For example, the structural similarity between ADP-binding site of human inositol (1,4,5)-triphosphate 3-kinase (PDB ID: 1W2D) and that of Archaeoglobus fulgidus Rio2 kinase (PDB ID: 1ZAR) was detected with a p-value of 8.1 × 10−17 (40 aligned atoms; RMSD = 0.75 Å)33 (Fig. 5). However, their surface similarity detected with PatSim with EPot is 0.118, but that detected with PatSim without Epot is 0.896. These patches had completely different EPots. The ADP-binding patch of 1W2D has a positive average EPot of 0.114 V, because its LBS is close to an inositol(1,3,4,5)-tetrakisphophate (4IP) binding site maintained by positively charged residues and with a higher positive EPot. Thus, we concluded that the atom position-based selection is not suitable for the selection of representative patches, because it ignored significant differences in EPots between such LBSs. The representative patches are available at eF-seek28 by selecting “rep_26059” from a drop-down list of the search database on the submission page.
Figure 5.
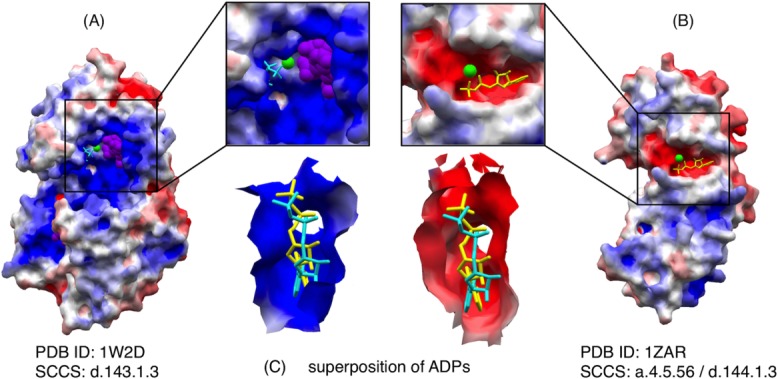
An example of similar ADP-binding patches with different EPots: (A) Human inositol (1,4,5)-triphosphate 3-kinase (PDB ID: 1W2D), which accommodates ADP (cyan), 4IP (inositol-(1,3,4,5)-tetrakisphosphate; purple) and MN (manganese (II) ion; green), (B) Archaeoglobus fulgidus Rio2 kinase (PDBID: 1ZAR) which accommodates ADP (yellow) and MN (green), (C) Superposition of these ADPs with the local molecular surface of 1W2D (left) and that of 1ZAR (right); PatSim score with EPot = 0.118 and that without EPot = 0.896.
To examine the search performance for surface similarity by eF-seek with rep_26059, functions of 53 apo-proteins taken from LigASiste47 were predicted, comparing with that using the original large patch dataset with 255,647 patches including small matches, and the results are shown in Supporting Information Table SIX. Consequently, eF-seek with rep_26059 has a performance similar to, but slightly lower than that with the original dataset. Thus, rep_26059 has an advantage of fast similarity search, without losing its search ability largely. The reason for the slightly lower prediction performance is probably because the large dataset, which contain a huge number of heterogenetic patches, could provide a lot of flexibility in the similarity search of eF-seek (see more detailed in the Supporting Information 3).
Previously, after comparing the shapes of protein binding pocket to the shapes of their ligands, Kahraman et al.48 concluded that shape complementarity in general is not sufficient to drive molecular recognition alone and requires additional physicochemical properties. As indicated in the Supporting Information 3, the larger patch dataset, which contains a huge number of heterogenetic patches for a specific ligand, achieved slightly higher overall accuracy for function prediction than the representative patches, which contains a relatively small number of canonical patches for a specific ligand. Namely, as the more variable structures of ligand binding sites are used, the more accurate predictions for the bound ligands can be possible. In addition, the positive effect of electrostatic potential distribution on molecular recognition was also confirmed. In fact, current PatSim score with EPot is more powerful than that without EPot for search of the similar molecular surfaces of the ligand binding sites.
More than 99% of the clusters (motifs), each with at least 10 atomic coordinates of LBSs, reportedly shared the same domains at family or superfamily level at an atomic level.33 In comparison with this previous report, the ratio of the clusters classified in the same SCOP levels was smaller at surface level; that is, about 63% (1,846/2,949 clusters; psize ≥200 Å2) of the clusters were classified into these levels. This could be the reason why the surface-based method is sensitive to differences in EPot and insensitive to differences in protein folds. As shown in Figure 5, it can recognize such differences in the EPots in LBSs. Furthermore, the similarity between patches for proteins with low SeqSim scores was detected, and such patches were included in the same cluster. For example, the similarity between two ADP-binding patches of two distantly related proteins with different folds and low SeqSim scores (PDB IDs: 1NNE and 3CF1) was detected with PatSim of 0.877 (a lower BLAST e-value or an e-value closer to zero implies a more significant SeqSim); however, the local structural similarities of these LBSs were not detected (Fig. 3-II). Another example is similarity between the 2′-deoxyadenosine-5′-monophosphate (DA) binding patch of the ribosome-inactivating protein alpha trichosanthin (PDB ID; 1GIS) and the ADP binding patch of the kinesin-like protein KAR3 (PDB ID: 1F9V) (Fig. 3-III). Although they have different SCOP folds, their PatSim score was 0.958, but structural similarity between their LBSs was never found by GIRAF.33 These examples suggested that the surface-based method has better potential to detect common patches shared by distantly related proteins and by proteins with cross-fold similarity. Thus, sensitivity of patches to EPot and insensitivity of them to protein folds lead to differences in the ratios of the clusters classified in family or superfamily levels.
The same analysis of patch clustering was carried out with CATH,49 which semiautomatically classifies protein domains to hierarchical groups. Consequently, overall results were not largely different from those with SCOP. In Table I, analysis with CATH topologies is also shown (see detailed analysis in the Supporting Information 4).
A comparison of PatSim score with similarity of protein functions revealed that patches with similar EPots tend to have more similar functions, especially in Biological process category of UFK, as shown in Figure 4. Since protein interactions with ligands, such as inhibitors, cofactors and substrates, underlie almost all biological processes that are series of chemical reactions or other molecular events, patches of LBSs in proteins need to be precisely conserved and correctly recognized by ligands to regulate the biological processes. In addition, EPot is fundamentally important in the chemical reaction and molecular recognition procedures in almost all living cells. This may be reflected by finding that PatSim score with EPot correlated strongly with similarity of protein functions, particularly for their biological processes.
It is interesting to examine how the current search method for similar molecular surfaces can be applied to function prediction for proteins of unknown functions. There are many hypothetical proteins, whose structures were determined during the structural genomics programs in the world. Previously, such an application was once made for the function prediction of TT1542 from Thermus thermopilus (PDBID: 1UAN), where we made prediction of its LBS and the sugar-like ligand,50 both of which were further confirmed by the distant homolog structure of MshB (deacetylase) from Mycobacterium tuberculosis (PDBID: 1q7t) and its function.51
In the same manner, we tried another function prediction for a hypothetical protein, pag5_736 from Pyrobaculum aerophilum (PDBID: 1RKI-B), whose structure was determined but function unknown, by the current search method for similar molecular surfaces of LBSs. Then, P6G (hexaethylene glycol) patch of the hypothetical protein was classified into the cluster, where all other patches accommodating different ligands had similar shape and similar EPot, and also their binding proteins all were hydrolases (Supporting Information Fig. S3 and Supporting Information Table SVII). Furthermore, we found that the triad of amino acid (His, Lys, and Phe) in the P6G-binding site was also used in other LBSs of proteins in the same cluster (Supporting Information Fig. S4). From these observations, 1RKI was strongly inferred as hydrolase. In addition, eF-seek23,28 could search similar patches of hydrolases to the P6G-binding patch of 1RKI with a similarity score >0.62, although similar structural motifs to it could not be found by GIRAF.24 More details are described in Supporting Information 2.
In addition, function prediction for 53 apo-proteins revealed that EPot contributes to narrow down candidate ligand that binds to target proteins as described above (see more details in the Supporting Information 3). These examples indicate that patches with EPot are useful and complementary tools to make novel function prediction for proteins of unknown functions.
We found that LBSs of specific ligands have diverse patches with different surface configurations and EPots, and that distantly related proteins or proteins with cross-fold similarity share common patches in many cases. This indicated that patches have unique characteristics that are independent of their sequences and local atomic positions. The results presented in this study will help to clarify various types of protein-ligand interactions at surface level and to predict protein functions, as a complement to conventional similarity searches based on SeqSim scores and local atomic positions.
Materials and Methods
Atomic coordinates of ligands
All of the atomic coordinates of ligands (nonpolymer molecules), except atoms of DNA, RNA, and unknown ligands, were obtained from PDBML data files52 in PDB.1 The atomic coordinates of monoatomic ions and single atom molecules were also excluded, because their patches are too tiny, flat, and indistinguishable (Supporting Information Table SI).
Molecular surfaces of proteins
The molecular surfaces of many proteins were obtained from eF-site (electrostatic surface of functional-site).34 At eF-site, they are generated using the molecular surface package (MSP)53 and represented by a set of vertices with their surface normals. In addition, for each vertex, EPot is calculated by solving the Poisson–Boltzmann equations numerically with a precise continuum model by the use of the SCB program,54 and maximum and minimum curvatures (MaxCuv, MinCuv) are calculated by rotating the normal plane from 0° to 180° with intervals of 5°.35
Local surfaces of ligand-binding sites
The local surface of LBS (patch) is defined as a set of vertices on the surface of single or multiple chains of a protein within 5 Å from any atoms of a ligand, provided that the angle between a normal vector at a vertex and a vector from the vertex to the nearest ligand atom should be ≤90°. Tiny patches constructed of ≤20 vertices were removed, because such patches are likely to match any local molecular surfaces with similar EPots.
The similarity search method
A similarity search method to compare two surfaces was developed using geometric hashing (GH) techniques55–57 to quickly find two similar objects (a model and a target) represented by a set of vertices. The conventional GH method requires large computation memory to store entries, each with a basis defined by a pair of vertices, into a hash table (HT) indexed by the coordinates of each of vertices on the transformed model.55–57 However, too much memory space would be required to store all of the entries of a large number of different patches into the HT all at once. Thus, a pair of two patches is repeated compared, where the matching is performed in the orthogonal coordinate system of the original model object. A detailed outline of the method is provided with a depiction of the flow in Supporting Information Figure S1.
Definition of patch similarity
The number of matching vertices must be normalized by the density of vertices on a patch, because of different densities of vertices on patches. When comparing patchB to patchA and the density of patchB (DB) is larger than that of patchA (DA), the number of vertices on patchB (NB), and that of the matching vertices (M) are normalized by DA/DB:
On the contrary, if DB is smaller than DA, then the number of vertices on patchA (NA) is normalized by DB/DA.
Then, PatSim score between two patches is calculated as:
In addition, the dissimilarity is defined as 1.0 – PatSim(patchA, patchB).
Selection of representative patches
To select representative patches, an all-against-all comparison among patches was performed for each ligand ID, and then a hierarchical clustering of patches was performed using the R statistical package. The distance between two clusters was computed using the group average method. In each cluster, a representative patch was determined as a patch with the minimum average mutual PatSim =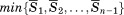 , where n is the number of patches in the same cluster and
, where n is the number of patches in the same cluster and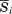 is the averaged mutual PatSim for a patchi given by:
is the averaged mutual PatSim for a patchi given by:
Hierarchical clustering of the representative patches
The representative patches were exhaustively compared, and were hierarchically clustered using R. Since the representatives of different ligand IDs are different in terms of their characteristics, we employed Ward's method (minimum variance method) to calculate the distance between two clusters. It minimizes the total sum of squares of the distance from each patch to the centroid of a cluster, and it produces compact clusters with clear separations between them.
Assignment of SCOP codes to each patch
Protein structures are hierarchically classified into class, fold, superfamily, and family in SCOP. In this study, we only considered seven classes: (a) all-α, (b) all-β, (c) α/β (parallel β sheet; β-α-β units), (d) α+β (antiparallel β sheets; segregated α+β regions), (e) multidomain, (f) membrane and cell surface proteins and peptides, and (g) small proteins, except for (h) coiled coil proteins, (i) low resolution protein structures, (j) peptides, or (k) designed proteins. A SCOP parseable file (version 1.75) was used for the assignment of a SCOP code(s) to each protein. For a patch extracted from multiple chains, SCOP code for a chain sharing the largest interface with a ligand was used.
Assignment of functional keywords to each patch
The UniProt function keywords (UFK)46 are tagged with each of UniProt43 entries to describe specific or more general properties of individual proteins. These UFKs are hierarchically organized and classified into 10 different categories, but we only used five categories: biological process, cellular component, ligand, molecular function, and post-translation modification (PTM), and not coding sequence diversity, developmental stage, disease, domain or technical term.
Acknowledgments
The authors thank Dr. Takeshi Kawabata (Institute for Protein Research, Osaka University, Japan) for kindly providing his KCOMBU program for matching chemical structures and his thoughtful comments.
Glossary
- EPot
electrostatic potentials
- LBS
ligand-binding site
- Patch
local molecular surface of LBS
- PatSim
patch similarity
- PDB
Protein Data Bank
- psize
patch area size
- SeqSim
sequence similarity
- UFK
UniProt functional keyword.
Supplementary material
Additional Supporting Information may be found in the online version of this article.
References
- 1.Berman H, Henrick K, Nakamura H, Markley JL. The worldwide Protein Data Bank (wwPDB): ensuring a single, uniform archive of PDB data. Nucleic Acids Res. 2007;35:D301–D303. doi: 10.1093/nar/gkl971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pearl F, Todd AE, Bray JE, Martin AC, Salamov AA, Suwa M, Swindells MB, Thornton JM, Orengo CA. Using the CATH domain database to assign structures and functions to the genome sequences. Biochem Soc Trans. 2000;28:269–275. doi: 10.1042/bst0280269. [DOI] [PubMed] [Google Scholar]
- 3.Devos D, Valencia A. Practical limits of function prediction. Proteins. 2000;41:98–107. [PubMed] [Google Scholar]
- 4.Wilson CA, Kreychman J, Gerstein M. Assessing annotation transfer for genomics: quantifying the relations between protein sequence, structure and function through traditional and probabilistic scores. J Mol Biol. 2000;297:233–249. doi: 10.1006/jmbi.2000.3550. [DOI] [PubMed] [Google Scholar]
- 5.Aloy P, Querol E, Aviles FX, Sternberg MJ. Automated structure-based prediction of functional sites in proteins: applications to assessing the validity of inheriting protein function from homology in genome annotation and to protein docking. J Mol Biol. 2001;311:395–408. doi: 10.1006/jmbi.2001.4870. [DOI] [PubMed] [Google Scholar]
- 6.Teichmann SA, Rison SC, Thornton JM, Riley M, Gough J, Chothia C. The evolution and structural anatomy of the small molecule metabolic pathways in Escherichia coli. J Mol Biol. 2001;311:693–708. doi: 10.1006/jmbi.2001.4912. [DOI] [PubMed] [Google Scholar]
- 7.Todd AE, Orengo CA, Thornton JM. Evolution of function in protein superfamilies, from a structural perspective. J Mol Biol. 2001;307:1113–1143. doi: 10.1006/jmbi.2001.4513. [DOI] [PubMed] [Google Scholar]
- 8.Ikura M, Ames JB. Genetic polymorphism and protein conformational plasticity in the calmodulin superfamily: two ways to promote multifunctionality. Proc Natl Acad Sci U S A. 2006;103:1159–1164. doi: 10.1073/pnas.0508640103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gifford JL, Walsh MP, Vogel HJ. Structures and metal-ion-binding properties of the Ca2+-binding helix-loop-helix EF-hand motifs. Biochem J. 2007;405:199–221. doi: 10.1042/BJ20070255. [DOI] [PubMed] [Google Scholar]
- 10.Campbell SJ, Gold ND, Jackson RM, Westhead DR. Ligand binding: functional site location, similarity and docking. Curr Opin Struct Biol. 2003;13:389–395. doi: 10.1016/s0959-440x(03)00075-7. [DOI] [PubMed] [Google Scholar]
- 11.Jones S, Thornton JM. Searching for functional sites in protein structures. Curr Opin Chem Biol. 2004;8:3–7. doi: 10.1016/j.cbpa.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 12.Watson JD, Laskowski RA, Thornton JM. Predicting protein function from sequence and structural data. Curr Opin Struct Biol. 2005;15:275–284. doi: 10.1016/j.sbi.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 13.Lee D, Redfern O, Orengo C. Predicting protein function from sequence and structure. Nat Rev Mol Cell Biol. 2007;8:995–1005. doi: 10.1038/nrm2281. [DOI] [PubMed] [Google Scholar]
- 14.Rosen M, Lin SL, Wolfson H, Nussinov R. Molecular shape comparisons in searches for active sites and functional similarity. Protein Eng. 1998;11:263–277. doi: 10.1093/protein/11.4.263. [DOI] [PubMed] [Google Scholar]
- 15.Schmitt S, Kuhn D, Klebe G. A new method to detect related function among proteins independent of sequence and fold homology. J Mol Biol. 2002;323:387–406. doi: 10.1016/s0022-2836(02)00811-2. [DOI] [PubMed] [Google Scholar]
- 16.Stark A, Sunyaev S, Russell RB. A model for statistical significance of local similarities in structure. J Mol Biol. 2003;326:1307–1316. doi: 10.1016/s0022-2836(03)00045-7. [DOI] [PubMed] [Google Scholar]
- 17.Wangikar PP, Tendulkar AV, Ramya S, Mali DN, Sarawagi S. Functional sites in protein families uncovered via an objective and automated graph theoretic approach. J Mol Biol. 2003;326:955–978. doi: 10.1016/s0022-2836(02)01384-0. [DOI] [PubMed] [Google Scholar]
- 18.Jambon M, Imberty A, Deleage G, Geourjon C. A new bioinformatic approach to detect common 3D sites in protein structures. Proteins. 2003;52:137–145. doi: 10.1002/prot.10339. [DOI] [PubMed] [Google Scholar]
- 19.Yao H, Kristensen DM, Mihalek I, Sowa ME, Shaw C, Kimmel M, Kavraki L, Lichtarge O. An accurate, sensitive, and scalable method to identify functional sites in protein structures. J Mol Biol. 2003;326:255–261. doi: 10.1016/s0022-2836(02)01336-0. [DOI] [PubMed] [Google Scholar]
- 20.Binkowski TA, Adamian L, Liang J. Inferring functional relationships of proteins from local sequence and spatial surface patterns. J Mol Biol. 2003;332:505–526. doi: 10.1016/s0022-2836(03)00882-9. [DOI] [PubMed] [Google Scholar]
- 21.Brakoulias A, Jackson RM. Towards a structural classification of phosphate binding sites in protein-nucleotide complexes: an automated all-against-all structural comparison using geometric matching. Proteins. 2004;56:250–260. doi: 10.1002/prot.20123. [DOI] [PubMed] [Google Scholar]
- 22.Shulman-Peleg A, Nussinov R, Wolfson HJ. Recognition of functional sites in protein structures. J Mol Biol. 2004;339:607–633. doi: 10.1016/j.jmb.2004.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kinoshita K, Nakamura H. Identification of the ligand binding sites on the molecular surface of proteins. Protein Sci. 2005;14:711–718. doi: 10.1110/ps.041080105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kinjo AR, Nakamura H. Similarity search for local protein structures at atomic resolution by exploiting a database management system. Biophysics. 2007;3:75–84. doi: 10.2142/biophysics.3.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sael L, Kihara D. Binding ligand prediction for proteins using partial matching of local surface patches. Int J Mol Sci. 2010;11:5009–5026. doi: 10.3390/ijms11125009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tu H, Shi T. Ligand binding site similarity identification based on chemical and geometric similarity. Protein J. 2013;32:373–385. doi: 10.1007/s10930-013-9494-1. [DOI] [PubMed] [Google Scholar]
- 27.Lee HS, Im W. Identification of ligand templates using local structure alignment for structure-based drug design. J Chem Inf Model. 2012;52:2784–2795. doi: 10.1021/ci300178e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kinoshita K, Murakami Y, Nakamura H. eF-seek: prediction of the functional sites of proteins by searching for similar electrostatic potential and molecular surface shape. Nucleic Acids Res. 2007;35:W398–W402. doi: 10.1093/nar/gkm351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kobayashi N, Go N. A method to search for similar protein local structures at ligand binding sites and its application to adenine recognition. Eur Biophys J. 1997;26:135–144. doi: 10.1007/s002490050065. [DOI] [PubMed] [Google Scholar]
- 30.Kinoshita K, Sadanami K, Kidera A, Go N. Structural motif of phosphate-binding site common to various protein superfamilies: all-against-all structural comparison of protein-mononucleotide complexes. Protein Eng. 1999;12:11–14. doi: 10.1093/protein/12.1.11. [DOI] [PubMed] [Google Scholar]
- 31.Gold ND, Jackson RM. Fold independent structural comparisons of protein-ligand binding sites for exploring functional relationships. J Mol Biol. 2006;355:1112–1124. doi: 10.1016/j.jmb.2005.11.044. [DOI] [PubMed] [Google Scholar]
- 32.Minai R, Matsuo Y, Onuki H, Hirota H. Method for comparing the structures of protein ligand-binding sites and application for predicting protein-drug interactions. Proteins. 2008;72:367–381. doi: 10.1002/prot.21933. [DOI] [PubMed] [Google Scholar]
- 33.Kinjo AR, Nakamura H. Comprehensive structural classification of ligand-binding motifs in proteins. Structure. 2009;17:234–246. doi: 10.1016/j.str.2008.11.009. [DOI] [PubMed] [Google Scholar]
- 34.Kinoshita K, Nakamura H. eF-site and PDBjViewer: database and viewer for protein functional sites. Bioinformatics. 2004;20:1329–1330. doi: 10.1093/bioinformatics/bth073. [DOI] [PubMed] [Google Scholar]
- 35.Kinoshita K, Nakamura H. Identification of protein biochemical functions by similarity search using the molecular surface database eF-site. Protein Sci. 2003;12:1589–1595. doi: 10.1110/ps.0368703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Murzin AG, Brenner SE, Hubbard T, Chothia C. SCOP: a structural classification of proteins database for the investigation of sequences and structures. J Mol Biol. 1995;247:536–540. doi: 10.1006/jmbi.1995.0159. [DOI] [PubMed] [Google Scholar]
- 37.Engh RA, Bossemeyer D. Structural aspects of protein kinase control-role of conformational flexibility. Pharmacol Ther. 2002;93:99–111. doi: 10.1016/s0163-7258(02)00180-8. [DOI] [PubMed] [Google Scholar]
- 38.Wierenga RK. The TIM-barrel fold: a versatile framework for efficient enzymes. FEBS Lett. 2001;492:193–198. doi: 10.1016/s0014-5793(01)02236-0. [DOI] [PubMed] [Google Scholar]
- 39.Orengo CA, Jones DT, Thornton JM. Protein superfamilies and domain superfolds. Nature. 1994;372:631–634. doi: 10.1038/372631a0. [DOI] [PubMed] [Google Scholar]
- 40.Nagano N, Orengo CA, Thornton JM. One fold with many functions: the evolutionary relationships between TIM barrel families based on their sequences, structures and functions. J Mol Biol. 2002;321:741–765. doi: 10.1016/s0022-2836(02)00649-6. [DOI] [PubMed] [Google Scholar]
- 41.Osadchy M, Kolodny R. Maps of protein structure space reveal a fundamental relationship between protein structure and function. Proc Natl Acad Sci U S A. 2011;108:12301–12306. doi: 10.1073/pnas.1102727108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Smith LJ, Kahraman A, Thornton JM. Heme proteins—diversity in structural characteristics, function, and folding. Proteins. 2010;78:2349–2368. doi: 10.1002/prot.22747. [DOI] [PubMed] [Google Scholar]
- 43.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 45.Rost B. Twilight zone of protein sequence alignments. Protein Eng. 1999;12:85–94. doi: 10.1093/protein/12.2.85. [DOI] [PubMed] [Google Scholar]
- 46.Magrane M, Consortium U. UniProt knowledgebase: a hub of integrated protein data. Database (Oxford) 2011;2011:bar009. doi: 10.1093/database/bar009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dessailly BH, Lensink MF, Orengo CA, Wodak SJ. LigASite—a database of biologically relevant binding sites in proteins with known apo-structures. Nucleic Acids Res. 2008;36:D667–D673. doi: 10.1093/nar/gkm839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kahraman A, Morris RJ, Laskowski RA, Thornton JM. Shape variation in protein binding pockets and their ligands. J Mol Biol. 2007;368:283–301. doi: 10.1016/j.jmb.2007.01.086. [DOI] [PubMed] [Google Scholar]
- 49.Sillitoe I, Cuff AL, Dessailly BH, Dawson NL, Furnham N, Lee D, Lees JG, Lewis TE, Studer RA, Rentzsch R, Yeats C, Thornton JM, Orengo CA. New functional families (FunFams) in CATH to improve the mapping of conserved functional sites to 3D structures. Nucleic Acids Res. 2013;41:D490–D498. doi: 10.1093/nar/gks1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Handa N, Terada T, Kamewari Y, Hamana H, Tame JR, Park SY, Kinoshita K, Ota M, Nakamura H, Kuramitsu S, Shirouzu M, Yokoyam S. Crystal structure of the conserved protein TT1542 from Thermus thermophilus HB8. Protein Sci. 2003;12:1621–1632. doi: 10.1110/gad.03104003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McCarthy AA, Peterson NA, Knijff R, Baker EN. Crystal structure of MshB from Mycobacterium tuberculosis, a deacetylase involved in mycothiol biosynthesis. J Mol Biol. 2004;335:1131–1141. doi: 10.1016/j.jmb.2003.11.034. [DOI] [PubMed] [Google Scholar]
- 52.Westbrook J, Ito N, Nakamura H, Henrick K, Berman HM. PDBML: the representation of archival macromolecular structure data in XML. Bioinformatics. 2005;21:988–992. doi: 10.1093/bioinformatics/bti082. [DOI] [PubMed] [Google Scholar]
- 53.Connolly ML. The molecular surface package. J Mol Graph. 1993;11:139–141. doi: 10.1016/0263-7855(93)87010-3. [DOI] [PubMed] [Google Scholar]
- 54.Nakamura H, Nishida S. Numerical calculations of electrostatic potentials of protein-solvent systems by the self consistent boundary method. J Phys Soc Jpn. 1987;56:1609–1622. [Google Scholar]
- 55.Fischer D, Lin SL, Wolfson HL, Nussinov R. A geometry-based suite of molecular docking processes. J Mol Biol. 1995;248:459–477. doi: 10.1016/s0022-2836(95)80063-8. [DOI] [PubMed] [Google Scholar]
- 56.Wolfson HJ, Nussinov R. From computer vision to protein structure and association. In: Salzberg S, Searls DB, Kasif S, editors. Computational methods in molecular biology. Amsterdam, Netherlands: Elsevier Science B.V; 1999. pp. 313–334. [Google Scholar]
- 57.Wolfson HJ, Rigoutsos I. Geometric hashing: an overview. Comp Sci Eng IEEE. 1997;4:10–21. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.