

Abstract
Many applications in biotechnology require human proteins generated from human cells. Stable cell lines commonly used for this purpose are difficult to develop, and scaling to large numbers of proteins can be problematic. Transient expression can circumvent this problem, but protein yields are generally too low for most applications. Here we report a novel 37-nucleotide leader sequence that promotes rapid and high transgene expression in mammalian cells. This sequence was identified by in vitro selection and functions in a transient vaccinia-based cytoplasmic expression system. Vectors containing this sequence produce microgram levels of protein in just 6 h from a small-scale expression in 106 cells. This level of protein synthesis is ideal for high throughput production of human proteins, and could be scaled to generate milligram quantities of protein. The technology is compatible with a broad range of cell lines, accepts plasmid and linear DNA, and functions with viruses that are approved for use under BSL1 conditions. We suggest that these advantages provide a powerful method for generating human protein in mammalian cells.
Keywords: mammalian cell culture, recombinant protein synthesis, human vaccinia virus, translation enhancing elements
Introduction
The synthesis of human proteins in human cells is necessary when properly modified protein is needed for biomedical assays.1–4 This requires developing stable cell lines or engineered viruses,2 which is technically challenging, because it requires integrating a foreign gene of interest into the genome of the host cell or virus.5,6 Even when properly constructed, stable cell lines are prone to contamination by viruses and microorganisms present in the laboratory environment. Consequently, human proteins are often synthesized in prokaryotic systems, even though these systems lack the capacity to produce post-translational modifications.7
Here, we describe a novel 37-nucleotide RNA sequence that promotes strong protein synthesis in a vaccinia virus (VACV)-based cytoplasmic expression system. This system is ideal because of its activity in a broad range of mammalian cell lines, high expression capacity, and rapid timeframe.8 Biochemical analysis of our novel leader sequence reveals an unusual dual activity that leads to enhanced expression and translation. As a proof-of-concept, we show that 12 arbitrarily chosen human proteins express without the need for optimization, suggesting a straightforward method for generating human proteins in human cells.
Results and Discussion
Discovery and characterization of a novel translation enhancing element
In a previous in vitro selection experiment, we isolated translation enhancing elements (TEEs) from the human genome.9 The selected TEEs were evaluated in a VACV cytoplasmic expression system (Fig. 1), and found to enhance translation by up to 100-fold when compared with unselected sequences from the naïve library or a traditional VACV synthetic late promoter (SLP) alone. Subsequent screening led us to identify one sequence, hTEE-658, with unusually high activity in our VACV system [Fig. 2(A)]. Comparative studies showed that hTEE-658 enhances translation more than 5,000-fold over a standard SLP VACV promoter. This observation suggested a possible strategy for increasing protein synthesis levels in mammalian cells.
Figure 1.
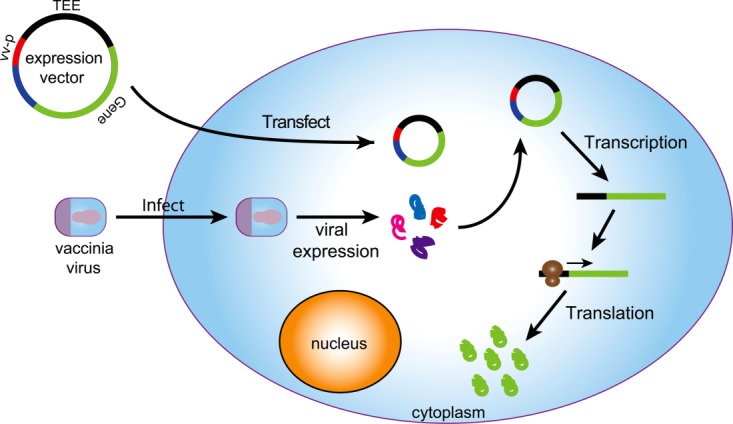
Vaccinia-based cytoplasmic expression of recombinant genes in mammalian cells. Cells transfected with a viral protein expression vector are infected with the vaccinia virus. Infected cells produce a viral RNA polymerase that recognizes a viral promoter in the protein expression vector and mediates the cytoplasmic transcription of gene-encoded RNA messages. Expressed mRNAs are translated using the translational machinery present inside the cell.
Figure 2.
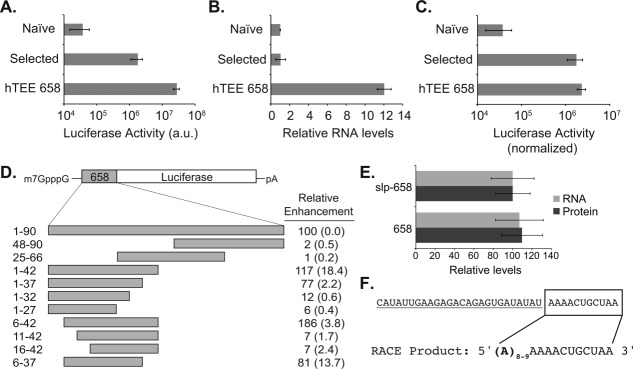
Functional characterization of hTEE-658. (A) Luciferase production driven by hTEE-658 compared to the average of 9 in vitro selected human TEEs and four randomly chosen human sequences from a naïve library. Results from the naïve library are equivalent to the SLP promoter alone. (B) Luciferase mRNA levels determined by qRT-PCR after normalization to HPRT. (C) Luciferase activity normalized to cellular mRNA. (D) Reporter constructs containing 5′ and 3′ deletions were used to identify the core functional domain of hTEE-658. Labels indicate the precise nucleotide fragment analyzed in vaccinia-infected cells. Relative enhancement is given as a percentage of full-length hTEE-658 with normalized percent error shown in parenthesis. (E) Luciferase mRNA and protein levels observed for vectors carrying and lacking the vaccinia SLP promoter upstream of hTEE-658. (F) 5′ RACE analysis was used to identify the viral promoter region (underlined) and ribosomal TEE (boxed) within the core functional region of hTEE-658.
To understand the function of hTEE-658, we used quantitative real-time PCR (qRT-PCR) to measure RNA levels from cells transfected with a luciferase reporter plasmid containing sequences from the naïve library, selection output, and hTEE-658. After normalization, the hTEE-658 plasmid produces ∼10-fold more RNA and leads to ∼5-fold more luciferase than the most active sequence previously identified from our selection [Fig. 2(B,C)]. We confirmed by qRT-PCR that plasmid copy number was not altered [Supporting Information Fig. S1], demonstrating that stronger mRNA expression and translation was not due to differences in plasmid replication by the virus. These results indicate that hTEE-658 enhances transcription and translation levels in the cell. The observation that a single sequence can affect both steps of protein synthesis is unusual, but not unprecedented. We are aware of at least one other RNA motif, the TISU element, which functions in this capacity.10
Next, we determined the minimal region required to achieve strong gene expression. A set of hTEE-658 variants were generated by first separating the parent sequence into the 5′ half, 3′ half, and central portion, which revealed that the functional region resided in the 5′ portion of the parent sequence [Fig. 2(D), Supporting Information Table S1]. We then performed an incremental deletion analysis on the 5′ half to identify the minimal sequence necessary for function. Sequential deletions from the 5′ and 3′ ends allowed us to identify a core functional region of 37-nts spanning a boundary from residues 6–42. This region is ∼2-fold more active than the full-length sequence and additional deletions that extend into either end led to significant drops in luciferase activity [Fig. 2(D)]. The remainder of our study focuses on the activity of the 37-nt core region of hTEE-658.
To verify that hTEE-658 functioned as a VACV promoter, we removed the vaccinia SLP promoter from the luciferase plasmid. Analysis of cellular RNA and luciferase activity values from vectors containing and lacking the SLP promoter showed no detectible difference in mRNA and protein levels [Fig. 2(E)], confirming that hTEE-658 functions as a VACV promoter. To discern which region of the sequence is responsible for promoter activity and which region is responsible for TEE activity, we sequenced the 5′ end of the luciferase mRNA by rapid amplification of cDNA ends (RACE). cDNA sequencing indicated that transcription initiated within the AAAACUGCUAA portion of the sequence, which was preceded by a stretch of 8 or 9 non-templated adenosine residues [Fig. 2(F)]. We anticipated the presence of short polyA ends since VACV encodes strong poly-adenylation enzymes that modify the 5′ and 3′ ends of primary transcripts.11 This analysis suggests that the first 26 nucleotides of hTEE-658 function as a VACV promoter, while the last 11 nucleotides function as a TEE.
Comparison of protein synthesis to known VACV promoters
We established the activity of hTEE-658 relative to known VACV promoters using viral vectors that contain the SLP and I1L promoters alone and in combination with hTEE-658 (Supporting Information Table S2). Vectors designed to express the luciferase and HIV-1 gag genes were tested in our cytoplasmic expression assay. After 6 h of expression, protein abundance was detected by western blot analysis using antigen specific antibodies. Analysis of the resulting gel indicates that vectors carrying hTEE-658, either alone or in tandem with SLP and I1L, produce substantial amounts of luciferase or HIV gag when compared with vectors containing only the SLP and I1L promoters alone (Fig. 3). This result is consistent with our quantitative luciferase measurements.
Figure 3.
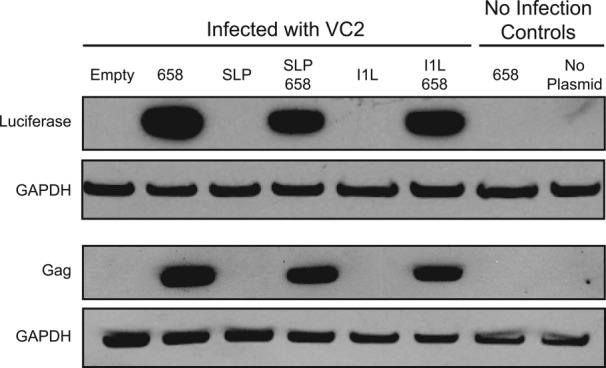
Western blot analysis confirms that hTEE-658 is a strong VACV promoter. Luciferase and HIV Gag proteins were produced in HeLa cells from vectors carrying hTEE-658, SLP, I1L or a combination of hTEE-658 in tandem with SLP or I1L. Western blot analysis was performed using antibodies directed against luciferase and HIV Gag proteins. GAPDH was used as a loading control. Empty refers to cells that were infected, but not transfected. No infection controls confirm that protein synthesis is VACV-driven. SLP and I1L protein is visible after prolonged exposure (data not shown).
Time course analysis of protein synthesis in mammalian cells
Next, we evaluated cell line and viral strain compatibility by measuring luciferase production in three different cell types using three different viral strains. In this case, HeLa, HEK, and BHK cells were chosen for analysis with the VACV strains VC2, vTF7-3, and MVA. VC2 is a wild-type Copenhagen strain, while vTF7-3 is an engineered VACV designed to express the T7 RNA polymerase.12 MVA is a highly attenuated VACV that is non-pathogenic to humans and compatible with biosafety level 1 (BSL1) conditions.13 Plasmids carrying an internal ribosomal entry site (IRES) from the encephalomyocarditis virus (EMCV), in combination with a T7 or SLP promoter, were used as controls. The EMCV IRES is a ∼500-nt noncoding RNA motif that is commonly used for protein synthesis in mammalian cells.14
Time-dependent measurements were collected over the course of 24 h. In nearly all cases, hTEE-658 proved superior to the EMCV IRES with luciferase expression following a general trend of early rapid expression that plateaued after 6–9 h (Supporting Information Fig. S2). While expression from the EMCV plasmid followed a similar trend, this plasmid generally required longer expression times and produced less overall protein (∼10-fold). In only two cases were the hTEE-658 and EMCV plasmids similar; however, this required the engineered VACV strain vTF7-3, an efficient virus optimized for EMCV. Among the three cell lines, BHK cells consistently produced the highest levels of luciferase, consistent with previous VACV expression results.15 These findings indicate that hTEE-658 vectors produce significant quantities of protein in a time frame competitive with most prokaryotic expression systems.
Broad antigen expression
To demonstrate the potential for broad protein synthesis, 12 human proteins of different sizes and functional categories were arbitrarily chosen for analysis (Supporting Information Table S3). In all cases, the gene encoding sequence was inserted into an expression vector containing hTEE-658 upstream of the coding region, and protein production levels were monitored after expression in HeLa cells using a common c-Myc epitope tag. Western blot analysis of cell lysates indicated that full-length proteins were obtained in all cases [Fig. 4(A)]. This result is important given the approximate 10-fold range in protein sizes. The ability of hTEE-658 to mediate the production of such a variety of proteins from a plasmid expression system conveys a significant advantage over prokaryotic and cell-free expression systems, where success rates for human proteins are highly variable and typically less than 50%.16 For example, we have found that six of the 12 human proteins analyzed above (PI3K, SRC, P53, MYOT, HADH, and HRAS) are undetectable or barely detectable in a coomassie stained gel after expression in E. coli (data not shown).
Figure 4.
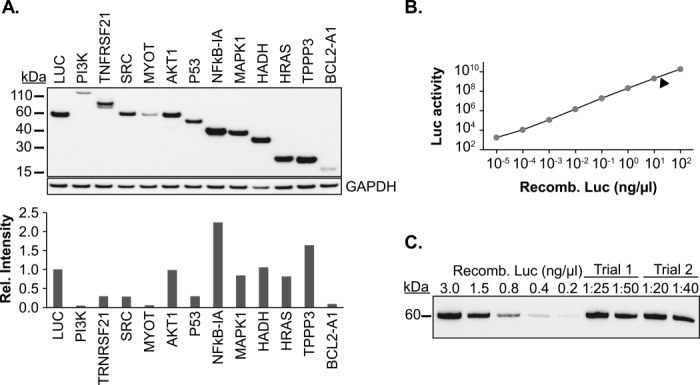
Synthesis of 12 human proteins in HeLa cells. (A) Twelve recombinant human proteins were generated from protein expression vectors engineered with hTEE-658. C-terminal myc-epitope tags were used to compare protein levels by Western blot analysis. Relative protein synthesis levels were determined by densitometry. (B) Quantification of luciferase production using a luciferase activity curve. The arrow indicates the average amount (20–50 ng/μL) of luciferase generated from 106 HeLa cells. This corresponds to 2–5 μg of total protein. (C) Western blot analysis showed strong concordance between the luciferase activity assay and protein synthesis levels from two independent trials. Protein samples were diluted to fit to the linear range of the Western blot.
Protein quantification
Two different assays were used to quantify protein production in our expression system. First, luciferase enzyme generated from hTEE-658-mediated expression in HeLa cells was quantified by linear calibration using known amounts of commercial recombinant luciferase to measure enzymatic activity [Fig. 4(B)]. Second, Western blot analysis was performed using the same protein standards and anti-luciferase antibody to measure protein production [Fig. 4(C)]. Both methods gave similar results, yielding 2–5 μg of luciferase protein from 106 HeLa cells. This result indicates that all or nearly all of the luciferase protein was properly folded and enzymatically active. Comparison of the luciferase levels to the 12 human proteins observed in the western blot indicates that protein expression levels ranged from 0.1 to 2-fold, with NFkB-IA showing the highest levels of expression. These results suggest that this transient cytoplasmic expression protocol could produce milligram quantities of protein by scaling the expression to 109 cells.
Protein synthesis from linear DNA
To further simplify our expression system, linear DNA was assayed for activity in the cytoplasmic expression assay. Overlap PCR was used to add the hTEE-658 sequence and the c-Myc tag to our set of 12 human proteins. The linear DNA was transfected into HeLa cells and protein levels were analyzed by western blot after overnight expression. Analysis of the cell lysates revealed the presence of all 12 full-length human proteins [Supporting Information Fig. S3]. Only one protein, TNFRSF21, showed a truncated product that was presumably due to incomplete translation. Quantification of protein levels using the luciferase activity assay indicates that linear DNA produces ∼10-fold less protein than plasmid DNA. Nevertheless, the simplicity of this approach makes it an attractive method for generating smaller amounts of protein for a large number of targets.
Applications in mammalian cell culture
The ability to produce significant quantities of human protein in mammalian cells without the need for stable cell lines or recombinant viruses is a major advantage of our translation enhancing technology. This advance is based on the discovery of hTEE-658 as a short genetic sequence capable of rapid and high transgene expression in a VACV cytoplasmic expression system. Relative to common IRESs, like EMCV, hTEE-658 is substantially shorter (37 vs. >500 nts), making it easy to engineer into vectors. hTEE-658 is also more effective than EMCV at engaging the ribosomal machinery, and functions with viruses that are non-pathogenic to humans. We suggest that this new technology provides a versatile platform for protein synthesis in mammalian cells. This could be especially useful in cases where prokaryotic and cell-free systems fail to produce protein or when post-translationally modified protein is needed for biological analysis. While further optimization, could lead to higher yields, the system is already ideal for routine protein synthesis.
Methods
Cell culture
All cells used in this study were obtained from the American Type Culture Collection (ATCC). HeLa and HEK293 cells were maintained in DMEM (Invitrogen), while BHK cells were maintained in MEM (Invitrogen). Media was supplemented with 5% fetal bovine serum (FBS, HyClone) and 5 mg/mL gentamicin (Invitrogen). Cells were kept at 37°C in a humidified atmosphere containing 5% CO2.
Vaccinia virus strains
The vaccinia virus Copenhagen (VC2) and vTF7-3 viral strains were obtained from Virogenetics and ATCC, respectively. The modified vaccinia virus Ankara (MVA) was obtained from Dr. Bernard Moss at the National Institute of Allergy and Infectious Diseases. VC2 is considered a wild-type vaccinia virus, MVA is an attenuated vaccinia virus strain that is non-pathogenic in humans, and vTF7-3 is a recombinant vaccinia virus strain derived from the Western Reserve (WR) strain that has been engineered to express T7 RNA polymerase. Viral stocks were stored in MEM with 2% FBS.
Cytoplasmic expression system
Cells were seeded 18 h before transfection according to Supporting Information Table S4. Transfections were carried out using Lipofectamine 2000 (Invitrogen). In brief, complexes containing either plasmid or linear DNA and Lipofectamine 2000 were formed in Opti-MEM (Invitrogen). During complex formation, culture media was removed from the cells and replaced with fresh Opti-MEM. Complexes were then carefully overlaid onto the cells. Plasmid DNA was obtained by standard mini or maxiprep (Qiagen), while linear DNA templates were generated by high fidelity PCR (accuprime taq, Invitrogen) using expression vectors as templates. Primers were designed so that the product included a T7 promoter, hTEE-658 core, gene of interest, c-Myc tag, and poly-adenosine track. Immediately following DNA transfections, cells were infected with VC2, MVA, or vTF7-3 at a multiplicity of infection (moi) of five plaque forming units (PFU)/cell for all 6 or 18 h assays and 30 PFU/cell for 24-h time course assays.
Luciferase activity assay
Post-transfect-infect cells were lysed using passive lysis buffer (Promega). Luciferase activity was determined by mixing a portion of the lysate with the Promega Luciferase Assay System and measuring light production with a Glomax microplate luminometer (Promega). Luciferase concentration was quantified by comparison to a standard curve of QuantiLum Recombinant luciferase (Promega) generated using the manufacturer's recommended protocol.
RNA characterization
RNA was isolated from HeLa cells 6-h post-infection. Lysate from 2-wells of a 96-well plate was pooled and RNA isolation was performed using the PerfectPure RNA cultured cell kit (5′) following the manufacturer's protocol. Isolated RNA was reverse transcribed with Superscript II (Invitrogen) using an oligo (dT) primer. Quantitative real-time PCR (iQ™ SYBR® Green Supermix, Bio-Rad) was used to measure luciferase mRNA levels, which were normalized to the housekeeping gene hypoxanthine-guanine phospho-ribosyltransferase (HPRT) using the ΔΔCt method.
End-mapping deletion analysis
To determine the core functional region of the 658 sequence, constructs were designed where various amounts of either the 5′ or 3′ end were removed. Each construct was built by Klenow extension of synthetic DNA oligos containing the desired fragment of hTEE-658 along with BamHI and NcoI restriction sites. The double-stranded DNA was restriction digested and ligated into a monocistronic firefly luciferase reporter plasmid carrying a vaccinia virus SLP upstream of the insert. Reporter plasmids containing truncated variants were assayed for activity by transfect-infect assay.
Expression vectors
Expression plasmids were obtained by engineering a monocistronic reporter vector with a leader sequence of interest inserted into the 5′ UTR. This vector contains a T7 RNA polymerase promoter site, a 5′ UTR which directly precedes an ORF containing the firefly luciferase gene followed by a poly-adenosine track. In order to test the expression of additional proteins, the luciferase was replaced with either HIV-1 Gag (a kind gift of Dr. Ralf Wagner of the University of Regensburg) or one of 12 human genes obtained from the DNASU Plasmid Repository (DNASU.asu.edu). A c-Myc tag was also inserted at the 3′ end of the human gene constructs to be used as an epitope tag for Western blotting. The full list of human genes is located in Supporting Information Table S3.
Western blotting
Proteins were expressed using the transfect-infect assay described above. After expression, HeLa cells were lysed with Passive Lysis Buffer (Promega) and cellular debris was removed by centrifugation. For protein analysis, samples were diluted with NuPage 4× LDS sample buffer (Invitrogen) and proteins were denatured by heating for 10 min at 95°C before being run on a NuPage 4–12% Bis-Tris gel (Invitrogen). Proteins were transferred to a nitrocellulose membrane using the iBlot Gel Transfer system (Invitrogen). After blocking for 1 h at 24°C in TBS-T (20 mM Tris, 125 mM NaCl, pH 7.5, and 0.05% Tween20) supplemented with 3% milk, the membrane was incubated with the appropriate primary antibody concentrations overnight at 4°C. Membranes were then incubated with appropriate concentrations of goat anti-mouse or goat anti-rabbit HRP conjugated secondary antibodies (Cell Signaling) for 1 h at room temperature. Chemiluminescent signal was visualized after reaction with SuperSignal West Pico or Dura Chemiluminescent Substrate (Pierce Biotechnology). Anti-luciferase antibody was obtained from AbDSerotec, anti-GAPDH from Abcam, anti-Myc Tag (clone 4A6) from Millipore and the HIV-1 Gag antibody was generously provided by Dr. Hohne at the Charite Institute for Biochemie in Berlin, Germany. Where possible, membranes were cut to immunoblot for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and proteins of interest separately. Alternatively, after the proteins of interest were detected the blots were stripped by incubating three times for 10 min with 0.2M glycine, 0.1% SDS, 2% Tween20, pH 2.2. After stripping, blots were washed twice for 10 min with phosphate-buffered saline (PBS), twice for 5 min with TBS-T and then placed back into block solution for 1 h before immunoblotting for GAPDH. Western blot signals were quantified using ImageJ to determine the relative intensity for bands of interest. Known quantities of QuantiLum Recombinant luciferase (Promega) were run as a standard curve to enable quantification of luciferase protein produced by transfect-infect assay.
RACE
RNA was isolated using the PerfectPure RNA cultured cell kit (5 Prime). RACE was performed with the 5′ RLM-RACE kit (Invitrogen) using total RNA following the small reaction protocol provided by the manufacturer with primers specific to the luciferase gene. RACE sequences were ligated into the pJET 1.2 vector (Fermentas), cloned, and sequenced at the ASU DNA Sequencing Facility.
DNA isolation and real-time PCR
Cellular and plasmid DNA was isolated from transfected HeLa cells 6-h post-infection with VC2 using the Trizol Reagent (Invitrogen) according to the manufacturer's protocol. Following isolation, DNA was ethanol precipitated and re-suspended in water. Quantitative real-time PCR (iQ™ SYBR® Green Supermix, Bio-Rad) was used to determine the levels of plasmid DNA as well as the housekeeping gene Ribonuclease P (RNase P) and normalized using the ΔΔCt method.
Acknowledgments
The authors thank Dr. Ralf Wagner of the University of Regensburg for kindly providing the vector from which the HIV-1 gag gene was extracted; and members of the Chaput laboratory for helpful discussions and comments on the article.
Glossary
- BSL
biosafety level
- EMCV
encephalomyocarditis virus
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- HPRT
hypoxanthine-guanine phospho-ribosyltransferase
- IRES
internal ribosomal entry site
- MVA
modified vaccinia virus Ankara
- PBS
phosphate-buffered saline
- PFU
plaque forming units
- qRT-PCR
quantitative real-time PCR
- RACE
rapid amplification of cDNA ends
- SLP
synthetic late promoter
- TEE
translation enhancing element
- WR
Western Reserve
Supplementary material
Additional Supporting Information may be found in the online version of this article.
References
- 1.Zhu J. Mammalian cell protein expression for biopharmaceutical production. Biotechnol Adv. 2012;30:1158–1170. doi: 10.1016/j.biotechadv.2011.08.022. [DOI] [PubMed] [Google Scholar]
- 2.Wurm FM. Production of recombinant protein therapeutics in cultivated mammalian cells. Nat Biotechnol. 2004;22:1393–1398. doi: 10.1038/nbt1026. [DOI] [PubMed] [Google Scholar]
- 3.Schmidt FR. Recombinant expression systems in the pharmaceutical industry. Appl Microbiol Biotechnol. 2004;65:363–372. doi: 10.1007/s00253-004-1656-9. [DOI] [PubMed] [Google Scholar]
- 4.Swiech K, Picanco-Castro V, Covas DT. Human cells: new platform for recombinant therapeutic protein production. Protein Expr Purif. 2012;84:147–153. doi: 10.1016/j.pep.2012.04.023. [DOI] [PubMed] [Google Scholar]
- 5.Colosimo A, Goncz KK, Holmes AR, Kunzelmann K, Novelli G, Malone RW, Bennett MJ, Gruenert DC. Transfer and expression of foreign genes in mammalian cells. BioTechniques. 2000;29:314–331. doi: 10.2144/00292rv01. [DOI] [PubMed] [Google Scholar]
- 6.Yin J, Li G, Ren X, Herrler G. Select what you need: a comparative evaluation of the advantages and limitations of frequently used expression systems for foreign genes. J Biotechnol. 2007;127:335–347. doi: 10.1016/j.jbiotec.2006.07.012. [DOI] [PubMed] [Google Scholar]
- 7.Demain AL, Vaishnav P. Production of recombinant proteins by microbes and higher organisms. Biotechnol Adv. 2009;27:297–306. doi: 10.1016/j.biotechadv.2009.01.008. [DOI] [PubMed] [Google Scholar]
- 8.Jacobs BL, Langland JO, Kibler KV, Denzler KL, White SD, Holechek SA, Wong S, Huynh T, Baskin CR. Vaccinia virus vaccines: past, present and future. Antiviral Res. 2009;84:1–13. doi: 10.1016/j.antiviral.2009.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wellensiek BP, Larsen AC, Stephens B, Kukurba K, Waern K, Briones N, Liu L, Snyder M, Jacobs BL, Kumar S, Chaput JC. Genome-wide profiling of human cap-independent translation enhancing elements. Nat Methods. 2013;10:747–750. doi: 10.1038/nmeth.2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Elfakess R, Dikstein R. A translation initiation element specific to mRNAs with very short 5′UTR that also regulates transcription. PLoS One. 2008;3:e3094. doi: 10.1371/journal.pone.0003094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schwer B, Visca P, Vos JC, Stunnenberg HG. Discontinuous transcription or RNA processing of vaccinia virus late messengers results in a 5′ poly(A) leader. Cell. 1987;50:163–169. doi: 10.1016/0092-8674(87)90212-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fuerst TR, Niles EG, Studier FW, Moss B. Eukaryotic transient-expression system based on recombinant vaccinia virus that synthesizes bacteriophage T7 RNA polymerase. Proc Natl Acad Sci USA. 1986;83:8122–8126. doi: 10.1073/pnas.83.21.8122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hebben M, Brants J, Birck C, Samama JP, Wasylyk B, Spehner D, Pradeau K, Domi A, Moss B, Schultz P, Drillien R. High level protein expression in mammalian cells using a safe viral vector: modified vaccinia virus Ankara. Protein Expr Purif. 2007;56:269–278. doi: 10.1016/j.pep.2007.08.003. [DOI] [PubMed] [Google Scholar]
- 14.Elroy-Stein O, Fuerst TR, Moss B. Cap-independent translation of mRNA conferred by encephalomyocarditis virus 5′ sequence improves the performance of the vaccinia virus/bacteriophage T7 hybrid expression system. Proc Natl Acad Sci USA. 1989;86:6126–6130. doi: 10.1073/pnas.86.16.6126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Drexler I, Heller K, Wahren B, Erfle V, Sutter G. Highly attenuated modified vaccinia virus Ankara replicates in baby hamster kidney cells, a potential host for virus propagation, but not in various human transformed and primary cells. J Gen Virol. 1998;79:347–352. doi: 10.1099/0022-1317-79-2-347. [DOI] [PubMed] [Google Scholar]
- 16.Büssow K, Scheich C, Sievert V, Harttig U, Schultz J, Simon B, Bork P, Lehrach H, Heinemann U. Structural genomics of human proteins—target selection and generation of a public catalogue of expression clones. Microbial Cell Factories. 2005;4:21. doi: 10.1186/1475-2859-4-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.