

Abstract
The obligate intracellular, gram-negative bacterium Rickettsia is the causative agent of spotted fevers and typhus in humans. Surface cell antigen (sca) proteins surround these bacteria. We recently reported the co-localization of one of these proteins, sca4, with vinculin in cells at sites of focal adhesions and demonstrated that two vinculin binding sites directed the sca4/vinculin interaction. Here we report the 2.2 Å crystal structure of the conserved N-terminal 38 kDa domain of sca4 from Rickettsia rickettsii. The structure reveals two subdomains. The first is an all-helical domain that is folded in a fashion similar to the dimeric assembly chaperone for rubisco, namely RbcX. The following and highly conserved β-strand domain lacks significant structural similarity with other known structures and to the best of our knowledge represents a new protein fold.
Keywords: Rickettsia rickettsia, vinculin, X-ray crystallography, novel fold
Introduction
Rickettsia species are strict intracellular gram-negative parasites that are transmitted to humans via arthropods. They are categorized into two groups depending on their antigenic characteristics: the spotted fever versus the typhus group. Rickettsia rickettsii belongs to the spotted fever group and causes one of the most severe diseases, Rocky Mountain spotted fever.1
Intracellular pathogenic bacteria (like Salmonella, Shigella, Listeria, and Rickettsia species) have developed various strategies for infecting the host cell and for cell-to-cell dissemination.2 For some, bacterial components function as structural mimics of normal ligand-receptor interactions that promote the bacterial adhesion and invasion of the host cell. For example, OmpB binds to the host cell Ku70 receptor to mediate the internalization of Rickettsia conorii.3,4 In the case of Yersinia, the outer surface protein invasin binds to β1 integrins on target cells and triggers phagocytic uptake by host cell.5 Finally, Listeria uses a protein called internalin, which binds to the adhesion molecule E-cadherin to promote entry into human epithelial cells.6
Once pathogens enter the host cell, they co-opt host cell actin filaments for motility, where the force generated by actin assembly propels bacteria through the cytoplasm and into neighboring cells.7–9 Actin assembly of Listeria and Rickettsia is mediated by the expression of ActA10 and RickA,11,12 respectively. These two proteins are structurally similar to WASP (Wiskott–Aldrich Syndrome Protein) family proteins that activate the Arp2/3 complex that nucleates and directs actin polymerization.13–15 On the other hand, Shigella expresses a surface protein called IcsA that recruits host N-WASP proteins to activate Arp2/3.16
In addition to WASP and the Arp2/3 complex, the cytoskeletal actin binding proteins VASP, α-actinin, cofilin, ezrin, and CapZ are found associated with intracellular pathogens.10 These findings suggest that intracellular pathogens may also co-opt a number of host proteins for their life cycle within the host.7,17,18 We have previously shown that Rickettsia cell surface antigen 4 (sca4) has two vinculin binding sites (VBSs) that are capable of activating vinculin,19 which provides essential links for cell–cell and focal adhesion complexes to the actin cytoskeleton.20,21 Our biochemical and structural studies have shown that full-length sca4 is sufficient to activate vinculin and identified unique features of the sca4/vinculin interaction. Finally, we showed that sca4 and vinculin co-localize at focal adhesions in cells.
To further define the functions of sca4, we determined the crystal structure of the N-terminal region of sca4, which is revealed to harbor two subdomains. The first subdomain is an all-helical domain that has features similar to a known chaperone, yet the other largely β-strand domain appears to represent a new protein fold in biology.
Results
The crystal structure of the amino-terminal domains of sca4
Secondary structure predictions using the Phyre server suggested that the N-terminal 20 residues of sca4 were unstructured (Supporting Information Fig. S1). We therefore focused our efforts on crystallizing the truncated N-terminal domain, residues 21–360 that harbors over 68% sequence identity across Rickettsia (Supporting Information Fig. S1). This domain crystallized in space group P65 with two polypeptide chains in the asymmetric unit and the structure was solved by single wavelength anomalous dispersion to 2.2 Å resolution. The final model contains residues 49–58 and 74–360 for chain A and residues 49–58, 63–72, and 79–359 for chain B. The final crystallographic and free R-factors are 0.18 and 0.216, respectively (Table I). The Molprobity23 score is in the 100th percentile with over 98% of all residues in the favored region of the Ramachandran plot. The two subunits in the asymmetric unit are very similar and can be superimposed with r.m.s.d. of 0.34 Å for 1,140 atoms. The most notable differences are in one loop region (residues 194–200), near the C-terminus of this portion of sca4, as well as the disordered region between residues 59 and 78.
Table I.
X-ray Data Reduction and Crystallographic Refinement Statistics
| Parameter | Native | SeMet |
|---|---|---|
| X-ray data reduction statistics | ||
| Space group | P65 | P65 |
| Unit cell dimensions | ||
| a, b | 98.2 Å | 98.2 Å |
| c | 155.5 Å | 156.3 Å |
| Resolution | 50 Å–2.20 Å | 50 Å–2.81 Å |
| Last shell | 2.26 Å–2.20 Å | 2.86 Å–2.81 Å |
| Total measurements | 145,037 | 134,973 |
| Number of unique reflections | 40,961 | 20,949 |
| Last shell | 3,135 | 1,039 |
| Wavelength | 0.9795 Å | 0.9795 Å |
| R-pima | 0.05 | 0.075 |
| Last shell | 0.37 | 0.383 |
| I/σ(I) | 16 | 12 |
| Last shell | 3.2 | 2.5 |
| Completeness | 0.946 | 1.0 |
| Last shell | 0.925 | 0.999 |
| Multiplicity | 3.5 | 6.4 |
| Last shell | 3.6 | 6.5 |
| Crystallographic refinement statistics | ||
| Resolution | 50 Å–2.20 Å | |
| Last shell | 2.26 Å–2.20 Å | |
| No. of reflections (working set) | 38,910 | |
| No. of reflections (test set) | 2,050 | |
| R-factorb | 0.178 | |
| Last shell | 0.221 | |
| R-freec | 0.214 | |
| Last shell | 0.268 | |
| No. of amino acid residues | 598 | |
| No. of atoms | 4,640 | |
| No. of solvent molecules | 259 | |
| Average B-factor | ||
| Protein | 37 Å2 | |
| Solvent | 44 Å2 | |
| R.m.s.d. (root-mean-square deviation) from ideal geometry | ||
| Bond lengths | 0.01 Å | |
| Bond angles | 1.08° | |
R-pim is the precision-indicating R-factor, which is related to the tradition R-merge (R-sym) but provides a better estimate of the data quality.22
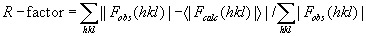 where the R-factor represents the relative disagreement between the observed structure factor amplitudes (Fobs) and the expectation values of the calculated structure factor amplitudes (<|Fcalc|>) under the conditional probability distribution on which the target likelihood function is based.
where the R-factor represents the relative disagreement between the observed structure factor amplitudes (Fobs) and the expectation values of the calculated structure factor amplitudes (<|Fcalc|>) under the conditional probability distribution on which the target likelihood function is based.
The free R-factor is a cross-validation residual calculated by using 5% reflections, which were randomly chosen and excluded from the refinement.
The N-terminus of sca4 is comprised of two distinct domains [Fig. 1(A–C)] that share 50% sequence identity across Rickettsia for residues 82–178 and 89% for residues 200–352 (Supporting Information Fig. S1). Residues 82–178 form an unusual helix bundle, where α-helices α1 (residues 84–111) and α2 (residues 115–131) are loosely packed in an up–down conformation and have inter-helical hydrophobic interactions without authentic coiled-coil packing. Inter-helical interactions are mediated by residues Ile102, Leu106, Leu109, Lys121, Ile125, and Lys129, and there is a steep inter-helical angle of ∼160° with only three residues forming the turn [Fig. 1(B)]. A four-residue turn connects α-helix α2 to a 310-helix (residues 136-140), where Phe138 and Phe139 engage in hydrophobic interactions with α-helices α1 (Lys98, Leu101, Ile102, and Ile105), α2 (Ile125), and α3 (residue Ile148). Residues 142–153 on α-helix α3 lie almost perpendicular to α-helices α1 and α2, where the Phe147 aromatic ring contributes to the hydrophobic interface by interacting with Leu112 and Leu109, and with Leu117 and Lys121, which reside on α-helices α1 and α2, respectively. Further, Lys154 and Asp108 engage in electrostatic interactions and this may stabilize the α3–α4 loop region. Finally, α-helix α4 is comprised of residues 155–176 and is anti-parallel to α-helix α1. These two helices have several hydrophobic interactions (involving Ile85, Ala88, Val89, Ile93, Gln97, and Leu101 with Leu157, Leu161, Ile164, Tyr169, Val172, and Phe176) as well as a hydrogen bond (Glu165–Gln97).
Figure 1.
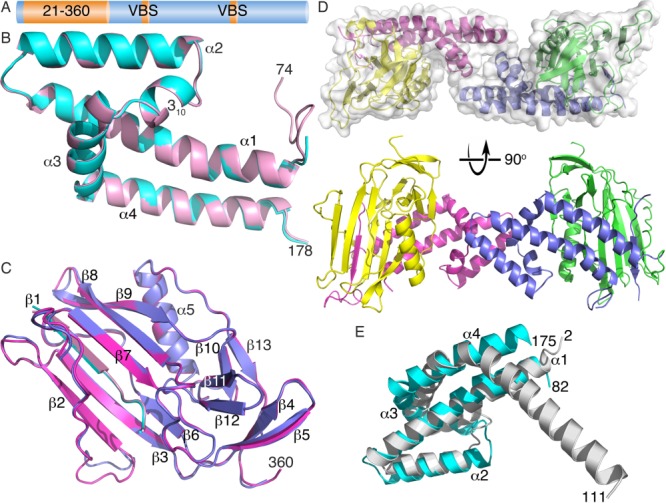
Crystal structure of the amino-terminal sca4 domains. (A) Schematic of the sca4 domain structure. Full-length sca4 (1,024 residues) harbors an N-terminal conserved 38 kDa domain (residues 21–360) as well as two vinculin binding sites (VBS), residues 413–431 and 814–832, respectively. Sequence identities across Rickettsia are as follows: 47% for residues 5–21, 68% for 21–360, 89% for 200–352, 89% for VBS 413–431, 53% for VBS 814–832, and 58% for 360–1,024. (B) Cartoon drawing of a superposition of the two helical domains (residues 74–178) in the asymmetric unit colored in cyan and pink for the two subunits. Secondary structural elements (α1–α2, 310-helix, α3–α4) are indicated. (C) Superposition of the two β-sheet subdomain of the N-terminus of sca4 in the asymmetric unit (colored in blue and magenta, respectively) Secondary structure elements (β1 from the helical domain shown in pink and cyan and β2–β12, α5, and β13 for the subsequent β-sheet domain) are labeled. (D) Orthogonal views (top, along the ∼160° rotation axis; bottom, the ∼160° rotation axis is vertical) of the dimer in the asymmetric unit (residues 49–178 are drawn in magenta or blue and residues 179–360 in yellow or green, respectively). (E) The helical domain has structural similarity to RbcX, the assembly chaperone for rubisco. Superposition of RbcX (residues 2–111, grey, PDB entry 2pei) onto sca4 (residues 82–175, cyan) shows that this sca4 domain has a fold similar to that of RbcX. The sca4 α-helices and all termini are labeled. Note that sca4 α-helices α1 and α2 are structurally similar with RbcX, and α-helix α3 of RbcX occupies a similar location as the sca4 310-helix. Sca4 α-helices α3 and α4 are replaced by the long RbcX α-helix α4 that has a ∼60° kink not seen in sca4.
Notably, the second domain is a β-strand domain (residues 200–352) that starts with an anti-parallel β-sheet (β-strand β2, residues 200–206; β3, 213–221; β6, 241–244) and a twisted β-hairpin formed by the last two β-strands (β4, 226–230, and β5, 233–238). A second anti-parallel β-sheet is also formed (β1, residues 53–57; β7, 256–262; β8, 278–286; and β9, 289–297), where the last two β-strands (β8 and β9) are twisted and engage in hydrogen bonding interactions with a well-defined β-strand (β1) N-terminal to the four-helix bundle [Fig. 1(C)]. β-Strands β10 (residues 302–305) and β13 (residues 349–352) are parallel, and β-strands β11 (residues 313–317) and β12 (residues 320–323) are anti-parallel. Finally, an α-helix (α5, residues 328–341) is found before the last (β13) β-strand. Several hydrophobic (including Ile85–Phe186, Leu101–Leu348, Phe139–Val346, Leu152–Pro301) and polar (Arg90–Lys294, Asp83–His258, Arg90–Glu295, Lys98–Gln344, Glu153–Gln231, Lys158–Glu316, Tyr169–Ser297, Lys170–Asp274) interactions are manifest at the interface of the helix bundle domain and β-strand domain (Supporting Information, Fig. S2).
The two molecules in the asymmetric unit are related by an ∼162° rotation [Fig. 1 (D)], where the intermolecular interface is comprised of hydrophobic (Leu117–Pro142 and Leu117–Ala143) and polar (Asp114–Arg145 and Asp116–Arg145) interactions involving just the N-terminal helical domain (residues 49–183); no residues of the β-strand domain (residues 184–359) engage in intermolecular interactions. This results in a relatively small buried surface area of about 750 Å2. Nevertheless, the shape correlation statistic derived using the CCP4 program SC is 0.731, which seems well within the range observed for oligomers where a value of 1 indicates perfect fit while 0.35 indicated the mismatch of an artificial association.24 However, purified 38 kDa sca4 protein elutes at a position equivalent to that of a globular ∼43 kDa protein (Fig. 2) suggesting this sca4 domain is a monomer in solution.
Figure 2.
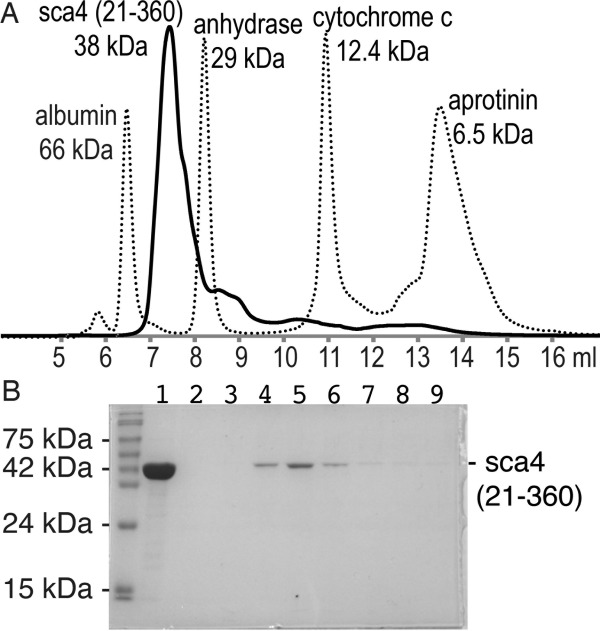
The N-terminal sca4 domain (residues 21-360) is a monomer in solution. (A) Size-exclusion chromatography profile (TSK-G2000SWxl column) of sca4 (residues 21–360). Globular protein standards (albumin, 66 kDa, Ve = 6.47 ml; carbonic anhydrase, 29 kDa, Ve = 8.21 ml; cytochrome C, 12.4 kDa, Ve = 10.93 ml; and aprotinin, 6.5 kDa, Ve = 13.49 ml; where Ve is the peak elution volume) are shown with a broken line and sca4 with a solid black line. The void volume of the column (Vo) was determined to be 5.43 mL using blue dextran (2,000 kDa; data not shown). The Ve/Vo values were used to generate a standard curve allowing the molecular mass of sca4 (residues 21–360) to be determined. The elution volume indicated that 38 kDa sca4 domain was a monomer in solution with an apparent molecular weight of 43.1 kDa (Ve = 7.38 ml). (B) Analysis of fractions from the sca4 analytical size exclusion chromatography experiment shown in panel A using 12% SDS-PAGE and Coomassie brilliant blue staining. Lane 1, purified sample that was loaded onto thecolumn; lane 2, fraction containing elution at 6.33–6.67 ml; lane 3, 6.67–7 ml; lane 4, 7–7.33 ml; lane 5, 7.33–7.67 ml; lane 6, 7.67–8 ml; lane 7, 8–8.33 ml; lane 8, 8.33–8.67 ml; lane 9, 8.67–9 ml.
Novel fold
There is some similarity of the N-terminal helical domain (residues 75–183) with a previously determined structure. Despite the low sequence homology of 28% (and a sequence identity of only 12%), RbcX, the assembly chaperone for rubisco (PDB entry 2pei),25 has structural similarity to the N-terminal domain of sca4 (Z-scores of 6.3) [Fig. 1(E)]. In particular α-helices α1 and α2 are structurally similar and α-helix α3 of RbcX occupies a similar location as the sca4 310-helix. Sca4 α-helices α3 and α4 are replaced by the long RbcX α-helix α4, which has a ∼60° kink not seen in sca4. RbcX facilitates correct folding of RbcL in cyanobacteria, and the N-terminal domain of our sca4 structure is homologous to the core structure of RbcX, which is comprised of a four-helix bundle and forms a homodimer.25 However, the intermolecular contacts and configurations of RbcX and sca4 are different: in RbcX, α-helices α1 engages in two-fold related interactions and the C-terminal α-helix α4 packs against the N-terminal α-helix α4 region of the other molecule in the asymmetric unit. In contrast, in sca4, α-helices α2 and α3 engage in interdomain contacts [Fig. 1(D)].
On the other hand, the β-strand domain of sca4 (residues 200–352) has no significant structural similarity with other known protein strctures as determined by the DALI26 server. Indeed, no structure having a Z score of greater than 6, the cutoff for structural homologs, was identified, indicating that the β-strand domain fold of sca4 is novel. The highest Z-score was 3.1, with a sequence identity of 6% for the vitamin B12 receptor structure from Escherichia coli (PDB entry 1nqe).
Discussion
We previously identified sca4 as an activator of vinculin, and shown that this occurs through the agency of VBSs that are conserved across Rickettsia19 with 79% sequence identity for residues 413–431 and 53% for residues 814–832 (Supporting Information Fig. S1). Most of the N-terminal region of sca4 is also highly conserved, in particular the second subdomain (residues 200–352). We therefore crystallized the N-terminal domain of sca4 (residues 21–360) and obtained clustered needles but these could not be improved, as the two smallest dimensions were always less than 20 μm. Nevertheless, using the microfocusing SSRL beamline 12-2 and careful dissection we succeeded in collecting a 2.2 Å native data set, as well as a 2.8 Å SAD data set from Selenomethionine-substituted crystals that arose in a different crystallization condition.
The crystal structure of this 38 kDa N-terminal domain of sca4 revealed two subdomains, a four-helix bundle domain (residues 84–176) that resembles the assembly chaperone of rubisco, RbcX, and a β-strand domain (residues 200–352) that appears to represent a new protein fold in biology, where residues 53–57 provide an additional β-strand. Given that the first subdomain resembles the RbcX chaperone, it follows that this sca4 domain may direct proper folding of this antigen in the infected host cell. Interestingly, the N-terminal region of sca4 engages in nearly two-fold interactions with the other molecule in the asymmetric unit yet these are distinct compared to those seen in the RbcX, which is a dimer in solution. Further, although the buried surface area across the dyad is rather modest, the shape correlation statistic suggests that sca4 could form dimer in solution yet size exclusion chromatography reveals that this sca4 domain is a monomer in solution. On the other hand, analytical gel filtration showed that the 108 kDa N-terminally truncated sca4 (residues 21–1,008) elutes at a position that corresponds to an apparent molecular mass of 367 kDa.19 Given that sca4 has at least two VBSs that can bind to two vinculin molecules, sca4 oligomerization would greatly amplify vinculin activation and the stabilization of adhesion-like complexes at sites of Rickettsia entry.
Materials and Methods
Cloning and protein expression and purification
Sca4 (residues 21–360) was cloned into a pET-28a vector (Novagen) and expressed in Escherichia coli BL21-DE3 (Invitrogen) to produce an N-terminal hexa-histidine-tagged protein with a thrombin cleavage site for removal of the His-tag. The expression construct was confirmed by DNA sequencing. Selenomethionine (SeMet)-labeled sca4 (21–360) protein was expressed in the methionine auxotroph E. coli B834 strain and the cells were grown using a SeMet media kit (Molecular Dimensions), according to the manufacturer's protocols. Transformed bacteria were grown at 37 °C in Luria Bertani medium containing 20 mg/L kanamycin. The bacterial cultures were induced at OD600 of ∼0.8 by adding isopropyl-1-thio-β-d-galactopyranoside to 0.5 mM and incubating at 30 °C for 24 h. Cell pellets were resuspended in lysis buffer (50 mM NaH2PO4, 300 mM NaCl, and 5 mM imidazole) and sonicated. The cell lysate was cleared by ultracentrifucation (100,000g for 30 min), loaded on a HisTrap chelating nickel affinity chromatography column (GE Healthcare), and washed with 25 column volumes of lysis buffer. The bound protein was then eluted over a gradient to 1 M imidazole (pH 8). After cleavage of the His-tag at 4 °C over night, the proteins were further purified on a Superdex-75 sizing chromatography column (Amersham) equilibrated in 20 mM Tris–HCl (pH 8) supplemented with 150 mM NaCl. Sca4 (residues 21–1,008) was prepared as described.19
Analytical size exclusion chromatography
Purified sca4 (residues 21–360) was loaded onto an TSK-G2000SWxl column (Tosoh Bioscience, Tokyo, Japan) equilibrated in 20 mM Tris–HCl (pH 8), 150 mM NaCl, and 1 mM DTT at a flow rate of 1 ml/min. The column was calibrated at 25 °C with globular protein standards that included albumin (66 kDa, Ve = 6.47 ml), carbonic anhydrase (29 kDa, Ve = 8.21 ml), cytochrome C (12.4 kDa, Ve = 10.93 ml), and aprotinin (6.5 kDa, Ve = 13.49 ml), where Ve is the peak elution volume. The void volume of the column (Vo) was obtained as 5.43 ml using blue dextran (2,000 kDa). Ve/Vo values were used to generate a standard curve and to determine the molecular mass of sca4 (residues 21–360).
Crystallization and structure determination
Native crystals were grown at 20 °C by hanging drop vapor diffusion by mixing an equal volume of the protein (117.3 mg/ml in 20 mM Tris–HCl, pH 8, 150 mM NaCl) with a reservoir solution containing 1.8M sodium malonate (pH 6) and 5% PEG 3350. A single needle-shaped crystal was frozen using mother liquor supplemented with 30% glycerol as a cryoprotectant. Native data were collected up to 2.2 Å Bragg spacings at the SSRL beamline 12-2 (wavelength of 0.9795 Å), integrated, and scaled using XDS27 and AIMLESS28 as implemented in autoPROC.29 SeMet labeled crystals were grown using hanging drop vapor diffusion at 20 °C. Drops comprised 1 μl of protein solution (110.5 mg/ml in 20 mM Tris–HCl, pH 8, 150 mM NaCl) and 1 μl of reservoir solution (0.1M MES, pH 6, and 2.2M ammonium sulfate). Cryoprotection was by successive transfers of the crystals into mother liquor supplemented with 10, 15, 20, and 30% glycerol. X-ray diffraction data of SeMet sca4 crystals were collected at the K-absorption edge to 2.81 Å Bragg spacings at the SSRL beam line 12-2 at a wavelength of 0.9795 Å and diffraction images were processed and scaled with autoPROC.29 Four of the 10 selenium sites were found by SHELXD.30 This model of the heavy-atom substructure was completed through inspection of LLG (log-likelihood gradient) maps as implemented in SHARP31 and analyzed within autoSHARP,32 leading to a total of eight sites. Phases using this heavy atom model were subjected to density-modification using SOLOMON33 and BUCCANNER34 was used to automatically build an initial model into the resulting electron density map. After initial crystallographic refinement using BUSTER35 and some initial model corrections using COOT36 the most complete polypeptide chain was used as a search model to solve the 2.2 Å native structure using MOLREP.37 Crystallographic refinement was performed using iterative rounds of BUSTER and manual rebuilding in COOT.36
Acknowledgments
We are indebted to Edith Gouin and Pascale Cossart (Institut Pasteur, France) for the full-length sca4 plasmid and to our colleagues at Scripps Florida: John Cleveland for discussions and critical review of the manuscript, Zhen Wu and Philippe Bois for sequencing, and Erumbi Rangarajan and Philippe Bois for fruitful discussions. Finally, we are grateful to the staff at the Stanford Synchrotron Radiation Laboratory (SSRL) for synchrotron support. JHL was a fellow of the American Heart Association. This is publication no. 24025 from the Scripps Research Institute. Sca4 (21–260) coordinates have been deposited in the Protein Data Bank with entry code 4lq8.
Glossary
- PDB
Protein Data Bank
- VBS
vinculin binding site
- sca
cell surface antigen
- WASP
Wiskott-Aldrich syndrome protein
Supplementary material
Additional Supporting Information may be found in the online version of this article.
References
- 1.Walker DH. Rickettsiae and rickettsial infections: the current state of knowledge. Clin Infect Dis. 2007;45(Suppl 1):S39–44. doi: 10.1086/518145. [DOI] [PubMed] [Google Scholar]
- 2.Pizarro-Cerda J, Cossart P. Bacterial adhesion and entry into host cells. Cell. 2006;124:715–727. doi: 10.1016/j.cell.2006.02.012. [DOI] [PubMed] [Google Scholar]
- 3.Martinez JJ, Seveau S, Veiga E, Matsuyama S, Cossart P. Ku70, a component of DNA-dependent protein kinase, is a mammalian receptor for Rickettsia conorii. Cell. 2005;123:1013–1023. doi: 10.1016/j.cell.2005.08.046. [DOI] [PubMed] [Google Scholar]
- 4.Chan YG, Cardwell MM, Hermanas TM, Uchiyama T, Martinez JJ. Rickettsial outer-membrane protein B (rOmpB) mediates bacterial invasion through Ku70 in an actin, c-Cbl, clathrin and caveolin 2-dependent manner. Cell Microbiol. 2009;11:629–644. doi: 10.1111/j.1462-5822.2008.01279.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Leong JM, Fournier RS, Isberg RR. Identification of the integrin binding domain of the Yersinia pseudotuberculosis invasin protein. EMBO J. 1990;9:1979–1989. doi: 10.1002/j.1460-2075.1990.tb08326.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lecuit M, Nelson DM, Smith SD, Khun H, Huerre M, Vacher-Lavenu MC, Gordon JI, Cossart P. Targeting and crossing of the human maternofetal barrier by Listeria monocytogenes: role of internalin interaction with trophoblast E-cadherin. Proc Natl Acad Sci USA. 2004;101:6152–6157. doi: 10.1073/pnas.0401434101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heinzen RA. Rickettsial actin-based motility: behavior and involvement of cytoskeletal regulators. Ann N Y Acad Sci. 2003;990:535–547. doi: 10.1111/j.1749-6632.2003.tb07424.x. [DOI] [PubMed] [Google Scholar]
- 8.Gouin E, Welch MD, Cossart P. Actin-based motility of intracellular pathogens. Curr Opin Microbiol. 2005;8:35–45. doi: 10.1016/j.mib.2004.12.013. [DOI] [PubMed] [Google Scholar]
- 9.Pollard TD, Borisy GG. Cellular motility driven by assembly and disassembly of actin filaments. Cell. 2003;112:453–465. doi: 10.1016/s0092-8674(03)00120-x. [DOI] [PubMed] [Google Scholar]
- 10.Welch MD, Rosenblatt J, Skoble J, Portnoy DA, Mitchison TJ. Interaction of human Arp2/3 complex and the Listeria monocytogenes ActA protein in actin filament nucleation. Science. 1998;281:105–108. doi: 10.1126/science.281.5373.105. [DOI] [PubMed] [Google Scholar]
- 11.Gouin E, Egile C, Dehoux P, Villiers V, Adams J, Gertler F, Li R, Cossart P. The RickA protein of Rickettsia conorii activates the Arp2/3 complex. Nature. 2004;427:457–461. doi: 10.1038/nature02318. [DOI] [PubMed] [Google Scholar]
- 12.Jeng RL, Goley ED, D'Alessio JA, Chaga OY, Svitkina TM, Borisy GG, Heinzen RA, Welch MD. A Rickettsia WASP-like protein activates the Arp2/3 complex and mediates actin-based motility. Cell Microbiol. 2004;6:761–769. doi: 10.1111/j.1462-5822.2004.00402.x. [DOI] [PubMed] [Google Scholar]
- 13.Pantaloni D, Le Clainche C, Carlier MF. Mechanism of actin-based motility. Science. 2001;292:1502–1506. doi: 10.1126/science.1059975. [DOI] [PubMed] [Google Scholar]
- 14.Takenawa T, Suetsugu S. The WASP-WAVE protein network: connecting the membrane to the cytoskeleton. Nat Rev Mol Cell Biol. 2007;8:37–48. doi: 10.1038/nrm2069. [DOI] [PubMed] [Google Scholar]
- 15.Stevens JM, Galyov EE, Stevens MP. Actin-dependent movement of bacterial pathogens. Nat Rev Microbiol. 2006;4:91–101. doi: 10.1038/nrmicro1320. [DOI] [PubMed] [Google Scholar]
- 16.Bernardini ML, Mounier J, d'Hauteville H, Coquis-Rondon M, Sansonetti PJ. Identification of icsA, a plasmid locus of Shigella flexneri that governs bacterial intra- and intercellular spread through interaction with F-actin. Proc Natl Acad Sci USA. 1989;86:3867–3871. doi: 10.1073/pnas.86.10.3867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haglund CM, Choe JE, Skau CT, Kovar DR, Welch MD. Rickettsia Sca2 is a bacterial formin-like mediator of actin-based motility. Nat Cell Biol. 2010;12:1057–1063. doi: 10.1038/ncb2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Riley SP, Goh KC, Hermanas TM, Cardwell MM, Chan YG, Martinez JJ. The Rickettsia conorii autotransporter protein Sca1 promotes adherence to nonphagocytic mammalian cells. Infect Immun. 2010;78:1895–1904. doi: 10.1128/IAI.01165-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Park H, Lee JH, Cossart P, Gouin E, Izard T. The Rickettsia surface cell antigen 4 applies mimicry to bind to and activate vinculin. J Biol Chem. 2011;286:35096–35103. doi: 10.1074/jbc.M111.263855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saunders RM, Holt MR, Jennings L, Sutton DH, Barsukov IL, Bobkov A, Liddington RC, Adamson EA, Dunn GA, Critchley DR. Role of vinculin in regulating focal adhesion turnover. Eur J Cell Biol. 2006;85:487–500. doi: 10.1016/j.ejcb.2006.01.014. [DOI] [PubMed] [Google Scholar]
- 21.Ziegler WH, Liddington RC, Critchley DR. The structure and regulation of vinculin. Trends Cell Biol. 2006;16:453–460. doi: 10.1016/j.tcb.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 22.Diederichs K, Karplus PA. Improved R-factors for diffraction data analysis in macromolecular crystallography. Nat Struct Biol. 1997;4:269–275. doi: 10.1038/nsb0497-269. [DOI] [PubMed] [Google Scholar]
- 23.Chen VB, Arendall WB, 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lawrence MC, Colman PM. Shape complementarity at protein/protein interfaces. J Mol Biol. 1993;234:946–950. doi: 10.1006/jmbi.1993.1648. [DOI] [PubMed] [Google Scholar]
- 25.Saschenbrecker S, Bracher A, Rao KV, Rao BV, Hartl FU, Hayer-Hartl M. Structure and function of Rbcx, an assembly chaperone for hexameric Rubisco. Cell. 2007;129:1189–1200. doi: 10.1016/j.cell.2007.04.025. [DOI] [PubMed] [Google Scholar]
- 26.Holm L, Rosenstrom P. Dali server: conservation mapping in 3D. Nucleic Acids Res. 2010;38:W545–W549. doi: 10.1093/nar/gkq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kabsch W. XDS. Acta Crystallogr D Biol Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Evans PR. An introduction to data reduction: space-group determination, scaling and intensity statistics. Acta Crystallogr D Biol Crystallogr. 2011;67:282–292. doi: 10.1107/S090744491003982X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vonrhein C, Flensburg C, Keller P, Sharff A, Smart O, Paciorek W, Womack T, Bricogne G. Data processing and analysis with the autoPROC toolbox. Acta Crystallogr D Biol Crystallogr. 2011;67:293–302. doi: 10.1107/S0907444911007773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schneider TR, Sheldrick GM. Substructure solution with SHELXD. Acta Crystallogr D Biol Crystallogr. 2002;58:1772–1779. doi: 10.1107/s0907444902011678. [DOI] [PubMed] [Google Scholar]
- 31.Bricogne G, Vonrhein C, Flensburg C, Schiltz M, Paciorek W. Generation, representation and flow of phase information in structure determination: recent developments in and around SHARP 2.0. Acta Crystallogr D Biol Crystallogr. 2003;59:2023–2030. doi: 10.1107/s0907444903017694. [DOI] [PubMed] [Google Scholar]
- 32.Vonrhein C, Blanc E, Roversi P, Bricogne G. Automated structure solution with autoSHARP. Methods Mol Biol. 2006;364:215–230. doi: 10.1385/1-59745-266-1:215. [DOI] [PubMed] [Google Scholar]
- 33.Abrahams JP, Leslie AG. Methods used in the structure determination of bovine mitochondrial F1 ATPase. Acta Crystallogr D Biol Crystallogr. 1996;52:30–42. doi: 10.1107/S0907444995008754. [DOI] [PubMed] [Google Scholar]
- 34.Cowtan K. The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr D Biol Crystallogr. 2006;62:1002–1011. doi: 10.1107/S0907444906022116. [DOI] [PubMed] [Google Scholar]
- 35.Bricogne G, Blanc E, Brandl M, Flensburg C, Keller P, Paciorek P, Roversi P, Sharff A, Smart OS, Vonrhein C, Womack TO. BUSTER version 2.9. Cambridge, United Kingdom: Global Phasing Ltd; 2011. [Google Scholar]
- 36.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vagin A, Teplyakov A. MOLREP: an automated program for molecular replacement. J Appl Cryst. 1997;30:1022–1025. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.