

A docking study of Mtb Eis with its substrate DUSP16/MKP-7 was performed. The docking model suggests dissociation of hexameric Mtb Eis into dimers or monomers.
Keywords: protein–protein docking, Eis, Mycobacterium tuberculosis, HADDOCK, comparative docking, docking study
Abstract
The intracellular pathogen Mycobacterium tuberculosis (Mtb) causes tuberculosis, and one of its secreted effector proteins, called enhanced intracellular survival (Eis) protein, enhances its survival in macrophages. Mtb Eis activates JNK-specific dual-specificity protein phosphatase 16 (DUSP16)/mitogen-activated protein kinase phosphatase-7 (MKP-7) through the acetylation on Lys55, thus inactivating JNK by dephosphorylation. Based on the recently reported crystal structure of Mtb Eis, a docking model for the binding of Mtb Eis to DUSP16/MKP-7 was generated. In the docking model, the substrate helix containing Lys55 of DUSP16/MKP-7 fits nicely into the active-site cleft of Mtb Eis; the twisted β-sheet of Eis domain II embraces the substrate helix from one side. Most importantly, the side-chain of Lys55 is inserted toward acetyl-CoA and the resulting distance is 4.6 Å between the NZ atom of Lys55 and the carbonyl carbon of the acetyl group in acetyl-CoA. The binding of Mtb Eis and DUSP16/MKP-7 is maintained by strong electrostatic interactions. The active-site cleft of Mtb Eis has a negatively charged surface formed by Asp25, Glu138, Asp286, Glu395 and the terminal carboxylic group of Phe396. In contrast, DUSP16/MKP-7 contains five basic residues, Lys52, Lys55, Arg56, Arg57 and Lys62, which point toward the negatively charged surface of the active-site pocket of Mtb Eis. Thus, the current docking model suggests that the binding of DUSP16/MKP-7 to Mtb Eis should be established by charge complementarity in addition to a very favorable geometric arrangement. The suggested mode of binding requires the dissociation of the hexameric Mtb Eis into dimers or monomers. This study may be useful for future studies aiming to develop inhibitors of Mtb Eis as a new anti-tuberculosis drug candidate.
1. Introduction
Tuberculosis, caused by pathogenic bacterium Mycobacterium tuberculosis (Mtb), is difficult to treat and increasing drug-resistance poses a serious global health threat (Dye & Williams, 2010 ▶; Chiang et al., 2010 ▶). This raises the urgent need for developing a new class of anti-tuberculosis drugs, which prompts the identification of novel drug target proteins of Mtb. Since one of survival strategies of Mtb is to inhibit the phagosomal maturation and autophagy in macrophages, Mtb effector proteins involved in these processes are of great interest for therapeutic intervention (Meena & Rajni, 2010 ▶). Eis (enhanced intracellular survival) protein secreted by Mtb was shown to enhance the survival of M. smegmatis (Msm) in the human macrophage-like cell line (Wei et al., 2000 ▶). The survival enhancement by Mtb Eis is achieved by down-regulating the JNK (c-Jun N-terminal kinase), which leads to the inhibition of inflammation and autophagic cell death (Shin et al., 2010 ▶). We recently revealed the molecular mechanism by which Mtb Eis down-regulates JNK and thereby enhances Mtb survival (Kim et al., 2012 ▶). In our recent study, Mtb Eis was shown to be an efficient N∊-acetyltransferase acting on Lys55 of DUSP16/MKP-7 (Kim et al., 2012 ▶), which is a JNK-specific phosphatase (Masuda et al., 2001 ▶). Mtb Eis activates DUSP16/MKP-7 through the acetylation, and the activated phosphatase inactivates JNK by dephosphorylation (Kim et al., 2012 ▶). The crystal structure determined in the same study revealed that the active-site cleft is formed in the interface between domains I and II (Kim et al., 2012 ▶). More importantly, the active-site cleft has a negatively charged surface, which seems suitable to elicit the binding of the positively charged substrate, Lys55 of DUSP16/MKP-7 (Kim et al., 2012 ▶).
To better understand the molecular details of the interaction between Mtb Eis and its substrate, DUSP16/MKP-7, we carried out the molecular docking with previously determined crystal structures of the both proteins [Protein Data Bank (PDB) accession numbers 3ryo for Mtb Eis (Kim et al., 2012 ▶) and 2vsw for DUSP16/MKP-7 (not published)]. The docked model suggests that the helix harboring Lys55 of DUSP16/MKP-7 fits into the active-site cleft of Mtb Eis and the binding may be established by not only the structural fit but also the electrostatic complementarity. Interestingly, however, the predicted binding mode requires dissociation of the hexameric Mtb Eis. This study may provide the structural basis for future research aiming to develop the inhibitor of Mtb Eis as a new tuberculosis drug.
2. Material and methods
We have used HADDOCK version 2.1 (de Vries et al., 2007 ▶; van Dijk et al., 2006 ▶) for the docking study. In HADDOCK, the overall docking is completed in three stages. It generates thousands of possible arrangements with starting structures and obtains hundreds of best structures using rigid-body docking for the next step. Then, these best structures are subject to semi-flexible docking, followed by water solvation. The semi-flexible docking employed in HADDOCK proceeds in four different stages as follows. The first and second stages are high-temperature rigid-body search and rigid-body simulated annealing (SA), respectively. The third and fourth stages are semi-flexible SA with flexible side-chains at the interface and semi-flexible SA with flexible backbone and side-chains, respectively. Here, the semi-flexible SA means SA in torsion-angle space. In this study, the values of the number of initial docked structure generations and the number of best structures from rigid-body docking were 2000 and 400, respectively. Among those 400 structures, we retained 200 best structures after the HADDOCK solvation analysis step.
Other than the above changes, we have used the default HADDOCK parameters with version 5.4 of protein and solvent topologies throughout the docking procedure. The starting structure of Mtb Eis with acetyl-CoA was obtained from our previous study (PDB code: 3ryo) and the DUSP16/MKP-7 structure was taken from the PDB (unpublished; PDB ID 2vsw). This is a three-body docking problem but we docked DUSP16/MKP-7 with Mtb Eis in the presence of acetyl-CoA inside Mtb Eis, reducing it to a usual two-body docking task effectively. We performed two sets of independent docking simulations, i.e. docking of single DUSP16/MKP-7 with Mtb Eis monomer and single DUSP16/MKP-7 with Mtb Eis dimer. The only condition we impose is an unambiguous distance requirement of 3.0 Å with a minimum 1.0 Å and a maximum 5.0 Å between the sulfur atom of acetyl CoA and the nitrogen atom of the Lys55 side-chain in DUSP16/MKP-7. In another trial, we imposed active (acetyl-CoA) and passive sites (Thr31, Asp32, Glu37, Arg43, Asp116, His125, Ser127, Glu128, Gly129, Tyr132, Arg134, Pro139, Leu143, Thr147, Val201, Lys211, Pro214, Thr237, Gln297, Trp301, Arg303, Met305, His318, Glu407 and Phe408) on Mtb Eis according to Kim et al. (2012 ▶) without distance restraints. For the HADDOCK passive site, we calculated the solvent accessibility of each residue using MOLMOL (Koradi et al., 1996 ▶) and residues near the active site with solvent accessibility greater than 40% were taken. However, the results were essentially the same. Therefore, we imposed a distance requirement only in subsequent docking with the Mtb Eis dimer. As for DUSP16/MKP-7, we used Lys55 as the active site with no passive site. For the analysis of the docked structures, we have used root-mean-square-deviation-(RMSD)-based cluster analysis as incorporated in HADDOCK (Fig. 1 ▶). The structure figures were plotted using the program PyMOL (http://pymol.sourceforge.net).
Figure 1.
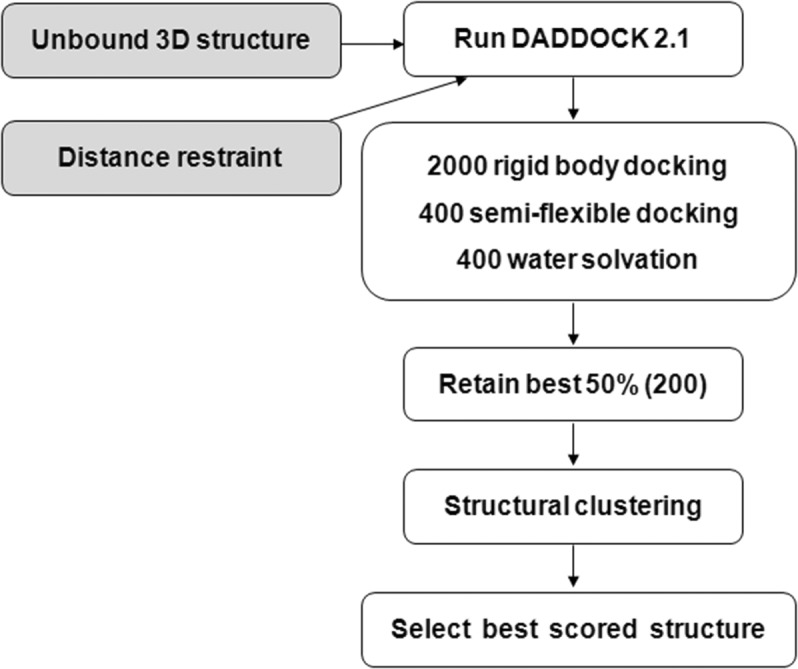
HADDOCK protocol flowchart employed in this study.
3. Results and discussion
3.1. Docking
In the crystal lattice, Mtb Eis forms a hexamer of 32 symmetry, in which three dimeric units are related by threefold symmetry. The active site is formed within each monomer, well separated from the subunit interface. However, the active site is blocked in the hexamer assembly by adjacent subunits related by the threefold symmetry. In contrast, the active site is fully accessible in the dimer assembly just as in an isolated monomer. This observation suggests the possibility that Eis would exist, transiently, as a monomer or a dimer, rather than as a hexamer, in order to bind and acetylate the substrate DUSP16/MKP-7. The dimer would be a more plausible unit than the monomer, because further dissociation of a dimeric unit into monomers is expected to be energetically less favorable. However, prediction of dominant species by considering the entropy alone is not straightforward. We performed the docking for both the dimer and monomer of Mtb Eis; the active-site cleft of Mtb Eis is fully accessible to the substrate, DUSP16/MKP-7, in both cases. For each docking study we have 200 possible docked structures after the water solvation step, since we retain the best 50% from 400 refined structures. As for the monomer, from RMSD-based structural clustering analysis with RMSD criteria ranging from 4.0 to 10.0 Å, the number of clusters with more than a single member in its cluster is only one. The number of structures in this single cluster is 45 regardless of RMSD criteria. This indicates that the final docked structures are structurally very diverse except structures within this single cluster, which has minimal structural diversity between them. We plotted the RMSD versus HADDOCK score of these 200 structures in Fig. 2 ▶. In fact, only these 45 structures within the cluster satisfied the distance restraint that we imposed. We also note that the best HADDOCK scored structure belongs to this cluster. We have represented the best score monomer (a) and dimer (b) structures in Fig. 3 ▶ along with Lys55 in DUSP16/MKP-7 and acetyl-CoA.
Figure 2.
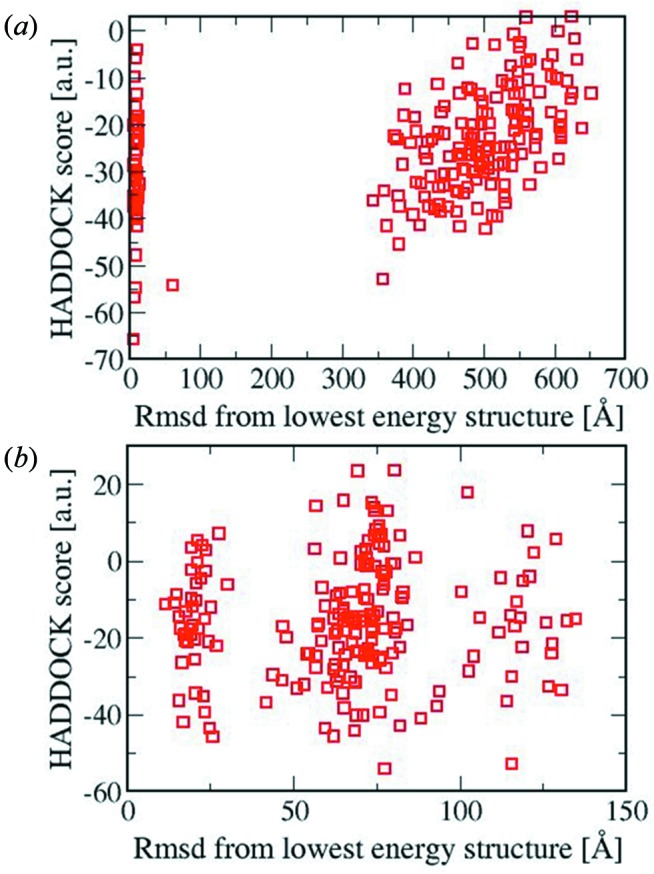
The RMSD versus HADDOCK score plot for the final 200 structures for the monomer (a) and dimer (b) of Mtb Eis. The reference structure for RMSD is the structure with the lowest HADDOCK score. The structures with RMSD > 50 Å do not meet the imposed distance restraint.
Figure 3.

Docking model of the Mtb Eis monomer with DUSP16/MKP-7. (a) Cartoon docking model of Mtb Eis monomer (PDB code 3ryo: green) and DUSP16/MKP-7 (PDB code 2vsw: yellow). Acetyl-CoA (blue) bound to Eis and Lys55 (magenta) of DUSP16/MKP-7 are shown as a ball-and-stick model. (b) Cartoon docking model of Mtb Eis dimer (PDB code 3ryo: green and cyan) and DUSP16/MKP-7 (PDB code 2vsw: yellow). Acetyl-CoA (blue) bound to Eis and Lys55 (red) of DUSP16/MKP-7 are shown as a ball-and-stick model. (c) The same monomer docking model is presented as an electrostatic surface diagram of Mtb Eis and a cartoon model of DUSP16/MKP-7. A black dotted circle indicates the active binding site of Mtb Eis. Both the cartoon model and electrostatic surface diagram of DUSP16/MKP-7 rotated by 180° are shown on a smaller scale. A red dotted circle indicates the binding site of DUSP16/MKP-7 with positive electrostatic potential. Blue and red colors of the electrostatic surface diagrams correspond to positive and negative electrostatic potentials at neutral pH, respectively.
Overall structures of both Mtb Eis and DUSP16/MKP-7 were maintained during the docking calculation. When we compare the initial structures of Mtb Eis (PDB ID 3ryo) and DUSP16/MKP-7 (PDB ID 2vsw) with those of Mtb Eis and DUSP16/MKP-7 from the best monomer Mtb Eis docking model, RMSDs are 1.19 Å and 1.15 Å, respectively. The largest Cα deviations occur at Mtb Eis Glu80 and Arg178, with 2.77 Å and 2.85 Å, respectively, and at DUSP16/MKP-7 Lys52 and Lys55, with 3.33 Å and 2.80 Å, respectively.
3.2. Structural features of the docking model
Lys55 of DUSP16/MKP-7, the target of the acetylation by Mtb Eis, is included in the helix spanning residues from Lys52 to Gln60, which is referred to as the substrate helix hereafter in this paper. The key issue of this docking study should be how the substrate helix of DUSP16/MKP-7 fits into the active-site cleft of Mtb Eis, which was described previously as a ‘deep and narrow channel’ (Kim et al., 2012 ▶), and thereby secures the structural environment for an acetylation reaction to occur. In our current docking model, the substrate helix fits nicely into the active-site cleft of Mtb Eis; the twisted β-sheet of Eis domain II embraces the substrate helix from one side [Figs. 3(a) and 3(c) ▶]. In particular, the side-chain of Lys55 is inserted toward acetyl-CoA, resulting in a distance of 4.6 Å between the NZ atom of Lys55 and the carbonyl carbon of the acetyl group (Fig. 4 ▶). The observed proximity here between the acetyl donor and the acceptor is close enough to fully facilitate the transfer of the acetyl group to occur.
Figure 4.
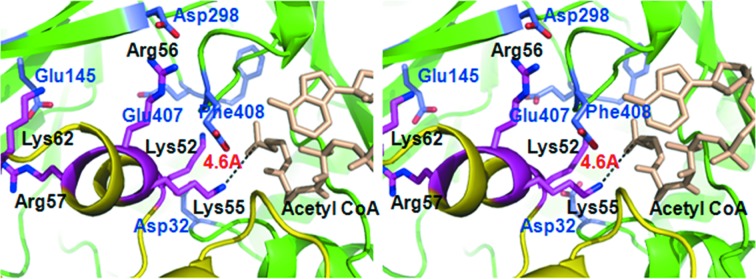
Enlarged stereoview of the active binding site in the docking model. Side-chains (in blue) and acetyl-CoA (in wheat color) of Mtb Eis (backbone in green and residues labeled in blue). Side-chains (in magenta) of DUSP16/MKP-7 (backbone in yellow and residues labeled in black) are shown as a ball-and-stick model.
Strikingly, the aforementioned structural fit between the active-site cleft of Mtb Eis and the substrate helix of DUSP16/MKP-7 is driven by favorable complementary charges at their interface. The active-site cleft of Mtb Eis has a negatively charged surface (Fig. 3c ▶) with Asp25, Glu138, Asp286, Glu395 and the terminal carboxylic group of Phe396 (Fig. 4 ▶), while the substrate helix of DUSP16/MKP-7 contains four basic residues, Lys52, Lys55, Arg56 and Arg57. In addition, the positively charged Lys62 is also located at the C-terminus of the helix (Fig. 4 ▶). All these five positively charged residues of the substrate helix of DUSP16/MKP-7 point toward the negatively charged surface of the active site of Mtb Eis. Acetylation is a crucial step for the enhanced survival capability of Msm against human autophagy. Understanding its action from a structural point of view is an important starting point for therapeutic purposes. The current docking model suggests that the binding of DUSP16/MKP-7 to Mtb Eis should be established by the charge complementarity, combined with favorable geometric shape complementarity between the substrate helix and the active-site cleft. We note that the present docking model requires the dissociation of hexameric Mtb Eis into dimers or monomers, at least transiently, for binding of DUSP16/MKP-7. Similar dissociation of other multimeric enzymes has been reported (Kernstock et al., 2012 ▶). We could detect only hexameric species of Mtb Eis in the presence of DUSP16/MKP-7 (residues 1–153) by size-exclusion chromatography. This result may be expected, because the dissociation of Mtb Eis hexamers would be only transient, if it occurs. Therefore, we plan to produce a fusion protein between Mtb Eis and DUSP16/MKP-7 to test the possibility. By presenting a putative docked model, we hope that this study will provide a useful basis for future efforts to characterize in more detail the binding interface between Mtb Eis and DUSP16/MKP-7, and to develop inhibitors of Mtb Eis as a new tuberculosis drug candidate.
Acknowledgments
This research was supported by a National Research Foundation of Korea (NRF) grant funded by the Korean government (20110013663) to HJY and (2012009242) to SJ. SJ acknowledges the financial support from the Ministry of Science, ITC and Future Planning, subjected to the project EDISON (EDucation-research Integration through Simulation On the Net; grant No. 2012M3C1A6051724).
References
- Chiang, C. Y., Centis, R. & Migliori, G. B. (2010). Respirology, 15, 413–432. [DOI] [PubMed]
- Dijk, M. van, van Dijk, A. D., Hsu, V., Boelens, R. & Bonvin, A. M. (2006). Nucl. Acids Res. 34, 3317–3325. [DOI] [PMC free article] [PubMed]
- Dye, C. & Williams, B. G. (2010). Science, 328, 856–861. [DOI] [PubMed]
- Kernstock, S., Davydova, E., Jakobsson, M., Moen, A., Pettersen, S., Maelandsmo, G. M., Egge-Jacobsen, W. & Falnes, P. Ø. (2012). Nat. Commun. 3, 1–11. [DOI] [PubMed]
- Kim, K. H., An, D. R., Song, J., Yoon, J. Y., Kim, H. S., Yoon, H. J., Im, H. N., Kim, J., Kim, do J., Lee, S. J., Kim, K. H., Lee, H. M., Kim, H. J., Jo, E. K., Lee, J. Y. & Suh, S. W. (2012). Proc. Natl Acad. Sci. USA, 109, 7729–7734.
- Koradi, R., Billeter, M. & Wüthrich, K. (1996). J. Mol. Graph. 14, 51–55, 29–32. [DOI] [PubMed]
- Masuda, K., Shima, H., Watanabe, M. & Kikuchi, K. (2001). J. Biol. Chem. 276, 39002–39011. [DOI] [PubMed]
- Meena, L. S. & Rajni (2010). FEBS J. 277, 2416–2427. [DOI] [PubMed]
- Shin, D. M., Jeon, B. Y., Lee, H. M., Jin, H. S., Yuk, J. M., Song, C. H., Lee, S. H., Lee, Z. W., Cho, S. N., Kim, J. M., Friedman, R. L. & Jo, E. K. (2010). PLoS Pathog. 6, e1001230. [DOI] [PMC free article] [PubMed]
- Vries, S. J. de, van Dijk, A. D., Krzeminski, M., van Dijk, M., Thureau, A., Hsu, V., Wassenaar, T. & Bonvin, A. M. (2007). Proteins, 69, 726–733. [DOI] [PubMed]
- Wei, J., Dahl, J. L., Moulder, J. W., Roberts, E. A., O’Gaora, P., Young, D. B. & Friedman, R. L. (2000). J. Bacteriol. 182, 377–384. [DOI] [PMC free article] [PubMed]