

Abstract
Objective:
To identify the cause of cervical dopa-responsive dystonia (DRD) in a Muslim Indian family inherited in an apparently autosomal recessive fashion, as previously described in this journal.
Methods:
Previous testing for mutations in the genes known to cause DRD (GCH1, TH, and SPR) had been negative. Whole exome sequencing was performed on all 3 affected individuals for whom DNA was available to identify potentially pathogenic shared variants. Genotyping data obtained for all 3 affected individuals using the OmniExpress single nucleotide polymorphism chip (Illumina, San Diego, CA) were used to perform linkage analysis, autozygosity mapping, and copy number variation analysis. Sanger sequencing was used to confirm all variants.
Results:
After filtering of the variants, exome sequencing revealed 2 genes harboring potentially pathogenic compound heterozygous variants (ATM and LRRC16A). Of these, the variants in ATM segregated perfectly with the cervical DRD. Both mutations detected in ATM have been shown to be pathogenic, and α-fetoprotein, a marker of ataxia telangiectasia, was increased in all affected individuals.
Conclusion:
Biallelic mutations in ATM can cause DRD, and mutations in this gene should be considered in the differential diagnosis of unexplained DRD, particularly if the dystonia is cervical and if there is a recessive family history. ATM has previously been reported to cause isolated cervical dystonia, but never, to our knowledge, DRD. Individuals with dystonia related to ataxia telangiectasia may benefit from a trial of levodopa.
Dopa-responsive dystonia (DRD) is an uncommon condition, with a prevalence of 0.5 to 1 individual per million. Nonetheless, it remains an important condition to recognize because it is, by definition, readily treatable by the oral administration of levodopa. Response to this treatment is often dramatic, sustained, and is not usually associated with the development of motor fluctuations or dyskinesias. To date, 3 genes have been shown to be responsible for most DRD: GCH1 (GTP cyclohydrolase 1), TH (tyrosine hydroxylase), and SPR (sepiapterin reductase).1–3 All 3 genes encode enzymes that are involved in the endogenous biosynthesis of dopamine.
We had previously identified a Muslim Indian kindred in which 3 of 5 siblings had presented with cervical DRD.4 Mutational screening had ruled out GCH1, TH, or SPR as the cause of the disorder.4 With the advent of exome sequencing technologies, we returned to this family to attempt to identify the genetic cause of the disorder.
METHODS
Standard protocol approvals, registrations, and patient consents.
Ethical approval for the study was obtained from the local ethics committee, and all subjects provided written consent for participation and a videoed examination, where applicable.
Subjects.
Participants were drawn from a multigenerational Muslim Indian family. The genetic pedigree of the family as originally supplied to us is shown in figure 1A. DNA was initially available for the 3 affected siblings as well as both parents. The pattern of transmission was most consistent with an autosomal recessive inheritance. The parents were neurologically normal and reported no consanguinity.
Figure 1. Original and updated genetic pedigree.

(A) Genetic pedigree as previously published. Individuals affected by cervical dopa-responsive dystonia are indicated by shaded symbols and those for whom DNA was available by “DNA.” (B) Updated genetic pedigree in light of the initial exome sequencing results. The individuals marked with a cross (III:6 and III:8) have clinically typical ataxia telangiectasia. Mutational status for ATM is marked below the relevant individuals as follows: wt = wild-type allele; fs = frameshift deletion (c.7886_7890del); and m = missense mutation (c.6154G>A). DNA availability is marked as previously. The marriage between individuals II:5 and II:6 is possibly consanguineous.
Individual II:1 developed jerky right-sided torticollis at the age of 15 years, which progressively worsened over the period of a year. Subsequently, laryngeal dystonia, clawing of his fingers, postural hand tremor, and difficulty writing also developed. Some of the movements in the hands when held outstretched appeared choreiform. Individuals II:2 and II:5 were similarly affected, developing cervical dystonia with associated side-to-side head tremor at ages 13 and 11 years, respectively. In addition, there was postural hand tremor.
Trihexyphenidyl and baclofen were ineffective. However, treatment with levodopa-carbidopa (100:25 mg 3 times daily) produced a dramatic improvement in the cervical dystonia in all 3 siblings, permitting them to return to work (see video on the Neurology® Web site at www.neurology.org). The laryngeal dystonia, postural hand tremor, and clawing of the fingers were, however, unaffected. The excellent response of the cervical dystonia to low doses of levodopa was sustained in all 3 cases. Withdrawal of levodopa resulted in the return of symptoms within 4 days and these resolved again after levodopa was readministered.
Genetic analyses.
DNA from individuals II:1, II:2, and II:5 (in figure 1A) was subjected to whole exome sequencing using Illumina's TruSeq (62-Mb) DNA Sample Prep Kit and Exome Enrichment Kit (Illumina, San Diego, CA). Sanger sequencing was used to confirm variants.
Linkage analysis, homozygosity mapping, and copy number variation analysis were also performed in this family. Details of the methods and results of these analyses can be found in the e-Methods, tables e-1 to e-3, and figure e-1.
RESULTS
Exome sequencing generated an average of 81,270,305 unique reads. Based on the CCDS hg19 definition of the exome, mean coverage across the exome was 66 reads and 91.2% of the exome was covered by at least 10 reads. Variant filtration was performed in a standard manner (described in the e-Methods) to isolate potentially pathogenic homozygous or compound heterozygous variants (figure 2). No shared, potentially pathogenic homozygous variants were identified by this method. We did, however, identify 2 genes with shared compound heterozygous variants: LRRC16A, on chromosome 6, which harbored 2 missense mutations (c.293C>T [p.A98V] and c.437A>G [p.D146G]); and ATM, on chromosome 11, which harbored a 4 base-pair, frameshift deletion (c.7886_7890del) and a missense mutation (c.6154 G>A [E2052K]).
Figure 2. Filtering of exome sequencing data.
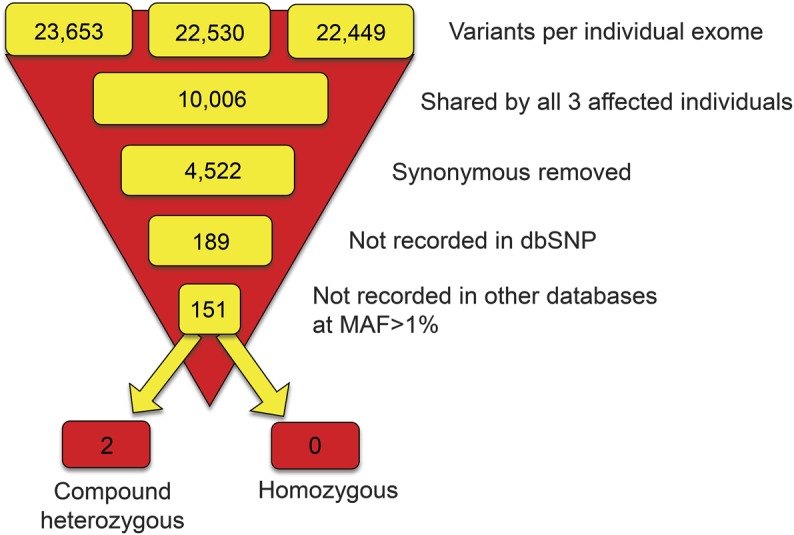
Graphic illustration of the filtering process applied to the exome datasets in order to select out likely candidate causal variants for this ostensibly autosomal recessive condition. Other databases = 1000 Genomes, NCHLBI Exome Sequencing Project, and the Complete Genomic 69 database. dbSNP = database of single nucleotide polymorphisms; MAF = minor allele frequency.
Segregation analysis by Sanger sequencing demonstrated that the variants in LRRC16A were present in the compound heterozygous form not just in the affected individuals but also in one unaffected sibling (II:3 in figure 1A), thus making LRRC16A an unlikely cause for the DRD. The variants in ATM, however, segregated perfectly with the DRD in the compound heterozygous state.
Moreover, the variants that we detected in ATM are both known to be pathogenic. The frameshift mutation in ATM is predicted to cause nonsense-mediated decay of the messenger RNA product and has been detected in our own patients with ataxia telangiectasia (AT). The missense mutation in ATM occurs at a conserved base (figure e-2), has previously been detected in AT in the homozygous state by others,5 and appears to cause skipping of exon 44.
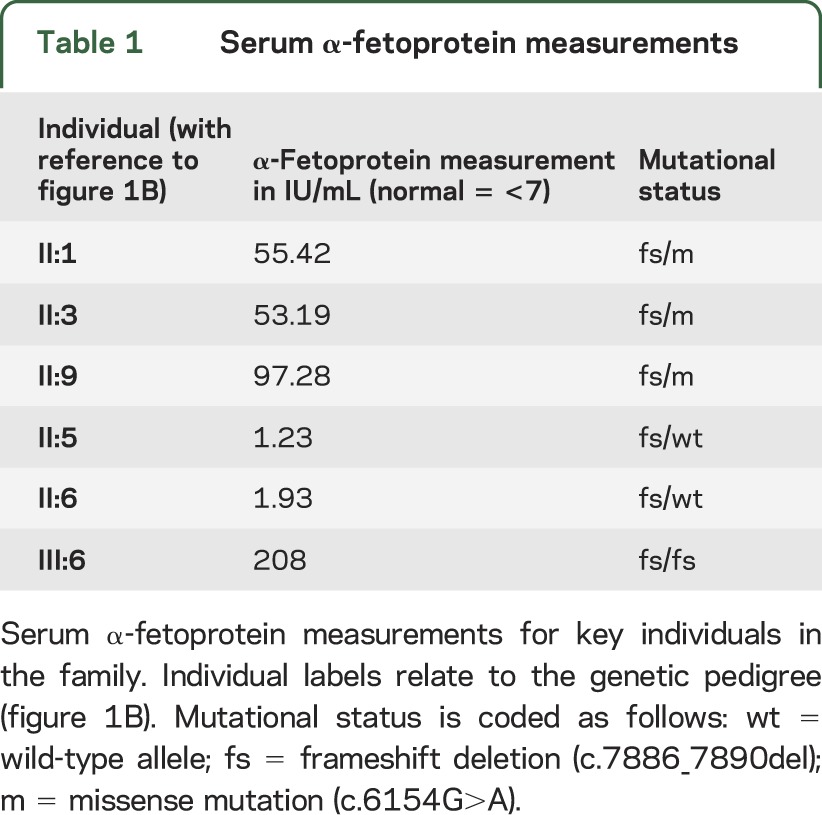
Given the finding of 2 pathologic mutations in ATM, we returned to the family in 2012 to reevaluate the pedigree and reexamine the index cases. Reevaluation of the pedigree revealed that one of the unaffected siblings has 2 daughters who are now affected by clinically typical AT (figure 1B). DNA samples were obtained for this branch of the family and the relevant exons of ATM sequenced. This revealed that the unaffected sibling and his wife (who may be related to him, although the family is unaware of this) both carry the 4 base-pair, frameshift deletion (but not the missense mutation) in the heterozygous state. One of his daughters, who has clinically typical AT, is homozygous for the same deletion; we were unable to obtain blood from the other affected daughter. Reexamination of the 3 siblings affected by DRD revealed conjunctival telangiectasia (figure e-3) but no ataxia or oculomotor signs. They remained responsive to levodopa. In addition, a fourth sibling now exhibited very mild cervical dystonia and dysarthria, but did not wish to participate in the genetic analysis and so his mutational status could not be ascertained. α-Fetoprotein, which is generally elevated in AT, was increased in all 3 siblings with DRD as well as the daughter of the unaffected sibling who has clinically typical AT (table 1).
Table 1.
Serum α-fetoprotein measurements
DISCUSSION
Biallelic mutations in ATM are generally associated with AT, an autosomal recessive form of early-onset ataxia associated with oculomotor apraxia, ocular and cutaneous telangiectasia, and variable immunodeficiency. Patients with classic disease generally develop an ataxic gait in their early childhood and are wheelchair-bound by early adolescence. Death often occurs in the second or third decade of life due to malignancies or respiratory failure.6,7 However, a milder variant form of the disease is recognized, and occasionally the cardinal features of the disease—ataxia, telangiectasia, and immunodeficiency—are completely absent.8 In these cases of variant AT, extrapyramidal disorders appear to dominate the clinical presentation: in one study, 60% of cases exhibited rest tremor, 60% dystonia, and 70% choreoathetosis.8
Notably, a recent publication identified members of 3 Canadian Mennonite families presenting with early-onset, isolated, predominantly cervical dystonia without frank ataxia or oculomotor apraxia, who harbored a homozygous c.6200C>A missense mutation in exon 43 of ATM.9 This is the same exon in which we detected a missense mutation (c.6154G>A) in our Indian Muslim kindred with DRD, suggesting that a missense mutation in this region of the gene may predispose to variant AT with a dystonic presentation. It is, nonetheless, difficult to conceive of the mechanism by which mutations in ATM might lead to DRD.
The identification of DRD as a manifestation of AT has a double significance. First, it suggests that at least a subset of the dystonic features seen in classic and variant AT may be improved by treatment with levodopa. Second, despite the milder phenotype, the rate of malignancy in variant AT appears, nonetheless, to be significantly elevated and patients remain at increased risk from exposure to radiation used diagnostically or as a treatment.8,10 Therefore, ATM should be considered in unexplained DRD, particularly if it involves the neck or is of young onset.
Supplementary Material
GLOSSARY
- AT
ataxia telangiectasia
- DRD
dopa-responsive dystonia
Footnotes
Supplemental data at www.neurology.org
AUTHOR CONTRIBUTIONS
Gavin Charlesworth: drafting/revising the manuscript for content, study concept or design, analysis or interpretation of data, acquisition of data. Mahavir D. Mohire: drafting/revising the manuscript for content, contribution of vital patients, acquisition of data. Susanne A. Schneider: drafting/revising the manuscript for content, original characterization of patients. Maria Stamelou: drafting/revising the manuscript for content, identification of patients. Nicholas W. Wood and Kailash P. Bhatia: drafting/revising the manuscript for content, study concept or design, study supervision, obtaining funding.
STUDY FUNDING
Supported by the Bachman-Strauss Dystonia and Parkinson Foundation and MRC/Wellcome Trust strategic award (WT089698/Z/09/Z). This work was undertaken at UCLH/UCL, who received a proportion of funding from the Department of Health's NIHR Biomedical Research Centre’s funding scheme.
DISCLOSURE
G. Charlesworth and M. Mohire report no disclosures. S. Schneider was the recipient of a Bosch fast-track stipend. She holds grants from the Eva Luise and Horst Köhler Foundation for rare diseases and the Novartis Foundation. She has received financial support to attend meetings from Teva and Ipsen Pharma. M. Stamelou received travel and speaker honoraria from Ipsen, Novartis, and the Movement Disorders Society. N. Wood holds grants from the Bachmann-Strauss Dystonia Parkinson Foundation, the MRC, and the Wellcome Trust. K. Bhatia has received honoraria/financial support to speak/attend meetings from GSK, Boehringer Ingelheim, Ipsen, Merz, and Orion Pharma companies. He holds grants from the Bachmann-Strauss Dystonia Parkinson Foundation, the Dystonia Society UK, and the Halley Stewart Trust. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Ichinose H, Ohye T, Takahashi E, et al. Hereditary progressive dystonia with marked diurnal fluctuation caused by mutations in the GTP cyclohydrolase I gene. Nat Genet 1994;8:236–242 [DOI] [PubMed] [Google Scholar]
- 2.Brautigam C, Wevers RA, Jansen RJ, et al. Biochemical hallmarks of tyrosine hydroxylase deficiency. Clin Chem 1998;44:1897–1904 [PubMed] [Google Scholar]
- 3.Bonafe L, Thony B, Penzien JM, Czarnecki B, Blau N. Mutations in the sepiapterin reductase gene cause a novel tetrahydrobiopterin-dependent monoamine-neurotransmitter deficiency without hyperphenylalaninemia. Am J Hum Genet 2001;69:269–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schneider SA, Mohire MD, Trender-Gerhard I, et al. Familial dopa-responsive cervical dystonia. Neurology 2006;66:599–601 [DOI] [PubMed] [Google Scholar]
- 5.Teraoka SN, Telatar M, Becker-Catania S, et al. Splicing defects in the ataxia-telangiectasia gene, ATM: underlying mutations and consequences. Am J Hum Genet 1999;64:1617–1631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chun HH, Gatti RA. Ataxia-telangiectasia, an evolving phenotype. DNA Repair 2004;3:1187–1196 [DOI] [PubMed] [Google Scholar]
- 7.Subramony SH, Durr A. Ataxic Disorders. Edinburgh: Elsevier; 2012 [Google Scholar]
- 8.Verhagen MM, Abdo WF, Willemsen MA, et al. Clinical spectrum of ataxia-telangiectasia in adulthood. Neurology 2009;73:430–437 [DOI] [PubMed] [Google Scholar]
- 9.Saunders-Pullman R, Raymond D, Stoessl AJ, et al. Variant ataxia-telangiectasia presenting as primary-appearing dystonia in Canadian Mennonites. Neurology 2012;78:649–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Renwick A, Thompson D, Seal S, et al. ATM mutations that cause ataxia-telangiectasia are breast cancer susceptibility alleles. Nat Genet 2006;38:873–875 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.