

Abstract
Objective:
The goal of this study was to examine the clinical presentation of chronic traumatic encephalopathy (CTE) in neuropathologically confirmed cases.
Methods:
Thirty-six adult male subjects were selected from all cases of neuropathologically confirmed CTE at the Boston University Center for the Study of Traumatic Encephalopathy brain bank. Subjects were all athletes, had no comorbid neurodegenerative or motor neuron disease, and had next-of-kin informants to provide retrospective reports of the subjects' histories and clinical presentations. These interviews were conducted blind to the subjects' neuropathologic findings.
Results:
A triad of cognitive, behavioral, and mood impairments was common overall, with cognitive deficits reported for almost all subjects. Three subjects were asymptomatic at the time of death. Consistent with earlier case reports of boxers, 2 relatively distinct clinical presentations emerged, with one group whose initial features developed at a younger age and involved behavioral and/or mood disturbance (n = 22), and another group whose initial presentation developed at an older age and involved cognitive impairment (n = 11).
Conclusions:
This suggests there are 2 major clinical presentations of CTE, one a behavior/mood variant and the other a cognitive variant.
Chronic traumatic encephalopathy (CTE) is a neurodegenerative disease marked by widespread accumulation of hyperphosphorylated tau (p-tau).1,2 To date, CTE has been documented in amateur and professional athletes involved in contact sports, military personnel exposed to explosive blast, and others subjected to repetitive brain trauma (RBT), including concussive and subconcussive injuries.1–5 All reported neuropathologically confirmed cases of CTE have had exposure to RBT. However, not all individuals with histories of RBT develop CTE, indicating that additional risk factors, including genetics, likely have a role in the neuropathogenesis of this disease. For example, it has been suggested that the APOE ε4 allele may increase susceptibility for CTE.6
Previously published descriptions of the clinical presentation of CTE vary. Case reports of presumptive CTE (formerly termed dementia pugilistica or “punch-drunk” when thought limited to boxers4) indicated a constellation of clinical features, including impairments in cognition, behavior, and mood, and in some cases, chronic headache and motor and cerebellar dysfunction. Several case reports of boxers suggested 2 forms of presentation: 1) younger onset, with initial behavioral and mood disturbance, but with minimal cognitive and motor features; and 2) older onset, with greater cognitive impairment and, often, motor disturbance.4,7–10 In advanced cases, CTE is associated with dementia, although it is unclear whether the clinical presentation of CTE dementia is different from that associated with Alzheimer disease (AD) or other age-related neurodegenerative disorders.11–13 Herein, we describe the clinical presentation, course, and APOE genotype of a sample of 36 athletes with neuropathologically confirmed CTE.
METHODS
Subjects.
The brains of 81 subjects in the Boston University Center for the Study of Traumatic Encephalopathy (CSTE) brain bank met recently published criteria for the neuropathologic diagnosis of CTE.1 For the current study, 45 cases were excluded because of 1) primary exposure to RBT from nonathletic activities; 2) inability to contact next-of-kin to conduct an interview; and 3) presence of comorbid motor neuron disease,14 neurodegenerative disease, or other significant neuropathology. Seven were military veterans with unknown or no athletic history, 10 had no next-of-kin contact, and 28 had comorbid neuropathologic disease. Of the 36 remaining subjects, 28 were included in a previous report1 and 8 were new cases.
CTE neuropathologic staging.
The cases were categorized into the 4-stage rating scale of CTE (I = least severe, IV = most severe) based on the severity of p-tau pathology, as previously reported.1 Diagnosis and staging were conducted blind to medical history, APOE genotype, and informant interview.
Interview and medical record review.
History and clinical presentation were obtained through postmortem telephone interviews with next-of-kin by a neuropsychologist (R.A.S.) blinded to neuropathologic findings and APOE genotype status. Medical records were available and reviewed for 23 cases. The semistructured interview was based on previous studies of postmortem dementia diagnosis made by interviews with family members.15,16 Information queried during the interview included the following: demographics; cause of death; and athletic, military, medical, neuropsychiatric, and social/occupational histories. The interview included specific questions regarding dementia, depression, changes in cognition, behavior, mood, and motor functioning, as well as instrumental activities of daily living. Responses were qualitatively summarized into an overall assessment of the subject's presentation and course of symptoms and functioning. The number of informants interviewed per case ranged from 1 to 7 (median = 2), with each interview lasting approximately 60 minutes. Interviews were conducted at a median time of 4 months after time of death.
APOE genotyping.
DNA was extracted from brain tissue samples using a Qiagen QIAamp DNA extraction kit (Qiagen, Valencia, CA). Two single nucleotide polymorphisms (National Center for Biotechnology Information SNPs rs429358 and rs7412) were examined using TaqMan assays (Applied Biosystems, Foster City, CA). Allelic discrimination was automated using the manufacturer's software. Positive controls, consisting of DNA of each of the 6 possible APOE genotypes (ε2/ε2, ε2/ε3, ε2/ε4, ε3/ε3, ε3/ε4, ε4/ε4), were included on each plate and genotyped with restriction isotyping.
Statistical analyses.
Between-group differences were examined by independent sample t tests. Chi-square analyses were used for between group comparisons for categorical data. APOE genotype analyses comparing CTE cases with population norms17 were conducted with the χ2 goodness-of-fit test. A probability level of p = 0.05 was used throughout. All statistical analyses were conducted with IBM SPSS Statistics, version 19.0 (IBM Corp., Armonk, NY).
Standard protocol approvals, registrations, and patient consents.
Approvals for brain donation, postmortem clinical record review, interviews with family members, and neuropathologic evaluation were provided by the Institutional Review Boards of Boston University Medical Center and the Bedford VA Hospital.
RESULTS
Table 1 summarizes the demographics, cause of death, athletic history, neuropathologic stage, and APOE genotypes of the sample. All subjects were male athletes, with 6 (17%) African American and 1 (3%) of Hispanic origin. There were 29 football players (22 who played professionally, 4 who only played through college, and 3 who only played through high school), 3 professional hockey players, 1 professional wrestler, and 3 boxers (1 professional, 2 amateur). Of the football players, the most common position played was lineman (48%), followed by running back (21%), linebacker (10%), and smaller numbers of other positions. There were no quarterbacks or kickers. Of the 36 subjects, 3 (8%) were asymptomatic. Tables 2 and 3 describe the clinical features and course of the remaining 33 subjects.
Table 1.
Description of sample by initial clinical presentation

Table 2.
Clinical features and course by initial clinical presentation
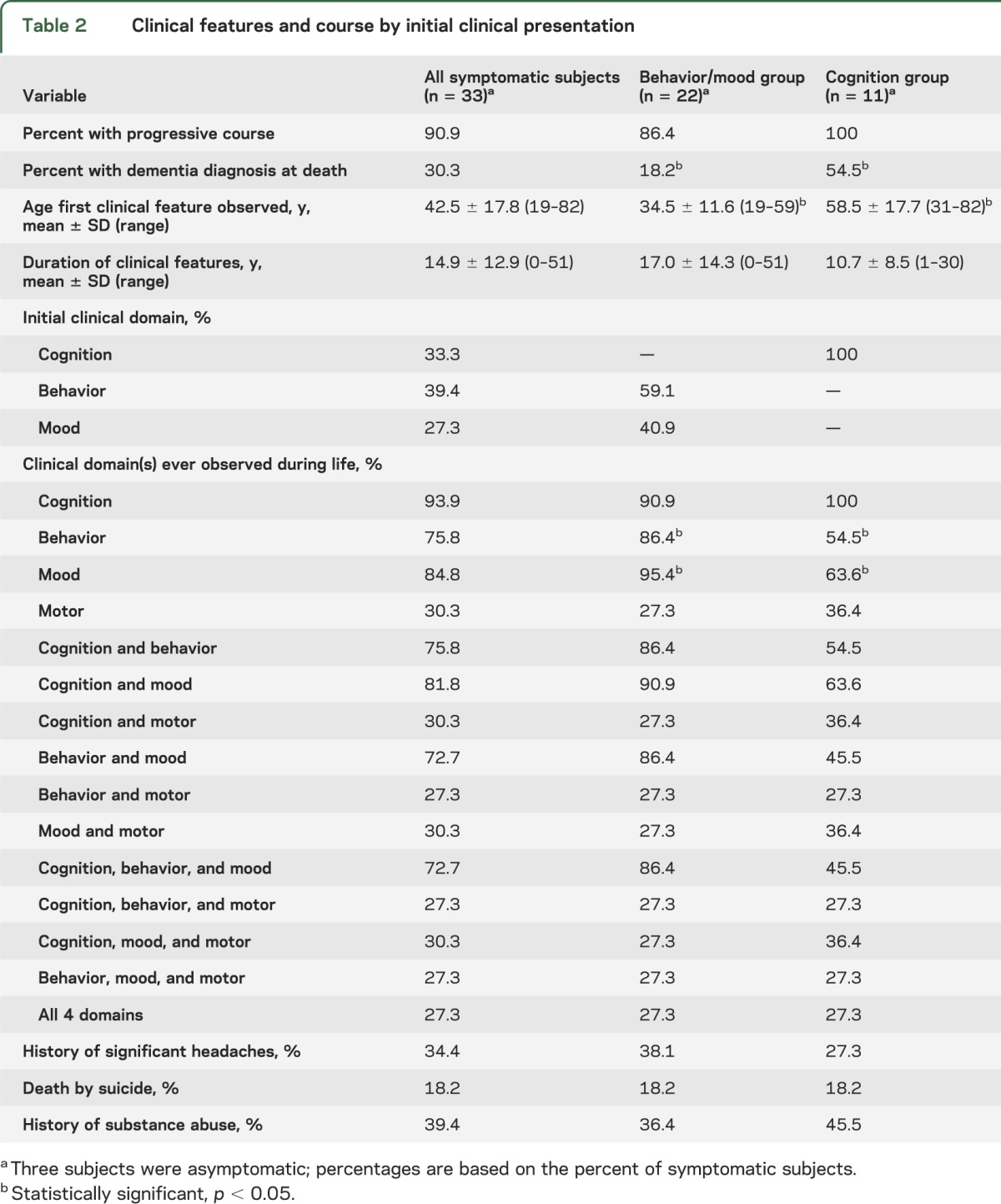
Table 3.
Specific clinical features by initial clinical presentation

Eleven of the symptomatic cases were reported to have initial changes in cognitive functioning (e.g., episodic memory impairment, executive dysfunction) before behavioral or mood disturbance. Initial changes in behavior (e.g., explosivity, impulsivity, violence) before mood or cognitive disturbance were reported in 13 subjects. Mood changes (e.g., depression, hopelessness) were reported as the initial feature in 9 subjects. None of the subjects had motor disturbance as their initial feature. The subgroups with initial behavioral symptoms and mood changes were similar in age of initial presentation, age of death, and neuropathologic stage, and were combined into a behavior/mood group (n = 22). Subjects whose initial difficulties involved cognitive functioning comprised a cognition group (n = 11). Tables 1–3 describe demographics and clinical features for the behavior/mood and cognition subgroups.
Ten subjects were diagnosed with dementia; 4 were clinically diagnosed with AD, 4 with “dementia pugilistica” or “football-related” dementia, and 2 with unspecified dementia. All had stage IV CTE. Of the 10, 7 exhibited cognitive symptoms initially, 2 exhibited mood symptoms initially, and 1 initially presented with behavior changes. The mean age of symptom onset for the dementia group was 57.7 years (SD = 18.3; range 25–82) and the mean age of dementia diagnosis was 72.6 years (SD = 8.5, range 56–83). The mean length of time between dementia diagnosis and death was 8.0 years (SD = 5.5, range <1–15). Four subjects with dementia had gait difficulties, 3 had a history of falls, and 1 had a history of tremor. Two subjects (20%) with dementia had a history of headaches, compared with 11 subjects (44%) without dementia. All 10 subjects had both memory and executive impairment, 7 had language deficits, and 2 had visuospatial difficulties. Six of the 10 were characterized by behavioral impairment, predominantly described as having a “short fuse” or being “out of control.” Four of the 10 were physically violent and 2 were verbally violent. Although one subject demonstrated disinhibited behavior, none of the subjects had disinhibited speech or socially inappropriate behaviors. Of the 7 who were reported to have mood disturbance, 2 had predominantly sadness/depressive symptoms and 2 had anxiety symptoms. The only 2 subjects in the entire sample reported to have had apathy were in the dementia group.
The proportions of APOE genotypes (i.e., ε4 homozygotes, combined ε4 homozygotes and heterozygotes, and ε4 noncarriers) in our CTE sample were significantly different from those found in an age-matched normative sample17 (χ2 [2] = 6.63, p < 0.05). A binomial test revealed that the primary difference between our CTE sample and population norms was a greater proportion of ε4 homozygotes in our sample (p < 0.05). When examining the 2 initial presentation groups, there were no differences between the behavior/mood group and the age-matched normative sample (χ2 [2] = 0.46, p > 0.05). However, there were proportionally more ε4 homozygotes in the cognition group than expected (χ2 [2] = 13.3, p < 0.05). The relative proportions of APOE genotypes in our 10 subjects with dementia were not significantly different from those seen in AD18 (χ2 [2] = 1.52, p > 0.05).
DISCUSSION
Consistent with earlier reports of boxers,4,7–10 our findings suggest that there may be 2 different clinical presentations of CTE, with one initially exhibiting behavioral or mood changes, and the other initially exhibiting cognitive impairment. The behavior/mood group demonstrated symptoms at a significantly younger age than the cognition group. Although almost all subjects in the behavior/mood group demonstrated cognitive impairments at some point, significantly fewer subjects in the cognition group demonstrated behavioral and mood changes during the course of their illness. There were distinctions between the 2 groups regarding specific features present in each domain. The behavior/mood group was significantly more explosive, out of control, physically and verbally violent, and depressed than the cognition group. Whereas all subjects in the cognition group were reported to have impaired episodic memory, approximately one-quarter of the behavior/mood group did not have memory difficulties. Subjects in the cognition group were significantly more likely to progress to dementia than those in the behavior/mood group but were also significantly older at the time of death. Given the small sample size in this study, however, it is unclear whether these 2 apparently distinct clinical subtypes are representative of all individuals with CTE. In addition, the subsample of cases with dementia is also small, thus limiting the generalization of the presentation of CTE dementia. Further research is needed to clarify and validate these findings.
We examined the potential role of the APOE ε4 allele as a susceptibility factor for CTE. Our findings indicate that there were significantly more ε4 homozygotes in the sample than expected in a normal, age-matched population. Furthermore, this effect was largely driven by the cognition group: 2 of 11 subjects in the cognition group and 1 of 22 subjects in the behavior/mood group were homozygous for the ε4 allele. In addition, 1 of the 10 CTE subjects diagnosed with dementia during life was ε4 homozygous. Although interpretation and generalization of these results is difficult because of the small sample, the proportion of ε4 homozygosity is in contrast to population norms in which ε4 homozygosity only occurs in 1% to 3% of the general population,17 and more consistent with the 10% of patients with AD who are ε4 homozygous.18 The APOE ε4 variant is the largest known genetic risk factor for sporadic AD.18 It has been associated with β-amyloid, but not tau, deposition in cognitively normal aging.19 APOE ε4 has also been associated with greater severity of cognitive deficits and longer recovery time after traumatic brain injury (TBI) and RBT in a variety of populations, including boxers and professional football players,20–24 and may increase the risk of clinical dementia after TBI.25 It has been hypothesized that the APOE ε4 isoform may have direct neurotoxic effects leading to mitochondrial dysfunction and cytoskeletal changes, resulting in increased risk of neurodegeneration.26 Despite the small sample size and other limitations in the current study, future research on the role of APOE in CTE risk appears warranted. However, other potential susceptibility genes also merit consideration, including mutations to the microtubule-associated protein tau (MAPT) gene, the progranulin (GRN) gene, and the chromosome 9 open reading frame 72 (C9ORF72) gene. Moreover, additional nongenetic risk factors for CTE should be examined in future research, including studies to determine what specific aspects of RBT exposure (e.g., types, severity, frequency, initial age, and duration of trauma) are associated with CTE, as well as what potential lifestyle variables (e.g., diet, exercise, obesity, steroid use) are associated with the disease initiation and variability in presentation.
It is noteworthy that motor features, including parkinsonism, were not common in our sample. This is in contrast to some earlier descriptions of CTE in boxers, in which these motor features were quite prominent.4 However, our findings are consistent with other case reports of predominantly younger onset boxers, in which motor disturbance was not common.4,7–10 It is not clear why some individuals with CTE develop motor features and others do not. One possibility may be the differences in the biomechanics of injury. For example, in boxing, angular acceleration and torsional injury involving the brainstem and cerebellum is thought to be a pathogenic mechanism of TBI after a hook or jab to the jaw, whereas transverse and linear acceleration and deceleration injury are more characteristic of football dynamics.27,28 As a result, degeneration of brainstem structures that produce parkinsonism, such as the substantia nigra, might occur earlier in the course of disease in boxers. In contrast, football players might develop substantia nigra degeneration later in the course of their disease, at a time when widespread cortical and basal ganglionic degeneration mask the development of motor disturbance. Related mechanisms of injury leading to CTE have been suggested by recent experimental studies of blast neurotrauma.3
Although many of the symptoms of CTE are similar to AD and other causes of dementia,11,29 there are factors that appear to clinically differentiate CTE from other age-related neurodegenerative diseases. For example, behavioral changes observed early in the course of CTE could be confused with the behavioral variant of frontotemporal dementia, especially in a patient in his or her 50s without any significant memory impairment. However, common changes in the behavioral variant of frontotemporal dementia typically include disinhibited and inappropriate behavior and speech, as well as apathy30; these symptoms were not frequent in our case series. In addition, the progressive memory impairment observed in more than 80% of our CTE cases, and in all 10 of the subjects with dementia, could lead to an inaccurate diagnosis of AD when the underlying disease is CTE.12
It is not clear what neuropathologic changes may lead to the 2 possible clinical presentations observed in this study. It is unlikely that the small, focal cortical p-tau lesions found in stage I and II CTE produce clinically meaningful behavioral and mood symptoms. However, these features may be associated with the neurofibrillary tangles in the locus coeruleus and amygdala found in younger subjects in a previous report.1 The memory and executive dysfunction in the older cognition group may be due to the more extensive degenerative changes in the hippocampus and frontal cortices seen in CTE stages III and IV.1 It is possible, however, that some of the features evident in the younger behavior/mood group were due to persistent postconcussion syndrome,31 with unresolved or even progressive32 axonal damage resulting from the initial traumas. Axonal injury has been shown in all neuropathologic stages of CTE, ranging from multifocal, perivascular axonal varicosities in the cortex and white matter in stages I and II, to more extensive, diffuse axonal loss in the cortex and white matter in stages III and IV.1 Recent reports have demonstrated that repetitive subconcussive trauma is associated with white matter abnormalities on diffusion tensor imaging33,34 and abnormal functional MRI tests.35 Additional findings indicate that there may be persistent and progressive inflammation and white matter degeneration after even a single TBI.36 Further research is required to delineate these clinicopathologic relationships.
Three subjects in our case series were asymptomatic. One of these cases was only 17 years old and had stage I neuropathology. Both of the other 2 cases were much older football players (one in 40s, one in 80s), had stage II neuropathology, and were homozygous for APOE ε3. Both also had advanced graduate degrees, were very successful in their professional careers, and were described as extremely intelligent. Although speculative, these findings raise the possibility that cognitive reserve37 may have a role in protecting against the clinical manifestations of CTE. A recent report suggests that cognitive reserve may mitigate cognitive decline in older individuals with earlier life TBI.38 Future research examining the roles of cognitive reserve, genetics, and environmental factors in determining resilience to clinical manifestations and the progression of p-tau pathology will help elucidate the pathobiology of CTE.
Although these findings are based on the largest cohort of subjects with neuropathologically confirmed CTE without comorbidities studied to date, interpretation and generalizability of these results are limited by several factors. First, the overall sample size is small, and caution should be taken when generalizing these results to the larger population of athletes or to the overall clinical presentation of CTE. In addition, there are inherent selection biases imposed in a postmortem brain donation study. For example, families choosing to donate may be more likely to have witnessed symptoms during life. This could lead to reports of more severe symptoms than a typical CTE population, and could account for only having 3 asymptomatic cases. From the broader CTE cohort in the CSTE brain bank, we selected a smaller sample by eliminating individuals with comorbid pathology and only including athletes; this restriction may further limit the generalizability of our findings. Results from this study should not be interpreted in terms of population prevalence or generalized to living athletes with CTE. In addition, there is the potential for reduced reliability and validity of retrospective reports from family members after the death of a loved one. However, several studies have demonstrated adequate reliability and validity of these verbal autopsies in a variety of patient populations, including those with dementia15,16 and psychiatric disorders.39 There also may be differences in the accuracy of informant reports when comparing younger and older subjects. That is, informants of older subjects were asked to recall early- or midlife events possibly resulting in reduced accuracy compared with the informants of younger subjects. Finally, there was no comparison group of former athletes without CTE. This may limit the ability to draw conclusions that the clinical presentation described is specifically due to the effects of CTE. In our available dataset of subjects whose tissue had been examined at the BU CSTE brain bank, there was not an adequate number of subjects without CTE to make such a comparison. For example, 34 of 35 former professional football players had neuropathologically confirmed CTE.1 Future research is needed to clarify the clinical presentation of CTE. The development of biomarkers (e.g., blood, CSF, neuroimaging, and tau-specific radiotracers) will result in the ability to detect and diagnose CTE during life and subsequent studies of risk factors, epidemiology, and treatment.40
GLOSSARY
- AD
Alzheimer disease
- CSTE
Center for the Study of Traumatic Encephalopathy
- CTE
chronic traumatic encephalopathy
- p-tau
hyperphosphorylated tau
- RBT
repetitive brain trauma
- TBI
traumatic brain injury
AUTHOR CONTRIBUTIONS
Dr. Stern was responsible for drafting the manuscript, study concept and design, and analysis and interpretation of data. He also conducted some of the statistical analyses and had a role in obtaining funding. Mr. Daneshvar participated in drafting the manuscript, as well as acquisition of data, statistical analysis, and interpretation of data. Ms. Baugh participated in drafting the manuscript, as well as study design and acquisition of data. Dr. Seichepine participated in drafting the manuscript, as well as analysis and interpretation of data. Mr. Montenigro participated in drafting the manuscript and in study design. Mr. Riley participated in revising the manuscript, study design, and acquisition of data. Mr. Fritts, Ms. Stamm, Mr. Robbins, and Ms. McHale participated in revising the manuscript and acquisition of data. Ms. Simkin participated in revising the manuscript as well as conducting the APOE genotyping. Dr. Stein and Dr. Alvarez participated in revising the manuscript, as well as acquisition and analysis of neuropathologic data. Dr. Goldstein and Dr. Budson participated in revising the manuscript and interpreting the data. Dr. Kowall participated in revising the manuscript, interpreting the data, and obtaining funding. Mr. Nowinski participated in revising the manuscript, study concept, acquisition of data, and obtaining funding. Dr. Cantu participated in drafting the manuscript, study design and concept, interpreting data, and obtaining funding. Dr. McKee participated in drafting the manuscript, study design and concept, acquiring, analyzing, and interpreting clinical data, acquiring, analyzing, and interpreting the neuropathologic data, and obtaining funding.
STUDY FUNDING
Supported by NIH (R01 NS078337, P30 AG13846), Department of Veterans Affairs (CSP 501, B6796-C), Sports Legacy Institute, National Operating Committee on Standards for Athletic Equipment, and unrestricted gifts from the National Football League and the Andlinger Foundation.
DISCLOSURE
R. Stern is funded by NIH grants R01 NS078337, R01 MH080295, R01 CA129769, P30 AG13846, U01 AG10483, and U01 AG015477; and has received research support from the Alzheimer's Association, the Andlinger Foundation, the National Operating Committee on Standards for Athletic Equipment, Janssen Alzheimer's Immunotherapy, Pfizer, and Medivation. He has been a paid consultant to Janssen Alzheimer's Immunotherapy, Outcome Science, and Elan Pharmaceuticals, and he has been a paid Expert Advisor to Eli Lilly. He receives royalties from Psychological Assessment Resources for the publication of neuropsychological tests. D. Daneshvar and C. Baugh report no disclosures. D. Seichepine receives funding from the Center for Integration of Medicine and Innovative Technology, as well as from NIH training grant T32 AG036697. P. Montenigro received support from Boston University School of Medicine for a summer research internship. D. Riley and N. Fritts report no disclosures. J. Stamm is supported by NIH grant F31NS081957. C. Robbins reports no disclosures. L. McHale is paid by Sports Legacy Institute for her work as Director of Family Relations. I. Simkin reports no disclosures. T. Stein is supported by NIH P30 AG13846 pilot grant. V. Alvarez is supported by the Department of Veterans Affairs. L. Goldstein is funded through grants from the NIH P30 AG13846, NASA SK-11-107, DOE DE-PS02-08ER08, and Cure Alzheimer's Fund. A. Budson is funded through the Department of Veterans Affairs. He receives royalties from Elsevier and Wiley-Blackwell for the publication of books. N. Kowall is funded by NIH grant P30 AG13846 and the Department of Veterans Affairs. C. Nowinski is supported by the Center for Integration of Medicine and Innovative Technology and the Andlinger Foundation. He receives consulting fees from MC10, and he receives royalties from the publication of his book, Head Games, and the documentary, “Head Games.” R. Cantu is Vice President of the National Operating Committee on Standards for Athletic Equipment, Cofounder and Medical Director of Sports Legacy Institute, and Senior Advisor to the NFL's Head, Neck and Spine Committee. He has received support from the Andlinger Foundation. He gave expert testimony in the trials of Carey vs Northwestern Memorial Hospital, Arbec vs Dr. Hardin and St. Joseph's, and Grane vs Methodist Medical Center of Illinois. He receives royalties from the publication of the books, Catastrophic Football Injuries, Diabetes and Exercise, Neurologic Head and Spine Injuries, and Concussions and Our Kids. A. McKee is funded through NIH grants P30 AG13846, R01 AG1649, and the Department of Veterans Affairs, and received research support for this work from the Department of Veterans Affairs; Veterans Affairs Biorepository (CSP 501); NIA supplement 0572063345-5, National Operating Committee on Standards for Athletic Equipment, the National Football League (unrestricted gift), and the Andlinger Foundation. Go to Neurology.org for full disclosures.
REFERENCES
- 1.McKee AC, Stern RA, Nowinski CJ, et al. The spectrum of disease in chronic traumatic encephalopathy. Brain 2013;136:43–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McKee AC, Cantu RC, Nowinski CJ, et al. Chronic traumatic encephalopathy in athletes: progressive tauopathy following repetitive concussion. J Neuropathol Exp Neurol 2009;68:709–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goldstein LE, Fisher AM, Tagge CA, et al. Chronic traumatic encephalopathy in blast-exposed military veterans and a blast neurotrauma mouse model. Sci Transl Med 2012;4:134ra60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Corsellis JA, Bruton CJ, Freeman-Browne D. The aftermath of boxing. Psychol Med 1973;3:270–303 [DOI] [PubMed] [Google Scholar]
- 5.Omalu BI, DeKosky ST, Minster RL, Kamboh MI, Hamilton RL, Wecht CH. Chronic traumatic encephalopathy in a National Football League player. Neurosurgery 2005;57:128–134 [DOI] [PubMed] [Google Scholar]
- 6.Gandy S, Dekosky ST. ApoE ε4 status and traumatic brain injury on the gridiron or the battlefield. Sci Transl Med 2012;4:134ed4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mawdsley C, Ferguson FR. Neurological disease in boxers. Lancet 1963;2:799–801 [DOI] [PubMed] [Google Scholar]
- 8.Soeder M, Arndt T. Affektive storungen und veréinderungen des hirnstrombildes bei boxern. Dtsch Med Wochenschr 1954;79:1792–1795 [PubMed] [Google Scholar]
- 9.Grahmann H, Ule G. Beitrag zur kenntis der chronischen cerebralen krankheitsbieder bei boxen. Psychiatr Neurol 1957;134:261–283 [PubMed] [Google Scholar]
- 10.Jokl E, Guttman E. Neurologisch-psychiatrische Untersuchung an boxern. Münch Med Woch 1931;1:560 [Google Scholar]
- 11.Shively S, Scher AI, Perl DP, Diaz-Arrastia R. Dementia resulting from traumatic brain injury: what is the pathology? Arch Neurol 2012;69:1245–1251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Areza-Fegyveres R, Rosemberg S, Castro RM, et al. Dementia pugilistica with clinical features of Alzheimer's disease. Arq Neuropsiquiatr 2007;65:830–833 [DOI] [PubMed] [Google Scholar]
- 13.Sayed N, Culver C, Dams-O'Connor K, Hammond F, Diaz-Arrastia R. Clinical phenotype of dementia after traumatic brain injury. J Neurotrauma 2013;30:1117–1122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McKee AC, Gavett BE, Stern RA, et al. TDP-43 proteinopathy and motor neuron disease in chronic traumatic encephalopathy. J Neuropathol Exp Neurol 2010;69:918–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barber R, Snowden JS, Craufurd D. Frontotemporal dementia and Alzheimer's disease: retrospective differentiation using information from informants. J Neurol Neurosurg Psychiatry 1995;59:61–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Davis PB, White H, Price JL, McKeel D, Robins LN. Retrospective postmortem dementia assessment: validation of a new clinical interview to assist neuropathologic study. Arch Neurol 1991;48:613–617 [DOI] [PubMed] [Google Scholar]
- 17.McKay GJ, Silvestri G, Chakravarthy U, et al. Variations in apolipoprotein E frequency with age in a pooled analysis of a large group of older people. Am J Epidemiol 2011;173:1357–1364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ward A, Crean S, Mercaldi CJ, et al. Prevalence of apolipoprotein E4 genotype and homozygotes (APOE e4/4) among patients diagnosed with Alzheimer's disease: a systematic review and meta-analysis. Neuroepidemiology 2012;38:1–17 [DOI] [PubMed] [Google Scholar]
- 19.Morris JC, Roe CM, Xiong C, et al. APOE predicts Aβ but not tau Alzheimer's pathology in cognitively normal aging. Ann Neurol 2010;67:122–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Friedman G, Froom P, Sazbon L, et al. Apolipoprotein E-epsilon4 genotype predicts a poor outcome in survivors of traumatic brain injury. Neurology 1999;52:244–248 [DOI] [PubMed] [Google Scholar]
- 21.Teasdale GM, Nicoll JA, Murray G, Fiddes M. Association of apolipoprotein E polymorphism with outcome after head injury. Lancet 1997;350:1069–1071 [DOI] [PubMed] [Google Scholar]
- 22.Jordan BD, Relkin NR, Ravdin LD, Jacobs AR, Bennett A, Gandy S. Apolipoprotein E epsilon4 associated with chronic traumatic brain injury in boxing. JAMA 1997;278:136–140 [PubMed] [Google Scholar]
- 23.Katzman R, Galasko DR, Saitoh T, et al. Apolipoprotein-epsilon4 and head trauma: synergistic or additive risks? Neurology 1996;46:889–891 [PubMed] [Google Scholar]
- 24.Kutner KC, Erlanger DM, Tsai J, Jordan B, Relkin NR. Lower cognitive performance of older football players possessing apolipoprotein E epsilon4. Neurosurgery 2000;47:651–657 [DOI] [PubMed] [Google Scholar]
- 25.Mayeux R, Ottman R, Maestre G, et al. Synergistic effects of traumatic head injury and apolipoprotein-epsilon 4 in patients with Alzheimer's disease. Neurology 1995;45:555–557 [DOI] [PubMed] [Google Scholar]
- 26.Mahley RW, Huang Y. Apolipoprotein E sets the stage: response to injury triggers neuropathology. Neuron 2012;76:871–885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Blennow K, Hardy J, Zetterberg H. The neuropathology and neurobiology of traumatic brain injury. Neuron 2012;76:886–899 [DOI] [PubMed] [Google Scholar]
- 28.Viano DC, Casson IR, Pellman EJ, et al. Concussion in professional football: comparison with boxing head impacts—part 10. Neurosurgery 2005;57:1154–1172 [DOI] [PubMed] [Google Scholar]
- 29.McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging–Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement 2011;7:263–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rascovsky K, Hodges JR, Knopman D, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 2011;134:2456–2477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bigler ED. Neuropsychology and clinical neuroscience of persistent post-concussive syndrome. J Int Neuropsychol Soc 2008;14:1–22 [DOI] [PubMed] [Google Scholar]
- 32.Johnson VE, Stewart W, Smith DH. Axonal pathology in traumatic brain injury. Exp Neurol 2013;246:35–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Koerte IK, Kaufmann D, Hartl E, et al. A prospective study of physician-observed concussion during a varsity university hockey season: white matter integrity in ice hockey players. Part 3 of 4. Neurosurg Focus 2012;33:E3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Koerte IK, Ertl-Wagner B, Reiser M, Zafonte R, Shenton ME. White matter integrity in the brains of professional soccer players without a symptomatic concussion. JAMA 2012;308:1859–1861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Breedlove EL, Robinson M, Talavage TM, et al. Biomechanical correlates of symptomatic and asymptomatic neurophysiological impairment in high school football. J Biomech 2012;45:1265–1272 [DOI] [PubMed] [Google Scholar]
- 36.Johnson VE, Stewart JE, Begbie FD, Trojanowski JQ, Smith DH, Stewart W. Inflammation and white matter degeneration persist for years after a single traumatic brain injury. Brain 2013;136:28–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stern Y. Cognitive reserve in ageing and Alzheimer's disease. Lancet Neurol 2012;11:1006–1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moretti L, Cristofori I, Weaver SM, Chau A, Portelli JN, Grafman J. Cognitive decline in older adults with a history of traumatic brain injury. Lancet Neurol 2012;11:1103–1112 [DOI] [PubMed] [Google Scholar]
- 39.Deep-Soboslay A, Akil M, Martin CE, et al. Reliability of psychiatric diagnosis in postmortem research. Biol Psychiatry 2005;57:96–101 [DOI] [PubMed] [Google Scholar]
- 40.Baugh CM, Stamm JM, Riley DO, et al. Chronic traumatic encephalopathy: neurodegeneration following repetitive concussive and subconcussive brain trauma. Brain Imaging Behav 2012;6:244–254 [DOI] [PubMed] [Google Scholar]