

Abstract
Objective:
To identify causative genes for centronuclear myopathies (CNM), a heterogeneous group of rare inherited muscle disorders that often present in infancy or early life with weakness and hypotonia, using next-generation sequencing of whole exomes and genomes.
Methods:
Whole-exome or -genome sequencing was performed in a cohort of 29 unrelated patients with clinicopathologic diagnoses of CNM or related myopathy depleted for cases with mutations of MTM1, DNM2, and BIN1. Immunofluorescence analyses on muscle biopsies, splicing assays, and gel electrophoresis of patient muscle proteins were performed to determine the molecular consequences of mutations of interest.
Results:
Autosomal recessive compound heterozygous truncating mutations of the titin gene, TTN, were identified in 5 individuals. Biochemical analyses demonstrated increased titin degradation and truncated titin proteins in patient muscles, establishing the impact of the mutations.
Conclusions:
Our study identifies truncating TTN mutations as a cause of congenital myopathy that is reported as CNM. Unlike the classic CNM genes that are all involved in excitation-contraction coupling at the triad, TTN encodes the giant sarcomeric protein titin, which forms a myofibrillar backbone for the components of the contractile machinery. This study expands the phenotypic spectrum associated with TTN mutations and indicates that TTN mutation analysis should be considered in cases of possible CNM without mutations in the classic CNM genes.
The centronuclear myopathies (CNM) are a group of clinically and genetically heterogeneous muscle disorders defined by the presence of one or more internalized nuclei in affected myofibers without excessive regeneration or additional structural abnormalities.1–3 The clinical presentation ranges from severe hypotonia with respiratory insufficiency at birth, to late-onset mild muscle weakness, with clinical findings largely limited to the skeletal musculature. To date, mutations in 4 genes involved in either the assembly or function of triads, the specialized membrane structures sustaining the excitation-contraction coupling,4 have been identified in CNM patients: MTM1 (myotubularin) in X-linked severe CNM (MIM 310400), DNM2 (dynamin 2) in autosomal dominant and sporadic cases (MIM 160150), BIN1 (amphiphysin 2) causing rare autosomal recessive disease (MIM 255200), and RYR1 (ryanodine receptor 1) associated with autosomal recessive and sporadic presentations.5 Nevertheless, the genetic basis for disease remains unknown in a significant proportion of patients with clinicopathologic diagnoses of CNM (∼20%),5 hampering a complete understanding of the molecular mechanisms underlying the pathogenesis and genetic counseling in such families. To identify additional genes involved in these conditions, we performed whole-exome sequencing, or in one case whole-genome sequencing, on DNA from a cohort of 29 unrelated patients who presented to the research study with clinicopathologic diagnoses of CNM.
METHODS
Standard protocol approvals, registrations, and patient consents.
This study was approved by the institutional review board of Boston Children's Hospital. Written informed consent was obtained from all participants.
Subjects.
The subjects were selected from a group of 121 neuromuscular patients with clinicopathologic diagnoses of CNM from referring neurologists and neuropathologists. Seventy of the patients were excluded for having known mutations in MTM1 (45 patients), DNM2 (21 patients), RYR1 (3 patients), or BIN1 (1 patient) identified by prior research or clinical testing. The remaining patients were prioritized based on the availability of sufficient DNA sample for NGS and follow-up analysis. After identification of putative gene mutations reported here, slides and images of muscle biopsies from the 5 patients with TTN mutations were reviewed in a blinded manner by one board-certified neuropathologist (M.W.L.) to independently establish a pathologic diagnosis for each case.
Whole-exome and whole-genome sequencing.
Exome sequencing of 28 probands was performed by the IDDRC Core Next-Gen Sequencing Facility of Boston Children's Hospital and Harvard Medical School, in collaboration with Axeq Technologies. Samples were enriched for exomic sequences using the Illumina Exome Enrichment protocol and captured libraries were sequenced using Illumina HiSeq 2000 Sequencers. The reads were mapped to the human genome assembly UCSC hg19 using Burrows-Wheeler Alignment (BWA version 0.5.8, http://bio-bwa.sourceforge.net/). Single-nucleotide polymorphisms and small insertions/deletions were called with SAMtools (version 0.1.7, http://samtools.sourceforge.net/). DNA from a 29th proband was sequenced by Complete Genomics Standard Sequencing Service for whole-genome sequencing through the Gene Partnership Program at Boston Children's Hospital.
Indirect immunofluorescence analysis of muscle biopsies.
Frozen muscle sections were fixed in ice-cold acetone for 10 minutes, followed by immunostaining with titin N-terminal (Sigma SAB1400284) (epitope: first 111 amino acids), A–I junction (titin MIR antibody6) (epitope: exons 256–258), C-terminal (Santa Cruz sc-271945) (epitope: last 300 amino acids), calpain 3 (Novocastra 2A12), ryanodine receptor (Sigma R128), aldolase A (AldoA) (Cell Signaling 3188), and anti-α-actinin (4D2) antibodies.
Hybrid minigene splicing assay.
Exons of interest were cloned with their flanking introns into the pZW4 splicing reporter construct7 between HpaI and KpnI restriction sites. The mutations at the donor or acceptor splice sites were introduced by site-directed mutagenesis. Wild-type or mutant hybrid minigene containing plasmids were transfected into HEK293 cells, followed by RNA extraction after 24 hours and reverse transcriptase–PCR using primers flanking the hybrid constructs.
Sodium dodecyl sulfate–gel electrophoresis of muscle proteins.
Protein extracts from patient and control muscles were prepared as described,8 separated by 1% agarose gel electrophoresis, and stained with Coomassie blue.
RESULTS
Identification of TTN mutations by next-generation sequencing.
On average, 88.7% of the total reads from whole-exome sequencing were uniquely aligned to the reference genome. The mean coverage rate of target regions was 94.47% and on average 86.77% of the target bases were covered by >10× reads for each exome. The annotated variants were filtered against variations reported on dbSNP132,9 the 1000 Genomes project November 2010 edition, and the National Heart, Lung, and Blood Institute Exome Sequencing Project (http://evs.gs.washington.edu/EVS/) databases. Remarkably, 5 individuals were notable for all having 2 or more potentially pathogenic variants in one common gene, TTN, which was an excellent candidate CNM gene because of its critical role in maintaining sarcomeric structure and function (figure e-1A on the Neurology® Web site at www.neurology.org). TTN encodes the giant sarcomeric protein titin that forms a myofibrillar backbone for the components of the contractile machinery, spanning the full distance from the Z-disk to the M-band in the sarcomere.10–12 Encompassing 363 exons, TTN includes several regions subject to extensive differential splicing, producing a number of different isoforms in cardiac and skeletal muscles.10,13,14 TTN mutations have previously been implicated in various cardiac and skeletal muscle diseases including adult-onset tibial muscular dystrophy,15–17 limb-girdle muscular dystrophy 2J,15,18 hereditary myopathy with early respiratory failure,19,20 and early-onset myopathy with fatal cardiomyopathy.21 Heterozygous truncating mutations in TTN have also been shown to be a common cause of dilated cardiomyopathy, accounting for ∼25% of cases in one large series.22–25 Mutations in various titin-associated proteins also disrupt the sarcomeric structure, leading to different neuromuscular diseases.11,26–31 Based on these reasons, we hypothesized that the TTN mutations present in our 5 patients with CNM were likely to be causative for the disease. Although occasional fibers with internal nuclei have been reported in other muscle diseases with TTN mutations,19,21 herein we describe TTN mutations presenting with histopathologic findings characterized as CNM by clinical neuropathology services.
Clinical phenotypes of patients with CNM and TTN mutations.
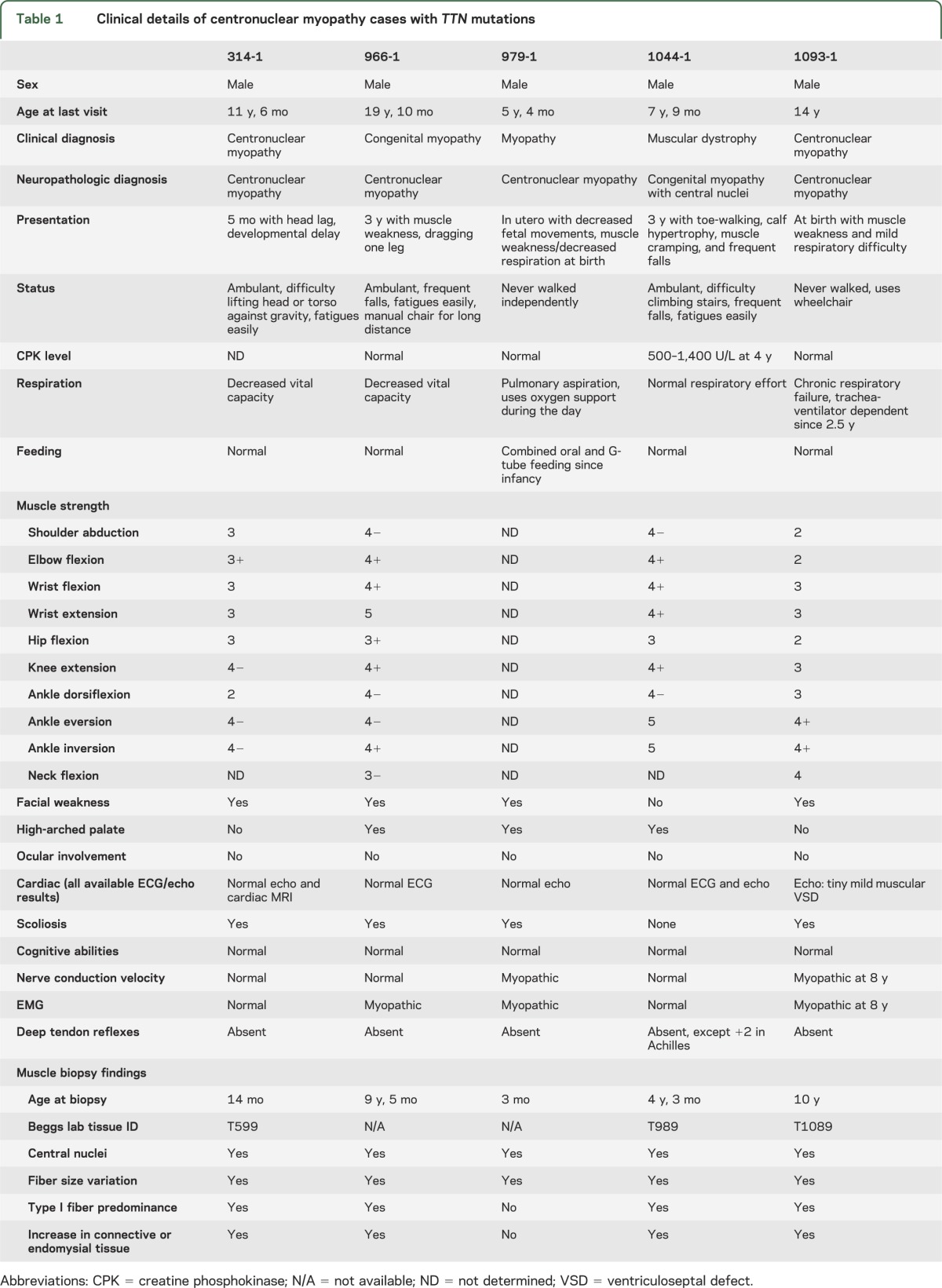
The clinical features of the 5 patients with TTN mutations are summarized in table 1. Diffuse muscle weakness from early childhood and decreased deep tendon reflexes were common presentations, while 4 individuals also had respiratory difficulty. The presence of high-arched palate suggests in utero muscle weakness in 3 of the patients. None of the patients had a noteworthy cardiac phenotype, although the ages at last examination only ranged from 5 to 19 years. In addition, ophthalmoplegia, a common finding in the previously defined CNM cases, was not present. One patient (1044-1) was found to have moderately elevated serum creatine kinase levels and was considered to carry a clinical diagnosis of possible muscular dystrophy, but his clinical course was nonprogressive, and muscle pathology was distinctly nondystrophic and consistent with CNM, leading to referral by his clinician to this research study focused on CNM.
Table 1.
Clinical details of centronuclear myopathy cases with TTN mutations
Histologic features of muscle biopsies.
A blinded evaluation of the muscle biopsy slides by a single neuropathologist (M.W.L.) confirmed that all 5 biopsies were consistent with a pathologic diagnosis of CNM. All biopsies showed marked increase in the number of fibers with internal and central nuclei, ranging from 29% to 86%, compared with approximately 3% observed in healthy muscles (figure 1, A–E). In contrast to the typical findings of a single centrally placed nucleus in myofibers of patients with MTM1 or DNM2 mutations, nearly all cells in the biopsy from patient 966-1 contained multiple internalized nuclei, with 3 to 5 internalized nuclei in the majority of the fibers. Many fibers had multiple central nuclei arranged in a row (figure 1G). Adenosine triphosphatase staining revealed marked type I fiber hypotrophy and predominance (65%–85% type I fibers) (figure 1H). Oxidative stains in one case (1044-1) demonstrated occasional fibers with core-like areas or sarcoplasmic clearing around centrally located nuclei, giving a “ring-like” appearance (figure 1, I and J). Central accumulation of oxidative stains, radial arrangements, or necklace fibers, which are often present in RYR1-,32 DNM2-,33,34 and MTM1-related35 CNM, respectively, were not observed. Moderate to focally severe increases in endomysial connective tissue and infiltrating adipose tissue, suggestive of a possible dystrophic etiology, were present particularly in the biopsy from patient 966-1, but these findings were associated with the presence of a myotendinous insertion, making the diagnostic relevance of that observation uncertain. No intracytoplasmic inclusions or subsarcolemmal depositions were identified by Gomori trichrome staining (figure 1K). In addition, alkaline phosphatase staining and immunolabeling with an embryonic myosin heavy chain antibody showed no evidence of regenerating fibers. Electron microscopy demonstrated severe myofibrillar disorganization in fibers with internal nuclei and varying degrees of Z-disk streaming (figure 1, L–Q).
Figure 1. Skeletal muscle histopathology in patients with centronuclear myopathy carrying TTN mutations.

(A–E) Hematoxylin & eosin (H&E) staining of transverse muscle sections from patients 314-1, 966-1, 979-1, 1044-1, and 1093-1 show multiple cells with internal and central nuclei. (F) Percentages of fibers with central nuclei (i.e., nuclei in the geometric center of the fiber), internal nuclei (i.e., nuclei anywhere in the cytoplasm but the geometric center), and peripheral nuclei (i.e., nuclei underneath the sarcolemma, at the cell periphery) were calculated based on a count of at least 200 fibers. (G–K) Histochemical staining of muscle from patient 1044-1. (G) H&E staining on longitudinal section shows several fibers with multiple internal nuclei arranged in a row (arrows). (H) Adenosine triphosphatase (ATPase) staining at pH 4.3 demonstrates predominance and hypotrophy of darkly stained type I fibers. (I) Cytochrome c oxidase–succinate dehydrogenase (CCO-SDH) and (J) nicotinamide adenine dinucleotide–tetrazolium reductase (NADH-TR) stains show several fibers with central core-like areas devoid of oxidative reaction (arrows). (K) No inclusions or depositions were detected in Gomori trichrome staining. (L–Q) Electron micrograph of muscle from patient 1044-1 (L–O) and from patient 314-1 (P, Q). (L) Fibers with central nuclei (asterisk) demonstrate varying degrees of myofibrillar disorganization, compared with the normal sarcomeres that lie in parallel in a fiber with peripheral nuclei (bottom left corner). (M) Two internal nuclei (asterisk) arranged in a row in a fiber with severe sarcomeric disorganization, and regions devoid of mitochondria (arrow). (N) Z-disk streaming (arrows). (O–Q) Focus on regions with disintegrated sarcomeres showing disrupted I- and A-band regions (arrowheads). Black scale bars = 40 μm; white scale bars = 10 μm. Pt = patient.
Mutation analysis.
Sanger sequencing in patients and parents validated that all 5 individuals were compound heterozygotes for different disruptive TTN mutations (table 2 and figure e-1B). Frameshift mutations in patients 966-1, 979-1, 1044-1, and 1093-1 introduce premature stop codons at p.32053, p.22897, p.33402, and p.16993, respectively, and are predicted to cause a loss of the last 1,370, 10,526, 21, and 16,430 amino acids of the protein in the absence of nonsense-mediated RNA decay. Four mutations, c.32854G>C, c.37112-1G>A, c.11764+1G>A, and c.18229G>A, present in patients 314-1, 979-1, and 1093-1 are at conserved splice sites and are predicted to produce splicing defects. Only patient 966-1 had an allele not predicted to truncate the protein as his paternal allele carried 2 indels that led to the deletion of 5 amino acids in exon 83 and insertion of a proline in exon 157 of N2A titin, the isoform expressed in skeletal muscle.
Table 2.
Summary of TTN mutations identified in patients with centronuclear myopathy
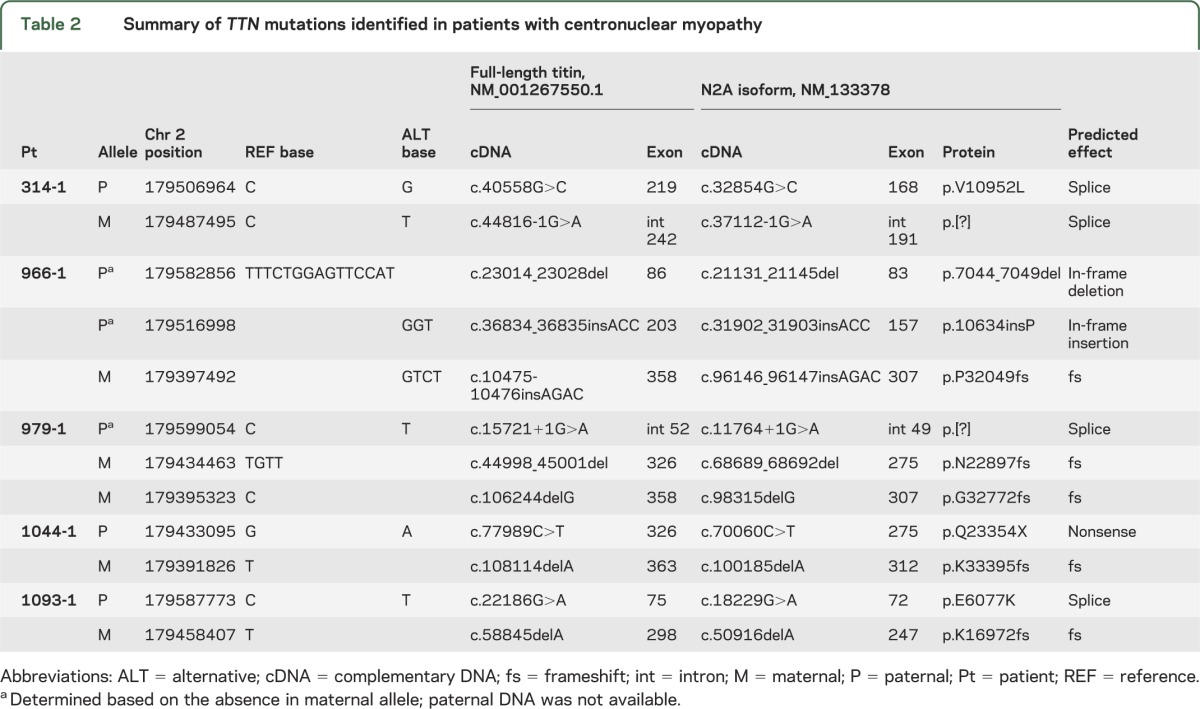
Two mutations identified in our patients, c.100185delA and c.32854G>C, have previously been associated with tibial muscular dystrophy and adult-onset cardiomyopathy, respectively, in the heterozygous state.16,25 Tibial muscular dystrophy presents in the fifth to sixth decades of life with preferential weakness in the tibialis anterior muscles.16 The mother of patient 1044-1, heterozygous for c.100185delA mutation, was adopted with no known family history, and had no clinical signs of muscle weakness at the age of 34 years. Extensive cardiac and muscle examination performed on the father of patient 314-1, heterozygous for c.32854G>C mutation, showed no evidence of cardiomyopathy or skeletal myopathy at the age of 46.5 years. However, similar clinical examinations of this patient's 44-year-old mother, heterozygous for c.37112-1G>A mutation, revealed mild subclinical cardiac and skeletal myopathies, suggesting that this mutation may have a dominant effect for both diseases. The parents heterozygous for the remaining mutations had no reported skeletal muscle or cardiac defects, although extensive cardiac examination has not been performed since identification of these mutations.
Immunofluorescence analysis on patient muscle biopsies.
To determine whether these mutations altered titin structure and its localization in the sarcomere, we performed indirect immunofluorescence analysis on muscle biopsies available from patients 314-1, 1044-1, and 1093-1 using 3 epitope-specific antibodies directed against the N-terminal, A–I junction, or C-terminal regions of titin (figure 2A). Cryosections from the 3 patients and 2 age-matched controls were immunostained with each of the titin antibodies and an α-actinin-2 antibody to visualize sarcomeric organization. All patient muscles showed normal sarcomeric labeling with the titin N-terminal and A–I junction antibodies, indicating that titin was integrated into the sarcomeres and its structure was intact up to the level of the A–I junction (figure 2C). On the other hand, the antibody that recognizes an epitope located at the C-terminal end of titin showed a complete loss of this site in all 3 patient muscles. The C-terminal region of titin contains 1 of the 2 binding sites for calpain 3 (CAPN3).11,21 Binding of calpain 3 to the M-line region of titin is required for its stabilization, as mutations in this region of titin cause a decrease in the amount of calpain 3 in the muscle.19,21,36 Patient muscles demonstrated reduced or absent immunostaining with a calpain 3 antibody, indicating a loss of the calpain 3 binding region on the mutant titin proteins (figure 2C). Altogether, these results suggested that all patient muscles analyzed had truncated titin proteins lacking a significant portion of their C-terminal regions.
Figure 2. TTN mutations associated with centronuclear myopathy cause a loss of titin C-terminal region in patient muscles.

(A) Locations of the antibody epitopes used in immunofluorescence analysis are depicted on the skeletal muscle titin isoform N2A (NM_133378). The Z-disk (red), I-band (yellow), A-band (dark blue), and M-band (light blue) regions of a titin molecule spanning halfway of a sarcomere are represented. (B) Locations of the TTN mutations in each patient are represented on the N2A isoform. Arrows depict the type of mutations: frameshift (green), nonsense (red), splicing (black), in-frame insertion/deletion (blue). (C) Immunofluorescence analysis of patient and age-matched healthy control muscles. Frozen muscle sections were stained with titin N-terminal, A–I junction, C-terminal, or calpain 3 antibody (red); and with anti-α-actinin antibody (green). Staining with the control anti-α-actinin antibodies are presented in the inset lower right panels where the titin C-terminal or calpain 3 antibodies showed no signal. (D) Overview of the hybrid minigene splicing assay. Wild-type exon 168 and 192 (numbering based on N2A isoform) were cloned with their flanking introns into the pZW4 splicing reporter construct between HpaI and KpnI restriction sites. The mutations at the donor or acceptor splice sites were introduced by site-directed mutagenesis. Wild-type or mutant hybrid minigene containing plasmids were transfected into HEK293 cells, followed by RNA extraction and reverse transcriptase–PCR using primers flanking the hybrid constructs. (E) Results of the hybrid minigene splicing assay. The minigenes containing wild-type exon 168 or exon 192 were spliced correctly. The c.32854G>C mutation at the donor splice site of exon 168 caused exon skipping (left panel). The c.37112-1G>A mutation at the acceptor splice site of exon 192 resulted in the majority of transcripts to remain unspliced, as well as exon skipping or intron retention in a subset of transcripts (right panel). Scale bar = 40 μm. For = forward; mut = mutation; Pt = patient; Rev = reverse; WT = wild-type.
Because calpain 3 is necessary for the proper recruitment of AldoA and ryanodine receptors to the triads,37 we also investigated the level and localization of AldoA and ryanodine receptors in patient muscles. While the labeling in muscle from patients 1044-1 and 1093-1 for both proteins was comparable with the controls, muscle from patient 314-1 showed reduced immunostaining for AldoA, consistent with the fact that mutations in this patient lie in the vicinity of the titin N2-line region, where AldoA-associated calpain 3 interacts with titin at the triads (figure e-2).37
Splicing assay.
To further support our interpretation of potential pathogenicity for several of these mutations, we examined the possible effects of TTN mutations that were predicted to cause splicing defects. Four mutations were found to reduce the consensus splice site agreement to background levels (table e-1). Two of these mutations were present in patient 314-1. Because we had a limited amount of tissue specimen available from this patient, we used an in vitro hybrid minigene splicing assay to test whether the mutations altered splicing (figure 2D). The c.32854G>C mutation within a donor splice site resulted in skipping of exon 168, whereas the c.37112-1G>A mutation disrupting the consensus acceptor splice site of exon 192 caused the majority of transcripts to remain unspliced, as well as skipping of exon 192 or retaining of intron 191 in a subset of transcripts (figure 2E). Because all of these resulting transcripts are out of frame, they would either become targets of nonsense-mediated decay or when translated produce truncated titin proteins.
Gel electrophoresis of patient muscle proteins.
To establish whether the mutations were indeed associated with reduced titin size and/or quantity, we analyzed titin proteins in patient muscles by sodium dodecyl sulfate–agarose gel electrophoresis (figure 3). While the size of the largest titin isoform in the muscle of patient 1044-1 was not significantly different from the full-length titin in the control human soleus muscle (3.8 MDa), the patient had a second protein band with an estimated molecular mass of 2.7 MDa (figure 3, left lane) and a minor amount of T2. T2 is one of the most frequently observed degradation products of titin that contains titin's A-band segment and a small proportion of the I-band segment with a total molecular mass of ∼2.1 MDa.38 We suspect that the ∼3.8-MDa isoform expressed in patient 1044-1 is likely the maternal allele product lacking only the last 21 amino acids, because the resulting protein size difference cannot be detected on the agarose gels, which have a resolution in the ∼4-MDa range of less than ∼0.1 MDa. The 2.7-MDa protein could be the paternal allele product with a nonsense mutation in exon 326, resulting in a truncated protein with a predicted molecular mass of 2.7 MDa. Patient 1093-1 had 2 titin bands that were significantly smaller than the full-length isoform, one with an estimated molecular mass of 3.5 MDa, and a second of ∼2.3 MDa. The maternal allele of patient 1093-1 has a frameshift mutation in exon 298 resulting in a predicted protein with a molecular mass of ∼2 MDa, and the paternal allele has a splice mutation in the I-band region of the molecule. We speculate that the paternal and maternal alleles produce the 3.5-MDa and the ∼2.3-MDa proteins, respectively, with the discordance between predicted and observed molecular masses possibly explained by the uncertainty in estimating the molecular mass of megadalton-sized proteins on gels. Interestingly, patient 1093-1 also had reduced nebulin levels, the second-largest sarcomeric protein and a key component of the skeletal muscle thin filament.39 Mutations in nebulin are known to cause nemaline myopathy, another form of congenital myopathy defined by muscle weakness and the presence of nemaline rods in the muscle biopsy. Patients with reduced nebulin levels have shorter thin filaments and altered sarcomeric organization.40 While the absence of nemaline rods in the muscle of patient 1093-1 excluded a diagnosis of nemaline myopathy, the disease may have been exacerbated by the secondary reduction of nebulin.
Figure 3. Reduced size and amount of titin in patients with centronuclear myopathy carrying TTN mutations.
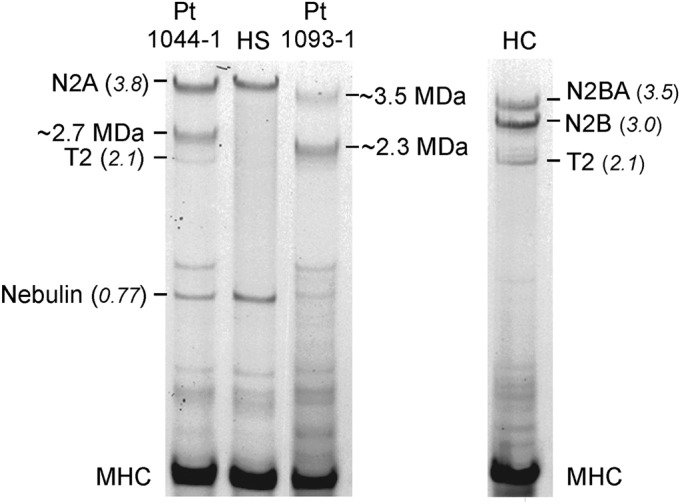
Gel electrophoresis of patient muscle proteins with control human soleus (HS) and cardiac (HC) muscle proteins. Both controls were run on the same gel to observe the various known titin isoforms present in the 2 muscle types that have been sequenced and therefore can serve as high-molecular-weight markers. Their molecular masses (in MDa) are shown in parentheses. The predominant titin isoforms expressed in skeletal (N2A, 3.8 MDa) and cardiac (N2BA, ∼3.5 MDa and N2B, 3.0 MDa) muscles are marked. The titin degradation product T2 (2.1 MDa) and nebulin (0.77 MDa) are shown as well. The known molecular masses were used to estimate the molecular masses of titin species present in the patients. Patient 1044-1: ∼3.8, ∼2.7, and ∼2.1 MDa; patient 1093-1: ∼3.5 and 2.3 MDa. Myosin heavy chain (MHC) was used as a loading control. Pt = patient.
DISCUSSION
We have identified compound heterozygous TTN mutations that all truncate or disrupt the predominant skeletal muscle isoforms of titin in 5 unrelated patients presenting with congenital myopathies diagnosed as CNM. Although mutations of MTM1 and DNM2 classically result in single and centrally placed nuclei, multiple internal nuclei, such as seen here, are more common in CNM due to RYR1 and BIN1 mutations. Thus, although one could retrospectively argue whether these 5 cases should really be classified as CNM, the fact remains that the referring neuropathologists had classified them as such, making this a useful categorization from a practical standpoint. Our results suggest that the different TTN mutations in our patients cause truncated proteins resulting in titin molecules that are devoid of the functions performed by titin's M-band segment, including structural and mechano-sensing roles. The discovery of TTN mutations associated with excessive central nucleation, reminiscent of CNM, does not necessarily indicate similar pathogenetic mechanisms. The 4 previously known CNM genes all encode proteins with a common functional theme: triad structure and function. On the other hand, titin is required for sarcomere assembly and keeping the contractile apparatus in place by providing binding sites for numerous components of the excitation-contraction coupling machinery. The electron micrographs of patient muscles show disintegrated sarcomeres with disrupted I- and A-band regions in fibers with central nuclei, suggesting that the lack of interaction sites for various titin-binding partners may cause failure to maintain the normal sarcomeric structure. Furthermore, we observed a reduction in the levels of calpain 3 and nebulin, 2 proteins that interact with titin, raising the possibility that the secondary reduction in their levels may contribute to the disease pathogenesis. Our results have important implications for guiding the clinical care of patients with CNM carrying TTN mutations. Mutations identified in various regions of TTN are known to cause adult-onset cardiomyopathy, with an average age of diagnosis at 38 years.25 The oldest patient in our study was 19 years old when last followed. Because many of the mutations identified in our patients with CNM are also present in cardiac titin isoforms, these patients, and their parents, should be closely monitored for potential cardiac issues in the future.
Supplementary Material
ACKNOWLEDGMENT
The authors thank the patients and families for their enrollment and their physicians for contributing the individuals to our study, and Dr. David Margulies and the Gene Partnership Program at Boston Children's Hospital for facilitating whole-genome sequencing of DNA from one proband.
GLOSSARY
- AldoA
aldolase A
- CNM
centronuclear myopathy
Footnotes
Editorial, page 1189
Supplemental data at www.neurology.org
AUTHOR CONTRIBUTIONS
O.C.-B., P.B.A., and A.H.B. conceived the project and performed next-generation sequencing analyses. O.C.-B. designed and performed most experiments and data analyses, with input from P.B.A. and A.H.B. C.H. and H.G. performed protein gel electrophoresis. E.T.D. and L.C.S. coordinated patient recruitment and collected medical history. R.S. and W.G.F. performed splicing assays. K.S.-A., K.M., N.V., and J.L. assisted with exome and genome sequencing analyses. S.T.I., P.B.S., N.S., and J.M.D. clinically examined patients. M.W.L reviewed the histopathology. O.C.-B. and A.H.B. wrote the manuscript with input from all authors.
STUDY FUNDING
Supported by the Dubai-Harvard Foundation for Medical Research postdoctoral fellowship and Schlumberger Foundation Faculty for the Future grant to O.C.-B., NIH R01 AR044345 to A.H.B., R01 AR05387 to H.G., the Muscular Dystrophy Association (United States) MDA201302 to A.H.B. and MDA186985 to J.L., K08 AR59750 and L40 AR057721 to M.W.L., Agence Nationale Recherche ANR-11-BSV1-026 and the Myotubular Trust to J.L., the Joshua Frase Foundation, and the Lee and Penny Anderson Family Foundation.
DISCLOSURE
The authors report no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Jungbluth H, Wallgren-Pettersson C, Laporte J. Centronuclear (myotubular) myopathy. Orphanet J Rare Dis 2008;3:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Romero NB, Bitoun M. Centronuclear myopathies. Semin Pediatr Neurol 2011;18:250–256 [DOI] [PubMed] [Google Scholar]
- 3.Romero NB. Centronuclear myopathies: a widening concept. Neuromuscul Disord 2010;20:223–228 [DOI] [PubMed] [Google Scholar]
- 4.Al-Qusairi L, Laporte J. T-tubule biogenesis and triad formation in skeletal muscle and implication in human diseases. Skelet Muscle 2011;1:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Biancalana V, Beggs AH, Das S, et al. Clinical utility gene card for: centronuclear and myotubular myopathies. Eur J Hum Genet Epub 2012 May 23. [DOI] [PMC free article] [PubMed]
- 6.Radke MH, Peng J, Wu Y, et al. Targeted deletion of titin N2B region leads to diastolic dysfunction and cardiac atrophy. Proc Natl Acad Sci USA 2007;104:3444–3449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang Z, Rolish ME, Yeo G, Tung V, Mawson M, Burge CB. Systematic identification and analysis of exonic splicing silencers. Cell 2004;119:831–845 [DOI] [PubMed] [Google Scholar]
- 8.Hudson B, Hidalgo C, Saripalli C, Granzier H. Hyperphosphorylation of mouse cardiac titin contributes to transverse aortic constriction-induced diastolic dysfunction. Circ Res 2011;109:858–866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sherry ST, Ward MH, Kholodov M, et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res 2001;29:308–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Granzier HL, Labeit S. Titin and its associated proteins: the third myofilament system of the sarcomere. Adv Protein Chem 2005;71:89–119 [DOI] [PubMed] [Google Scholar]
- 11.Linke WA. Sense and stretchability: the role of titin and titin-associated proteins in myocardial stress-sensing and mechanical dysfunction. Cardiovasc Res 2008;77:637–648 [DOI] [PubMed] [Google Scholar]
- 12.van der Ven PF, Bartsch JW, Gautel M, Jockusch H, Furst DO. A functional knock-out of titin results in defective myofibril assembly. J Cell Sci 2000;113(pt 8):1405–1414 [DOI] [PubMed] [Google Scholar]
- 13.Bang ML, Centner T, Fornoff F, et al. The complete gene sequence of titin, expression of an unusual approximately 700-kDa titin isoform, and its interaction with obscurin identify a novel Z-line to I-band linking system. Circ Res 2001;89:1065–1072 [DOI] [PubMed] [Google Scholar]
- 14.Granzier HL, Labeit S. The giant protein titin: a major player in myocardial mechanics, signaling, and disease. Circ Res 2004;94:284–295 [DOI] [PubMed] [Google Scholar]
- 15.Hackman P, Vihola A, Haravuori H, et al. Tibial muscular dystrophy is a titinopathy caused by mutations in TTN, the gene encoding the giant skeletal-muscle protein titin. Am J Hum Genet 2002;71:492–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hackman P, Marchand S, Sarparanta J, et al. Truncating mutations in C-terminal titin may cause more severe tibial muscular dystrophy (TMD). Neuromuscul Disord 2008;18:922–928 [DOI] [PubMed] [Google Scholar]
- 17.Haravuori H, Makela-Bengs P, Udd B, et al. Assignment of the tibial muscular dystrophy locus to chromosome 2q31. Am J Hum Genet 1998;62:620–626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Udd B, Vihola A, Sarparanta J, Richard I, Hackman P. Titinopathies and extension of the M-line mutation phenotype beyond distal myopathy and LGMD2J. Neurology 2005;64:636–642 [DOI] [PubMed] [Google Scholar]
- 19.Pfeffer G, Elliott HR, Griffin H, et al. Titin mutation segregates with hereditary myopathy with early respiratory failure. Brain 2012;135:1695–1713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ohlsson M, Hedberg C, Bradvik B, et al. Hereditary myopathy with early respiratory failure associated with a mutation in A-band titin. Brain 2012;135:1682–1694 [DOI] [PubMed] [Google Scholar]
- 21.Carmignac V, Salih MA, Quijano-Roy S, et al. C-terminal titin deletions cause a novel early-onset myopathy with fatal cardiomyopathy. Ann Neurol 2007;61:340–351 [DOI] [PubMed] [Google Scholar]
- 22.Itoh-Satoh M, Hayashi T, Nishi H, et al. Titin mutations as the molecular basis for dilated cardiomyopathy. Biochem Biophys Res Commun 2002;291:385–393 [DOI] [PubMed] [Google Scholar]
- 23.Gerull B, Atherton J, Geupel A, et al. Identification of a novel frameshift mutation in the giant muscle filament titin in a large Australian family with dilated cardiomyopathy. J Mol Med 2006;84:478–483 [DOI] [PubMed] [Google Scholar]
- 24.Gerull B, Gramlich M, Atherton J, et al. Mutations of TTN, encoding the giant muscle filament titin, cause familial dilated cardiomyopathy. Nat Genet 2002;30:201–204 [DOI] [PubMed] [Google Scholar]
- 25.Herman DS, Lam L, Taylor MR, et al. Truncations of titin causing dilated cardiomyopathy. N Engl J Med 2012;366:619–628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ottenheijm CA, Granzier H. Role of titin in skeletal muscle function and disease. Adv Exp Med Biol 2010;682:105–122 [DOI] [PubMed] [Google Scholar]
- 27.Richard I, Roudaut C, Marchand S, et al. Loss of calpain 3 proteolytic activity leads to muscular dystrophy and to apoptosis-associated IkappaBalpha/nuclear factor kappaB pathway perturbation in mice. J Cell Biol 2000;151:1583–1590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Salmikangas P, Mykkanen OM, Gronholm M, Heiska L, Kere J, Carpen O. Myotilin, a novel sarcomeric protein with two Ig-like domains, is encoded by a candidate gene for limb-girdle muscular dystrophy. Hum Mol Genet 1999;8:1329–1336 [DOI] [PubMed] [Google Scholar]
- 29.Richard I, Broux O, Allamand V, et al. Mutations in the proteolytic enzyme calpain 3 cause limb-girdle muscular dystrophy type 2A. Cell 1995;81:27–40 [DOI] [PubMed] [Google Scholar]
- 30.Meredith C, Herrmann R, Parry C, et al. Mutations in the slow skeletal muscle fiber myosin heavy chain gene (MYH7) cause Laing early-onset distal myopathy (MPD1). Am J Hum Genet 2004;75:703–708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moreira ES, Wiltshire TJ, Faulkner G, et al. Limb-girdle muscular dystrophy type 2G is caused by mutations in the gene encoding the sarcomeric protein telethonin. Nat Genet 2000;24:163–166 [DOI] [PubMed] [Google Scholar]
- 32.Wilmshurst JM, Lillis S, Zhou H, et al. RYR1 mutations are a common cause of congenital myopathies with central nuclei. Ann Neurol 2010;68:717–726 [DOI] [PubMed] [Google Scholar]
- 33.Bitoun M, Maugenre S, Jeannet PY, et al. Mutations in dynamin 2 cause dominant centronuclear myopathy. Nat Genet 2005;37:1207–1209 [DOI] [PubMed] [Google Scholar]
- 34.Bohm J, Biancalana V, Dechene ET, et al. Mutation spectrum in the large GTPase dynamin 2, and genotype-phenotype correlation in autosomal dominant centronuclear myopathy. Hum Mutat 2012;33:949–959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bevilacqua JA, Bitoun M, Biancalana V, et al. “Necklace” fibers, a new histological marker of late-onset MTM1-related centronuclear myopathy. Acta Neuropathol 2009;117:283–291 [DOI] [PubMed] [Google Scholar]
- 36.Haravuori H, Vihola A, Straub V, et al. Secondary calpain3 deficiency in 2q-linked muscular dystrophy: titin is the candidate gene. Neurology 2001;56:869–877 [DOI] [PubMed] [Google Scholar]
- 37.Kramerova I, Kudryashova E, Wu B, Ottenheijm C, Granzier H, Spencer MJ. Novel role of calpain-3 in the triad-associated protein complex regulating calcium release in skeletal muscle. Hum Mol Genet 2008;17:3271–3280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huff-Lonergan E, Parrish FC, Jr, Robson RM. Effects of postmortem aging time, animal age, and sex on degradation of titin and nebulin in bovine longissimus muscle. J Anim Sci 1995;73:1064–1073 [DOI] [PubMed] [Google Scholar]
- 39.Labeit S, Ottenheijm CA, Granzier H. Nebulin, a major player in muscle health and disease. FASEB J 2011;25:822–829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ottenheijm CA, Witt CC, Stienen GJ, Labeit S, Beggs AH, Granzier H. Thin filament length dysregulation contributes to muscle weakness in nemaline myopathy patients with nebulin deficiency. Hum Mol Genet 2009;18:2359–2369 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.