

Abstract
Objective:
To 1) determine, using contemporary recombinant antigen–based assays, the aquaporin-4 (AQP4)–immunoglobulin G (IgG) detection rate in sequential sera of patients assigned a clinical diagnosis of neuromyelitis optica (NMO) but initially scored negative by tissue-based indirect immunofluorescence (IIF) assay; and 2) evaluate the impact of serostatus on phenotype and outcome.
Methods:
From Mayo Clinic records (2005–2011), we identified 163 patients with NMO; 110 (67%) were seropositive by IIF and 53 (33%) were scored seronegative. Available stored sera from 49 “seronegative” patients were tested by ELISA, AQP4-transfected cell-based assay, and in-house fluorescence-activated cell sorting assay. Clinical characteristics were compared based on final serostatus.
Results:
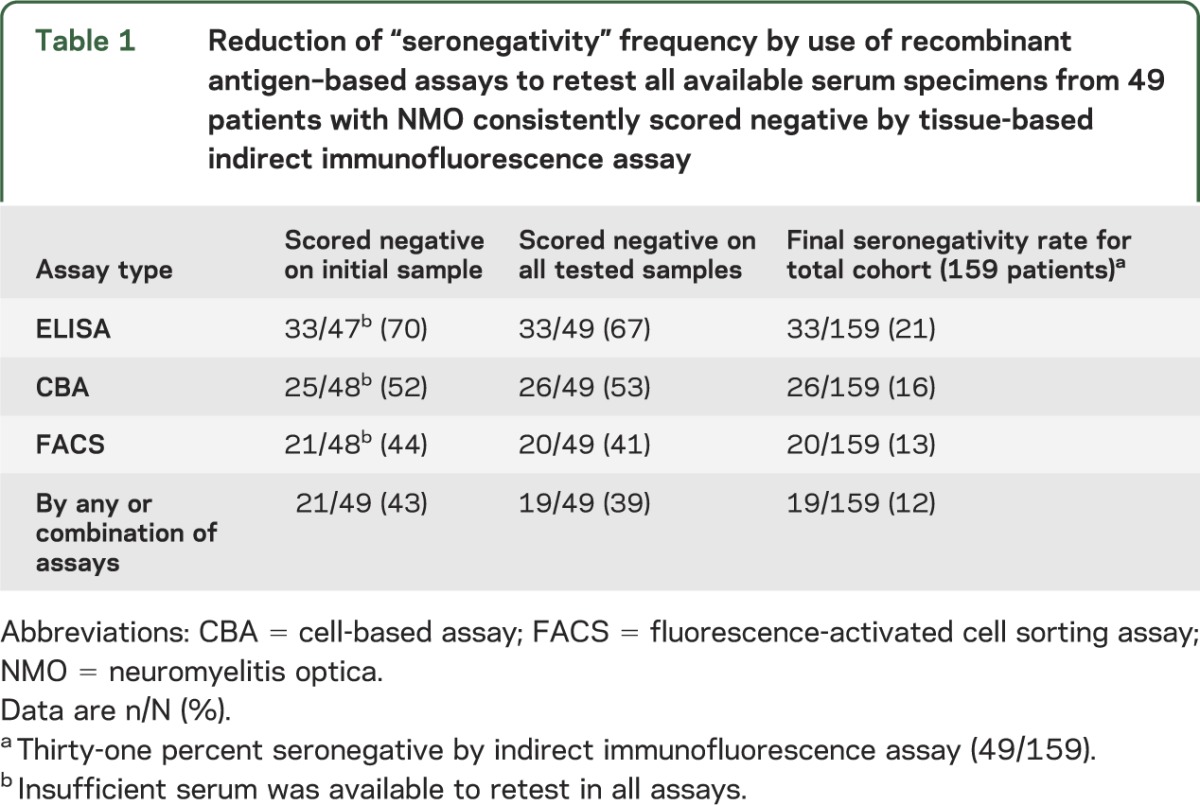
Thirty of the 49 IIF-negative patients (61%) were reclassified as seropositive, yielding an overall AQP4-IgG seropositivity rate of 88% (i.e., 12% seronegative). The fluorescence-activated cell sorting assay improved the detection rate to 87%, cell-based assay to 84%, and ELISA to 79%. The sex ratio (female to male) was 1:1 for seronegatives and 9:1 for seropositives (p < 0.0001). Simultaneous optic neuritis and transverse myelitis as onset attack type (i.e., within 30 days of each other) occurred in 32% of seronegatives and in 3.6% of seropositives (p < 0.0001). Relapse rate, disability outcome, and other clinical characteristics did not differ significantly.
Conclusions:
Serological tests using recombinant AQP4 antigen are significantly more sensitive than tissue-based IIF for detecting AQP4-IgG. Testing should precede immunotherapy; if negative, later-drawn specimens should be tested. AQP4-IgG–seronegative NMO is less frequent than previously reported and is clinically similar to AQP4-IgG–seropositive NMO.
Neuromyelitis optica (NMO) is an autoimmune inflammatory disease of the CNS that is characterized by recurrent episodes of optic neuritis (ON) and transverse myelitis (TM). Morbidity (blindness, paraplegia, and respiratory failure) is cumulative.1,2 An aquaporin-4 (AQP4)-specific autoantibody (AQP4–immunoglobulin G [IgG]) distinguishes NMO and partial or inaugural forms (NMO spectrum disorders [NMOSD]) from multiple sclerosis.2,3
A majority of patients with NMO are AQP4-IgG seropositive. Seronegativity rates vary among reported studies because of diagnostic inaccuracy, differing sensitivities of serological assays, and immunosuppressant therapy.4–6 A recent international collaborative comparison of the sensitivities of currently used assay methodologies (indirect immunofluorescence [IIF], cell-based assay [CBA], ELISA, immunoprecipitation assay, and fluorescence-activated cell sorting [FACS] assay) confirmed on a blinded basis that IIF assay was less sensitive than second-generation recombinant antigen–based assays.7 Assay insensitivity overestimates the frequency of seronegativity and invalidates phenotypic comparisons.
Herein, we report results of retesting, by recombinant antigen–based assays, stored original serum specimens (and subsequently available specimens) obtained from Mayo Clinic patients with NMO diagnosis and negative tissue-based IIF assay results. We also evaluated factors potentially contributing to false-negative results, and compared clinical characteristics and NMO phenotype of patients according to final serostatus.
METHODS
Standard protocol approvals, registrations, and patient consents.
The study protocol was reviewed and approved by the Mayo Clinic Institutional Review Board (IRB 08-006647, IRB 08-007846). Only patients providing consent for research studies were included.
Patients.
From October 1, 2005 to November 30, 2011, the Mayo Clinic Neuroimmunology Laboratory was the only facility offering, on a service basis, a validated IIF assay for AQP4-IgG. Sera from 5,349 Mayo Clinic patients were tested. By searching the Mayo Clinic computerized central diagnostic index, we ascertained that 699 patients were assigned in that period a diagnosis of “NMO,” “NMOSD,” “Devic disease,” “Devic syndrome,” “myelitis,” “myelopathy,” “optic neuritis,” “optic neuropathy,” “clinical isolated syndrome (CIS),” “acute disseminated encephalomyelitis (ADEM),” or “CNS demyelinating disease.” Among those patients, we identified 164 who fulfilled Wingerchuk diagnostic criteria (either 1999 or 2006 [excluding antibody status]).1,8 One patient refused consent for research studies.
AQP4-IgG assays.
Serum samples were collected at clinic visits, particularly at acute exacerbations. All testing was performed in blinded conditions. The IIF substrate was a composite cryosection of normal adult mouse brain, kidney, and gut tissues.3,5 Patients whose sera were scored positive by IIF were not retested by other assays because of the 99% specificity of IIF for NMO. All serial serum samples yielding a negative IIF result were retested using commercial recombinant human AQP4 ELISA kits (M1 isoform; RSR/Kronus, Ltd.), M1 transfected CBA (immunofluorescence slides; Euroimmun, Lubeck, Germany), and an in-house FACS assay (M23 transfected cells). ELISA assay was scored positive when the result was ≥5 U/mL; CBA was scored positive or negative.
For FACS assay, human embryonic kidney cells (HEK 293) were transfected transiently with a plasmid encoding both green fluorescent protein (GFP) and the M23 isoform of human AQP4. After 36 hours, FcR Blocking Reagent (Miltenyi Biotec cat. no. 130-059-901) was added to the mixed cell population (nontransfected and transfected [expressing surface AQP4 and cytoplasmic GFP] and nontransfected). Next, patient serum was added (1:5 dilution; heat-inactivated 56°C, 30 minutes) to 100,000 cells (100 µL). After incubation (4°C, 15 minutes) and washing, AlexaFluor 647–tagged anti-human IgG was added (Invitrogen cat. no A21445, 1:500 dilution). The cells were washed, fixed, and examined by flow cytometer (BD FACS Canto; Becton, Dickinson and Company, San Jose, CA). Results were computed using acquisition/analysis software. Binding of a patient's IgG to the AQP4-transfected (GFP-positive) cells was measured in terms of the intensity of AlexaFluor 647 fluorescence. The median fluorescence intensity (MFI) value for AlexaFluor 647 corresponding to IgG bound to AQP4-transfected cells was compared with the MFI value for that patient's IgG binding to nontransfected control cells in the same aliquot. Resulting ratios (MFI AQP4-transfected cells/MFI nontransfected cells) were reported as AQP4-IgG Binding Index; values of ≥3 were considered positive.
Statistical analyses.
Characteristics were compared between patient groups (i.e., seropositive vs seronegative) using χ2 (or Fisher exact) tests for categorical data and 2-sample t tests (or Wilcoxon rank sum) for continuous data. Disease characteristics over the course per patient were compared using regression models with generalized estimating equations—logistic regression for binary data and linear regression for continuous data. Therapy “intervals” were defined for each patient to correspond to periods during which long-term immunosuppressive therapy was (or was not) being used. Relapse rates (number of attacks per year) were compared between on-therapy vs not on-therapy intervals using linear regression models with generalized estimating equations. Disability and blindness outcomes were compared between groups using log-rank tests, and the risk of these outcomes was estimated using the Kaplan-Meier method. Adjusted associations of patient characteristics with these outcomes were assessed with Cox proportional hazards regression models. All statistical testing was considered as exploratory data analysis; no adjustments for multiple testing have been made. Data were analyzed using SAS version 9 statistical software (SAS Institute Inc., Cary, NC) and figures were created using R (http://www.R-project.org/). Values of p < 0.05 were considered statistically significant.
RESULTS
Two-thirds of initially “seronegative” patients with NMO were reclassifiable as “seropositive” after testing by recombinant antigen–based assays.
By IIF assay, 110 (67%) of the 163 patients with NMO were seropositive and 53 (33%) were seronegative. Stored sera were available to retest 49 of the 53 apparently seronegative cases; 30 of the 49 sera (61%) yielded positive results in one or more of the second-generation assays (figure 1; figure e-1A on the Neurology® Web site at www.neurology.org). The overall AQP4-IgG–seronegative rate (table 1) decreased from 33% to 12% (19/159); by ELISA to 21% (33/159), by CBA to 16% (26/159), and by FACS to 13% (20/159).
Figure 1. Flowchart of the NMO cohort.

Of 163 patients whose serum was tested (one or more sequential specimens) by tissue-based IIF, 53 were scored negative. For 49 with available stored specimens, 30 (61%) were found to be AQP4-IgG positive when retested by recombinant antigen–based assays. AQP4 = aquaporin-4; CBA = cell-based assay; FACS = fluorescence-activated cell sorting assay; IgG = immunoglobulin G; IIF = indirect immunofluorescence; NMO = neuromyelitis optica; QNS = insufficient serum for further evaluation.
Table 1.
Reduction of “seronegativity” frequency by use of recombinant antigen–based assays to retest all available serum specimens from 49 patients with NMO consistently scored negative by tissue-based indirect immunofluorescence assay
Testing serial specimens may increase AQP4-IgG detection rates.
Patients whose initial serum specimens are scored negative by IIF may convert to positive when subsequent samples are tested by IIF: among the 110 patients, positive by IIF, the serostatus of 7 patients changed from an initial negative result to positive after subsequent serum specimens were tested by IIF.
Patients whose initial serum specimens are scored negative by recombinant antigen–based assays may convert to positive when subsequent samples are tested: among 30 patients initially scored negative by IIF but subsequently classified as seropositive using recombinant antigen–based assays, all except 2 patients were seropositive by one or more assays on initial serum testing. For the 2 patients testing negative by all assays on the initial serum, the first was receiving immunosuppressant therapy (mycophenolate mofetil [MMF]) when the initial sample was drawn. The fourth of 6 serial specimens yielded a positive FACS result (no clinical information available for that time point). The second patient's third, fourth, and fifth of 5 specimens were positive by FACS; his second serum was drawn after 6 plasma exchanges and IV methylprednisolone (IVMP) (1 g daily, 5 consecutive days; figure e-1B).
Serostatus may convert from positive to negative with immunotherapy.
Among the 30 patients reclassified as seropositive, 20 had more than one specimen available for testing (median 2, range 2–6; intervals greater than 1 month). Five of these 20 patients (17%) converted to seronegative status during immunotherapy (IVMP [n = 1], azathioprine [n = 1], MMF [n = 3]) (see, e.g., figure e-1C).
AQP4-IgG–negative and AQP4-IgG–positive patients differ demographically.
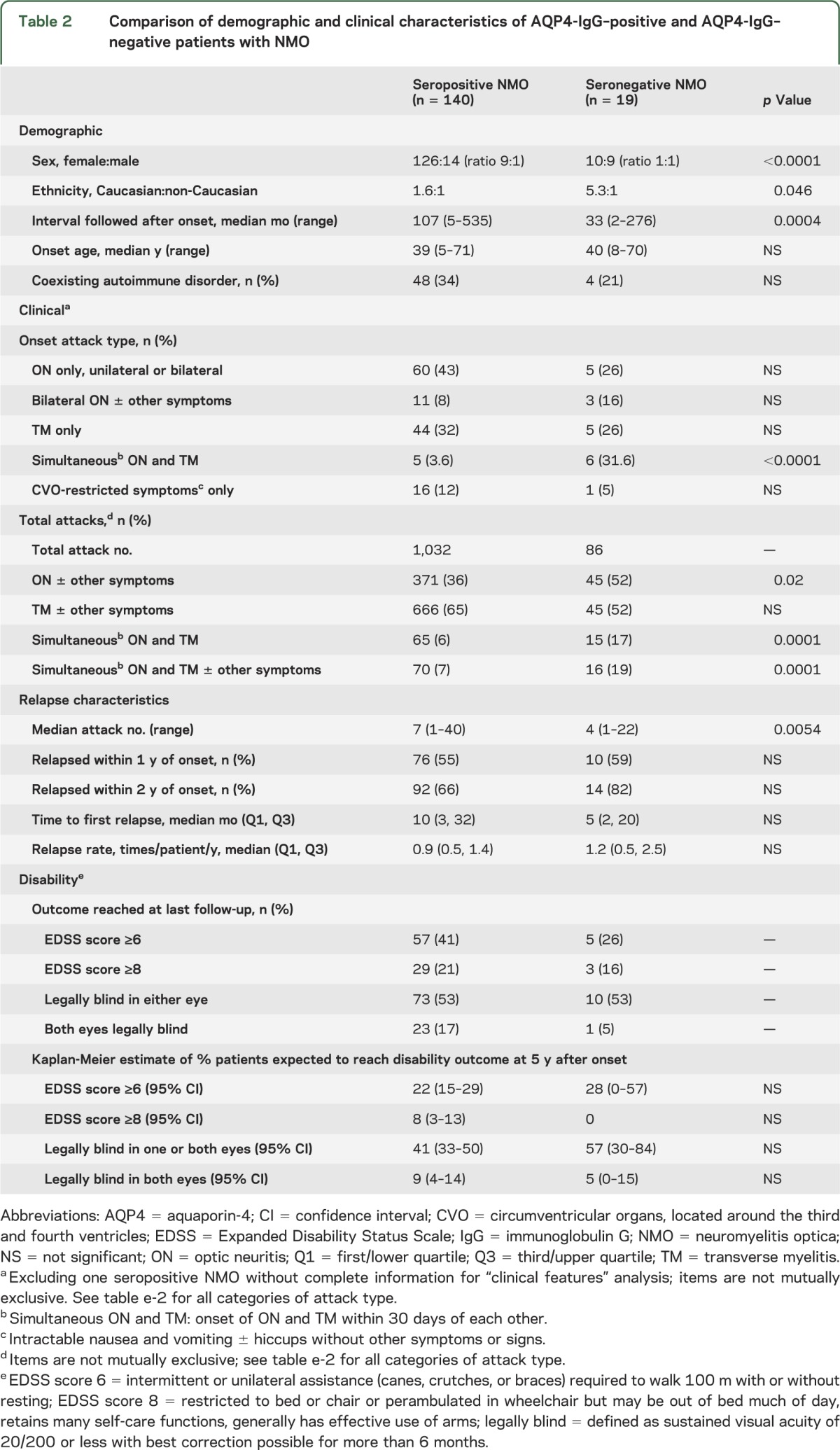
Sexes were equally represented among seronegative cases; among AQP4-IgG–positive cases the female to male ratio was 9:1 (p < 0.0001). The Caucasian to non-Caucasian ratio was higher in the seronegative group (table 2). The frequency of coexisting autoimmune diseases was similar in both groups (table e-1).
Table 2.
Comparison of demographic and clinical characteristics of AQP4-IgG–positive and AQP4-IgG–negative patients with NMO
AQP4-IgG–negative and AQP4-IgG–positive patients differ phenotypically.
Simultaneous occurrence of ON and TM at disease onset (i.e., within 30 days) was nearly 10-fold more frequent in AQP4-IgG–negative cases than in AQP4-IgG–positive cases (31.6% vs 3.6%, p < 0.0001). Four of the 5 seropositive and 4 of the 6 seronegative cases relapsed within 2 years; only 3 (1 seropositive and 2 seronegative) patients with simultaneous ON and TM at disease onset had not relapsed after less than 36 months of follow-up. Over the clinical course of the disease, ON was encountered more frequently in persistently AQP4-IgG–negative cases than in AQP4-IgG–positive cases (55% vs 36%, p = 0.02). TM was encountered more frequently, but not significantly so, in the AQP4-IgG–positive cases than in AQP4-IgG–negative cases (65% vs 55%, p = 0.35). The types of subsequent attacks (ON vs TM) were independent of the initial attack type. For example, the frequency of subsequent ON attacks was 34% for ON-onset and 33% for TM-onset patients (p = 0.95). The frequency of subsequent TM attacks was 70% for ON-onset and 71% for TM-onset patients (p = 0.66).
Serostatus did not significantly affect the interval to relapse or the relapse rate.
Three patients with only a single attack of simultaneous ON and TM (1 AQP4-IgG positive and 2 AQP4-IgG negative) had less than 3 years of follow-up from disease onset. For the remaining patients, the interval between the first and second attacks, annualized relapse rate, and frequency of relapse within 1 or 2 years of onset did not differ significantly between seropositive and seronegative patients (table 2). All seronegative patients who had at least one relapse had their first relapse within 8 years. The interval between the onset attack and the second attack exceeded 10 years for 11 (17%) of 63 seropositive patients with disease duration exceeding 10 years. None was receiving immunotherapy except for one who received IV immune globulin monthly for the first 3 years.
Serostatus does not affect attack severity or disability outcome.
Onset symptoms were deemed severe (inability to walk at nadir for TM and visual acuity 20/200 or worse in affected eye at nadir for ON) in 58% of seronegative (11/19) and 46% of seropositive (63/137) cases (p = 0.33). To minimize confounding effects of immunosuppressant therapies, we analyzed severity of the first 3 attacks. For seronegative and seropositive patients, the proportion of attacks considered severe was similar (47% vs 43%, p = 0.69).
Using Kaplan-Meier analyses, no significant difference was found between seronegative and seropositive patients regarding time to motor or visual disability. For the entire NMO cohort (n = 159), the median time to Expanded Disability Status Scale (EDSS) score of 6 is estimated to be 17 years (figure 2A). At 5 years after disease onset, 28% of seronegative and 22% of seropositive patients were expected to require a cane to walk (EDSS 6), and no seronegative and 8% of seropositive cases would be restricted to wheelchair (EDSS 8) (figure 2B). At 5 years after disease onset, approximately half the patients with NMO (57% of seronegative and 41% of seropositive patients) were expected to be legally blind (sustained visual acuity of 20/200 or less for more than 6 months, with best possible correction) in at least one eye (figure 2C), with a smaller proportion (5% of seronegative and 9% of seropositive patients) expected to be legally blind in both eyes (figure 2D). Furthermore, for seropositive NMO patients, motor disability tended to be more frequent in patients with TM-onset than in those with ON-onset (p = 0.07, figure 2E); conversely, visual disability tended to be more frequent in patients with ON-onset than in those with TM-onset (p = 0.09, figure 2F).
Figure 2. Kaplan-Meier estimates of time to motor and visual disability by AQP4-IgG serostatus.

(A) Years from onset to use of a cane (p = 0.43): at 5 years after onset, 28% of seronegative patients and 22% of seropositive patients were expected to need a cane to walk (EDSS score 6). (B) Years from onset to need for a wheelchair (p = 0.10): at 5 years after onset, no seronegative and 8% of seropositive patients were expected to be unable to walk and to need a wheelchair (EDSS score 8). (C) Years from onset to legal blindness in at least one eye (p = 0.34): at 5 years after onset, 57% of seronegative and 41% of seropositive patients were expected to be legally blind in at least one eye. (D) Years from onset to legal blindness in both eyes (p = 0.64): at 5 years after onset, 5% of seronegative and 9% of seropositive patients were expected to be legally blind in both eyes. (E) Years from onset to use of a cane for seropositive NMO (p = 0.07): at 5 years after disease onset, 36% TM-onset vs 14% ON-onset patients were expected to need a cane. (F) Years from onset to legal blindness in at least one eye for seropositive NMO (p = 0.09): at 5 years after disease onset, 39% of TM-onset vs 55% of ON-onset patients were expected to be legally blind in at least one eye. EDSS score 6 = intermittent or unilateral assistance (canes, crutches, or braces) required to walk 100 m with or without resting; EDSS 8 = restricted to bed or chair or perambulated in wheelchair but may be out of bed much of day, retains many self-care functions, generally has effective use of arms; legal blindness = sustained visual acuity of 20/200 or less with best correction possible for more than 6 months. AQP4 = aquaporin-4; EDSS = Expanded Disability Status Scale; IgG = immunoglobulin G; NMO = neuromyelitis optica; ON = optic neuritis; TM = transverse myelitis.
Predictors of outcome in NMO.
We investigated whether type of attack at onset, sex, age at onset, ethnicity, severity of onset attack, and numbers of attacks within the first year and 2 years after onset affected development of motor or visual disability. The small sample size of the seronegative cohort precluded adjusted comparison stratified by serostatus, thus analysis was restricted to the entire NMO cohort. The risk of requiring a cane to walk increased with onset age: for every 10 years’ increase in age at onset, the risk of needing a cane increased by 32% (hazard ratio [HR] = 1.32, p = 0.006). This effect remained strong even after adjusting for the other characteristics listed above. The risk for developing legal blindness in at least one eye was higher in those with severe ON at onset (HR = 1.92 as compared with nonsevere, p = 0.004) and non-Caucasian ethnicity (HR = 1.67 as compared with Caucasian, p = 0.02). These effects also remained strong after adjusting for the other patient characteristics listed above.
Immunosuppressant therapy is associated with lower relapse rate.
A total of 1,118 attacks were recorded among the 140 seropositive and 19 seronegative patients with NMO. Immunosuppressant therapy (azathioprine, MMF, oral prednisone, monthly IVMP, rituximab, eculizumab, or combinations) was administered for half the cumulative disease duration (mean 50.3% and 48.0% of disease duration for seronegative and seropositive cases, respectively). The average annualized relapse rate for seropositive cases was 2.2 (SD: 2.7) while not on immunosuppressant therapy vs 0.7 (SD: 0.9) on therapy (p < 0.0001). The average annualized relapse rate for seronegative cases was 2.1 (SD: 2.7) while not on therapy vs 1.0 (SD: 1.6) on therapy (p = 0.44). Comparison of the effect of immunosuppressant therapy on relapse rate revealed no significant difference between AQP4-IgG–seropositive and –seronegative patients with NMO (p = 0.82).
DISCUSSION
The key observations in this study were that for patients with NMO: 1) recombinant antigen–based assays increase AQP4-IgG detection rate to approximately 90%; 2) AQP4-IgG–negative NMO is uncommon (12%); 3) timing of blood draw and immunosuppressant therapy influences serostatus; 4) sexes are equally represented among seronegative cases; 5) simultaneous TM and ON is a more common initial presentation in seronegative than in seropositive cases; 6) serostatus is not a significant predictor of relapse likelihood, attack frequency, severity, or long-term disability outcome; and 7) immunosuppressant therapy reduces relapse rate regardless of serostatus.
Serological assay methodologies currently validated for detecting AQP4-IgG have high specificity for diagnosis of NMO spectrum disorders but vary in sensitivity. Other factors contributing to different seronegativity rates reported for NMO include diagnostic inaccuracy, clinical and demographic differences, and timing of blood draws regarding disease course and immunotherapy. Using 3 different recombinant antigen–based assays, we detected AQP4-IgG in nearly two-thirds of patients with NMO initially deemed “seronegative” by IIF, reducing the seronegativity rate from 33% to 12%. This revised “seronegative” NMO rate is lower than has previously been reported.3,4,7,9–11 Both the timing of blood draw and immunosuppressant therapy affect AQP4-IgG detection. It is common for an individual's AQP4-IgG status to convert from positive to negative when sequential serum samples are tested in the setting of immunotherapy. This may be due to a reduction in antibody titer below an assay's detectable limit. Even with the most sensitive tests, the initial sample occasionally yielded a negative result, while a subsequent sample yielded a positive result. Thus, when clinical suspicion for NMO is high, retesting using recombinant antigen–based assays is warranted. The fact that fewer serial samples were available from seronegative cases than from seropositive cases in this study suggests that the value of serial testing in follow-up of apparently seronegative cases is unappreciated.
The equal sex ratio we documented for seronegative cases sharply contrasts with the 9:1 excess of women among seropositive cases. A female to male ratio of approximately 2:1 has been reported previously for seronegative NMO. It is likely that lower assay sensitivities may have overestimated the prevalence of seronegativity. Contamination of the seronegative group with “false-negative cases” would compromise the validity of phenotype comparisons.10–12 The small sample number of our patients ultimately classified as seronegative precludes definitive conclusions regarding phenotype.
Simultaneous occurrence of ON and TM at disease onset (i.e., within 30 days of each other) was more common in the seronegative group, and most patients relapsed during the follow-up period regardless of serostatus; those that did not relapse had a short duration of disease (<3 years). These data suggest that with adequate follow-up, truly monophasic NMO is rare. Contrary to an earlier report,11 our seronegative group did not have a higher frequency of bilateral ON at onset. Over the course of disease, attacks of ON (either unilateral or bilateral) occurred more frequently in seronegative cases than in seropositive cases.
Consistent with an earlier report,11 serostatus did not significantly affect motor or visual disability, but in contrast to other reports,10,12 we did not observe a difference in median relapse rate or disability outcome between seronegative and seropositive groups. Other investigators have reported that relapses were more frequent in seropositive NMO, but relapse rate data were lacking for seronegative patients with NMO.12 Phenotypic comparisons based on serostatus should be interpreted with care as the numbers of seronegative patients with NMO were small and their phenotype may be prone to selection bias for more severe or recurrent disease.
After a median follow-up of 8.3 years (mean 10.4) after disease onset, more than half of our patients were legally blind in one eye, 15% were blind in both eyes, and more than one-third could not walk without unilateral assistance. Although severe, the outcome data we now report appear more favorable than was reported by Wingerchuk et al.1 in 1999 (47% required unilateral assistance and 60% were legally blind in at least one eye after a mean interval of 7.7 years after onset). Our disability outcome estimates suggest more favorable prognosis for patients ascertained in the “post–AQP4-IgG era,” both motor and visual, and is more favorable than recently reported.13 Because disability accrual in NMO is attack-dependent,14,15 the administration of immunosuppressive therapy for at least half the cumulative disease duration of our entire patient cohort plausibly explains the more favorable outcome.
NMO is generally considered a severe inflammatory demyelinating disease, but the favorable course observed in 11 of our NMO-IgG-positive patients who did not receive long-term immunosuppressant therapies raises the question of whether a “benign” form of the disease exists. The delay between first and second attacks was 10 or more years for those patients. Furthermore, of 63 patients with disease duration exceeding 10 years, 16% had EDSS score ≤3 at last follow-up. A French study recently reported that 12% of patients with NMO followed for more than 10 years with EDSS score ≤3 had “good outcome.”16 Firm conclusions cannot be reached from clinic-based data. A population-based study is needed. Severe or extremely disabled cases may have been selectively lost to follow-up. Nevertheless, our findings agree with the French experience16 that some patients with NMO may have a favorable clinical course and remain free of significant disability for a long period. Unfortunately, because there are no reliable prognostic biomarkers in NMO, it is not possible to predict outcome or clinical course at any stage of the disease. Therefore, attack prevention therapies should be initiated as early as possible regardless of the initial presentation, course of disease, or degree of disability at the time of diagnosis.
Supplementary Material
ACKNOWLEDGMENT
The authors thank John Schmeling and Eric Jedynak for assistance with serological assays and data analysis and Karen Brekke for providing study coordination.
GLOSSARY
- AQP4
aquaporin-4
- CBA
cell-based assay
- EDSS
Expanded Disability Status Scale
- FACS
fluorescence-activated cell sorting
- GFP
green fluorescent protein
- HR
hazard ratio
- IgG
immunoglobulin G
- IIF
indirect immunofluorescence
- IVMP
IV methylprednisolone
- MFI
median fluorescence intensity
- MMF
mycophenolate mofetil
- NMO
neuromyelitis optica
- NMOSD
neuromyelitis optica spectrum disorder
- ON
optic neuritis
- TM
transverse myelitis
Footnotes
Editorial, page 1186
Supplemental data at www.neurology.org
AUTHOR CONTRIBUTIONS
Study design and conceptualization: Y.J., S.J.P. Drafting of manuscript: Y.J., S.J.P. Acquisition, analysis, and interpretation of data: Y.J., J.P.F., V.A.L., A.M.L.Q., A.M., C.C., R.I., B.G.W., D.M.W., E.A.S., C.F.L., S.J.P. Statistical analysis: Y.J., S.M.J., C.Y.S. Critical revision of the manuscript: Y.J., V.A.L., S.J.P. Obtained funding: S.J.P.
STUDY FUNDING
Supported by the Mayo Clinic Foundation, the Guthy-Jackson Charitable Foundation, and the NIH (R01-NS065829).
DISCLOSURE
Y. Jiao and J. Fryer report no disclosures. V. Lennon is a named inventor on 2 patent applications filed by Mayo Foundation for Medical Education and Research that relate to aquaporin-4 (AQP4) autoantibody and its application to cancer and functional assays for its detection. She shares in royalties from marketing of kits for detecting AQP4-IgG. Royalties received to date by Dr. Lennon and Mayo Clinic exceed the federal threshold for significant financial interest. Serological testing for neural autoantibodies is offered on a service basis by Mayo Collaborative Service, Inc., an agency of Mayo Foundation. Neither Dr. Lennon nor her laboratory benefit financially from this testing. Dr. Lennon has received research support from the Guthy-Jackson Charitable Foundation and the NIH (R01-DK71209, P01-DK068055, and R01-NS065829). S. Jenkins and A. Quek report no disclosures. C. Smith received financial support for research activities from Abbott Laboratories. A. McKeon receives research support from the Guthy-Jackson Charitable Foundation. C. Costanzi and R. Iorio report no disclosures B. Weinshenker serves on data safety monitoring boards for Novartis, Biogen Idec, and Mitsubishi Pharmaceuticals; serves on the editorial boards of the Canadian Journal of Neurological Sciences and the Turkish Journal of Neurology; receives license royalties (<$5,000 to date) from RSR Ltd. for marketing of kits for the detection of AQP4 antibodies as a diagnostic aid for neuromyelitis optica. He has received consulting fees from Asahi Kasei Medical Company, GlaxoSmithKline, Ono Pharmaceuticals, and Elan Pharmaceuticals. D. Wingerchuk has received research support from Genentech, Genzyme, Alexion, and CaridianBCT. E. Shuster reports no disclosures C. Lucchinetti shares in royalties from marketing of kits for detecting AQP4 autoantibody and from the sale of Blue Books of Neurology: Multiple Sclerosis 3 (Saunders Elsevier, 2010); she receives research support from the NIH (RO1-NS49577), the Guthy-Jackson Charitable Foundation, and the National Multiple Sclerosis Society (RG 3185-B-3). S. Pittock has received no royalties to date but may accrue revenue for patents relating to AQP4 antibodies for diagnosis of neuromyelitis optica and AQP4 autoantibody as a cancer marker. He has received research support from the Guthy-Jackson Charitable Foundation, Alexion Pharmaceuticals, Inc., and the NIH (R01-NS065829). Go to Neurology.org for full disclosures.
REFERENCES
- 1.Wingerchuk DM, Hogancamp WF, O'Brien PC, Weinshenker BG. The clinical course of neuromyelitis optica (Devic's syndrome). Neurology 1999;53:1107–1114 [DOI] [PubMed] [Google Scholar]
- 2.Wingerchuk DM, Lennon VA, Lucchinetti CF, Pittock SJ, Weinshenker BG. The spectrum of neuromyelitis optica. Lancet Neurol 2007;6:805–815 [DOI] [PubMed] [Google Scholar]
- 3.Lennon VA, Wingerchuk DM, Kryzer TJ, et al. A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet 2004;364:2106–2112 [DOI] [PubMed] [Google Scholar]
- 4.Hayakawa S, Mori M, Okuta A, et al. Neuromyelitis optica and anti-aquaporin-4 antibodies measured by an enzyme-linked immunosorbent assay. J Neuroimmunol 2008;196:181–187 [DOI] [PubMed] [Google Scholar]
- 5.McKeon A, Fryer JP, Apiwattanakul M, et al. Diagnosis of neuromyelitis spectrum disorders: comparative sensitivities and specificities of immunohistochemical and immunoprecipitation assays. Arch Neurol 2009;66:1134–1138 [DOI] [PubMed] [Google Scholar]
- 6.De Vidi I, Boursier G, Delouche N, et al. Strategy for anti-aquaporin-4 auto-antibody identification and quantification using a new cell-based assay. Clin Immunol 2011;138:239–246 [DOI] [PubMed] [Google Scholar]
- 7.Waters PJ, McKeon A, Leite MI, et al. Serologic diagnosis of NMO: a multicenter comparison of aquaporin-4-IgG assays. Neurology 2012;78:665–671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wingerchuk DM, Lennon VA, Pittock SJ, Lucchinetti CF, Weinshenker BG. Revised diagnostic criteria for neuromyelitis optica. Neurology 2006;66:1485–1489 [DOI] [PubMed] [Google Scholar]
- 9.Bichuetti DB, Oliveira EM, Souza NA, Rivero RL, Gabbai AA. Neuromyelitis optica in Brazil: a study on clinical and prognostic factors. Mult Scler 2009;15:613–619 [DOI] [PubMed] [Google Scholar]
- 10.Akman-Demir G, Tuzun E, Waters P, et al. Prognostic implications of aquaporin-4 antibody status in neuromyelitis optica patients. J Neurol 2011;258:464–470 [DOI] [PubMed] [Google Scholar]
- 11.Jarius S, Ruprecht K, Wildemann B, et al. Contrasting disease patterns in seropositive and seronegative neuromyelitis optica: a multicentre study of 175 patients. J Neuroinflammation 2012;9:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang Y, Huang DH, Wu WP, Wu L, Chen LF, Wu Q. The role of aquaporin-4 antibodies in Chinese patients with neuromyelitis optica. J Clin Neurosci 2013;20:94–98 [DOI] [PubMed] [Google Scholar]
- 13.Collongues N, Marignier R, Zephir H, et al. Neuromyelitis optica in France: a multicenter study of 125 patients. Neurology 2010;74:736–742 [DOI] [PubMed] [Google Scholar]
- 14.Morrow MJ, Wingerchuk D. Neuromyelitis optica. J Neuroophthalmol 2012;32:154–166 [DOI] [PubMed] [Google Scholar]
- 15.Wingerchuk DM, Weinshenker BG. Neuromyelitis optica: clinical predictors of a relapsing course and survival. Neurology 2003;60:848–853 [DOI] [PubMed] [Google Scholar]
- 16.Collongues N, Cabre P, Marignier R, et al. A benign form of neuromyelitis optica: does it exist? Arch Neurol 2011;68:918–924 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.