

Abstract
FMN adenylyltransferase (FMNAT) is an essential enzyme catalyzing the last step of a two-step pathway converting riboflavin (vitamin B2) to FAD, the ubiquitous flavocoenzyme. A structure-based mutagenesis and steady-state kinetic analysis of yeast FMNAT unexpectedly revealed that mutant D181A had a much faster turnover rate than the wild type enzyme. Product inhibition analysis showed that wild type FMNAT is strongly inhibited by FAD, whereas D181A mutant enzyme has an attenuated product inhibition. These results provide a structural basis for the product inhibition of the enzyme and suggest that product release may be the rate-limiting step of the reaction.
Flavocoenzymes, including flavin mononucleotide (FMN) and flavin adenine dinucleotide (FAD), are versatile redox cofactors involved in many fundamental cellular processes in all living organisms. They participate in energy production, metabolism, light sensing, DNA repair, chromatin remodeling, protein folding, neural development, and circadian rhythm regulation (1–4). In mammals, FAD is synthesized from riboflavin (vitamin B2) obtained from the diet (5, 6) via two enzymatic steps catalyzed by riboflavin kinase (RFK, EC 2.7.1.26) and FMN adenylyltransferase (FMNAT,EC 2.7.7.2). Phosphorylation of riboflavin by RFK is crucial for specific absorption of the vitamin and is the physiologically rate-limiting step in the biosynthesis of flavocoenzymes(7–9), whereas product (FAD) feedback inhibition was observed for mammalian FMNAT, suggesting that biosynthesis of FAD is also regulated at the FMNAT reaction step (10, 11).
We have previously determined crystal structures of a eukaryotic FMNAT from a yeast species Candida glabrata (CgFMNAT) and its complexes with substrates (FMN and ATP) and products (FAD and pyrophosphate) (12). Eukaryotic FMNAT is related to phosphoadenosine phosphosulfate (PAPS) reductase family proteins (13, 14) and contains a core domain with a modified Rossman-fold topology and a C-terminal extension (12). The substrate binding and catalytic site is located at the interface of the two domains. Most prominently, the isoalloxazine ring of FMN substrate binds to one face of the central β-sheet on the same side as the adenine moiety of ATP, constituting a novel flavin-binding site unseen in any other flavin binding proteins (12, 15). Steady-state kinetics analysis of CgFMNAT showed that although the enzyme apparently binds its substrates with high affinity (the Km’s for FMN and ATP are 0.76 ± 0.15 μM and 10.7 ± 2.3 μM, respectively), the overall turn-over rate is very slow (kcat of 0.087 s−1). Previous kinetic studies on endogenous mammalian FMNAT indicated that the enzyme is product inhibited, and the Ki values of FAD (0.75–1.30 μM) are close to the estimated free FAD concentration (0.4 μM) in the cell (10). It has been suggested that biosynthesis of FAD is regulated at the FMNAT reaction step through a product feedback inhibition mechanism (10).
In the present study, we carried out structure-based mutagenesis and kinetics analysis of CgFMNAT to investigate the role of a number of active site residues and to understand the catalytic mechanism of eukaryotic FMNAT. The crystal structures of CgFMNAT revealed details of the unique active site arrangement and substrate binding mode of the enzyme (12, 16) (Figure 1A). Several structural elements, collectively termed “flavin motif”, are involved in flavin ligand binding. In particular, the isoalloxazine ring is sandwiched between the indole ring of Trp184 and the planar guanidinium group of Arg189, while the hydrophilic pyrimidine ring forms two specific hydrogen bonds between its C4 carbonyl and the main chain amide of Asp181, and between its N3 amide and the side chain of Asp181, respectively. Additionally, residues Asn62, Asp66, Asp168, and Arg297 are found to interact either with ATP phosphate groups, or to coordinate the catalytic Mg2+ ion either directly or indirectly through water molecules (Figure 1A).
Figure 1.
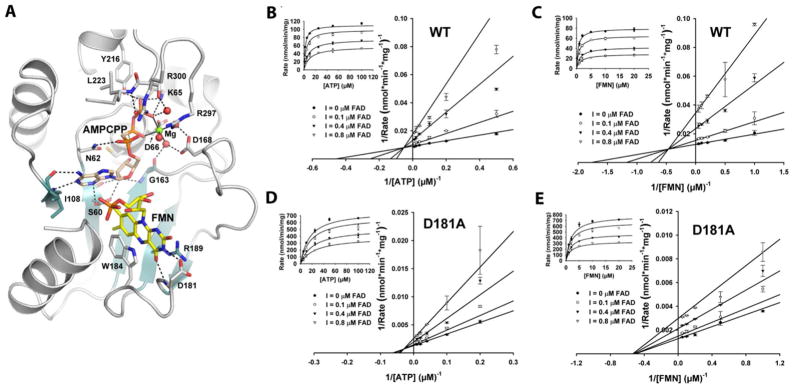
Structure-based mutagenesis and product inhibition analysis of CgFMNAT. (A) Active site configuration and interactions with substrate FMN, and ATP analog AMPCPP. The bound Mg2+ ion is shown as a green sphere and water ligands are shown as red spheres. Hydrogen bonds are shown as dash lines and metal ligand interactions are shown as solid lines. (B) and (C) Steady-state kinetics of product inhibition of wt CgFMNAT by FAD against ATP and FMN. (D) and (E) Steady-state kinetics of product inhibition of CgFMNAT D181A mutant by FAD against ATP and FMN. The rates were globally fitted to the velocity equation for general inhibition mode (Eq.(2) in the Supporting information).
Based on these structural observations, we selected a set of active site residues for mutagenesis studies to investigate their roles in substrate binding and/or catalysis (Table S1 of the Supporting Information). Under the wild-type (wt) CgFMNAT saturation condition, the specific activities of the mutants were determined and are summarized in Table S2 of the Supporting Information. Mutation of the PP-loop motif residue Asn62 to either Ala or Ser resulted in a moderate decrease of activity, while mutation of the Mg2+ ion coordinating Asp66 to an Ala resulted in a practically inactive enzyme, indicating the essential role of Asp66 to directly ligate to the catalytic Mg2+ ion. Similar to D66A but to a lesser extent, the specific activity of D168A mutant is reduced by over 90%, indicating that the interaction with the two Mg2+ ion coordinating waters by Asp168 is important for the catalytic activity of the enzyme. Interestingly, mutations of the two Arg residues, Arg297 and adjacent Arg300 near the C-terminus of the protein produced opposite results. While the specific activity of R300A is reduced to only ~7% of that of the wt enzyme, R297A mutant is nearly twice as active, indicating that both residues are involved in enzyme reaction but in different ways.
To explore roles of the flavin motif residues in FMN substrate binding, both W184A and D181A mutants were designed to weaken the interaction with FMN and were expected to have higher Km,FMN. These mutants have even more dramatic and unexpected outcomes. The loss of the stacking interaction with the isoalloxazine ring of FMN in W184A mutant reduced specific activity to ~13% of that of wt enzyme. However, the specific activity of D181A is about 8-fold higher than wt CgFMNAT. Due to the fact that D181A and R297A have increased specific activity, both are referred to as “super mutants”. Lastly, removing the last three positively charge lysine residues (Δ3K) resulted in no appreciable difference in specific activity, indicating that these residues are not involved in either substrate binding or catalysis.
To understand the effect of the active site mutants on substrate binding and turnover rate (kcat) of CgFMNAT, the apparent steady-state kinetic parameters were determined for all mutants except for D66A, which had no measureable activity. The results are listed in Table 1. As predicted, N62S mutant has a decreased kcat (0.017 vs. 0.89 s−1) accompanied by an increase in Km,ATP (from 2.0 to 5.7μM), while Km,FMN of this mutant is unchanged, consistent with the role of N62 in ATP binding (Figure 1). Interestingly, N62A mutant has a lower apparent Km,ATP than that of wt CgFMNAT (0.5 vs. 2.0μM), which was unexpected, as Asn62 interacts with ATP and N62A mutation was predicted to increase the apparent Km,ATP. It is possible that while N62A is able to bind ATP tightly, the conformation of ATP substrate may not be as optimal as that bound to the wild-type (wt) enzyme, resulting in a slightly slower kcat (0.042 vs. 0.089 s−1). D168A mutant has a significantly decreased kcat and a much higher apparent Km,ATP (Table 1), consistent with its role in coordinating the catalytic metal ion and ATP substrate interactions (Figure 1A). Arg297 was predicted to interact with either ATP or FMN phosphates (12). The apparent Km,ATP and Km,FMN of R297A mutant increased ~5 times and ~3 times, respectively (Table 1), supporting such a proposal. Despite an increase in the apparent Km’s, R297A mutant has a kcat two-times faster than the wt CgFMNAT.
Table 1.
Steady-state kinetic parameters of CgFMNAT mutants
| Protein | Km,ATP (μM) | Km,FMN (μM) | kcat (s−1) |
|---|---|---|---|
| Wild-type | 2.0 ± 0.3 | 1.0 ± 0.4 | 0.089 |
| N62A | 0.5 ± 0.2 | < 0.50 | 0.042 |
| N62S | 5.7 ± 0.9 | 1.1 ± 0.1 | 0.017 |
| D168A | 28.1 ± 7.2 | 7.1 ± 2.2 | 0.017 |
| D181A | 6.4 ± 0.4 | 1.5 ± 0.4 | 0.88 |
| W184A | 23.6 ± 4.6 | 199.4 ± 27.9 | 0.26 |
| R297A | 10.3 ± 1.6 | 3.0 ± 0.5 | 0.21 |
| R300A | 68.3 ± 11.4 | 2.3 ± 0.6 | 0.010 |
| R297A,R300A | 123.5 ± 24.0 | 9.3 ± 2.2 | 0.012 |
For the flavin motif mutants, FMN binding of W184A mutant is significantly weakened as demonstrated by a 200-fold increase of Km,FMN. The Km,ATP of the mutant is also increased (from 2.0 to 24 μM), indicating that ATP binding is also affected. Unexpectedly, the kcat of W184A mutant is not affected and even increased by about 2-folds. Most surprisingly, mutant D181A has a much increased apparent kcat, about 10 times of wt CgFMNAT, and a relatively unchanged apparent Km,FMN. It appears that Asp181, though observed to interact with FMN or FAD at the outer edge of the isoalloxazine ring, does not contribute to the initial binding of the FMN substrate, but may act as a “gate keeper” to stabilize enzyme bound FMN, as well as product FAD.
Because Asp181 is involved in the interaction with the iso-alloxazine ring of FAD or FMN and is distant from the catalytic site of the enzyme where adenylyltransfer occurs, we hypothesized that the rate-limiting step of CgFMNAT could be product release, and D181A mutant would have a less pronounced product inhibition, thus a faster turn-over rate. To test this hypothesis, the effect of FAD product inhibition on the initial rates was investigated. The results show that wt CgFMNAT is strongly inhibited by the product FAD. The Ki is 0.10 μM against ATP and 0.12 μM against FMN (Figure 1B and 1C). In the case of super mutant D181A, the Ki values of FAD against ATP and FMN are 0.39 μM and 0.67 μM (Figure 1D and 1E), respectively, which are markedly higher than that of the wt enzyme. Fitting of the general inhibition equation gives α = 8.6, suggesting that FAD acts as a mixed-type inhibitor with respect to both ATP and FMN.
These results revealed several interesting aspects of CgFMNAT that were not obvious from the crystal structures of. First, the binding of the two substrates ATP and FMN appears to be synergistic. For example, mutating residue Asp168 disrupted the coordination of the catalytic Mg2+ ion, which leads to a significant increase of Km, ATP and a decrease of kcat. Although Asp168 does not interact with FMN substrate directly, the Km, FMN also increased 7-fold, indicating that FMN substrate binding is affected by this mutation as well. Conversely, residue Trp184 stabilizes the bound FMN through stacking interaction between its indole ring and the isoalloxizine moiety of the substrate, and does not make any direct contact with the ATP substrate. While mutating Trp184 to Ala severely compromised FMN binding as the Km,FMN increased 200-fold, the Km,ATP of the mutant also increased >10-fold. Apparently, the effect of W184A mutation on the binding of ATP is indirect and likely a consequence of destabilization of FMN. From the structure of the ternary CgFMNAT-substrate complex, the binding sites for ATP and the isoalloxizine ring of FMN form a continuous channel (12). The C8 methyl group of FMN is in direct contact with the adenine ring of ATP (Figure 1A), providing additional stabilizing interactions for ATP. Likewise, the bound ATP molecule forms part of the isoalloxazine ring binding pockets and would restrict its potential movement.
The crystal structure of CgFMNAT-product complex revealed the proximity of Arg297 side chain to both phosphates of the FAD product, which comes from the ATP α-phosphate and FMN phosphate (12, 16). We speculate that Arg297 could be involved in the interaction with the phosphate groups of both substrates, and helps in their positioning for the nucleophilic attack in the adenylyltransfer reaction. Additionally, the crystal structure of Saccharomyces cerevisae FMNAT•FAD complex (16) indicates that the nearby Arg300 (Arg303 of S. cerevisae FMNAT) could also be involved in the interaction with the ATP phosphates in a similar manner as that observed in bacterial adenosine 5′-phosphosulfate (APS) reductase (17). Steady-state kinetics analysis supports the involvement of Arg297 and Arg300 in binding ATP, probably through positioning the α- or/and β-phosphate for the adenylyltransfer reaction. Interestingly, although R297A mutant has slightly weaker affinities toward both ATP and FMN, its kcat is about two-fold faster than the wt enzyme, suggesting that it also likely plays a role in the release of FAD product.
The most unexpected results are obtained with CgFMNAT mutant D181A. In wt CgFMNAT, Asp181 forms two hydrogen bonds with the hydrophilic pyrimidine ring of the bound flavin (Figure 1A). There is no conformational difference between wt CgFMNAT and D181A mutant (See Supporting Information and Table S3) except that the loss of the Asp181 side chain eliminated one of these two hydrogen bonds. However, mutant D181A results in no appreciable change in the apparent Km,FMN. Instead the kcat of D181A increased about 10-fold. Because Asp181 is distant from the catalytic site where adenylyltransfer occurs and does not markedly affect substrates binding, we thus propose that D181A mutant would affect product release and that product release is the rate limiting step of CgFMNAT-catalyzed reaction. Indeed, product inhibition analysis showed that product inhibition in D181A mutant is significantly weakened compared to wt CgFMNAT. Our data suggest that Asp181 plays a role primarily in regulating FAD product release rather than FMN substrate binding.
Supplementary Material
Acknowledgments
Funding Sources
This work was supported by a grant from American Heart Association (10GRNT4310090 to H.Z.) and from Welch Foundation (I-1505 to N.V.G.). C.H. was supported by National Institutes of Health training grant T32 GM008297.
We thank Dr. Nian Huang for help with the malachite green endpoint assay, Dr. Margret Philips for advice on product inhibition analysis.
ABBREVIATIONS
- Wt
wild-type
- AMPCPP
α, β-methyleneadenosine 5′-triphosphate
- ATP
adenosine 5′-triphosphate
- CgFMNAT
Candida glabrata FMNAT
- FAD
flavin adenine dinucleotide
- FADS
FAD synthetase
- FMN
flavin mononucleotide
- FMNAT
FMN adenylyltransferase
- RFK
riboflavin kinase
Footnotes
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interests.
ASSOCIATED CONTENT
Details of the experimental procedures and Table S1 and S2. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Müller F. Chemistry and Biochemistry of Flavoenzymes. I. CRC Press; Boca Raton, FL: 1991. [Google Scholar]
- 2.Macheroux P, Kappes B, Ealick SE. FEBS J. 2011;278:2625–2634. doi: 10.1111/j.1742-4658.2011.08202.x. [DOI] [PubMed] [Google Scholar]
- 3.Mansoorabadi SO, Thibodeaux CJ, Liu HW. J Org Chem. 2007;72:6329–6342. doi: 10.1021/jo0703092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Joosten V, van Berkel WJ. Curr Opin Chem Biol. 2007;11:195–202. doi: 10.1016/j.cbpa.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 5.Fischer M, Bacher A. Nat Prod Rep. 2005;22:324–350. doi: 10.1039/b210142b. [DOI] [PubMed] [Google Scholar]
- 6.Merrill AH, Jr, Lambeth JD, Edmondson DE, McCormick DB. Annu Rev Nutr. 1981;1:281–317. doi: 10.1146/annurev.nu.01.070181.001433. [DOI] [PubMed] [Google Scholar]
- 7.Lee SS, McCormick DB. J Nutr. 1983;113:2274–2279. doi: 10.1093/jn/113.11.2274. [DOI] [PubMed] [Google Scholar]
- 8.Kasai S, Nakano H, Maeda K, Matsui K. J Biochem (Tokyo) 1990;107:298–303. doi: 10.1093/oxfordjournals.jbchem.a123042. [DOI] [PubMed] [Google Scholar]
- 9.McCormick DB. In: Riboflavin. Rivlin RS, editor. Plenum Press; New York: 1975. pp. 153–198. [Google Scholar]
- 10.Yamada Y, Merrill AH, Jr, McCormick DB. Arch Biochem Biophys. 1990;278:125–130. doi: 10.1016/0003-9861(90)90240-y. [DOI] [PubMed] [Google Scholar]
- 11.Torchetti EM, Bonomi F, Galluccio M, Gianazza E, Giancaspero TA, Iametti S, Indiveri C, Barile M. FEBS J. 2011;278:4434–4449. doi: 10.1111/j.1742-4658.2011.08368.x. [DOI] [PubMed] [Google Scholar]
- 12.Huerta C, Borek D, Machius M, Grishin NV, Zhang H. J Mol Biol. 2009;389:388–400. doi: 10.1016/j.jmb.2009.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Savage H, Montoya G, Svensson C, Schwenn JD, Sinning I. Structure. 1997;5:895–906. doi: 10.1016/s0969-2126(97)00244-x. [DOI] [PubMed] [Google Scholar]
- 14.Bork P, Koonin EV. Proteins. 1994;20:347–355. doi: 10.1002/prot.340200407. [DOI] [PubMed] [Google Scholar]
- 15.Dym O, Eisenberg D. Protein Sci. 2001;10:1712–1728. doi: 10.1110/ps.12801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leulliot N, Blondeau K, Keller J, Ulryck N, Quevillon-Cheruel S, van Tilbeurgh H. J Mol Biol. 2010;398:641–646. doi: 10.1016/j.jmb.2010.03.040. [DOI] [PubMed] [Google Scholar]
- 17.Chartron J, Carroll KS, Shiau C, Gao H, Leary JA, Bertozzi CR, Stout CD. J Mol Biol. 2006;364:152–169. doi: 10.1016/j.jmb.2006.08. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.