

Abstract
Numerous nanocarriers of small molecules depend on either non-specific physical encapsulation or direct covalent linkage. In contrast, this manuscript explores an alternative encapsulation strategy based on high-specificity avidity between a small molecule drug and its cognate protein target fused to the corona of protein polymer nanoparticles. With the new strategy, the drug associates tightly to the carrier and releases slowly, which may decrease toxicity and promote tumor accumulation via the enhanced permeability and retention effect. To test this hypothesis, the drug Rapamycin (Rapa) was selected for its potent anti-proliferative properties, which give it immunosuppressant and anti-tumor activity. Despite its potency, Rapa has low solubility, low oral bioavailability, and rapid systemic clearance, which make it an excellent candidate for nanoparticulate drug delivery. To explore this approach, genetically engineered diblock copolymers were constructed from elastin-like polypeptides (ELPs) that assemble small (<100 nm) nanoparticles. ELPs are protein polymers of the sequence (Val-Pro-Gly-Xaa-Gly)n, where the identity of Xaa and n determine their assembly properties. Initially, a screening assay for model drug encapsulation in ELP nanoparticles was developed, which showed that Rose Bengal and Rapa have high non-specific encapsulation in the core of ELP nanoparticles with a sequence where Xaa = Ile or Phe. While excellent at entrapping these drugs, their release was relatively fast (2.2 h half-life) compared to their intended mean residence time in the human body. Having determined that Rapa can be non-specifically entrapped in the core of ELP nanoparticles, FK506 binding protein 12 (FKBP), which is the cognate protein target of Rapa, was genetically fused to the surface of these nanoparticles (FSI) to enhance their avidity towards Rapa. The fusion of FKBP to these nanoparticles slowed the terminal half-life of drug release to 57.8 h. To determine if this class of drug carriers has potential applications in vivo, FSI/Rapa was administered to mice carrying a human breast cancer model (MDA-MB-468). Compared to free drug, FSI encapsulation significantly decreased gross toxicity and enhanced the anti-cancer activity. In conclusion, protein polymer nanoparticles decorated with the cognate receptor of a high potency, low solubility drug (Rapa) efficiently improved drug loading capacity and its release. This approach has applications to the delivery of Rapa and its analogues; furthermore, this strategy has broader applications in the encapsulation, targeting, and release of other potent small molecules.
Keywords: nanoparticulate drug delivery, protein polymer, elastin-like polypeptide, Rapamycin encapsulation, high-specificity avidity
1. Introduction
Rapamycin (Rapa) is a cyclic and hydrophobic macrolide antibiotic which was discovered from a product of Streptomyces hygroscopicus in a sample of soil from Easter Island [1]. Because Rapa has great potency in suppressing immune response by inhibiting proliferation of lymphocytes, its clinical applications have shifted from anti-fungal to anti-transplant rejection formulations such as Rapamune [2]. Recently, Rapa’s anti-proliferation properties have been explored, which have led to the clinical observation of anti-tumor efficacy in malignancy of the breast, prostate, and colon [3–6]. Rapa’s anti-proliferation mechanism has also been revealed inhibition of mTOR (mammalian target of rapamycin) pathway. When bound to its cognate receptor FKBP (Kd = 0.2 nM) [7], Rapa inhibits the mTOR pathway and then sequesters cancer cells in G1 phase [8]. mTOR has essential functions in cell proliferation and growth. Screening studies confirmed that cancer cell lines having overexpression of S6K1 and expression of phosphorylated Akt e. g. MDA-MB-468 breast cancer cell are sensitive to Rapa treatment [9].
Although Rapa is extremely potent in cancer treatment, a number of drawbacks such as severe cytotoxicity, low bioavailability and rapid clearance limit wider usage of free Rapa. Recent studies have shown that Rapa and other macrolide mTOR inhibitors have serious lung toxicity by causing interstitial pneumonitis [10]. Free Rapa has poor bioavailability because of its high hydrophobicity and low water solubility (ca. 2.6 μg/mL) [11]. As result, organic solvents such as DMSO, polyethylene glycol (PEG) 400 and ethanol are presently used to deliver free Rapa [12]. However, most of these organic solvents are cytotoxic to the liver and kidney, and they may also cause hemolysis and acute hypersensitivity reactions [13, 14]. It has also been determined that free Rapa has high tendency to partition into the erythrocytes which makes it more difficult to reach intratumoral targets [15]. Therefore, a well-designed Rapa formulation is currently in demand to overcome the limitations of this potent drug.
Derived from human tropoelastin, elastin-like polypeptides (ELPs) are repetitive protein polymers with the sequence of (Val-Pro-Gly-Xaa-Gly)n, where Xaa is the guest residue and n is the length of the repetitive units [16]. ELPs undergo an inverse phase transition, which can be used to promote temperature-dependent self-assembly [17]. Below a tunable transition temperature (Tt), these ELPs are highly soluble. Above Tt they coacervate into a secondary aqueous phase, akin to a lower critical solution temperature. This phase separation can be used to purify ELPs and their fusion proteins by a process named inverse transition cycling (ITC). Here we explore two ELP diblock copolymers with a hydrophobic to hydrophilic length of n = 1:1 that form stable nanoparticles e.g. G(Val-Pro-Gly-Ile-Gly)48 (Val-Pro-Gly-Ser-Gly)48Y and G(Val-Pro-Gly-Phe-Gly)24(Val-Pro-Gly-Ser-Gly)24Y, which are named I48S48 and F24S24 respectively. These diblock copolymers form nanoparticles that are potentially excellent drug carriers because: i) they are genetically engineered, which enables precise modification and fusion to proteins; ii) they can be biosynthesized efficiently in E. coli; iii) they form monodisperse nanoparticles that do not require electrostatic stabilization; iv) they are biodegradable into non-cytotoxic amino acid components. For example, the ELP nanoparticle I48S48 is effectively biodegraded in murine hepatocytes by elastase and collagenase endopeptidases without any obvious cytotoxicity [18]. With these advantages, we raised the hypothesis that drug (Rapa) encapsulation and release from ELP nanoparticles can be modulated by high-specificity avidity binding to the cognate protein receptor for the drug (FKBP) decorated at the nanoparticle surface. In such a formulation the drug can be encapsulated into the nanoparticle core via hydrophobic interactions, similar to other micelle delivery systems; however, it would also have specific drug binding capacity at its corona. The FKBP-bound Rapa was expected to remain tightly associated with decorated nanoparticles, which may promote tumor accumulation via the enhanced permeability and retention effect. To discover the capability of drug encapsulation using ELP nanoparticles, four different fluorescent small molecules were first screened for efficient encapsulation into the hydrophobic core of the I48S48 nanoparticle. Next, encapsulation and release experiments of the model drug Rose Bengal and the clinically-approved drug Rapa were performed using ELP micelles I48S48 and F24S24. Rapa has been previously demonstrated to bind competitively to an FKBP domain on an FKBP-ELP fusion protein. [19] To further enhance drug-specific encapsulation and improve the drug release, the FKBP domain was genetically fused onto the corona of the nanoparticles formed from SI. This optimized construct FSI was then examined for Rapa encapsulation and release. Finally, cell proliferation assays and in vivo tumor regression studies were performed using FSI with encapsulated Rapa (FSI Rapa) and free Rapa in solvent (DMSO) to evaluate their relative toxicity and anti-tumor efficacy. These studies reveal an exciting new strategy for drug delivery and targeted encapsulation using genetically engineered nanoparticles.
2. Material and Methods
2.1 Materials and reagents
Rose Bengal, copper chloride, phosphate buffered saline (PBS) tablets, polyethylenimine (PEI), Congo Red, Thioflavin and Erythrosin were purchased from Sigma-Aldrich (St. Louis, MO). Rapamycin was ordered from LC Laboratories (Woburn, MA). TB dry growth medium was obtained from MO BIO Laboratories, Inc. (Carlsbad, CA). pET25b(+) vector and BLR (DE3) E-coli cell were ordered from Novagen Inc. (Madison, WI). MDA-MB-468 and MDA-MB-231 cells were purchased from the American Type Tissue Culture Collection. MDA-MB-468 cells were cultured at 37°C humidified in 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM)/F12 medium with 10% fetal bovine serum. MDA-MB-231 cells were cultured at 37°C humidified in 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal bovine serum.
2.2 Biosynthesis of ELPs
To generate the ELPs evaluated during this study, synthetic genes encoding for both ELP mono and diblock copolymers were constructed (Table 1). BLR (DE3) E-coli cells were transformed with recombinant pET25b(+) vectors containing ELP genes [19, 20]. The E-coli cells were incubated using 50mL of TB dry growth medium with 100 μg/mL ampicillin overnight at a 37°C orbital shaker. The culture was then centrifuged at 4000 rpm for 10min and the pellet was re-suspended with 5mL fresh TB dry growth medium. 500μL of the re-suspended culture media was inoculated in a 1L TB dry growth medium with 100 μg/mL ampicillin and then incubated for 24h at a 37°C orbital shaker. The E-coli cells were harvested by being centrifuged at 4000rpm for 15min and then resuspended in PBS and lysed by ultrasonication. The lysate was centrifuged at 12000rpm for 15min to remove insoluble cells debris, and nucleic acids were precipitated by PEI (0.5% w/v final concentration) and removed by 12000rpm 15min centrifugation. Inverse transition cycling (ITC), which has been described previously, was used to further purify the cell lysate containing ELPs [19, 20]. In brief, ELP was heated to 37°C and supplemented with NaCl (~ 1–3 M) to trigger phase separation. ELP aggregates were isolated by 4000rpm 10min centrifugation at 37°C. The ELP pellet was re-suspended with PBS on ice. After ELPs re-dissolved (usually takes about 5 to 20 min with gentle agitation), other insoluble proteins and contaminants were removed by 4000rpm 10min centrifugation at 4°C. Generally two to six cycles of ITC are needed to obtain pure ELP samples [21–23]. After purification, about 30 – 50mg of ELP was obtained from 1L of BLR (DE3) E-coli culture. The purity and molecular weight of ELP were examined by SDS-PAGE 4 – 20% gradient gel. 20–40 μg of ELP samples were loaded onto an SDS-PAGE gel and then stained by 10% (w/v) CuCl2 staining solution.
Table 1.
ELP protein polymers evaluated in this article
| ELP Nomenclature | a Amino acid sequence | bTt (°C) | c Calculated ELP MW (Da) | d Observed ELP MW (Da) |
|---|---|---|---|---|
| I48 | G(VPGIG)48Y | 22.0 | 20,566.9 | 20,309.9 |
| S192 | G(VPGSG)192Y | 56.5 | 80,000.8 | 79,779.5 |
| I48S48 | G(VPGIG)48(VPGSG)48Y | 27.0 | 39,643.6 | 39,435.5 |
| F24S24 | G(VPGFG)24(VPGSG)24Y | n.a. | 20,757.3 | 20,493.9 |
| SI | G(VPGSG)48(VPGIG)48Y | 27.0 | 39,643.6 | 39,445.0 |
| FSIe | FKBP-G(VPGSG)48(VPGIG)48Y | 24.5 | 51,445.2 | 51,446.8 |
ELP gene sequences confirmed by DNA sequencing from N and C terminal and diagnostic digestions.
Transition temperature (Tt) (25μM, pH 7.4) for I48, S192; Critical micelle temperature (CMT) for I48S48, SI, and FSI; The CMT of F24S24 was not applicable (n.a.) as nanoparticles form below 4 °C (Fig. 1d).
Estimated from open reading frame excluding methionine start codon
Results from matrix assisted laser desorption ion time of flight (MALDI-ToF) mass spectrometry
FKBP amino acid sequence: “MGVQVETISPGDGRTFPKRGQTCVVHYTGMLEDGKKFDSSRDRNKPFKFMLGKQEVIRGWEEGVAQMSVGQRAKLTISPDYAYGATGHPGIIPPHATLVFDVELLKLE”
2.3 Optical density measurement of ELP phase transition
Optical density was used to monitor the thermal phase behavior for all ELPs evaluated as a function of concentration. The optical density was measured by absorbance at 350nm in DU800 UV-vis spectrophotometer (Beckman Coulter, CA) under a temperature gradient of 1°C/min. The ELP transition temperature was defined at the maximum first derivative of the optical density at 350 nm. For ELP diblock copolymers, two phase transitions were observed. The first transition temperature at which small nanostructures assemble was defined as critical micelle temperature (CMT) and the second transition temperature at which the second major phase transition occurs was defined as bulk transition temperature.
2.4 Dynamic light scattering (DLS) analysis and zeta potential measurement
To estimate nanoparticle hydrodynamic radii and stability, pure 25μM ELP samples in PBS were passed through 20 nm membrane filters at 4°C. 80μL of sample was applied to a 384 pre-chilled well plate and covered with 20μL of mineral oil. Samples were measured by Wyatt Dynapro plate reader (Santa Barbara, CA) at a temperature interval of 1°C. The experiment was performed in triplicate and the data was presented as mean ± SD. To evaluate the surface charge of ELP nanoparticles, zeta potentials of FSI and SI were measured using a Malvern Zetasizer Nano ZS90 (Worcs, U.K.). Pure 100 μM FSI and SI samples in low salt PBS (10 mM NaCl, 1 mM Na2HPO4) were passed through 20 nm membrane filters at 4 °C and measured for their zeta potentials at 20 °C and 37 °C. The experiment was performed in triplicate and the data was presented as the mean value of the three measurements.
2. 5 Transmission electron microscopy (TEM) and cryogenic-transmission electron microscopy (Cryo-TEM)
TEM was used to observe the dominant nanoparticle morphology for ELP nanoparticles. For TEM procedure, 5μL of sample was pipetted on a Ted Pella carbon/formvar grid (Redding, CA). 1 % uranyl acetate was added to stain the sample and the excess liquid was removed by filter paper. The grid was dried in an incubator at 37°C and placed into a JEOL JEM 2100 laB6 microscope (Tokyo, Japan). All images were captured under 200 kV accelerating voltage. In order to observe particle morphology in solution, cryo-TEM specimens were prepared using an FEI Vitrobot (Hillsboro, OR). ELP solutions were kept in an ice bath (4°C) before processing and then raised to 37 °C immediately prior to blotting. A typical procedure involves first loading ~6 μL of the sample on a TEM grid coated with a lacey carbon film (LC325-Cu, Electron Microscopy Sciences). Then, the specimen was carefully blotted under 95% humidity following blotting parameters that were preset depending on the viscosity and concentration of the studied sample. The blotted grid was immediately transferred into liquid ethane, and stored in liquid nitrogen environment. Micrographs were acquired using FEI Tecnai 12 TWIN transmission electron microscope equipped with 16 bit 2Kx2K FEI eagle bottom mount camera (Hillsboro, OR). All images were captured under 100 kV accelerating voltage and processed using ImageJ (NIH, USA).
2.6 ELP-mediated encapsulation of a model drug, Rose Bengal
Having first determined that Rose Bengal associates with the core ELP, I48 (Supplemental Fig. S1), Rose Bengal was then encapsulated in ELP diblock copolymers, I48S48 and F24S24 (250μM) by mixing at a 1:1 ratio by mol and dialyzed at a MWCO = 10000g/mol cassette (Pierce Inc.). Rose Bengal without ELP was also added as a control. Dialysis cassettes were divided into two groups: the first group was placed in 2L PBS (pH = 7.4) and kept at 37°C and the second group were kept at 4°C. The cassettes were slowly stirred and samples were withdrawn during release. The dialysis buffer (PBS) was changed every two hours to maintain the sink conditions. The concentrations of Rose Bengal were determined by absorbance at 559nm (MEC = 90,400 M−1 cm−1). The experiment was performed in triplicate and the data was presented as mean ± SD.
2. 7 Thin-film encapsulation of an anti-proliferative drug, Rapa
To study the encapsulation of a clinically-relevant drug, diblock copolymers I48S48 and F24S24 or a soluble control S192 were desalted by dialysis in deionized H2O. Samples were lyophilized, and mixtures of ELP and Rapa molar ratios were obtained by co-dissolving ELP and Rapa in acetonitrile. Thin films were prepared by evaporating acetonitrile at 50°C using a rotary evaporator. Films were re-hydrated using sterile deionized H2O. Insoluble Rapa was removed by 10min 13000rpm centrifugation and the samples were then filtered (0.2μm membrane) before analytical Reverse Phase HPLC analysis. Samples were injected into a C-18 analytical reverse phase HPLC column (Waters Inc.) and run at a H2O: acetonitrile gradient from 30% to 100% acetonitrile. The amount of Rapa entrapped into ELP samples was determined using a standard curve. Statistical two-way ANOVA analysis was performed to compare the experimental groups (F24S24 and I48S48 with Rapa) with the control (S192 with Rapa). The experiment was performed in triplicate and the data was presented as mean ± SD.
2. 8 Rapa encapsulation and release using FSI nanoparticles
Thin film encapsulation was not feasible due to precipitation of the FKBP domain in response to drying; therefore, a two-phase solvent evaporation method was developed to encapsulate Rapa into FSI nanoparticles. An aqueous phase PBS (sterile-filtered) containing FSI was mixed with an organic phase hexane/EtOH containing Rapa in a glass vial (Molar ratio of FSI: Rapa = 1:1). The vial was stirred and heated to about 5 °C higher than the CMT. Anhydrous nitrogen gas was applied to facilitate the evaporation of the hexane/EtOH phase. A 13000rpm 10min centrifugation (above the CMT) was performed to remove any insoluble Rapa from the remaining aqueous phase. 100μL of the sample was filtered (0.2μm membrane) and injected into a C-18 reverse phase HPLC column (Waters Inc.) to analyze the amount of the Rapa in each solution. Rapa release experiments were performed by dialysis. Samples were collected in the dialysis cassette from 0 to 48 hours. Prism software was used to plot Rapa release from FSI and SI, identify different Rapa release phases, and calculate the half-life of each phase using a two-phase exponential decay model. The experiment was performed in triplicate and the data was presented as mean ± SD.
2.9 Cell proliferation assay
Cell proliferation was assessed by using 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxyme thoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazoli um (MTS) cell proliferation assay. Cells (~3000 cells per well) were plated in 96 well plates with 100μL culture medium. The cells were cultured for 24 h to adhere, and Rapa encapsulated by FSI or free Rapa (0–10μM) were added to the culture medium and incubated for 3 days. Cell proliferation was then examined by following standard protocol of CellTiler-96 Non-Radioactive Cell Proliferation Assay Kit (Promega, Madison, WI). IC50 of FSI Rapa and free Rapa in MDA-MB-468 cells were evaluated using a non-linear regression model in software Prism. The experiment was performed with N = 6 per group, and the data was presented as mean ± SD.
2.10 In vivo evaluation of FSI/Rapa in human breast cancer xenografts
All the animal experiments have been performed according to the guidelines of the American Association of Laboratory Animal Care under an approved protocol. 3×106 MDA-MB-468 cells were injected into the mammary fat pads of 7-week-old female athymic nude (nu/nu) mice (Harlan, Inc.) First treatment started on the 11th day after tumor implantation when the average tumor size reached 40mm3. The mice were randomly divided into three groups. Group 1 and 2 were received PBS and FSI Rapa (0.75mg/kg) three times a week intravenously. Group 3 was initially received free Rapa (DMSO, 0.75mg/kg) three times a week; however, due to severe weight loss after the first two treatments, free Rapa was then dosed only once a week for the rest of the study. Tumors were measured with an electronic caliper, and the sizes were calculated based on the formula a2 × b × π / 6 (a, b are the width and length of the tumor respectively). The experiment was performed with N = 9 per group. The data of tumor sizes was presented as median ± interquartile range, and the data of body weights was presented as mean ± SD.
3. Results and discussion
3.1 Characterization of ELP diblock copolymer nanoparticles
This study successfully developed a new approach to encapsulate drugs in proteinacious nanoparticles through high-avidity interactions with their cognate-human drug target decorated at their surface. To accomplish this, ELP diblock copolymers were selected to assemble these nanoparticles (Fig. 1a). To determine the critical parameters necessary for this approach, a small library of ELPs were successfully biosynthesized (Fig. 1b). ELP diblock copolymers such as I48S48 and F24S24 contain approximately 50% by mass hydrophilic blocks and 50% hydrophobic blocks. Optical density measurements and DLS analysis were performed to examine their thermal and concentration dependent assembly. I48S48 displays two different phase transitions: i) the formation of nanoparticles at the first transition at 27°C; ii) the formation of larger aggregates at the second transition at 85°C (Fig. 1c). The lower temperature transition is defined as the CMT. Depending on concentration, the CMT varies from 22°C (250 μM) to 27°C (25 μM), which is well below physiological temperatures. Alternatively, F24S24 was used to explore the properties of an ELP nanoparticle that remains assembled at all temperatures observed. The hydrophobic core ELP of F24S24 contains phenylalanine, which gives it a CMT below 4°C (Fig. 1d). To further confirm the size of the nanoparticles, DLS analysis was used to estimate the hydrodynamic radius (Fig. 1d). For I48S48, unimeric ELP molecules were present at temperatures below the CMT; however, above its CMT (27°C) 24 nm radius nanoparticles assembled. F24S24 nanoparticles were assembled even at 4°C; furthermore, their hydrodynamic radii were similar to that observed for I48S48. To decorate the surface of the nanoparticles with the FKBP domain, it was necessary to genetically engineer the folded protein domain at the amino terminal end of the ELP diblock copolymer, (FSI). An additional control ELP diblock SI was prepared as a control (Table 1). Surprisingly, neither orientation nor fusion to the FKBP domain had a substantial effect on the CMT or nanoparticle radius compared with I48S48 (Fig. 1e). To study the surface charge of FSI and SI, zeta potentials of these two nanoparticles were measured at 20 °C (not assembled) and 37 °C (assembled). The zeta potentials of FSI were 0.41mV (20 °C) and 1.97mV (37 °C) while the zeta potentials of SI were −4.31mV (20 °C) and −3.74mV (37 °C). Even in buffer with low ionic strength, the zeta potentials of both ELP nanoparticles were close to neutral in charge.
Figure 1. Design of ELP nanoparticles that carry anti-proliferative drugs-- decoration with protein drug receptors minimally influences assembly. a.

Schematic showing high-avidity interaction between a drug and its cognate human target decorated at the nanoparticle surface, while the nanoparticle core may facilitate lower-avidity drug affinity. b. SDS-PAGE of ELP library stained with copper chloride (Table 1). c. Representative optical density (I48S48) used to determine ELP-mediated assembly as a function of temperature and concentration. At 25 μM, the CMT is 27 °C, while a bulk phase transition occurs at 85 °C. d. DLS analysis of I48S48 and F24S24. Above CMT (27°C for 25 μM), I48S48 forms stable micelles with a hydrodynamic radius of 24 nm. For F24S24, nanoparticles have already assembled below 4°C. (Mean ± SD, N=3) e. DLS analysis of SI and FSI. Above their CMT, Both SI and FSI form stable nanoparticles with similar hydrodynamic radii of 24 nm (Mean ± SD, N=3)
3.2 Nanoparticle assembly is required for entrapment of Rose Bengal into plain ELP nanoparticles
Prior to the decoration of nanoparticles with FKBP, we explored how model small molecules associate with the core of plain ELP nanoparticles. Rose Bengal was selected as a model drug because it showed high association with ELP nanoparticle core I48 (Supplemental Fig. S1). Therefore, it seemed plausible that Rose Bengal can be effectively entrapped into ELP nanoparticles. To confirm this hypothesis, ELP diblock copolymers I48S48 and F24S24 were mixed with Rose Bengal at different ratios and dialyzed at 37°C and 4°C. Both I48S48 and F24S24 efficiently encapsulated Rose Bengal (Fig. 2a); however, I48S48 nanoparticles only retained the drug in the dialysis cassette when incubated at 37°C, above their CMT. The release of Rose Bengal from I48S48 at 4°C was similar to that observed when no ELP was added (Fig. 2a). This indicates that ELP nanoparticle assembly is essential for Rose Bengal encapsulation. For F24S24, nanoparticles are present at both temperatures. Therefore, the time-absorbance curves of 37°C and 4°C showed minimal differences between each other (Fig. 2a). After 6 hours of dialysis, 48% of Rose Bengal was retained inside the I48S48 nanoparticles at temperature above its CMT; however, when incubated below its CMT, less than 10% of the Rose Bengal remained (Fig. 2b). For F24S24, 76% of Rose Bengal remained entrapped by the nanoparticles, and this level was unaffected by low temperature dialysis (Fig. 2b). While both nanoparticles were able to encapsulate Rose Bengal, F24S24 nanoparticles has slower release (50% loss after 24 hours) compared to I48S48 nanoparticles (50% loss after 6 hours). One possibility is that the higher hydrophobicity of F24S24 nanoparticle core might slow drug release due to an increased affinity for the drug. Therefore, optimization of the core of ELP nanoparticles may be a possible strategy to control the rate of drug release.
Figure 2. Assembly of ELP nanoparticles slows the release of a water soluble model drug, Rose Bengal.
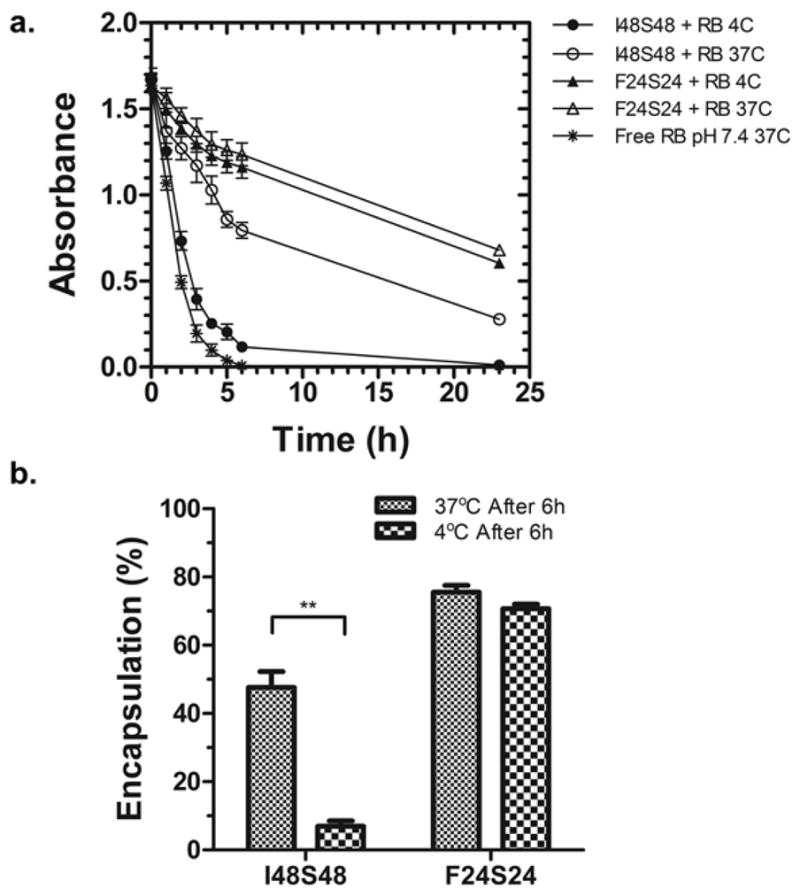
a. The absorbance was measured over time at 37°C and 4°C. F24S24 forms nanoparticles at both temperatures; however, I48S48 only forms nanoparticles at 37°C. Nanoparticle formation was associated with significantly delayed release of drug. Mean ± SD, N=3. b. Comparison of Rose Bengal encapsulation after 6h dialysis. I48S48 retained significantly more Rose Bengal at 37°C than 4°C (**p = 0.005) while no difference observed in F24S24. Mean ± SD, N=3.
3.3 The water insoluble drug, Rapa encapsulates into unmodified ELP diblock copolymer nanoparticles
After identifying the high efficiency encapsulation of the water soluble small molecule, Rose Bengal (Fig. 2), we investigated the encapsulation of a clinically viable drug with low water solubility, Rapa. Unfortunately, the encapsulation method used for Rose Bengal could not be used for Rapa because it has poor water solubility (~2.8 μM) [11]. As an alternative, film hydration was optimized for Rapa encapsulation into plain ELP diblock copolymers I48S48 and F24S24 with monomer S192 as the control (Fig. 3). In the case of F24S24: Rapa = 1:1, 50.6% Rapa was encapsulated after the film hydration. Following hydration, dialysis was used to exam the stability of the formulation. The results show that after 6 hours, 22.9% Rapa remained associated with the nanoparticles, while the soluble ELP control S192 had undetectable levels of drug (Fig. 3a). When the ratio of F24S24 to Rapa was increased to 5:1, 58.8% initial Rapa encapsulation was obtained. A two-way ANOVA was performed to examine the differences between F24S24 groups and S192 control at 2 h. There was a significant difference between F24S24: Rapa of 1:1 and 5:1 and the S192 control respectively (p<0.0001). The failure of S192 to solubilize and retain Rapa strongly suggests the necessity of a hydrophobic ELP core to encapsulate Rapa. The same experiment was carried for I48S48 nanoparticles formed and dialyzed above their CMT (Fig. 3b). Similar to F24S24, I48S48 nanoparticles display reasonably good encapsulation of Rapa. By increasing the molar ratio of I48S48 to Rapa to 10:1, the initial Rapa encapsulation reached 92.9%. This encapsulation was stable against 6 h of dialysis, whereby 66.1% of the initial drug was retained (Fig. 3b). ANOVA demonstrated that Rapa encapsulation in I48S48 was significantly higher than for S192. (p<0.0001) In conclusion, similarly to Rose Bengal, Rapa can be efficiently entrapped into the nanoparticles of ELP I48S48 and F24S24.
Figure 3. Thin film hydration with ELP nanoparticles promotes encapsulation of the hydrophobic drug, Rapa.

Different ELP: Rapa ratios were assayed for Rapa encapsulation using thin film hydration. Time 0h depicts the initial percent of encapsulated Rapa in the supernatant. S192 was used as a control because it cannot form nanoparticles. a. Two-way ANOVA demonstrated statistically significant differences between F24S24 and S192 groups. Mean ± SD, N=3. b. Alternatively, the diblock copolymer I48S48 was used to entrap Rapa at 37 °C, above its CMT. At high ratios of ELP: Rapa, drug encapsulation in the nanoparticle approaches 90%. Two-way ANOVA manifested statistically significant differences between I48S48 and S192 groups. Mean ± SD, N=3. (****p = 0.0001)
3.4 Decoration of ELP nanoparticles with FKBP protein and loading with Rapa minimally influences nanoparticle dimensions
Severe side effects of Rapa are thought to result from rapid partitioning into erythrocytes and endothelial cells [15]. Therefore, we hypothesized that ELP nanoparticles with an enhanced avidity for Rapa might prevent the rapid diffusion of drug into non-target intracellular environments, thereby increasing the tolerated dose for this drug. As a unique solution to this problem, the human cognate binding domain of Rapa FKBP was genetically fused onto the corona of SI nanoparticles (Fig. 1a), which are similar in composition to I48S48 (Table 1). The DNA sequence of FKBP was inserted to the N-terminus of the ELP, where superior protein activity has been reported [24]. DLS demonstrated that FSI fusion proteins assembled particles with a 23.8 nm hydrodynamic radius (Fig. 1e) above their CMT (24.5°C, 25μM) (Table 1). Compared to SI, the CMT of FSI shifted slightly from 27 to 24.5 °C due to the FKBP fusion; however, hydrodynamic radius and stability appeared to be nearly unaffected by the fusion of FKBP. Cryo-TEM imaging verified that the radius of FSI nanoparticles (18.5 ± 1.3 nm) was slightly larger than SI nanoparticles (15.0 ± 2.3 nm) (Fig. 4a, b). The morphology of both FSI and SI is consistent with I48S48 (Supplemental Fig. S2). Similar to plain SI or I48S48 nanoparticles, FSI nanoparticles have a hydrophobic core (isoleucine ELP) and a hydrophilic corona (serine ELP), in addition to their FKBP domains. Thus, we hypothesized FSI nanoparticles may have two distinct sites for Rapa encapsulation, at their core and at their corona (Fig. 1a).
Figure 4. Morphology of ELP nanoparticles is minimally influenced by fusion of FKBP or encapsulation of Rapa.
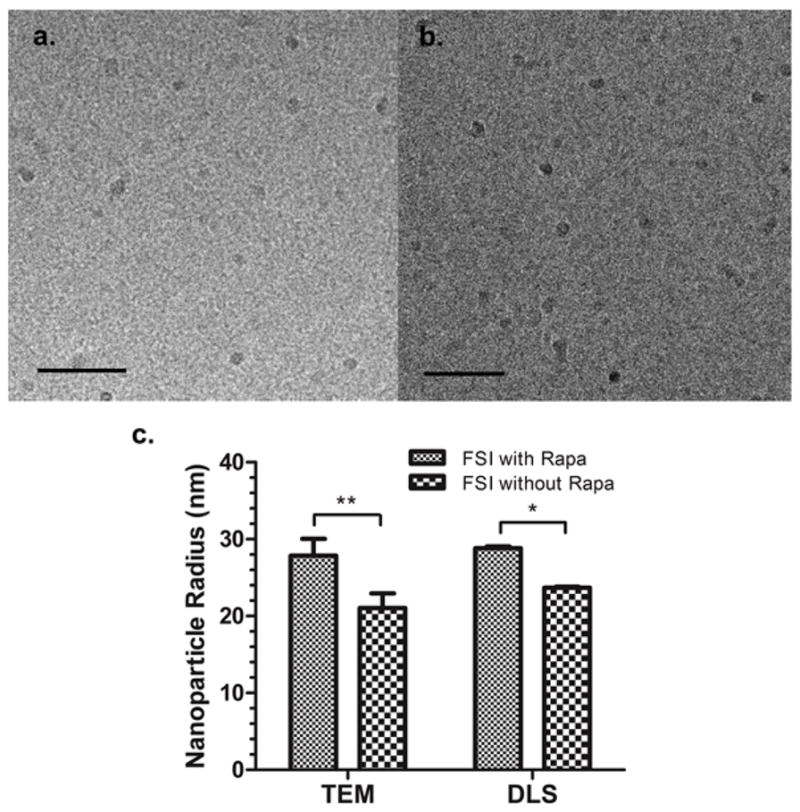
a, b. Cryo-TEM imaging of ELP nanoparticles with and without the FKBP domain. a. The average radius of FSI is 18.5 ± 1.3 nm. b. The average radius of SI nanoparticles is 15.0 ± 2.3 nm. Mean ± SD (n = 10). Bar length = 200nm. c. A comparison of FSI nanoparticle radius determined using TEM and DLS before and after Rapa encapsulation suggests that presentation and loading of the FKBP domain minimally affects nanoparticle morphology. Mean ± SD (n = 10). (**p=0.005 and *p=0.05)
Unlike plain ELP diblock copolymers (SI, I48S48), FSI has limited stability in organic solvents, which is presumably due to denaturation of the folded FKBP domain. For this reason, the film hydration method could not be used for FSI Rapa encapsulation. Instead, a new method of two-phase solvent evaporation was developed specifically for FSI Rapa encapsulation (Section 2.8). To confirm that drug loading did not influence particle morphology, FSI before and after Rapa encapsulation were imaged by TEM (Supplemental Fig. S3). Similarly, their hydrodynamic radii were measured by DLS. Both TEM and DLS revealed that the radii of FSI nanoparticles after Rapa encapsulation were only slightly larger than those before encapsulation (Fig. 4c). These observations suggest that Rapa encapsulation only minimally influences the particle size and morphology of FSI.
3.5 Decoration of ELP nanoparticles with the FKBP domain prolongs the release of Rapa
Having confirmed that Rapa encapsulation does not greatly affect the properties of the FSI nanoparticles, FSI, SI and S192 were evaluated for encapsulation and release (Fig. 5). Using the two-phase solvent evaporation method to load these three formulations, both FSI and SI nanoparticles have about 75% efficiency encapsulation of Rapa; however, S192 a linear ELP mono-block that does not form nanostructures had very low efficiency (less than 10%) of Rapa association (Fig. 5a). Release experiments were conducted to compare the release of drug from ELP nanoparticles with and without the FKBP domain (Fig. 5b). Again, dialysis under sink conditions was used to track the release of Rapa from FSI and SI nanoparticles. Encouragingly, FSI and SI nanoparticles exhibited significantly different profiles of Rapa release. For FSI, there were two exponential decay phases of Rapa release: a fast phase with a half-life of 1.9 h and a slow phase with a half-life of 57.8 h. In contrast only one exponential decay phase was observed for SI-Rapa. This release had a half-life of 2.2 h, which is similar to the fast phase of FSI Rapa release (Fig. 5b) and Rapa release from I48S48 mixed at a 1:1 ratio (Fig. 3b). After 12 hours of dialysis, SI-Rapa samples retained undetectable levels of Rapa. In contrast, approximately 30% of the drug remained associated with FSI nanoparticles. Because the association between FKBP and Rapa is much stronger than that between ELP nanoparticle core and Rapa, we propose that the population of Rapa that is encapsulated into ELP nanoparticle core mainly contributes the fast phase of FSI Rapa release while the population of Rapa that is bound to FKBP primarily contributes the slow phase. Therefore, it might be inferred that about 70% Rapa is associated into the FSI nanoparticle core, which displays rapid release. In contrast, the remaining 30% is bound to FKBP and releases much slower (Fig. 1a). With a longer terminal release half-life, Rapa may remain associated with FSI nanoparticles more stably, promoting its in vivo tolerability at higher doses and potentially better anti-tumor efficacy.
Figure 5. The FKBP domain prolongs the release of Rapa from FSI nanoparticles.

a. Both FSI and SI nanoparticles efficiently encapsulate Rapa using a two-phase solvent evaporation method. In contrast, an ELP S192, which does not form nanoparticles, was unable to encapsulate the drug. (***p = 0.001) Mean ± SD (n=3) b. Dialysis was used to track the loss of Rapa from the nanoparticles. FSI-Rapa exhibited bi-exponential drug release (Half-life fast = 1.9 h, Half-life slow = 57.8 h). SI-Rapa release followed a mono-exponential release curve (Half-life = 2.2 h) down to the limit of detection of 2 μM. After 12 hours, only background was detected for SI-Rapa; however, approximately 30% of the drug remained associated with FSI. Mean ± SD (n=3).
3.6 FSI Rapa is as potent as free Rapa in MTS cell proliferation assay
To test and compare in vitro anti-proliferative efficacy of FSI Rapa and free Rapa, the MTS cell proliferation assay was performed using two breast cancer cell lines. MDA-MB-468 cells are Rapa sensitive, while MDA-MB-231 cells are Rapa insensitive (Fig. 6). In Rapa sensitive MDA-MB-468 cells, both FSI Rapa and free Rapa efficiently decreased cell viability. In addition, the additional solubility of FSI Rapa enabled a higher dose at 10 μM and a further reduction in cell viability to 15%. Free Rapa could not reach such a high concentration because of its limited water solubility. In contrast, in Rapa insensitive MDA-MB-231 cells, neither FSI Rapa nor free Rapa reduced cell viability more than 50%, even at the highest concentrations. The inhibitory concentration yielding 50% viability, IC50, of FSI Rapa and free Rapa in MDA-MB-468 was estimated using non-linear regression. The IC50 of FSI Rapa was 0.28 nM and IC50 of free Rapa was 0.27 nM. The critical micelle concentration (CMC) for FSI was estimated at 0.17 nM (Supplemental Fig. S4); furthermore, since the ratio of Rapa to FSI is approximately 1:1, then above 1 nM Rapa the nanoparticles remain intact. Even though the nanoparticles remain assembled, the similar viabilities for FSI and Free Rapa reflect the assay’s three-day incubation period. Fig. 5b shows that after 48 hours, over 80% of the FSI Rapa has been released, which allows released Rapa to influence cell viability similar to free Rapa. These results demonstrate that the Rapa encapsulated in FSI nanoparticles was as potent as free drug in decreasing the viability of MDA-MB-468 breast cancer cells. In addition, the FSI nanoparticle had the additional benefit of extending the solubility of Rapa at least 10 fold compared to free Rapa, which could facilitate their utility in vivo.
Figure 6. FSI-encapsulated Rapa inhibits cell viability in a mTOR dependent cell line MDA-MB-468 but not in an insensitive cell line, MDA-MB-231.
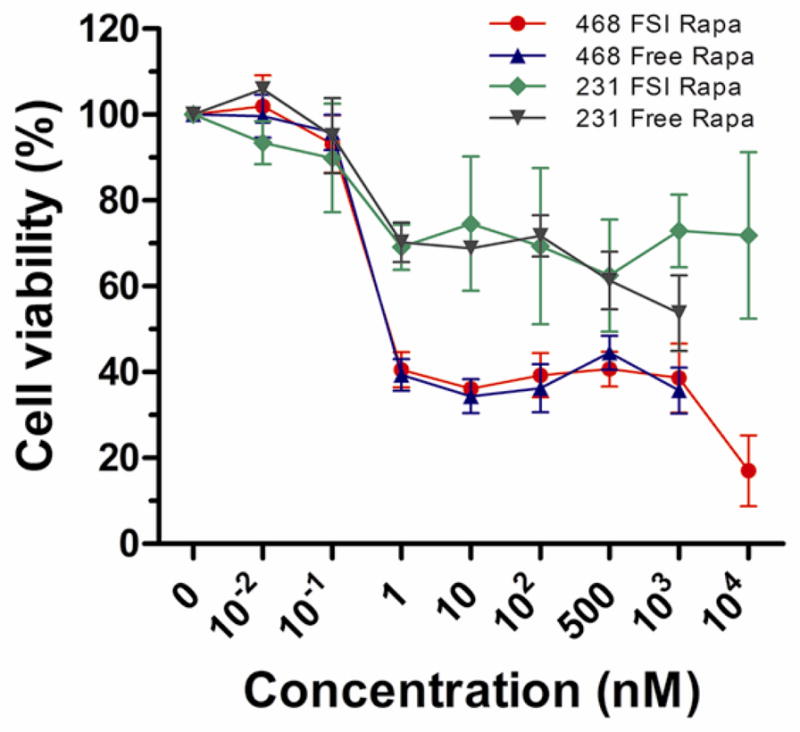
FSI Rapa is as potent as free Rapa in decreasing the viability of Rapa sensitive MDA-MB-468 cells; however, FSI encapsulation increased the Rapa solubility at least 10 fold, to about 10 μM. Mean ± SD (n=6).
3. 7 FSI Rapa nanoparticles have greater anti-tumor efficacy and lower toxicity than free drug
Having optimized the ELP nanoparticle formulation to improve Rapa solubility and extend drug release through combined encapsulation in both the core and corona (Fig. 1a), this formulation was compared head-to-head with free Rapa in the MDA-MB-468 breast cancer xenograft mouse model (Fig. 7a). The reported maximum tolerated dose for free drug 0.75 mg/kg body weight was chosen for initial dosing in both FSI Rapa group and free drug groups three times a week. After the first two injections, subjects in the free Rapa group lost almost 10% body weight (Fig. 7b). Due to this toxicity, the administration frequency of free drug was then reduced to once a week. Even after reducing the dose frequency, free Rapa still showed strong accumulative toxicity. 23 days after the first treatment, all mice administered free Rapa lost more than 15% of their body weight and were removed from the study. In contrast, the FSI Rapa group showed no signs of behavioral changes or body weight loss (Fig. 7b). Compared to the PBS treatment group, FSI Rapa effectively halted tumor growth (Fig. 7a, c). In summary, compared to free Rapa group, FSI Rapa nanoparticles showed better anti-tumor efficacy and much lower overt toxicity in treating this mTOR dependent breast cancer xenograft mouse model.
Figure 7. FSI-encapsulated Rapa has better anti-tumor efficacy and lower toxicity than free Rapa in the mTOR dependent MDA-MB-468 breast cancer xenograft.
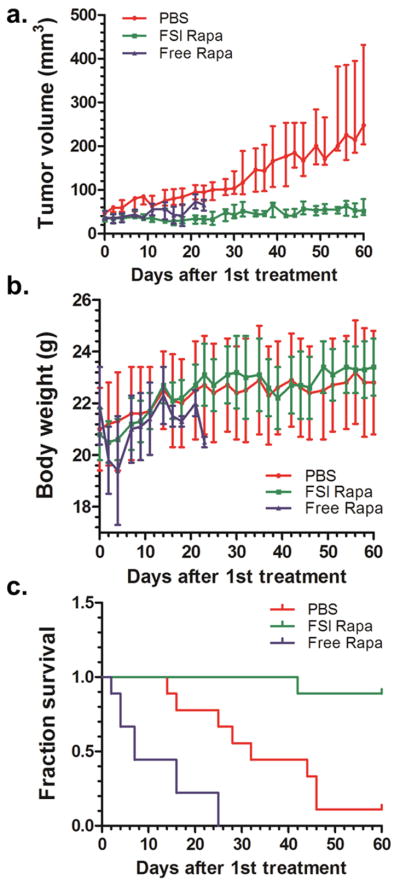
a. Tumor volumes of FSI Rapa group are significantly smaller than PBS group (Median ± Interquartile Range p<0.0001, N = 9). b. FSI Rapa group also shows less body weight loss than free Rapa group. c. Kaplan-Meier survival analysis demonstrates that FSI Rapa has better anti-tumor efficacy than free Rapa. (p=0.004)
Based on these findings, we propose that Rapa delivered by FSI nanoparticles more effectively retains in the central blood compartment. These particles have several potentially beneficial effects: i) increased aqueous solubility for Rapa; ii) interaction with the FKBP domain provides a slow off-rate on the order of a 60 hour half-life; iii) the slow off-rate enables this formulation to passively accumulate in the tumor; and iv) the protein polymer is entirely composed from biodegradable polypeptides, which may release active drug following cellular internalization in the tumor [25]; and v) upon proteolysis, the free Rapa relocates to the cytoplasm and inhibits mTOR-dependent cell proliferation. Despite these encouraging findings, this study has several limitations. First, it remains unknown how effectively the Rapa is retained by the nanoparticles in vivo. It is encouraging to note that the FSI Rapa formulation retains its cargo under in vitro sink conditions (Fig. 5b) for at least as long as the ELP nanoparticles have been reported to circulate in mice [26]. Second, it remains unknown how much the dose can be decreased, while maintaining the same level of efficacy. Third, it remains unknown whether the current levels of efficacy result from the fast Rapa release, the slow Rapa release, or some optimal combination of the two.
The level of Rapa efficacy towards mTOR dependent tumors is consistent with that observed using synthetic polymeric systems, such as Poly (lactic-co-glycolic acid) (PLGA) [27], Poly (ethylene glycol)-block-poly (2-methyl-2-benzoxycarbonyl-propylene carbonate) (PEG-b-PBC) [28], poly (ethylene glycol)-b-poly (caprolactone) (PEG PCL) [29] and acetylated dextran microparticles [30]. Compared with these delivery systems, ELP nanoparticles have unique advantages. First, they can be engineered consisting of only five naturally occurring amino acids, Val, Pro, Gly, Xaa and Gly derived from human tropoelastin [31]. Unlike the byproducts of some synthetic polymers, ELP components are non-toxic and biodegradable in vivo. Second, the size of ELP nanoparticles is optimal compared to other delivery systems. The hydrodynamic radii of I48S48, F24S24, SI, and FSI nanoparticles are all less 30 nm. This size range brings them significantly above the renal filtration cutoff without producing a significant population of particles greater than 50 nm in diameter where hepatic uptake accelerates. This property has enabled other ELP nanoparticles to display pharmacokinetics in a mouse on the order of a 6 hour half-life [26]. Third, ELP nanoparticles can be easily modified to display functional folded proteins via biosynthesis. This avoids the common requirement of optimizing bioconjugation chemistry, which can reduce the activity of sensitive protein domains. Fourth, the purity and homogeneity of ELP protein polymers are among the best achievable using synthetic polymerization strategies [32, 33]. Lastly, the FSI nanoparticle has been engineered both at its core and corona for specific encapsulation and delivery of Rapa one of FKBP’s natural ligands. FSI may be useful to deliver other Rapa analogues; furthermore, by changing the fusion domain decorating these nanoparticles, this strategy may enable the specialized engineering of sustained release nanocarriers for other classes of drugs.
Having obtained evidence that the release of drug from an ELP nanoparticle can be modulated by incorporation of its target protein, FKBP, there remain unknowns to this approach. First, the relationship between the pharmacokinetic clearance of FSI and the release of Rapa from FSI remains unknown. It was previously found that other ELP nanoparticles have a blood half-life in mice in the 6 hour range [26]; however, the effect of FKBP modification on nanoparticle clearance is unknown. For FSI, the nanoparticle corona remains hydrophilic and neutral in zeta potential, which may promote the steric stabilization of these nanoparticles. During circulation, these properties may maintain low clearance of FSI from the central blood pool. While Rapa remains bound, its clearance may also be reduced. Interestingly, FSI Rapa has both a fast release half-life of 1.9h (core) and a slow release half-life of 57.8h (corona). While Rapa released quickly may undergo rapid re-distribution in the body, the significant fraction of drug that is released slowly is expected to remain associated with FSI during tumor accumulation. Based on the current study, it is unknown if both slow and fast release is required for a therapeutic response (Fig. 7), and this needs to be assessed in future studies. Second, the mechanism for transfer of Rapa to the tumor remains unclear. The FSI formulation is capable of releasing Rapa both prior to and after accumulation in a tumor; furthermore, both free and encapsulated drug may be acting to halt tumor proliferation. Since this formulation is effective without targeting cell-surface receptors, tumor targeting of FSI is most likely via passive tumor accumulation. The size of the tumors treated (around 40 mm3) in this study suggests that they are undergoing angiogenesis, which initiates when the size of the tumor reaches 2 mm3 [34, 35]. During the formation of new blood vessels, remodeling of immature vasculature often results in irregular shaped, dilated, tortuous and leaky tumor blood vessels, which has been demonstrated for various drug carriers using multiple in vivo tumor models. Our group has previously obtained evidence that these ELP nanoparticles are biodegradable [18]; therefore, it is feasible that intra or extracellular proteases act upon FSI as a mechanism to release active Rapa. Despite these unknowns, this novel formulation poses a new approach to drug encapsulation that is effective in vivo and warrants additional study.
4. Conclusions
ELP diblock copolymers form stable, monodisperse and biocompatible nanoparticles. For the first time, we report successful drug encapsulation into both the core and corona of these emerging drug carriers. Both Rose Bengal and Rapa have high association with the cores of I48S48 and F24S24 nanoparticles. With the genetic fusion of Rapa’s cognate receptor FKBP to nanoparticle corona, the terminal release half-life of encapsulated Rapa has been prolonged from 2.2 h to 57.8h. With a long Rapa terminal half-life, FSI nanoparticles may stably retain a significant fraction of drug during the circulation in the bloodstream, which may reduce toxicity and promote delivery to tumors. This study shows that FSI Rapa achieves greater anti-tumor efficacy and lower cytotoxicity than free Rapa in treating a breast cancer xenograft mouse model. Moreover, Rapa-specific encapsulation and delivery using FKBP modified nanoparticles is proof-of-concept for a new strategy to develop biodegradable drug-specific carriers.
Supplementary Material
Acknowledgments
This work was made possible by the University of Southern California, the National Institute of Health R21EB012281 to J.A.M., and P30 CA014089 to the Norris Comprehensive Cancer Center, the USC Molecular Imaging Center, the USC Nanobiophysics Core Facility, the Translational Research Laboratory at the School of Pharmacy, the American Cancer Society IRG-58-007-48, the Stop Cancer Foundation, the USC Ming Hsieh Institute, and the USC Whittier Foundation. Dr. Herbert Meiselman of USC generously made available his instrument for making Zeta Potential measurements.
Abbreviations
- Rapa
Rapamycin
- ELPs
elastin-like polypeptides
- FKBP
FK506 binding protein 12
- mTOR
mammalian target of rapamycin
- ITC
inverse transition cycling
- CMT
critical micelle temperature
- CMC
critical micelle concentration
- DLS
dynamic light scattering
- TEM
transmission electron microscopy
- cryo-TEM
cryogenic-transmission electron microscopy
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Vezina C, Kudelski A, Sehgal SN. Rapamycin (AY-22,989), a new antifungal antibiotic. I. Taxonomy of the producing streptomycete and isolation of the active principle. The Journal of antibiotics. 1975;28:721–726. doi: 10.7164/antibiotics.28.721. [DOI] [PubMed] [Google Scholar]
- 2.Ho S, Clipstone N, Timmermann L, Northrop J, Graef I, Fiorentino D, Nourse J, Crabtree GR. The mechanism of action of cyclosporin A and FK506. Clinical immunology and immunopathology. 1996;80:S40–45. doi: 10.1006/clin.1996.0140. [DOI] [PubMed] [Google Scholar]
- 3.Yu K, Toral-Barza L, Discafani C, Zhang WG, Skotnicki J, Frost P, Gibbons JJ. mTOR, a novel target in breast cancer: the effect of CCI-779, an mTOR inhibitor, in preclinical models of breast cancer. Endocrine-related cancer. 2001;8:249–258. doi: 10.1677/erc.0.0080249. [DOI] [PubMed] [Google Scholar]
- 4.Seeliger H, Guba M, Koehl GE, Doenecke A, Steinbauer M, Bruns CJ, Wagner C, Frank E, Jauch KW, Geissler EK. Blockage of 2-deoxy-D-ribose-induced angiogenesis with rapamycin counteracts a thymidine phosphorylase-based escape mechanism available for colon cancer under 5-fluorouracil therapy. Clinical cancer research : an official journal of the American Association for Cancer Research. 2004;10:1843–1852. doi: 10.1158/1078-0432.ccr-1176-3. [DOI] [PubMed] [Google Scholar]
- 5.Majumder PK, Febbo PG, Bikoff R, Berger R, Xue Q, McMahon LM, Manola J, Brugarolas J, McDonnell TJ, Golub TR, Loda M, Lane HA, Sellers WR. mTOR inhibition reverses Akt-dependent prostate intraepithelial neoplasia through regulation of apoptotic and HIF-1-dependent pathways. Nat Med. 2004;10:594–601. doi: 10.1038/nm1052. [DOI] [PubMed] [Google Scholar]
- 6.Luan FL, Ding R, Sharma VK, Chon WJ, Lagman M, Suthanthiran M. Rapamycin is an effective inhibitor of human renal cancer metastasis. Kidney international. 2003;63:917–926. doi: 10.1046/j.1523-1755.2003.00805.x. [DOI] [PubMed] [Google Scholar]
- 7.Bierer BE, Mattila PS, Standaert RF, Herzenberg LA, Burakoff SJ, Crabtree G, Schreiber SL. Two distinct signal transmission pathways in T lymphocytes are inhibited by complexes formed between an immunophilin and either FK506 or rapamycin. Proc Natl Acad Sci U S A. 1990;87:9231–9235. doi: 10.1073/pnas.87.23.9231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bjornsti MA, Houghton PJ. The TOR pathway: a target for cancer therapy. Nature reviews Cancer. 2004;4:335–348. doi: 10.1038/nrc1362. [DOI] [PubMed] [Google Scholar]
- 9.Noh WC, Mondesire WH, Peng J, Jian W, Zhang H, Dong J, Mills GB, Hung MC, Meric-Bernstam F. Determinants of rapamycin sensitivity in breast cancer cells. Clinical cancer research : an official journal of the American Association for Cancer Research. 2004;10:1013–1023. doi: 10.1158/1078-0432.ccr-03-0043. [DOI] [PubMed] [Google Scholar]
- 10.Chhajed PN, Dickenmann M, Bubendorf L, Mayr M, Steiger J, Tamm M. Patterns of pulmonary complications associated with sirolimus. Respiration; international review of thoracic diseases. 2006;73:367–374. doi: 10.1159/000087945. [DOI] [PubMed] [Google Scholar]
- 11.Simamora P, Alvarez JM, Yalkowsky SH. Solubilization of rapamycin. International journal of pharmaceutics. 2001;213:25–29. doi: 10.1016/s0378-5173(00)00617-7. [DOI] [PubMed] [Google Scholar]
- 12.Yanez JA, Forrest ML, Ohgami Y, Kwon GS, Davies NM. Pharmacometrics and delivery of novel nanoformulated PEG-b-poly(epsilon-caprolactone) micelles of rapamycin. Cancer chemotherapy and pharmacology. 2008;61:133–144. doi: 10.1007/s00280-007-0458-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hennenfent KL, Govindan R. Novel formulations of taxanes: a review. Old wine in a new bottle? Annals of oncology : official journal of the European Society for Medical Oncology / ESMO. 2006;17:735–749. doi: 10.1093/annonc/mdj100. [DOI] [PubMed] [Google Scholar]
- 14.Gaucher G, Marchessault RH, Leroux JC. Polyester-based micelles and nanoparticles for the parenteral delivery of taxanes. Journal of controlled release : official journal of the Controlled Release Society. 2010;143:2–12. doi: 10.1016/j.jconrel.2009.11.012. [DOI] [PubMed] [Google Scholar]
- 15.Yatscoff RW, Wang P, Chan K, Hicks D, Zimmerman J. Rapamycin: distribution, pharmacokinetics, and therapeutic range investigations. Therapeutic drug monitoring. 1995;17:666–671. doi: 10.1097/00007691-199512000-00020. [DOI] [PubMed] [Google Scholar]
- 16.Mackay JA, Chilkoti A. Temperature sensitive peptides: engineering hyperthermia-directed therapeutics, International journal of hyperthermia : the official journal of European Society for Hyperthermic Oncology. North American Hyperthermia Group. 2008;24:483–495. doi: 10.1080/02656730802149570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Urry DW. Physical Chemistry of Biological Free Energy Transduction As Demonstrated by Elastic Protein-Based Polymers†. The Journal of Physical Chemistry B. 1997;101:11007–11028. [Google Scholar]
- 18.Shah M, Hsueh PY, Sun G, Chang HY, Janib SM, MacKay JA. Biodegradation of elastin-like polypeptide nanoparticles. Protein science : a publication of the Protein Society. 2012;21:743–750. doi: 10.1002/pro.2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dhandhukia J, Weitzhandler I, Wang W, Mackay JA. Switchable Elastin-Like Polypeptides that Respond to Chemical Inducers of Dimerization. Biomacromolecules. 2013 doi: 10.1021/bm301558q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sun G, Hsueh PY, Janib SM, Hamm-Alvarez S, Andrew MacKay J. Design and cellular internalization of genetically engineered polypeptide nanoparticles displaying adenovirus knob domain. Journal of controlled release : official journal of the Controlled Release Society. 2011;155:218–226. doi: 10.1016/j.jconrel.2011.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Banki MR, Feng L, Wood DW. Simple bioseparations using self-cleaving elastin-like polypeptide tags. Nature methods. 2005;2:659–661. doi: 10.1038/nmeth787. [DOI] [PubMed] [Google Scholar]
- 22.Meyer DE, Chilkoti A. Purification of recombinant proteins by fusion with thermally-responsive polypeptides. Nature biotechnology. 1999;17:1112–1115. doi: 10.1038/15100. [DOI] [PubMed] [Google Scholar]
- 23.Wu WY, Mee C, Califano F, Banki R, Wood DW. Recombinant protein purification by self-cleaving aggregation tag. Nature protocols. 2006;1:2257–2262. doi: 10.1038/nprot.2006.314. [DOI] [PubMed] [Google Scholar]
- 24.Christensen T, Amiram M, Dagher S, Trabbic-Carlson K, Shamji MF, Setton LA, Chilkoti A. Fusion order controls expression level and activity of elastin-like polypeptide fusion proteins. Protein Sci. 2009;18:1377–1387. doi: 10.1002/pro.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shah M, Hsueh PY, Sun G, Chang HY, Janib SM, MacKay JA. Biodegradation of elastin-like polypeptide nanoparticles. Protein Sci. 2012;21:743–750. doi: 10.1002/pro.2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Janib SM, Liu S, Park R, Pastuszka MK, Shi P, Moses AS, Orosco MM, Lin YA, Cui H, Conti PS, Li Z, MacKay JA. Kinetic quantification of protein polymer nanoparticles using non-invasive imaging. Integrative Biology. 2013;5:183–194. doi: 10.1039/c2ib20169k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zou J, Zhang X, Yang H, Zhu Y, Ma H, Wang S. Rapamycin-loaded nanoparticles for inhibition of neointimal hyperplasia in experimental vein grafts. Ann Vasc Surg. 2011;25:538–546. doi: 10.1016/j.avsg.2011.01.003. [DOI] [PubMed] [Google Scholar]
- 28.Lu W, Li F, Mahato RI. Poly(ethylene glycol)-block-poly(2-methyl-2-benzoxycarbonyl-propylene carbonate) micelles for rapamycin delivery: in vitro characterization and biodistribution. J Pharm Sci. 2011;100:2418–2429. doi: 10.1002/jps.22467. [DOI] [PubMed] [Google Scholar]
- 29.Forrest ML, Won CY, Malick AW, Kwon GS. In vitro release of the mTOR inhibitor rapamycin from poly(ethylene glycol)-b-poly(epsilon-caprolactone) micelles. Journal of controlled release : official journal of the Controlled Release Society. 2006;110:370–377. doi: 10.1016/j.jconrel.2005.10.008. [DOI] [PubMed] [Google Scholar]
- 30.Kauffman KJ, Kanthamneni N, Meenach SA, Pierson BC, Bachelder EM, Ainslie KM. Optimization of rapamycin-loaded acetalated dextran microparticles for immunosuppression. International journal of pharmaceutics. 2012;422:356–363. doi: 10.1016/j.ijpharm.2011.10.034. [DOI] [PubMed] [Google Scholar]
- 31.Dreher MR, Simnick AJ, Fischer K, Smith RJ, Patel A, Schmidt M, Chilkoti A. Temperature triggered self-assembly of polypeptides into multivalent spherical micelles. Journal of the American Chemical Society. 2008;130:687–694. doi: 10.1021/ja0764862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Koehl P, Delarue M. Polar and nonpolar atomic environments in the protein core: implications for folding and binding. Proteins. 1994;20:264–278. doi: 10.1002/prot.340200307. [DOI] [PubMed] [Google Scholar]
- 33.Rabotyagova OS, Cebe P, Kaplan DL. Protein-based block copolymers. Biomacromolecules. 2011;12:269–289. doi: 10.1021/bm100928x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Danhier F, Feron O, Preat V. To exploit the tumor microenvironment: Passive and active tumor targeting of nanocarriers for anti-cancer drug delivery. Journal of controlled release : official journal of the Controlled Release Society. 2010;148:135–146. doi: 10.1016/j.jconrel.2010.08.027. [DOI] [PubMed] [Google Scholar]
- 35.Bergers G, Benjamin LE. Tumorigenesis and the angiogenic switch. Nature reviews Cancer. 2003;3:401–410. doi: 10.1038/nrc1093. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.