

Abstract
Background
Alloantibody can contribute significantly to rejection of heart transplants by activation of complement and interactions with a variety of effector cells, including macrophages and monocytes through activating FcγRI, FcγRIII, FcγRIV, the inhibitory FcγRIIB and complement receptors. These receptors link cellular and humoral immunity by bridging the antibody specificity to effector cells. Activating FcγRs are also involved in serum amyloid P component (SAP)-mediated clearance of apoptotic bodies.
Methods
B10.A (H-2a) hearts were transplanted into WT or FcγRIII-KO C57BL/6 (H-2b) mouse recipients. Levels of alloantibodies and SAP in the circulation were determined by flow cytometry and ELISA, respectively. Intragraft cytokine mRNA expression was measured by real-time PCR. Intragraft deposition of C4d, von-Willebrand factor (vWF), SAP and activated caspase 3 was visualized by immunochemistry.
Results
B10.A hearts in C57BL/6 FcγRIII-KO recipients were rejected acutely within 6-8 days as compared to 10-14 days in WT. The rejection in FcγRIII-KO was accompanied by higher levels of circulating IgM/IgG alloantibodies and SAP than in WT recipients. Histology in FcγRIII-KO cardiac allograft recipients indicated: perivascular margination of monocytes and neutrophils, vascular endothelial cell injury, intense vasculocentric infiltrates with extensive apoptosis. Higher numbers of apoptotic cells, stronger C4d and SAP deposition and extensive activated caspase 3 were found in areas of dense pockets of apoptotic blebs in FcγRIII-KO.
Conclusions
We propose that absence of FcγRIII is associated with the lack of efficient SAP-mediated clearance of apoptotic cells through FcγRs. Apoptotic cells become immunogenic, induce enhanced inflammation, AlloAb production and complement activation leading to accelerated cardiac allograft rejection.
Keywords: heart transplantation, Fc receptor, serum amyloid P component (SAP), apoptosis, alloantibody, complement
Introduction
Transplant rejection is a complex process that usually involves multiple innate and adaptive immune responses. Alloantibody mediated organ rejection (AMR) has become clinically critical because this form of rejection is unresponsive to conventional anti-rejection therapy. Therefore AMR has become a therapeutic challenge in transplantation. Alloantibody can contribute significantly to rejection of heart transplants by activation of complement (C) and interactions with a variety of effector cells, including macrophages and monocytes through Fcγ (FcγR) and complement receptors. These receptors link cellular and humoral immunity by bridging antibody specificity to effector cells.
Data from several transplantation centers (1, 2) indicate that complement split products such as C4d and C3d are deposited on the vascular endothelium in a significant number of acute antibody-mediated rejections of renal and cardiac transplants. This finding is frequently associated with marginated neutrophils or monocytes and donor-specific antibodies in the circulation.
The recent growing interest in antibody-mediated rejection has stimulated the development of in vivo and in vitro experimental models to study antibody and complement in acute and chronic rejection. These experiments have demonstrated multiple mechanisms by which antibodies and complement can intensify macrophage, B cell and T cell responses (3, 4).
We developed a mouse model of antibody- and C-mediated rejection. In this model, B10.A hearts are transplanted to Ig deficient C57BL/6 recipients that receive passively transferred alloantibodies to MHC class I antigens (5-7). We documented that non-complement-activating IgG1 in combination with low doses of complement-activating IgG2b alloantibody caused irreversible rejection of cardiac allografts that was accompanied by linear deposits of C4d on endothelium. In parallel in vitro experiments, we demonstrated that IgG1 alloantibodies to class I MHC in the absence of complement stimulate production of pro-inflammatory cytokines by endothelial cells. This response was increased in the presence of macrophages through a mechanism that was dependent on stimulatory FcγRIII.
FcγR provide a critical link between specific humoral responses and the cellular pathways of the immune system (8). Alloantibodies interact with effector cells through activating (FcγRI, FcγRIII, FcγRIV) and inhibitory (FcγRIIB) Fc receptors. These two classes of receptors function in concert and are usually co-expressed on the cell surface (8). FcγRI, FcγRIIB, FcγRIII and FcγRIV are expressed by variety of leukocytes: macrophages, monocytes, NK, PMNs and small number of T cells, whereas FcγRIIB are expressed on both myeloid and lymphoid lineages. They mediate effector functions, including phagocytosis, ADCC (9, 10) and the release of pro- and anti-inflammatory mediators (11). Antibodies also provide powerful feedback through Fc receptors to increase complement production (12, 13), and complement split products can modulate the expression and function of FcR for antibodies.
In addition, Du Clos, Mold and colleagues identified FcγRs as the major receptors for C-reactive protein (CRP) and serum amyloid P component (SAP) and implicated their involvement in the process of phagocytosis (14-17). Based on analysis of pentraxin interactions with FcγRs this group unraveled the crystal structure of human SAP interacting with FcγRIIa (18). CRP and SAP are members of pentraxin family of proteins that are evolutionary highly conserved and characterized by a pentameric structure (19). They both have important functions in innate host defense (20), clearance of phospholipids and nuclear components from the late apoptotic and necrotic cells (21-23), and regulation of the inflammatory response (20). While CRP is an acute-phase protein in humans, SAP plays the same role in the mouse. Recently both pro- and anti-inflammatory functions of CRP and SAP were identified. These functions depend on differential interactions of both pentraxins with complement, FcγRs and complement regulatory proteins (24, 25).
Mice with a genetic mutation of the γ chain (FcRγ-KO) have impaired expression of FcγRI and FcγRIII. They exhibit impaired antibody-mediated responses, including loss of NK cell-mediated ADCC, macrophage phagocytosis, and mast cell degranulation in response to FcR cross-linking (26, 27).
In this study we investigated the mechanism of accelerated acute cardiac allograft rejection in recipients deprived of functional FcγRIII. We provided evidence that in the absence of activating FcγRIII cardiac allograft rejection is associated with increased alloantibody production, activation of complement (C4d deposition) and widespread apoptosis.
Results
B10.A cardiac allograft survival in C57BL/6 FcγRIII-KO recipients. In these studies we documented that cardiac allografts from B10.A (H-2a) hearts transplanted to C57BL/6 FcγRIII-KO (H-2b) recipients were rejected acutely within 6-8 days after transplantation (n=20). In contrast, hearts in their WT counterparts survived longer and were rejected within 10-14 days after transplantation (n=22). This difference in survival was significant (P<0.0001) by log-rank test in Kaplan-Meier plot analysis (Fig 1).
Fig 1. Cardiac allograft survival in FcγRIII-KO and WT.

Cardiac allografts from B10.A (H-2a) hearts transplanted to C57BL/6 FcγRIII-KO (H-2b) recipients were rejected acutely within 6-8 days after transplantation (n=20). In contrast hearts in their WT counterparts survived longer and were rejected within 10-14 days after transplantation (n=22). This difference in survival was significant (P<0.0001) by log-rank test in Kaplan-Meier plot analysis.
Vascular injury is increased in B10.A cardiac allografts transplanted to C57BL/6 FcγRIII-KO vs. WT recipients
In preliminary experiments H&E staining of B10.A hearts transplanted to FcγRIII-KO recipients performed 7 days after transplantation indicated that accelerated acute rejection was manifested by prominent perivascular margination of monocytes and neutrophils, vascular endothelial cell injury, and intense vasculocentric infiltrates with extensive apoptosis. Macrophages were the predominant cell type in the infiltrates (SDC Fig 1) as has been described in the Banff criteria for antibody mediated rejection (1). At the same time hearts transplanted to WT recipients demonstrated only mild cellular infiltration and vascular endothelial cell injury with only few apoptotic cells. High levels of DNA fragmentation of apoptotic cells was visualized by Tunel assay (data not shown). Since the interpretation and specificity of this assay is controversial (28), we confirmed the findings with immunohistology for activated caspase 3 and caspase-clevead cytokeratin. We found extensive areas containing apoptotic blebs that stained for activated caspase 3 in cardiac allografts transplanted to FcγRIII-KO (Fig 2A). In contrast, in WT recipients DNA fragmentation was minimal and activated caspase 3 was expressed in small numbers of apoptotic cells (Fig 2B).
Fig 2. Apoptotic cells visualized by staining for activated caspase 3 and caspase-cleaved cytokeratin.

Both nuclei and nuclear fragments are stained for activated caspase 3 in larger numbers in cardiac allgrafts to FcγRIII KO recipients (A) compared to WT recipients (B). Arterial and capillary deposits of C4d were more intense in cardiac allgrafts to FcγRIII KO recipients (C) compared to WT recipients (D).
The extent of apoptosis was quantified on 5 separate fields in the central myocardium distant from areas associated with epicardial and endocardial inflammation. In allografts approaching complete rejection, areas that displayed advanced rejection and myocyte necrosis were excluded. To avoid multiple apoptotic bodies from single cells contributing to the count disproportionately (SDC Fig 2), a grid of 50 microns square was superimposed on digital images captured on medium power (SDC Fig 3) and the number of squares containing apoptotic cells were counted. This method documented that apoptosis was much more widespread in cardiac grafts to FcγRIII-KO than wild type recipients (Fig 3A). Apoptosis was sparse in allografts to wild type recipients at the initial stages of rejection (6-8 days) and in the terminal phases of rejection (9-11 days). This suggests that the difference in apoptosis was not simply related to the stage of rejection, but to an alternate mechanism. The stains also demonstrated that donor cells including cardiac myocytes underwent apoptosis (SDC Fig 2). Excess apoptotic myocytes could be a source of antigen processed in antigen presentation.
Fig 3. Measurements of apoptosis and C4d deposition.
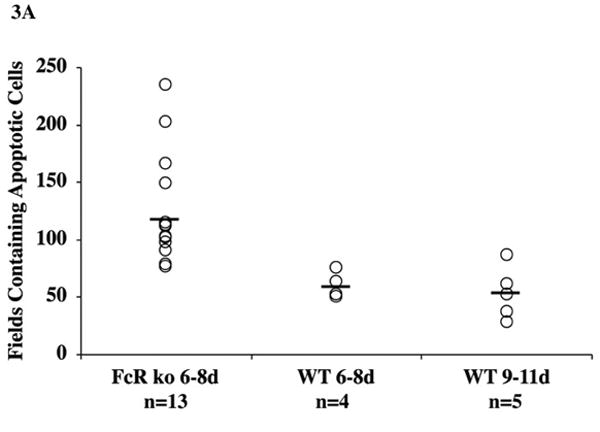
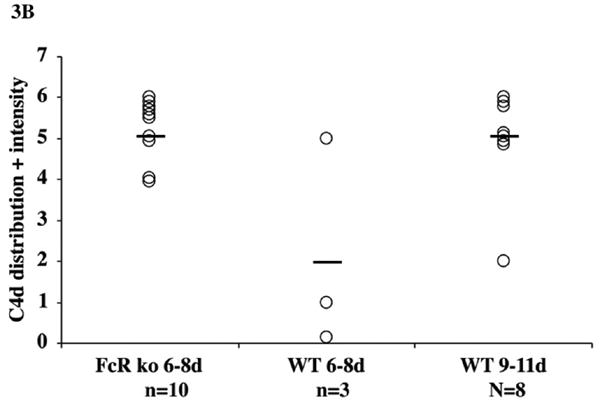
The extent of apoptotic cells and nuclear fragments stained for activated caspase 3 was quantified in grids of 50 microns square superimposed on digital images captured on medium power. The number of squares containing apoptotic cells was much more widespread in cardiac grafts to FcγRIII-KO than WT recipients (A). Apoptosis was sparse in allografts to wild type recipients at the initial stages of rejection (6-8 days) and in the terminal phases of rejection (9-11 days). C4d deposition graded on the clinical criteria of distribution (focal versus diffuse) and intensity with each parameter scored on a 0 to 3+ scale demonstrated diffuse and intense deposits occurred more acutely in cardiac grafts to FcγRIII-KO than WT recipients (B). The more acute deposition of C4d correlated with more rapid generation of alloantibodies responses to cardiac grafts to FcγRIII-KO than WT recipients.
Immunohistochemical staining demonstrated arterial and capillary deposition of C4d that was more diffuse and intense at 6 – 8 days in FcγRIII-KO than in WT recipients (Figs 2C, 2D, 3B). C4d deposition became more diffuse and intense in the terminal phases of rejection in WT recipients (Figs 2D and 3B). The vascular injury in acutely rejected B10.A cardiac allografts in FcγRIII-KO recipients was accompanied by intense intravascular release of von-Willebrand factor (vWf) and formation of platelet aggregates that occluded capillaries. Immunohistological staining demonstrated abundant vWf associated with these platelet aggregates (SDC Fig 4). In FcγRIII-KO recipients more vWf was associated with platelets and less vWf was confined to cytoplasmic storage granules of endothelial cells than in hearts transplanted to WT recipients.
Fig 4. IgM, IgG and IgG subclasses of alloantibodies to MHC class I antigens measured by flow cytometry in serum from C57BL/6 WT (square symbols) and FcγRIII-KO recipients (circle symbols) 7 days after transplantation.

FcγRIII-KO demonstrated higher levels of IgM (A) and IgG (B) alloantibodies to MHC class I antigens in the circulation than in WT recipients (titer >64). IgG was mainly composed of C-activating IgG2a and IgG2b subclasses (C, D). The level of non-C activating IgG1 was very low in both FcγRIII-KO and WT recipients (E). Alloantibodies from FcγRIII-KO mice showed significantly higher ability to activate C and deposit C4d on target cells than their WT counterparts (F). Each point represents the average mode channel fluorescence staining +SD (n>10). Statistically significant differences (p<0.05) for IgG, IgM, IgG2a, IgG2b and C4d are indicated with *.
Alloantibody responses to class I MHC in C57BL/6 FcγRIII-KO and WT recipients elicited by cardiac allografts from B10.A
The level of alloantibodies in the circulation was measured by two-color flow cytometry using donor specific lymph node lymphocytes as target cells. Rejection of B10.A cardiac allografts in C57BL/6 FcγRIII-KO was accompanied by significantly higher levels of IgM and IgG alloantibodies to MHC class I antigens in the circulation than in WT recipients (titer >64) measured 7 days after transplantation (Figs 4A, 4B). Interestingly, IgG was mainly composed of complement-activating IgG2a and IgG2b subclasses (Figs 4C, 4D). The level of non-complement-activating IgG1 was very low in both FcγRIII-KO and WT recipients (Fig 4E).
To further confirm the ability of these antibodies to activate complement we performed a flow cytometry experiment using donor-specific lymphocytes sensitized with alloantibodies in the presence of 15% fresh mouse sera as the source of complement. Alloantibodies from FcγRIII-KO mice showed significantly higher ability to activate complement and deposit C4d on target cells than their WT counterparts (Fig 4F).
SAP-opsonizes apoptotic bodies and cardiomycytes in transplanted hearts
Because SAP is known to bind to apoptotic bodies and potentiate their clearance by FcγRIII-bearing cells in non-transplanted tispplees, we stained cardiac allograft tissues for the presence of intragraft SAP. Immunohistological stains indicated that SAP bound not only apoptotic bodies, but also nuclei of injured myocytes (SDC Fig 5). This observation indicates that SAP can opsonize cardiac myocytes in the early apoptotic process as well as late apoptotic bodies.
Fig 5. The level of SAP in the circulation measured by ELISA and intragraft expression of cytokines of cardiac allograft recipients measured by Real-Time-PCR.

The levels of SAP in sera of FcγRIII-KO recipients were significantly higher (p<0.0001) than in their WT counterparts (A). In FcγRIII-KO recipients, the intragraft level of IFN-γ RNA transcripts was significantly higher (p<0.01) than in WT recipients, while IL-10 transcripts were significantly downregulated in FcγRIII-KO hearts (p<0.01), (B). The levels of IL-12 and TNF- α in WT and FcγRIII-KO hearts were statistically insignificant. Each bar represents the average value of SAP (mg/ml) +SD or relative gene expression +SD (n>15) calculated using a comparative ddCT formula (see Materials and Methods). Statistically significant differences are indicated with *.
The level of SAP in the circulation of cardiac allograft recipients
Knowing that SAP is an acute phase reactant, the levels of SAP were measured by ELISA as an indicator of the extent of pro-inflammatory responses in the FcγRIII-KO recipients. The levels of SAP in sera of FcγRIII-KO recipients were significantly higher (p<0.0001) than in their WT counterparts (Fig 5A). These findings were confirmed in FcγRIII-KO recipients of B10.A skin grafts (data not shown).
The pattern of intragraft expression of pro- and anti-inflammatory cytokines
We tested whether the lack of effective clearance of apoptotic bodies affects the intragraft production of cytokines. To this end we analyzed the profile of intragraft mRNA transcripts for pro-inflammatory (IFN-γ, IL-12, and TNF-α) and anti-inflammatory (IL-10) cytokines using a quantitative Real-Time PCR method (Fig 5B). While the expression of mRNA transcripts for IL-12 and TNF-α was not statistically different in FcγRIII-KO vs WT recipients, we found significant differences in the expression of IFN-γ and IL-10. In FcγRIII-KO recipients, the intragraft level of IFN-γ RNA transcripts was significantly higher (p<0.01) than in WT recipients, while IL-10 transcripts were significantly downregulated in FcγRIII-KO hearts (p<0.01) (Fig 5B).
Discussion
In the present studies, we found B10.A hearts transplanted to C57BL/6 FcγRIII-KO recipients were rejected faster than in WT recipients (6-8 vs 10-14 days, respectively). The apparent paradoxical finding that the lack of activating FcγRIII contributes to acceleration of acute cardiac allograft rejection may be attributable to another function of FcR, namely the efficient clearance of apoptotic cells. Rejection in FcγRIII-KO recipients was associated with extensive accumulation of apoptotic cells compared with that seen in WT. Furthermore, pockets of apoptotic blebs were opsonized with SAP, but in the absence of FcγRIII they were not cleared by phagocytic cells.
Previous studies have reported that acute and chronic rejection is associated with apoptosis of cardiac myocytes and vascular endothelial cells (29-32). Our immunohistological stains for activated caspase 3 clearly demonstrated apoptosis of cardiac myocytes. In the absence of normal clearance, these apoptotic myocytes would present added donor antigen.
The acceleration of acute rejection in FcγRIII-KO was also accompanied by intense perivascular margination of monocytes and neutrophils and increased IgM and IgG alloantibody production. The IgG alloantibodies were mainly represented by C-activating IgG2b and IgG2a subclasses that activated C4 as measured by its final product C4d deposited on donor specific lymphocytes in vitro and intragraft C4d deposition. Endothelial injury was indicated by intravascular release of vWf and abundant formation of platelet aggregates. Systemic inflammation was indicated by elevated SAP levels in the circulation, while intragraft inflammation was accompanied by SAP bound to apoptotic bodies, a significant increase of intragraft IFN-γ and decrease of IL-10 mRNA expression.
Our data support the concept that accelerated acute cardiac allograft rejection in the absence of FcγRIII in vivo results from vigorous pro-inflammatory immune responses involving FcR-dependent functions through complement, antibodies and SAP. There are a few reports in the literature indicating that the lack of FcγRIII might contribute to development of autoimmunity (33, 34). However, no similar studies have been reported in models of allograft rejection.
In this study we found high levels of complement-activating IgG2a and IgG2b alloantibodies in the circulation as well as strong perivascular deposition of C4d in grafts of FcγRIII-KO recipients. In our model intragraft deposition of C4d might reflect activation of C4 that is initiated by binding of C1q to alloantibodies and SAP or directly to apoptotic bodies (35), and in addition by MBL binding to alloantibodies or apoptotic bodies (36, 37).
Similar to other molecules of innate immune system SAP recognizes a wide variety of ligands from apoptotic cells, damaged or altered tissues as well as determinants on microorganisms. The SAP ligand-binding site is calcium-dependent and recognizes specifically membrane phospholipids containing phosphoethanolamine/phosphocholine (38), heparan sulfate (39), laminin (40), fibronectin and C4b binding protein (41). DNA, chromatin and histones serve also as common physiological ligands for SAP (42, 43).
CRP, expressed in high levels in humans, binds to phospholipids on apoptotic blebs and activates complement through binding of C1q. CRP also interacts with FcγRs on macrophages, PMNs and NK cells (17). Similarly, in mice, apoptotic cells and bodies are opsonized by SAP and phagocytosed through interaction with FcγRs on macrophages and neutrophils. However, when SAP-opsonized apoptotic bodies are not engaged by FcγR they become immunogenic leading to an increase of pro-inflammatory immune responses. Opsonization of apoptotic blebs by SAP followed by sufficient elimination of apoptotic blebs through interaction with activating FcγRs prevents development of uncontrolled allo- and autoimmunity (44).
Du Clos, Mold and colleagues (14, 15) documented that bone marrow macrophages from FcR-γ-chain-deficient mice did not phagocytose SAP- and CRP-opsonized zymosan. While in this study FcγRI was identified as a high affinity and the only receptor for CRP, both FcγRI and low affinity FcγRIII were identified as receptors for SAP in the mouse. CRP and SAP binding to apoptotic cells has been well documented in experiments in vitro.
Our study provides in vivo evidence that accelerated cardiac rejection in the absence of FcγRIII is associated with the lack of efficient SAP-mediated clearance of apoptotic cells through interaction with FcγRs. We suggest that apoptotic cells that are not sufficiently cleared become immunogenic and induce enhanced inflammation that is mediated in part by complement activation. Similarly, in some models of autoimmune diseases (e.g., in SLE nephritis) genetically determined low levels of CRP have been associated with the severity of the disease (45). Nuclear antigens released from apoptotic cells that were not cleared properly promoted autoimmunity, an increase of pro-inflammatory responses that were associated with the expression of high levels of IFN-γ. In our model of cardiac allograft rejection in FcγRIII-KO recipients we found similar type of pro-inflammatory phenotype associated with high levels of intragraft m-RNA transcripts for IFN-γ.
Regulatory role of IL-10 produced by macrophages has been implicated in FcR/CRP/SAP interactions in different mouse models in vivo (44, 46-49). Du Clos' and Mold's group showed that CRP-mediated protection from LPS-challenged mice required the presence of functional FcγR and was associated with downregulation of proinflammatory response and enhanced production of IL-10 (46). In the absence of FcγRs the profile of cytokine expression was reversed and the protective effect of CRP was abolished (46).
Taken together, the data presented in our study indicate that SAP-opsonized apoptotic bodies are not engaged by FcγR in FcγRIII-KO recipients and consequently not sufficiently cleared. Therefore we suggest that the severely impaired process of clearance of SAP-opsonized apoptotic bodies in FcγRIII-KO recipients is one of the potential mechanisms that contribute to rapidly developed pro-inflammatory immune responses leading to accelerated rejection of mouse cardiac allografts.
Materials and Methods
Mice
Male C57BL/6 (H-2b), B10.A (H-2a) and FcγR3 tm1Sjv (FcγRIII-KO) inbred mice were purchased from Jackson Laboratories (Bar Harbor, ME) and used at 8-12 weeks of age. This genetic deletion has been backcrossed onto C57BL/6 for 12 generations. By the 12th generation, the backcrossed mice are genetically identical to the parental strain (50).
Heterotopic Heart Transplantation
B10.A hearts were transplanted into wild type (WT) or FcγRIII-KO C57BL/6 recipients using the standard technique with some modifications as previously described (6). Briefly, the donor aorta and pulmonary artery were anastomosed to the recipient abdominal aorta and inferior vena cava, respectively. Graft function was monitored by abdominal palpation daily until rejection, which was defined as total cessation of contractions.
Histology
Full cross sections through the center of cardiac grafts obtained at the time of sacrifice were frozen in OCT. Adjacent cross-sections were fixed in 60% methanol/10% glacial acetic acid solution, embedded in paraffin, and sectioned at 7 microns. Rejection was assessed on sections that were stained with hematoxylin and eosin (H&E).
Immunohistochemistry
High-temperature antigen retrieval and paraffin removal was performed by immersing the slides in Trilogy (Cell Marque, Hot Springs, AR) in a pressure cooker until the chamber reached 125°C and 23 psi. Endogenous peroxidase activity was blocked for 18 min in 0.3% H2O2 in methanol. Slides were incubated for 30 minutes in a serum free blocking solution (Dako, Carpenteria, CA) followed by an overnight incubation with a monoclonal antibody rat anti-mouse Mac2 marker of “inflammatory macrophages” (Cedarlane Laboratories, Burlington, NC), a polyclonal rabbit antibody to either human cleaved caspase-3 (Cell Signaling Technology, Boston, MA), vWf (Dako) or mouse C4d (developed in our laboratory (7), now available from Hycult, Biotech, Plymouth Meeting, PA). Sections were then incubated with biotinylated polyclonal donkey anti-rat or donkey anti-rabbit antibodies (Jackson Labs, West Grove, PA). For detection of SAP a polyclonal biotinylated sheep antibody to mouse SAP was used (R&D Systems). Immunoperoxidase staining was performed using Vectastain ABC Elite (Vector Labs, Burlingame, CA). The avidin-biotin complex was visualized using Impact DAB substrate (Vector Labs). Sections were counterstained in hematoxylin (Richard-Allen, Kalamazoo, MI).
Measurement of extent of apoptosis and C4d deposition
The extent of apoptotic cells and nuclear fragments marked by staining for activated caspase 3 was quantified on digital images captured with a 20x magnification objective. Five separate images were captured from fields in the central myocardium distant from areas associated with epicardial and endocardial inflammation. In allografts approaching complete rejection, areas that displayed advanced rejection and myocyte necrosis were excluded. Counting was performed by superimposing a grid of 50 microns square on the captured image (SDC Fig 3) and counting the number of squares containing apoptotic cells. This method avoided multiple apoptotic bodies from single cells contributing to the count disproportionately (SDC Fig 2).
C4d deposition was graded on the clinical criteria of distribution (focal versus diffuse) and intensity. Each parameter was scored on a 0 to 3+ scale and with a maximum total score of 6.
Measurement of alloantibodies in sera
Alloantibodies were measured by flow cytometry on single cell suspensions of cervical lymph nodes from donor B10.A mice using standard technique described previously (6). Briefly, 50 μl aliquots containing 1.5×105 B10.A lymph node lymphocytes were incubated with 50 μl of diluted sera from transplanted mice (1:4, 1:16, 1:64). To stain for IgM and IgG alloantibody, the washed cells were reacted with 50 μl of PBS + 0.5% BSA + 0.02% sodium azide (PBA) containing a mixture of FITC-conjugated goat antibody specific for the Fc portion of mouse IgG and PE-conjugated goat antibody specific for the μ chain of mouse IgM (Jackson Immunoresearch Laboratories). After staining, the cells were washed, fixed, and analyzed by flow cytometry. To analyze the IgG subclass of alloantibodies, cells were washed after the first incubation with diluted sera and then reacted with 50 μl of PBA containing an optimal dilution of FITC-conjugated rat mAb against mouse IgG1, IgG2a and IgG2b subclasses (Pharmingen, CA). After two washes, the cells were fixed in PBS/1% formaldehyde, and analyzed using FACScan (Becton Dickinson, Mountain View, CA). IgM, IgG and IgG subclass antibodies to class I MHC antigens on B10.A target cells were expressed as the mode channel fluorescent (MCF). The average MCF value for control sera was subtracted from the MCF value for experimental sera.
Measurement of C4d deposition on sensitized lymphocytes by flow cytometry
Activation of complement by IgM and IgG alloantibodies was measured by flow cytometry according to method developed in our laboratory and previously described (7). Briefly, 50 μl aliquots containing 2×105 B10.A (H-2a) lymph node lymphocytes were incubated with 50 μl of diluted sera from transplanted mice (1:4, 1:16, 1:64) for 45 min in +4°C. The cells were washed twice in Gelatin Veronal Buffer 2+ (GVB2+), (Sigma-Aldrich, St. Louis, MO), then incubated for 30 minutes at 37°C in GVB2+ containing 15% fresh sera from normal mouse as the source of complement. The cells were washed twice in PBA, then stained with the polyclonal rabbit anti-mouse C4d antibody for 30 min at +4°C and washed twice in PBA. Finally, the cells were stained with a goat anti-rabbit-IgG-FITC (Jackson ImmunoResearch), incubated for 30 min at +4°C and washed twice in PBA. The cells were fixed in PBS/1% formaldehyde, then analyzed on a FACScan cytometer.
Measurement of SAP in sera by ELISA
The levels of SAP in sera from heart recipients were assessed using ELISA detection assay for mouse SAP (Kamiya Biomedical, Seattle, WA) according to the manufacture's recommendations. The intensity of the enzymatic reaction was measured at 450 nm in an E Max Microplate Reader 2100R (Molecular Devices Corporation, Sunnyvale, CA).
Preparation of RNA
Total cellular RNA was isolated from snap frozen heart tissues using TRIZOL (Invitrogen, CA) according to the manufacture's guidelines and the method described in our previous studies (5, 6).
Real-Time-PCR
Total RNA from heart tissues was reverse transcribed into cDNA as described in our previous studies (6). The cDNA was then analyzed for cytokine expression by Real-Time-PCR method using Pre-Developed TaqMan Reagents containing specific target primers and probes for mouse IFN-γ, IL-10, IL-12, TNF- α and GAPDH (PE Applied Biosystems, Foster City, CA). Data were collected with Sequence Detector software (Bio-Rad MyIQ thermocycler, Hercules, CA) from which an amplification plot was generated. From this plot the threshold value (CT) was calculated, representing the PCR cycle value at which fluorescence was detectable above an arbitrary threshold. Relative gene expression within each group was calculated using a comparative ddCT method. Target gene levels are expressed as a ratio of CT for a target gene relative to the endogenous housekeeping gene GAPDH (dCT) and to calibrator sample using the formula 2-ddCT.
Statistical analysis
Survival time data in WT and FcγRIII-KO recipients was analyzed using log-rank test in Kaplan-Meier plot analysis. All other data were analyzed using TT-Student's test.
Supplementary Material
Acknowledgments
Support received by authors of the study: Barbara A. Wasowska was supported by: ROTRF Research Grant #513443778 American Heart Association Grant-in-Aid, Grifols Therapeutics, Inc., Talent Program research grant. William M. Baldwin, III, was supported by NIH, P01AI087586 grant.
We thank Nina Dvorina, MD, from the Department of Immunology Lerner Research Institute, Cleveland Clinic, for providing immuno-histochemical stains with mAb rat-anti-mouse Mac2 marker of “inflammatory macrophages”. This work was supported by the following grants: ROTRF Research Grant #513443778, American Heart Association Grant-in-Aid, Grifols Therapeutics, Inc., Talent Program research grant for Barbara A. Wasowska; NIH, P01AI087586 grant for William M. Baldwin, III.
Footnotes
None of the authors declared potential conflict of interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Racusen LC, Colvin RB, Solez K, Mihatsch MJ, Halloran PF, Campbell PM, et al. Antibody-mediated rejection criteria - an addition to the banff 97 classification of renal allograft rejection. Am J Transplant. 2003;3(6):708–14. doi: 10.1034/j.1600-6143.2003.00072.x. [DOI] [PubMed] [Google Scholar]
- 2.Feucht HE. Complement C4d in graft capillaries - the missing link in the recognition of humoral alloreactivity. Am J Transplant. 2003;3(6):646–52. doi: 10.1034/j.1600-6143.2003.00171.x. [DOI] [PubMed] [Google Scholar]
- 3.Baldwin WM, III, Larsen CP, Fairchild RL. Innate immune responses to transplants: a significant variable with cadaver donors. Immunity. 2001;14:369–76. doi: 10.1016/s1074-7613(01)00117-0. [DOI] [PubMed] [Google Scholar]
- 4.Sacks SH, Zhou W. Locally produced complement and its role in renal allograft rejection. Am J Transplant. 2003;3(8):927–32. doi: 10.1034/j.1600-6143.2003.00175.x. [DOI] [PubMed] [Google Scholar]
- 5.Wasowska BA, Qian Z, Cangello DL, Behrens E, Van Tran K, Layton J, et al. Passive transfer of alloantibodies restores acute cardiac rejection in IgKO mice. Transplantation. 2001;71(6):727–36. doi: 10.1097/00007890-200103270-00007. [DOI] [PubMed] [Google Scholar]
- 6.Rahimi S, Qian Z, Layton J, Fox-Talbot K, Baldwin WM, Wasowska BA. Non-complement and complement activating antibodies synergize to cause rejection of cardiac allografts. American J Transpl. 2004;4:326–34. doi: 10.1111/j.1600-6143.2004.00334.x. [DOI] [PubMed] [Google Scholar]
- 7.Murata K, Fox-Talbot K, Qian Z, Takahashi K, Stahl GL, Baldwin WM, 3rd, et al. Synergistic Deposition of C4d by Complement-Activating and Non-activating Antibodies in Cardiac Transplants. Am J Transplant. 2007;7(11):2605–14. doi: 10.1111/j.1600-6143.2007.01971.x. [DOI] [PubMed] [Google Scholar]
- 8.Nimmerjahn F, Ravetch JV. Fc-receptors as regulators of immunity. Adv Immunol. 2007;96:179–204. doi: 10.1016/S0065-2776(07)96005-8. [DOI] [PubMed] [Google Scholar]
- 9.Macdermott RP, Nash GS, Merkle NS, Weinrieb IJ, Bertovich MJ, Formeister jF. Further evidence that antibody-dependent and spontaneous cell-mediated cytotoxicity are mediated by different processes or cell types. Immunology. 1980;41(2):439–47. [PMC free article] [PubMed] [Google Scholar]
- 10.Miltenburg AMM, Meijer-Paape ME, Weening JJ, Daha MR, van Es LA, van der Woude FJ. Induction of antibody-dependent cellular cytotoxicity against endothelial cells by renal transplantation. Transplantation. 1989;48:681. [PubMed] [Google Scholar]
- 11.Marsh CB, Wewers MD, Tan LC, Rovin BH. Fc(gamma) receptor cross-linking induces peripheral blood mononuclear cell monocyte chemoattractant protein-1 expression: role of lymphocyte Fc(gamma)RIII. J Immunol. 1997;158(3):1078–84. [PubMed] [Google Scholar]
- 12.Shushakova N, Skokowa J, Schulman J, Baumann U, Zwirner J, Schmidt RE, et al. C5a anaphylatoxin is a major regulator of activating versus inhibitory FcgammaRs in immune complex-induced lung disease. J Clin Invest. 2002;110(12):1823–30. doi: 10.1172/JCI200216577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kumar V, Ali SR, Konrad S, Zwirner J, Verbeek JS, Schmidt RE, et al. Cell-derived anaphylatoxins as key mediators of antibody-dependent type II autoimmunity in mice. J Clin Invest. 2006;116(2):512–20. doi: 10.1172/JCI25536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stein MP, Mold C, Du Clos TW. C-reactive protein binding to murine leukocytes requires Fc gamma receptors. J Immunol. 2000;164(3):1514–20. doi: 10.4049/jimmunol.164.3.1514. [DOI] [PubMed] [Google Scholar]
- 15.Mold C, Gresham HD, Du Clos TW. Serum amyloid P component and C-reactive protein mediate phagocytosis through murine Fc gamma Rs. J Immunol. 2001;166(2):1200–5. doi: 10.4049/jimmunol.166.2.1200. [DOI] [PubMed] [Google Scholar]
- 16.Bharadwaj D, Mold C, Markham E, Du Clos TW. Serum amyloid P component binds to Fc gamma receptors and opsonizes particles for phagocytosis. J Immunol. 2001;166(11):6735–41. doi: 10.4049/jimmunol.166.11.6735. Epub 2001/05/22. [DOI] [PubMed] [Google Scholar]
- 17.Bharadwaj D, Stein MP, Volzer M, Mold C, Du Clos TW. The major receptor for C-reactive protein on leukocytes is fcgamma receptor II. J Exp Med. 1999;190(4):585–90. doi: 10.1084/jem.190.4.585. Epub 1999/08/17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lu J, Marnell LL, Marjon KD, Mold C, Du Clos TW, Sun PD. Structural recognition and functional activation of FcgammaR by innate pentraxins. Nature. 2008;456(7224):989–92. doi: 10.1038/nature07468. Epub 2008/11/18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pepys MB, Baltz ML. Acute phase proteins with special reference to C-reactive protein and related proteins (pentaxins) and serum amyloid A protein. Adv Immunol. 1983;34:141–212. doi: 10.1016/s0065-2776(08)60379-x. Epub 1983/01/01. [DOI] [PubMed] [Google Scholar]
- 20.Bottazzi B, Doni A, Garlanda C, Mantovani A. An integrated view of humoral innate immunity: pentraxins as a paradigm. Annu Rev Immunol. 2010;28:157–83. doi: 10.1146/annurev-immunol-030409-101305. Epub 2009/12/09. [DOI] [PubMed] [Google Scholar]
- 21.Poon IK, Hulett MD, Parish CR. Molecular mechanisms of late apoptotic/necrotic cell clearance. Cell Death Differ. 2010;17(3):381–97. doi: 10.1038/cdd.2009.195. Epub 2009/12/18. [DOI] [PubMed] [Google Scholar]
- 22.Du Clos TW. The interaction of C-reactive protein and serum amyloid P component with nuclear antigens. Mol Biol Rep. 1996;23(3-4):253–60. doi: 10.1007/BF00351177. Epub 1996/01/01. [DOI] [PubMed] [Google Scholar]
- 23.Du Clos TW, Mold C, Stump RF. Identification of a polypeptide sequence that mediates nuclear localization of the acute phase protein C-reactive protein. J Immunol. 1990;145(11):3869–75. Epub 1990/12/01. [PubMed] [Google Scholar]
- 24.Du Clos TW, Mold C. Pentraxins (CRP, SAP) in the process of complement activation and clearance of apoptotic bodies through Fcgamma receptors. Curr Opin Organ Transplant. 2010 doi: 10.1097/MOT.0b013e32834253c7. Epub 2010/12/15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bottazzi B, Garlanda C, Salvatori G, Jeannin P, Manfredi A, Mantovani A. Pentraxins as a key component of innate immunity. Curr Opin Immunol. 2006;18(1):10–5. doi: 10.1016/j.coi.2005.11.009. [DOI] [PubMed] [Google Scholar]
- 26.Takai T, Li M, Sylvestre D, Clynes R, Ravetch JV. FcR gamma chain deletion results in pleiotrophic effector cell defects. Cell. 1994;76(3):519–29. doi: 10.1016/0092-8674(94)90115-5. Epub 1994/02/11. [DOI] [PubMed] [Google Scholar]
- 27.Takai T. Roles of Fc receptors in autoimmunity. Nat Rev Immunol. 2002;2(8):580–92. doi: 10.1038/nri856. [DOI] [PubMed] [Google Scholar]
- 28.Duan WR, Garner DS, Williams SD, Funckes-Shippy CL, Spath IS, Blomme EA. Comparison of immunohistochemistry for activated caspase-3 and cleaved cytokeratin 18 with the TUNEL method for quantification of apoptosis in histological sections of PC-3 subcutaneous xenografts. J Pathol. 2003;199(2):221–8. doi: 10.1002/path.1289. Epub 2003/01/21. [DOI] [PubMed] [Google Scholar]
- 29.Koglin J, Granville DJ, Glysing-Jensen T, Mudgett JS, Carthy CM, McManus BM, et al. Attenuated acute cardiac rejection in NOS2 -/- recipients correlates with reduced apoptosis. Circulation. 1999;99(6):836–42. doi: 10.1161/01.cir.99.6.836. Epub 1999/02/17. [DOI] [PubMed] [Google Scholar]
- 30.Szabolcs M, Michler RE, Yang X, Aji W, Roy D, Athan E, et al. Apoptosis of cardiac myocytes during cardiac allograft rejection. Relation to induction of nitric oxide synthase. Circulation. 1996;94(7):1665–73. doi: 10.1161/01.cir.94.7.1665. Epub 1996/10/01. [DOI] [PubMed] [Google Scholar]
- 31.Le Bas-Bernardet S, Hourmant M, Coupel S, Bignon JD, Soulillou JP, Charreau B. Non-HLA-type endothelial cell reactive alloantibodies in pre-transplant sera of kidney recipients trigger apoptosis. Am J Transplant. 2003;3(2):167–77. doi: 10.1034/j.1600-6143.2003.00021.x. Epub 2003/02/27. [DOI] [PubMed] [Google Scholar]
- 32.Cailhier JF, Laplante P, Hebert MJ. Endothelial apoptosis and chronic transplant vasculopathy: recent results, novel mechanisms. Am J Transplant. 2006;6(2):247–53. doi: 10.1111/j.1600-6143.2005.01165.x. Epub 2006/01/24. [DOI] [PubMed] [Google Scholar]
- 33.Clynes R. IVIG therapy: interfering with interferon-gamma. Immunity. 2007;26(1):4–6. doi: 10.1016/j.immuni.2007.01.006. [DOI] [PubMed] [Google Scholar]
- 34.Araujo LM, Chauvineau A, Zhu R, Diem S, Bourgeois EA, Levescot A, et al. Cutting edge: intravenous Ig inhibits invariant NKT cell-mediated allergic airway inflammation through FcgammaRIIIA-dependent mechanisms. J Immunol. 2011;186(6):3289–93. doi: 10.4049/jimmunol.1003076. Epub Epub 2011 Feb 11. [DOI] [PubMed] [Google Scholar]
- 35.Korb LC, Ahearn JM. C1q binds directly and specifically to surface blebs of apoptotic human keratinocytes: complement deficiency and systemic lupus erythematosus revisited. J Immunol. 1997;158(10):4525–8. Epub 1997/05/15. [PubMed] [Google Scholar]
- 36.Nauta AJ, Raaschou-Jensen N, Roos A, Daha MR, Madsen HO, Borrias-Essers MC, et al. Mannose-binding lectin engagement with late apoptotic and necrotic cells. Eur J Immunol. 2003;33(10):2853–63. doi: 10.1002/eji.200323888. [DOI] [PubMed] [Google Scholar]
- 37.Nauta AJ, Castellano G, Xu W, Woltman AM, Borrias MC, Daha MR, et al. Opsonization with C1q and mannose-binding lectin targets apoptotic cells to dendritic cells. J Immunol. 2004;173(5):3044–50. doi: 10.4049/jimmunol.173.5.3044. Epub 2004/08/24. [DOI] [PubMed] [Google Scholar]
- 38.Thompson D, Pepys MB, Wood SP. The physiological structure of human C-reactive protein and its complex with phosphocholine. Structure. 1999;7(2):169–77. doi: 10.1016/S0969-2126(99)80023-9. Epub 1999/06/16. [DOI] [PubMed] [Google Scholar]
- 39.Hamazaki H. Ca2+-mediated association of human serum amyloid P component with heparan sulfate and dermatan sulfate. J Biol Chem. 1987;262(4):1456–60. Epub 1987/02/05. [PubMed] [Google Scholar]
- 40.Zahedi K. Characterization of the binding of serum amyloid P to laminin. J Biol Chem. 1997;272(4):2143–8. Epub 1997/01/24. [PubMed] [Google Scholar]
- 41.de Beer FC, Baltz ML, Holford S, Feinstein A, Pepys MB. Fibronectin and C4-binding protein are selectively bound by aggregated amyloid P component. J Exp Med. 1981;154(4):1134–9. doi: 10.1084/jem.154.4.1134. Epub 1981/10/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pepys MB, Butler PJ. Serum amyloid P component is the major calcium-dependent specific DNA binding protein of the serum. Biochem Biophys Res Commun. 1987;148(1):308–13. doi: 10.1016/0006-291x(87)91111-9. Epub 1987/10/14. [DOI] [PubMed] [Google Scholar]
- 43.Bickerstaff MC, Botto M, Hutchinson WL, Herbert J, Tennent GA, Bybee A, et al. Serum amyloid P component controls chromatin degradation and prevents antinuclear autoimmunity. Nat Med. 1999;5(6):694–7. doi: 10.1038/9544. Epub 1999/06/17. [DOI] [PubMed] [Google Scholar]
- 44.Castano AP, Lin SL, Surowy T, Nowlin BT, Turlapati SA, Patel T, et al. Serum amyloid P inhibits fibrosis through Fc gamma R-dependent monocyte-macrophage regulation in vivo. Sci Transl Med. 2009;1(5):5ra13. doi: 10.1126/scitranslmed.3000111. Epub 2010/04/07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jonsen A, Gunnarsson I, Gullstrand B, Svenungsson E, Bengtsson AA, Nived O, et al. Association between SLE nephritis and polymorphic variants of the CRP and FcgammaRIIIa genes. Rheumatology (Oxford) 2007;46(9):1417–21. doi: 10.1093/rheumatology/kem167. Epub 2007/06/29. [DOI] [PubMed] [Google Scholar]
- 46.Mold C, Rodriguez W, Rodic-Polic B, Du Clos TW. C-reactive protein mediates protection from lipopolysaccharide through interactions with Fc gamma R. J Immunol. 2002;169(12):7019–25. doi: 10.4049/jimmunol.169.12.7019. Epub 2002/12/10. [DOI] [PubMed] [Google Scholar]
- 47.Padigel UM, Farrell JP. Control of infection with Leishmania major in susceptible BALB/c mice lacking the common gamma-chain for FcR is associated with reduced production of IL-10 and TGF-beta by parasitized cells. J Immunol. 2005;174(10):6340–5. doi: 10.4049/jimmunol.174.10.6340. Epub 2005/05/10. [DOI] [PubMed] [Google Scholar]
- 48.Singh U, Devaraj S, Dasu MR, Ciobanu D, Reusch J, Jialal I. C-reactive protein decreases interleukin-10 secretion in activated human monocyte-derived macrophages via inhibition of cyclic AMP production. Arterioscler Thromb Vasc Biol. 2006;26(11):2469–75. doi: 10.1161/01.ATV.0000241572.05292.fb. Epub 2006/08/19. [DOI] [PubMed] [Google Scholar]
- 49.Rodriguez W, Mold C, Kataranovski M, Hutt JA, Marnell LL, Verbeek JS, et al. C-reactive protein-mediated suppression of nephrotoxic nephritis: role of macrophages, complement, and Fcgamma receptors. J Immunol. 2007;178(1):530–8. doi: 10.4049/jimmunol.178.1.530. Epub 2006/12/22. [DOI] [PubMed] [Google Scholar]
- 50.Sechler JM, Yip JC, Rosenberg AS. Genetic variation among 129 substrains: practical consequences. J Immunol. 1997;159(12):5766–8. Epub 1998/04/29. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.