

Abstract
Leptin a regulator of body weight is involved in reproductive and developmental functions. Leptin promoter DNA methylation (LEP) regulates gene expression in a tissue-specific manner and has been linked to adverse pregnancy outcomes. In non-pathologic human pregnancies, we assessed LEP methylation, genotyped the single nucleotide polymorphism (SNP) rs2167270 in placental (n=81), maternal and cord blood samples (n=60), and examined the association between methylation, genotype, and perinatal factors. Maternal blood LEP methylation was lower in pre-pregnancy obese women (P=0.01). Cord blood LEP methylation was higher in small for gestational age (SGA) (P=4.6×10−3) and A/A genotype (P=1.6×10−4), lower (−1.47, P=0.03) in infants born to pre-pregnancy obese mothers and correlated (P=0.01) with maternal blood LEP. Gender was associated with placental LEP methylation (P=0.05). These results suggest that LEP epigenetic control may be influenced by perinatal factors including: maternal obesity, infant growth, genotype and gender in a tissue-specific manner and may have multigenerational implications.
Keywords: leptin, epigenetics, DNA methylation, rs2167270, pregnancy, maternal obesity, small for gestational age
1. Introduction
Obesity is a global public health concern, responsible for substantial morbidity, mortality and increasing health-care costs (Swinburn et al., 2011). In the United States, it is estimated that one in three adults are obese (Flegal et al., 2012); women of reproductive age do not escape this epidemic (Lawlor et al., 2012). Obesity during pregnancy and increased gestational weight gain (GWG) have been linked to obstetric pathology and early and adult life offspring outcomes (Oken, 2009; Poston et al., 2011). Simultaneously, small for gestational age (SGA) or large for gestational age (LGA) infants are at increased risk of obesity and other chronic diseases (Mehta et al., 2011; Rinaudo and Wang, 2012; Saenger et al., 2007). The relation between birthweight and adult disease gave rise to the “thrifty phenotype” hypothesis that posits that a suboptimal in utero environment “programs” the organism to adapt to low-resource conditions that if mismatched in adult life can result in metabolic disease (Hales and Barker, 2001). Later, this evolved into the developmental origins of adult health and disease (DOHaD) paradigm (Gluckman et al., 2005). Hence, it is possible that metabolic fetal programming of adult disease could contribute to the rising obesity epidemic.
Recent evidence has identified genetic and epigenetic factors could be involved in fetal programming; since it requires plasticity, epigenetic mechanisms are likely candidates, as increasing evidence has demonstrated their susceptibility to modification by environmental cues but their relative persistence following development (Christensen and Marsit, 2011; Michels and Waterland, 2012; Novakovic and Saffery, 2012). DNA methylation is an epigenetic modification that involves the addition of methyl group to a cytosine base in the context of a CpG dinucleotide, these to cluster in high density regions, often in gene promoters where increased methylation has been linked to decreased expression (Jones, 2012). DNA methylation marks are reset during embryonic development, and are then critical in establishing cellular differentiation through their restitution in a tissue-specific manner (Novakovic and Saffery, 2012).
Leptin, a 16 kD peptide hormone encoded by the gene LEP, predominantly produced by adipose tissue, is involved in food intake, energy expenditure, and reproduction amongst many other functions. Serum leptin correlates with adipose mass and body mass index (BMI) and induces satiety. Obesity is usually associated with hyperleptinemia, but obese women commonly have satiety deregulation because of a concomitant leptin resistance (Caprio et al., 2001; Friedman, 2011). Serum leptin increases during pregnancy (Lepercq et al., 2001) when it is produced by the placenta (Masuzaki et al., 1997) as well as both maternal and fetal adipose tissues. Leptin placental production starts early in development where it has paracrine and endocrine functions (Ashworth et al., 2000). DNA methylation at the LEP promoter follows an inverse relationship with LEP tissue expression in human cells in vitro (Melzner et al., 2002; Noer et al., 2006) and in primary tissue (Bouchard et al., 2010; Marchi et al., 2011). Placental LEP promoter methylation has been associated with pregnancy complications such as impaired glucose metabolism (Bouchard et al., 2010) and early onset pre-eclampsia (Hogg et al., 2013). During development, both excess and reduced nutrient availability could increase obesity risk, possibly by disruption of leptin-regulated food intake behaviors programmed in utero, but the mechanisms by which this happens are not understood (Vickers and Sloboda, 2012). In summary, LEP is epigenetically regulated and exerts metabolic and reproductive functions. Thus, LEP DNA methylation is a plausible factor to be involved in fetal metabolic programming. Using a quantitative methylation technique, the present study aimed to investigate the associations between LEP promoter DNA methylation in placental tissue, maternal blood, and infant cord blood, and perinatal maternal and infant factors in a population of healthy term infants.
2. Materials and methods
2.1. Study population and design
The subjects involved in this study are part of the ongoing Rhode Island Child Health Study (Marsit et al., 2012) that recruits mother-infant pairs following delivery at Women and Infants Hospital in Providence Rhode Island, USA starting in March 2009 and continuing to enroll. Briefly, term infants born small (≤10th percentile) and large (≥90th percentile) for gestational age, according to the Fenton growth charts (Fenton, 2003) were gender and gestational age matched with an infant appropriate for gestational age (AGA). Post-recruitment all infants were re-classified into birthweight categories using recently developed growth charts (Fenton and Kim, 2013). All subjects provided written informed consent following protocols approved by the Institutional Review Board at Women and Infants Hospital and Dartmouth College. Exclusion criteria included non-singleton pregnancy, maternal age <18 years, maternal life-threatening complications and infant congenital or chromosomal abnormalities. Clinical information was collected using a structured chart review form, followed by an interviewer-administered questionnaire to gather information on socio-demographic variables, personal and family medical, lifestyle and exposure history. Maternal pre-pregnancy body mass index (BMI), tobacco use during pregnancy and pregnancy hypertension were obtained from medical charts records and maternal GWG was self-reported in the study questionnaire.
2.2. Sample collection and nucleic acid extraction
Fetal placental samples were collected from all subjects within two hours following delivery; twelve fragments, three from each quadrant were obtained two centimeters (cm) from the umbilical cord and free of maternal decidua. Collected tissue was immediately placed on RNAlater solution (Life Technologies, Grand Island, NY, USA) and stored at 4°C. Subsequently, tissue segments from each placental region were blotted dry, snap frozen in liquid nitrogen, homogenized by pulverization using a stainless steel cup and piston unit (Cellcrusher, Cork, Ireland) and stored at −80°C until needed. When available, paired maternal and infant cord blood was obtained from residual samples collected for clinical purposes, making them suitable for total genomic DNA extraction but not for mRNA or protein measurements. Maternal blood was collected via venipuncture at the time of admission for delivery, and infant blood was obtained with a syringe from the umbilical cord after delivery. DNA was extracted from homogenized placental samples and from blood using the DNAeasy Blood & Tissue Kit (Qiagen, Inc, Valencia, CA, USA). The resulting DNA was quantified using the ND 2000 spectrophotometer (Thermo Fisher Scientific Inc., Watham, MA, USA). All procedures were performed following manufacturer’s protocols.
2.3. DNA Bisulfite Modification and Pyrosequencing Analysis
DNA samples (500 ng) were sodium bisulfite-modified using the EZ DNA methylation Kit (Zymo Research, Irvine, CA, USA), following the manufacturer’s protocols. For methylation detection, bisulfite pyrosequencing was employed. Primers (Integrated DNA Technologies, Inc, Coralville, IA) were designed using the PyroMark Assay Design software version 2.0.1.15 (Qiagen) in a region previously associated with leptin expression (Bouchard et al., 2010; Melzner et al., 2002; Yokomori et al., 2002). The PyroMark PCR kit (Qiagen) and PCR primers (Table S1) were used to amplify a 383 base pair region in the LEP promoter; cycling conditions were 94°C for 15 min followed by 50 cycles of 94°C for 1 min, 56°C for 1 min and 72°C for 1 min with a final extension of 10 min at 72°C. Pyrosequencing was performed in triplicate using the Pyromark MD (Qiagen) instrument with two forward assays covering a total of 23 CpG loci (Tables S1 and S2). Non-CpG cytosines within each read served as internal controls to verify bisulfite DNA modification efficiency (≥95% in all samples) and each pyrosequencing run included a no template control; all samples were sequenced by the same operator. DNA methylation results were analyzed with the PyroMark CpG software, version 1.0.11 (Qiagen).
2.4. SNP Genotyping
Samples were genotyped for rs2167270 (+19 G>A), a common SNP in the LEP promoter region studied. Genotypes calls were made by analyzing the pyrograms and comparing peak heights for each allele; triplicate wells were called independently and compared for quality control. Additionally, we confirmed the pyrosequencing genotypes by performing an allelic discrimination Taqman (Life Technologies) assay (ID:C__15966471_20) for maternal and cord blood DNA samples, using the CFX Connect™ Real-Time PCR Detection System (Bio-Rad Laboratories, Inc., Hercules, CA, USA). Genotype calls from the two methods were equivalent in all samples compared.
2.5. Statistical Analysis
The LEP promoter mean methylation distributions were assessed with Shapiro-Wilk test, confirming normality in the analyzed tissues. Pairwise Pearson correlations were used to compare methylation between each of the 23 CpG loci within each sample type. Bivariate associations between methylation extent and covariates were initially compared with a Student’s t-test or ANOVA. Fisher exact or χ2 tests were used accordingly to evaluate frequency differences between groups. Models were constructed with methylation as the dependent variable and either birthweight group, maternal pre-pregnancy BMI category or maternal blood LEP methylation as the independent variable. Covariates evaluated as possible confounders included variables that could potentially affect leptin methylation or birthweight (cord blood and placenta models) and/or maternal BMI (maternal blood models). Infant birthweight was evaluated as a categorical variable because of the known association of serum leptin with birthweight group, the clinical relevance of these groupings, and as the relationship between cord blood LEP methylation and infant weight was not linear. Self-reported maternal GWG data was combined with pre-pregnancy BMI to construct a categorical variable (adequate, inadequate or excessive GWG) following the Institute of Medicine (IMO) cutoffs (Rasmussen et al., 2010). The final adjusted regression models were corrected for maternal factors (pre-pregnancy BMI, GWG, race, tobacco use during pregnancy, pregnancy hypertension, rs2167270 genotype) and or infant factors (birthweight group, gender, infant genotype, delivery method). The maternal blood LEP regression model was adjusted only for the mentioned maternal factors and the cord blood and placental LEP methylation models we adjusted for both maternal and infant factors). Sensitivity analyses were performed to test that associations obtained from the linear models were not driven by outlying subjects. All analyses were conducted in RStudio version 0.97.314 (RStudio, Boston, MA, USA). All tests were two-sided and statistical significance was determined at P-value <0.05.
3. Results
3.1. Socio-demographic characteristics
The maternal and infant characteristics of the study population (n=81) are described in Table 1. The mean gestational age was 39 weeks with a similar proportion of female and male infants. In accordance with the planned composition of the cohort, our sample is overrepresented for LGA (28%, n=23) and SGA (27%, n=22) infants. The average maternal age was 29 years with the majority (73%) of the participants reporting Caucasian race. Only 5% reported tobacco use during pregnancy. Mothers were grouped according to World Health Organization classification of adult BMI categories: normal (18.5–24.9 kg/m2), overweight (25–29.9 kg/m2) and obese (≥30 kg/m2); the average BMI was 25.5 kg/m2 (standard deviation (SD) 5.9) and ranged from 17–41 kg/m2. Only five women had BMI <18.5 kg/m2 and were included in the normal category because of the low prevalence and their maternal blood LEP methylation values did not differ from those considered normal BMI (P=0.64, data not shown). In this study, we did not have any participants with gestational, type I or type II diabetes. Of note, in this population we observed a positive correlation between pre-pregnancy maternal BMI and infant birthweight (r =0.37, P=8.4×10−4).
Table 1.
Study population characteristics
Clinical and socio-demographic characteristic of the infants and mothers enrolled in the study (n=81) with placental LEP methylation information. Infant and maternal characteristic are described for specific parameters. Infants were classified according to gestational age and birthweight into three groups: adequate for gestational age (AGA), large for gestational age (LGA) and small for gestational age (SGA). SD: standard deviation.
| n | % | Mean | SD | |
|---|---|---|---|---|
| Infant variables | ||||
| Birthweight group | 81 | |||
| AGA | 36 | 44 | ||
| LGA | 23 | 28 | ||
| SGA | 22 | 27 | ||
| Birthweight (g) | 81 | 3406 | 714 | |
| Gestational age (weeks) | 81 | 39 | 1.1 | |
| Gender | 81 | |||
| Female | 40 | 49 | ||
| Male | 41 | 51 | ||
| IUGR | 77 | |||
| No | 67 | 87 | ||
| Yes | 10 | 13 | ||
| Maternal variables | ||||
| Age (yrs) | 81 | 29 | 6.2 | |
| Pre-pregnancy BMI (kg/m2) | 81 | 25.5 | 5.9 | |
| Normal | 47 | 58 | ||
| Overweight | 19 | 23 | ||
| Obese | 15 | 19 | ||
| Pregnancy Weight Gain | 80 | 14.1 | 5.9 | |
| Inadequate | 18 | 22.5 | ||
| Adequate | 22 | 27.5 | ||
| Excessive | 40 | 50 | ||
| Maternal race | 81 | |||
| Other | 22 | 27 | ||
| White | 59 | 73 | ||
| Tobacco during pregnancy | 81 | |||
| No | 77 | 95 | ||
| Yes | 4 | 5 | ||
| Delivery Method | 81 | |||
| Vaginal | 46 | 57 | ||
| C-Section | 35 | 43 | ||
| Pregnancy Hypertension | 80 | |||
| No | 74 | 92.5 | ||
| Yes | 6 | 7.5 | ||
SD: standard deviation; AGA: adequate for gestational age; LGA: large for gestational age; SGA: small for gestational age
3.2. LEP methylation distributions across tissues
We examined LEP promoter methylation using pyrosequencing of 23 CpG sites across different tissues: maternal blood (n=60), cord blood (n=60) and placentas (n=81). A group of 39 mother-infant pairs had matched maternal, cord blood and placental data, the remaining (n=42) had placental with either cord or maternal blood LEP methylation data. We observed a moderate correlation between all the CpG sites surveyed across the region in each of the examined tissues (Fig. S1, S2, S3). Due to this level of correlation and a prior report which utilized the mean methylation across the whole region (Bouchard et al., 2010), we utilized the mean of these 23 sites in subsequent analysis. The mean LEP methylation distribution from each tissue is displayed in Fig. 1. Placental methylation showed higher variability across samples than cord and maternal blood methylation. The average LEP mean promoter methylation across tissues differed significantly (P < 5×10−16); cord blood methylation was the lowest (14.1%, SD 1.9) followed by maternal blood (20.1%, SD 3.1) and placenta (23.7%, SD 5.5) (Table S3).
Fig. 1. LEP promoter methylation by tissue.
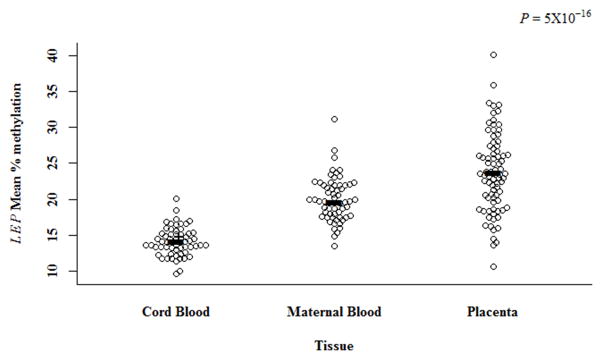
Distribution of LEP methylation (y-axis) in samples by tissue (x-axis): cord blood (n=60), maternal blood (n=60) and placenta (n=81). The black bars represents the mean within each tissue; cord blood (14.1%), maternal blood (20.1%) and placenta (23.7%) The P-value is the result of ANOVA test.
3.3. rs2167270 genotype, birthweight and maternal BMI
To examine if a common genetic variant (rs2167270) in the LEP promoter region analyzed was associated with methylation, we genotyped infant and maternal samples. Genotype frequencies in both mothers and infants were in Hardy-Weinberg equilibrium. Overall, 35% of the infants were homozygous for the wild type allele (G/G), 52% heterozygous (G/A) and 13% homozygous for the variant (A/A). Infant genotype frequencies did not differ between birthweight groups (Fisher’s exact test, P=0.23). For the subset of participants with maternal blood samples (n=60) we observed the following maternal genotype frequencies: 43% (G/G), 45% (G/A) and 12% (A/A), and these did not differ between BMI categories (P=1).
3.4. Maternal blood LEP DNA methylation
Considering the association of serum leptin and body weight during pregnancy (Highman et al., 1998; Misra and Trudeau, 2011), we performed multivariable linear regression analysis to model the association of maternal blood LEP promoter methylation and pre-pregnancy BMI category in the subset of participants with available maternal blood samples (Table 2, Fig. S4). The population characteristics of this group are shown in Table S4. We observed differences in maternal blood LEP methylation between BMI categories, which remained significant after adjusting for potential confounders including maternal age, genotype, ethnicity, tobacco use during pregnancy and pregnancy weight gain. Obese women had 2.95% (P=0.01) lower maternal blood LEP promoter methylation compared to normal weight women, controlling for confounders. In addition, although not statistically significant (P=0.17), we observed that overweight women had 1.65% lower methylation in blood, and a test for trend suggested significantly less methylation with increasing BMI class (P<0.01). The mean maternal blood LEP methylation was not different between rs2167270 genotypes as demonstrated in the model or in specific analyses comparing each genotype: A/A (21%), G/A (19.8%) and G/G (20.1%) (P=0.68, Table S5).
Table 2.
Multivariable linear regression models of maternal blood LEP methylation
Multivariable linear regression models of maternal blood LEP methylation as the dependent variable and pre-pregnancy BMI category as the independent variable. Models are provided unadjusted and adjusted for possible confounder variables included in the table. * P for trend<0.01.a Data missing on gestational weight gain and pregnancy hypertension.
| Unajusted (n=60) | P | Ajusted (n=58)a,b | P | |||
|---|---|---|---|---|---|---|
| Estimate | Std.Error | Estimate | Std.Error | |||
| Maternal Fators | ||||||
| Pre-pregnancy BMI (kg/m2) category* | ||||||
| Normal | Reference | Reference | ||||
| Overweight | −1.74 | 1.06 | 0.11 | −1.65 | 1.18 | 0.17 |
| Obese | −2.75 | 0.92 | 4.1×10−3 | −2.95 | 1.15 | 0.01 |
| Gestational weight gain | ||||||
| Adequate | Reference | |||||
| Inadequate | - | - | - | −0.23 | 1.17 | 0.84 |
| Excessive | - | - | - | 0.46 | 1.03 | 0.66 |
| Tobacco use during pregnancy | ||||||
| No | Reference | |||||
| Yes | - | - | - | 1.57 | 2.36 | 0.51 |
| Pregnancy hypertension | ||||||
| No | Reference | |||||
| Yes | - | - | - | −0.96 | 1.37 | 0.49 |
| Race | ||||||
| White | Reference | |||||
| Other | - | - | - | 0.81 | 0.96 | 0.38 |
| Meternal age (yrs) | - | - | - | 0.02 | 0.08 | 0.80 |
| Genotype rs2167270 | ||||||
| G/G and G/A | Reference | |||||
| A/A | - | - | - | 0.84 | 1.28 | 0.51 |
Model adjusted all variables included in table.
Data missing on gestational weight gain and pregnancy hypertension.
P <0.05 are shown in bold,
P for trend <0.01
3.5. Cord blood LEP DNA methylation
Cord blood leptin has previously been shown to be associated with infant birthweight, and SGA infants have lower serum leptin (Karakosta et al., 2011; Ren and Shen, 2010; Schubring et al., 1997). Therefore, we sought to investigate possible relationships between LEP promoter methylation in cord blood and infant birthweight in the group of participants with available data (Table S4). First, we examined the effect of genotype on cord blood LEP DNA methylation. The mean extent of methylation was higher in A/A infants (16.5%) compared to G/A (13.8) or G/G (13.7%) (ANOVA, P=6.13×10−4)(Table S5). Since the cord blood methylation values between the G/G and G/A genotypes did not differ (Tukey’s test, P=0.98), we grouped the homozygous (G/G) and heterozygous (G/A) and compared those to the homozygous variant group (A/A) in all further analyses (A/A= 16.6% versus G/G and G/A 13.8%, Student’s t-test, P=5×10−3, Figure 2A). We did not observe a significant difference in LEP methylation extent by birthweight group (ANOVA, P=0.29, Figure 2B), but this analysis suggested a trend of higher LEP methylation in SGA (14.8%) infants compared to AGA (13.8%) infants and LGA (14.1%). Of note, in our sample we did not have any infants in the SGA group with the A/A genotype. Because we observed higher cord blood LEP methylation in the A/A genotype and the SGA group, we repeated the bivariate analyses between birthweight group and cord blood LEP excluding the A/A infants. In this analysis we detected significantly higher LEP methylation in SGA (14.8%) infants (ANOVA, P=0.02) compared to AGA (13.5%, Tukey’s test, P=0.04) or LGA (13.2%, Tukey’s test, P=0.02). In multivariable linear regression analyses (Table 3) adjusted for possible confounders (maternal and infant factors), we observed a positive association between cord blood LEP methylation and birthweight group; SGA infants had 1.78% greater methylation than AGA infants (P=4.6×10−3). Similarly, infants with A/A genotype at rs2167270 variant had significantly higher methylation (2.68%) levels compared to those with at least one G allele (P=1.2×10−4) independent of birthweight status and other factors included in the model. Interestingly, cord blood LEP methylation was significantly lower (−1.47%, P=0.03) in infants born to obese mothers and higher (1.06%, P=0.04) in women with excessive GWG. In our study, obese and overweight women were more likely to gain excessive weight during pregnancy than normal BMI women (73%, 68% and 35%, respectively; Fisher’s exact test, P=0.007). Additionally, we observed that infants born to women who reported smoking during pregnancy had higher (2.22%, P=0.03) cord blood LEP methylation than infants born to non-smokers, but this result should be interpreted with caution as we only have three mothers that reported smoking during pregnancy in this data set. Because of the known association between SGA and cigarette smoking, we repeated the regression analysis excluding the mothers that reported tobacco use during pregnancy; the observed effects of SGA birth weight group, infant genotype, pre-pregnancy obese BMI and excessive GWG remain similar in size and statistically significant (data not shown).
Fig. 2. LEP promoter methylation in cord blood.
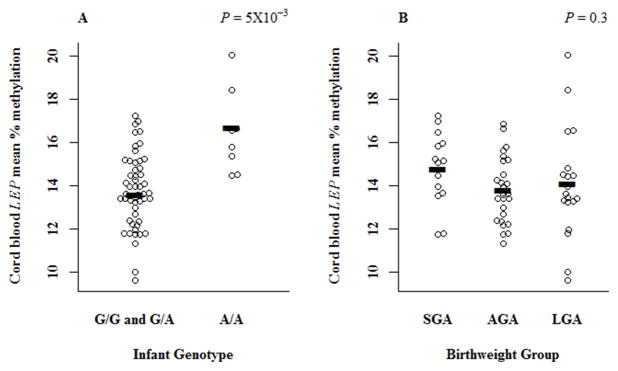
(A) Cord blood LEP promoter methylation by rs2167270 genotype. Distribution of LEP methylation (y-axis) in cord blood samples by genotype (x-axis): G/G and G/A (n=52) and A/A (n=8). Black bars represent the mean value for each group; G/G/and G/A (13.8%) and A/A (16.6%). The P-value is the result of Student’s t-test.
(B). Cord blood LEP promoter methylation by birthweight group. Distribution of LEP methylation (y-axis) in study samples by birthweight (x-axis): SGA (n=15), AGA (n=26) and LGA (n=19). Black bars represent the mean value for each group; SGA (14.8%), AGA (13.8%) and LGA (14.1%). The P-value is the result of ANOVA.
Table 3.
Multivariable linear regression models of cord blood and placental LEP methylation
Multivariable linear regression models of cord blood LEP methylation and placental LEP methylation as the dependent variable and birthweight group as the independent variable. Models are provided unadjusted and adjusted for possible confounder variables included in the table. AGA: adequate for gestational age; LGA: large for gestational age; SGA: small for gestational age; Std. Error: standard error, a Data missing on gestational weight gain and pregnancy hypertension.
| Cord Blood Methylation | Placental Methylation | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Unadjusted (n=60) | P | Adjusted(n=58)a,b | P | Unadjusted (n=81) | P | Adjusted (n=79)a,b | P | |||||
| Estimate | Std.Error | Estimate | Std.Error | Estimate | Std.Error | Estimate | Std.Error | |||||
| Infant Factors | ||||||||||||
| Birthweight Group | ||||||||||||
| AGA | Reference | Reference | Reference | Reference | ||||||||
| LGA | 0.11 | 0.49 | 0.82 | 0.48 | 0.53 | 0.31 | 0.17 | 1.48 | 0.91 | −0.63 | 1.77 | 0.72 |
| SGA | 1.47 | 0.54 | 8.3×10−3 | 1.78 | 0.60 | 4.6×10−3 | 2.56 | 1.50 | 0.09 | 2.85 | 1.79 | 0.12 |
| rs2167270 | ||||||||||||
| G/G and G/A | Reference | Reference | Reference | Reference | ||||||||
| A/A | 3.11 | 0.64 | 9.8×10−6 | 2.68 | 0.63 | 1.2×10−4 | 0.14 | 1.68 | 0.94 | 0.45 | 1.83 | 0.81 |
| Infant Gender | ||||||||||||
| Female | Reference | Reference | ||||||||||
| Male | - | - | - | −0.42 | 0.43 | 0.33 | - | - | - | 2.91 | 1.44 | 0.05 |
| Maternal Factors | ||||||||||||
| Pre-pregnancy BMI (kg/m2) category | ||||||||||||
| Normal | Reference | Reference | ||||||||||
| Overweight | - | - | - | −0.32 | 0.49 | 0.51 | - | - | - | 0.48 | 1.68 | 0.78 |
| Obese | - | - | - | −1.47 | 0.65 | 0.03 | - | - | - | 2.30 | 1.93 | 0.24 |
| Pregnancy weight gain | ||||||||||||
| Adequate | Reference | Reference | ||||||||||
| Inadequate | - | - | - | −0.03 | 0.65 | 0.97 | - | - | - | 1.11 | 2.07 | 0.59 |
| Excessive | - | - | - | 1.06 | 0.50 | 0.04 | - | - | - | 0.02 | 1.69 | 0.99 |
| Race | ||||||||||||
| White | Reference | Referent | ||||||||||
| Other | - | - | - | 0.41 | 0.48 | 0.40 | - | - | - | 0.67 | 1.54 | 0.67 |
| Tobacco use during pregnancy | ||||||||||||
| No | Reference | Reference | ||||||||||
| Yes | - | - | - | 2.22 | 0.99 | 0.03 | - | - | - | −1.77 | 3.15 | 0.58 |
| Pregnancy hypertension | ||||||||||||
| No | Reference | Reference | ||||||||||
| Yes | - | - | - | 1.18 | 0.84 | 0.17 | - | - | - | |||
| Maternal age (yrs) | 0.05 | 0.04 | 0.17 | - | - | - | −0.84 | 2.54 | 0.74 | |||
| Delivery method | ||||||||||||
| Vaginal | Reference | Reference | ||||||||||
| C-section | - | - | - | −0.39 | 0.45 | 0.39 | - | - | - | −0.16 | 1.41 | 0.91 |
Model adjusted for all variables included in table.
Data missing on gestational weight gain and pregnancy hypertension, P <0.05 are shown in bold.
3.6. Placental LEP DNA methylation
The placenta produces leptin and this adipokine has shown to have a variety of roles in this tissue (Gambino et al., 2012). In contrast to the results obtained in cord blood, we did not observe a significant association between birthweight and placental LEP DNA methylation in either bivariate or multivariable linear regression models controlled for confounders (Table 3). Likewise, we did not observe an association between infant genotype and mean placental LEP methylation (Table S5); A/A (23.7%), G/A (24.2%) and G/G (22.9%) (ANOVA, P=0.67). However, we detected a significant association between gender and LEP methylation (P=0.05) in this tissue after adjusting for possible confounders (infant and maternal factors). Male infants had 2.91% higher placental LEP methylation compared to females. Additionally, SGA status and infant A/A genotype showed higher placental LEP methylation similar to the results obtained in cord blood, but without statistical significance. Interestingly and although not significant, we observed opposite effect of pre-pregnancy obesity; obese women have infants with higher placental LEP methylation. Nonetheless, in this study infant gender was the only covariate that we could identify that significantly predicted LEP DNA methylation in placental tissue.
3.7. LEP DNA methylation between tissues
Placental LEP methylation was not correlated with maternal or cord blood methylation (r= −0.04, P=0.79; r=0.04, P=0.75), respectively. In contrast, maternal blood was positively correlated with cord blood methylation (r=0.4, P=0.01) (Table S3, Fig. 3). In a linear regression model of cord blood methylation as the dependent variable and maternal LEP methylation and genotype as independent variables, we identified a significant association after adjusting for possible infant and maternal confounders (Table S6). In the 37 mother-infant pairs examined, a 1% increase in maternal blood methylation is associated with a 0.25% increase in infant blood methylation (β=0.25; Std. Error=0.08; P=0.01). Similar to our previous result, infants with A/A genotype had a significantly higher cord blood LEP methylation compared to infants with at least one G allele and a we observed a marginally significant effect of obesity on cord blood LEP methylation, despite the sample size reduction (Table S6).
Fig. 3. Maternal and cord blood LEP promoter methylation.

Scatterplot of maternal blood LEP methylation (y-axis) and cord blood LEP methylation (x-axis) in 39 mother-infant pairs. Depicted is r and the Pearson correlation P-value.
4. Discussion
In this study, we found evidence of associations between perinatal maternal and infant factors and LEP promoter methylation. Specifically, maternal blood methylation was associated with pre-pregnancy obesity. Cord blood methylation was associated with infant SGA status, A/A (rs2167270) genotype, excessive maternal GWG and pregnancy smoking and negatively associated with pre-pregnancy obesity. In placental tissue, LEP DNA methylation was associated with infant gender. Finally, we found that maternal blood LEP DNA methylation correlates with infant blood LEP methylation.
We detected a moderate correlation between methylation at individual CpGs and the overall mean LEP promoter methylation for the analyzed region. Similar results were obtained in a recent study (Bouchard et al., 2010) that assessed LEP promoter methylation in human placental biopsies. Correspondingly, the average methylation (24%) values we observed in placenta resemble those obtained in the prior study (≈28%); the slight difference in the results could be due to partial overlap in the examined region, a different analysis technique, and different population characteristics. Our findings indicate that there are differences between mean and distribution of methylation values between the analyzed tissues; this complements previous reports that described tissue-specific LEP DNA methylation (Marchi et al., 2011; Stöger, 2006). DNA methylation extent in the examined region of the LEP promoter has been previously associated with leptin expression in mouse (Yokomori et al., 2002), human adipocytes (Melzner et al., 2002), and human placenta (Bouchard et al., 2010). Moreover, DNA binding proteins such as specificity protein (SP1) and CAAT-enhancer binding protein (C/EBP α) (Mason et al., 1998) are known to bind to this area, and C/EBPα binding affinity is reduced in methylated probes compared to non-methylated ones as assessed by electrophoretic mobility shift assay in LiSa-2 cells nuclear extracts (Melzner et al., 2002).
Studies from the Dutch Famine Birth Cohort have found higher blood LEP methylation levels in famine-exposed adult men compared to unexposed siblings, although they did not comment on the association with current BMI (Tobi et al., 2009); whereas, other studies from the same cohort have observed that famine exposure increases the risk of obesity later in life (Ravelli et al., 1976; Roseboom et al., 2006). Additionally, previous studies (Highman et al., 1998; Misra and Trudeau, 2011) have reported associations between serum leptin and maternal weight and adiposity, but the association between maternal blood LEP methylation extent and maternal BMI category has not been described before. To our knowledge this is the first report that has established an association between pre-pregnancy obesity and maternal blood LEP methylation at term. Our results showed lower blood LEP methylation amongst obese women, thus we can hypothesize that peripheral blood cells may also contribute to increased serum leptin in obesity, and that peripheral blood cells may mimic the metabolic changes occurring in adipocytes. This would need to be more thoroughly examined in samples appropriate to examine leptin expression in peripheral blood cells. Prior studies have not specifically correlated blood LEP DNA methylation with serum leptin or measurements of leptin in adipocytes in humans, and the samples collected for our study could not appropriately address this issue, thus highlighting a need for future studies to assess this connections. Our results should be replicated in different population studies to further evaluate the observed association, and if possible, the relationship between adipocyte and peripheral blood LEP methylation and serum levels specifically interrogated.
Cord blood methylation is higher in SGA infants and those with the A/A (homozygous variant) genotype (rs2167270) in the LEP promoter, and our multivariable model suggests these are independent associations. Serum cord blood leptin has been consistently correlated with birthweight in different populations (Ren and Shen, 2010), moreover SGA infants have lower leptin levels (Ren and Shen, 2010) and adipose tissue mass (Martinez-Cordero et al., 2006). Hence, we can hypothesize that the higher LEP promoter methylation observed in SGA infants reflect lower infant body fat, similar to maternal blood LEP DNA methylation levels reflecting maternal adiposity. Further research is needed to assess the connections between fetal adipose mass at birth and LEP DNA methylation in cord blood. In addition, it is recognized that being born SGA carries a higher risk of metabolic disease such as obesity later in life (Saenger et al., 2007), hence it would be interesting to investigate LEP DNA methylation at different time points over the life course to assess if there are differences between SGA individuals that eventually develop obesity and those who do not. Additionally, our results show an association of maternal pre-pregnancy obesity and lower cord blood methylation levels, results that are consistent with those observed in maternal blood. We can hypothesize that this association is derived from the known relationship between maternal weight and birthweight (Frederick et al., 2008; Hull et al., 2008). Obese mothers have larger infants and this resulted in an opposite association on cord blood LEP from the observed effects of SGA. Concomitantly, the adjusted regression model for cord blood LEP methylation shows a direct association of GWG and LEP methylation; excessive GWG increases LEP methylation. This result is unexpected as it has been reported that women with excessive GWG have infants with increased adiposity and LGA (Johnson et al., 2013; Poston et al., 2011). However, we can hypothesized that this results from an infant adaptive response to the augmented maternal adiposity levels. Nonetheless, from our results the effect size of SGA on cord blood LEP methylation was higher than that of excessive GWG. Hence, specific mechanistic research is needed to address these adaptive mechanisms in utero, while further population-based research is needed to confirm and extend these associations.
Interestingly, we observed an association between genotype and LEP DNA methylation in cord blood that was not detected between genotype and methylation in maternal blood or placental tissue. DNA sequence variation acting in cis can exert effects on DNA methylation across the genome, and allele specific methylation has been reported to differ between tissues within loci (Schalkwyk et al., 2010). This may explain both the association between genotype and methylation in cord blood as well as the lack of association between genotype and methylation in the other tissues examined. These associations should be explored further, and in a larger population which is more statistically powered to examine interactions between birthweight group and genotype on LEP promoter methylation.
Placental DNA methylation in other loci has been previously associated with infant birthweight (Filiberto et al., 2011; Hogg et al., 2012; Marsit et al., 2012), hence, we hypothesized that placental LEP DNA methylation would be associated with birthweight. Surprisingly, we did not find evidence of a significant association. Our findings indicate an association between gender and placental LEP methylation, which was not detected in cord blood, further supporting the concept of placenta as a unique pregnancy tissue that can be influenced by fetal factors, but one that is epigenetically distinct from cord blood. Gender differences in LEP DNA methylation have been observed previously (Tobi et al., 2009) in peripheral blood of adults exposed to wartime famine; men had higher methylation than women. Additionally, a different study (Chan et al., 2006) found elevated leptin levels in amniotic fluid of female fetuses compared to males in normal and pre-eclamptic pregnancies. This observation supports our LEP methylation finding; since lower methylation levels in placentas from female infants could account for these increases in this hormone in amniotic fluid. Moreover, 17β-estradiol increases leptin expression in placental cells through actions on the estrogen receptor-α (Maymo et al., 2011), although, we are aware that this can occur through other mechanisms of gene regulation. Our findings, while plausible, need to be validated in other studies, especially as a recent study (Hogg et al., 2013) did not observe differences in placental LEP methylation by infant gender.
In the 39 mother-infant pairs for which we have paired cord and maternal blood information, we detected a significant, moderate, positive correlation between cord LEP methylation and maternal blood LEP. This result is likely related to the positive correlation between infant birthweight and pre-pregnancy maternal BMI observed in this study and other reports (Frederick et al., 2008; Hull et al., 2008). However, this association persisted after adjusting for pre-gestational BMI and birthweight group. Overall, our findings from maternal and cord blood suggest that pre-pregnancy obesity influences not only maternal blood LEP methylation but perhaps also infant cord blood LEP methylation. Interestingly, evidence from a murine study (Jousse et al., 2011) demonstrated a similar effect, but with opposite exposure; maternal undernutrition results in LEP hypomethylation in offspring adipose tissue. The results from our study suggest a potential for multigenerational epigenetic inheritance in humans which has been proposed (Daxinger and Whitelaw, 2012), and add to the increasingly strong evidence (Hogg et al., 2012) of associations between in utero environmental influences and altered DNA methylation.
Our study has a number of strengths: we have mother-infant paired LEP methylation data in different tissues and we are the first study to evaluate LEP methylation in infant and maternal blood and placental tissue in non-pathologic pregnancies. Notably, this study is also the first to find a correlation between maternal and infant DNA methylation in humans. However, this work has several limitations: (1) our sample size is small, (2) we lack serum leptin measurements, (3) we did not directly measure anthropometric data and this could potentially introduce self-report bias and (4) we studied only one gene in a myriad of possible candidates that contribute to complex phenotypes such as maternal obesity and SGA birthweight. In addition, like all population-based studies we are unable to elucidate the exact cellular mechanisms that link perinatal maternal and infant factors studied with epigenetic alterations.
5. Conclusion
In summary, in this study we demonstrated that LEP DNA methylation is associated in a tissue-specific manner with maternal (pre-pregnancy obesity, pregnancy smoking and GWG) and infant factors (SGA, genotype and gender) in non-pathological pregnancies. We also provide evidence for an association between maternal and infant LEP DNA methylation. Future research in larger studies is needed to further elucidate and confirm the detected associations. However, this investigation contributes to the increasing number of studies that have identified the importance of leptin in pregnancy and to the small number of studies in LEP DNA promoter methylation. Identifying maternal factors that can produce potentially life-long epigenetic alterations in offspring is of crucial importance, as this supports the need for perinatal intervention studies to decrease adverse infant and adult-life outcomes such as obesity and cardiovascular disease.
Supplementary Material
Highlights.
Maternal and infant blood LEP methylation are associated with pre-pregnancy obesity.
SGA and A/A (rs2167270) infants have higher cord blood LEP DNA methylation.
LEP DNA methylation is higher in placentas from male infants.
Maternal blood LEP DNA methylation correlates to infant cord blood LEP methylation.
Acknowledgments
Thanks to Joyce Lee for her hard work in recruitment of subjects into this study and the support of the staff of the Brown Center for the Study of Children at Risk for their efforts. This work was funded by the National Institute of Health (NIH) through the following grants: R01MH094609 (NIH-NIMH), P20GM103537 (NIH-NIGMS) and P30CA23108 (NIH-NCI). Its contents are the responsibility of the authors and do not necessarily represent the official views of the funding institutions.
Abbreviations
- LEP
leptin promoter DNA methylation
- SGA
small for gestational age
- SNP
single nucleotide polymorphism
- LGA
large for gestational age
- AGA
adequate for gestational age
- GWG
gestational weight gain
- DOHaD
developmental origins of adult health and disease
- LGA
large for gestational age, BMI, body mass index
- SD
standard deviation
- Std.Error
standard error
- SP1
specificity protein
- C/EBP α
CAAT-enhancer binding protein alpha
Footnotes
Conflict of interest: The authors declare there are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Corina Lesseur, Email: corina.lesseur.perez.GR@dartmouth.edu.
David A. Armstrong, Email: David.Armstrong@dartmouth.edu.
Alison G. Paquette, Email: Alison.G.Paquette.GR@dartmouth.edu.
Devin C. Koestler, Email: Devin.C.Koestler@Dartmouth.edu.
James F. Padbury, Email: James_Padbury@Brown.EDU.
References
- Ashworth CJ, Hoggard N, Thomas L, Mercer JG, Wallace JM, Lea RG. Placental leptin. Reproduction. 2000;5:18. doi: 10.1530/ror.0.0050018. [DOI] [PubMed] [Google Scholar]
- Bouchard L, Thibault S, Guay SP, Santure M, Monpetit A, St-Pierre J, Perron P, Brisson D. Leptin gene epigenetic adaptation to impaired glucose metabolism during pregnancy. Diabetes Care. 2010;33:2436–41. doi: 10.2337/dc10-1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caprio M, Fabbrini E, Isidori AM, Aversa A, Fabbri A. Leptin in reproduction. TRENDS in Endocrinology and Metabolism. 2001;12:65–72. doi: 10.1016/s1043-2760(00)00352-0. [DOI] [PubMed] [Google Scholar]
- Chan TF, Su JH, Chung YF, Hsu YH, Yeh YT, Yuan SSF. Elevated amniotic fluid leptin levels in pregnant women who are destined to develop preeclampsia. Acta Obstetricia et Gynecologica Scandinavica. 2006;85:171–174. doi: 10.1080/00016340500460615. [DOI] [PubMed] [Google Scholar]
- Christensen BC, Marsit CJ. Epigenomics in environmental health. Front Genet. 2011;2:84. doi: 10.3389/fgene.2011.00084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daxinger L, Whitelaw E. Understanding transgenerational epigenetic inheritance via the gametes in mammals. Nature Reviews Genetics. 2012;13:153–162. doi: 10.1038/nrg3188. [DOI] [PubMed] [Google Scholar]
- Fenton T. A new growth chart for preterm babies: Babson and Benda’s chart updated with recent data and a new format. BMC pediatrics. 2003;3:13. doi: 10.1186/1471-2431-3-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenton TR, Kim JH. A systematic review and meta-analysis to revise the Fenton growth chart for preterm infants. BMC pediatrics. 2013;13:59. doi: 10.1186/1471-2431-13-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filiberto AC, Maccani MA, Koestler D, Wilhelm-Benartzi C, Avissar-Whiting M, Banister CE, Gagne LA, Marsit CJ. Birthweight is associated with DNA promoter methylation of the glucocorticoid receptor in human placenta. Epigenetics. 2011;6:566–572. doi: 10.4161/epi.6.5.15236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flegal KM, Carroll MD, Kit BK, Ogden CL. Prevalence of obesity and trends in the distribution of body mass index among US adults, 1999–2010. JAMA. 2012;307:491–7. doi: 10.1001/jama.2012.39. [DOI] [PubMed] [Google Scholar]
- Frederick IO, Williams MA, Sales AE, Martin DP, Killien M. Pre-pregnancy body mass index, gestational weight gain, and other maternal characteristics in relation to infant birth weight. Matern Child Health J. 2008;12:557–67. doi: 10.1007/s10995-007-0276-2. [DOI] [PubMed] [Google Scholar]
- Friedman JM. Leptin and the regulation of body weigh. The Keio Journal of Medicine. 2011;60:1–9. doi: 10.2302/kjm.60.1. [DOI] [PubMed] [Google Scholar]
- Gambino YP, Maymo JL, Perez Perez A, Calvo JC, Sanchez-Margalet V, Varone CL. Elsevier Trophoblast Research Award lecture: Molecular mechanisms underlying estrogen functions in trophoblastic cells--focus on leptin expression. Placenta. 2012;33(Suppl):S63–70. doi: 10.1016/j.placenta.2011.12.001. [DOI] [PubMed] [Google Scholar]
- Gluckman PD, Hanson MA, Pinal C. The developmental origins of adult disease. Maternal & child nutrition. 2005;1:130–141. doi: 10.1111/j.1740-8709.2005.00020.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hales CN, Barker D. The thrifty phenotype hypothesis. British medical bulletin. 2001;60:5. doi: 10.1093/bmb/60.1.5. [DOI] [PubMed] [Google Scholar]
- Highman TJ, Friedman JE, Huston LP, Wong WW, Catalano PM. Longitudinal changes in maternal serum leptin concentrations, body composition, and resting metabolic rate in pregnancy. American Journal of Obstetrics and Gynecology. 1998;178:1010–1015. doi: 10.1016/s0002-9378(98)70540-x. [DOI] [PubMed] [Google Scholar]
- Hogg K, Blair JD, von Dadelszen P, Robinson WP. Hypomethylation of the LEP gene in placenta and elevated maternal leptin concentration in early onset pre-eclampsia. Mol Cell Endocrinol. 2013 doi: 10.1016/j.mce.2012.12.018. [DOI] [PubMed] [Google Scholar]
- Hogg K, Price EM, Hanna CW, Robinson WP. Prenatal and perinatal environmental influences on the human fetal and placental epigenome. Clin Pharmacol Ther. 2012;92:716–26. doi: 10.1038/clpt.2012.141. [DOI] [PubMed] [Google Scholar]
- Hull HR, Dinger MK, Knehans AW, Thompson DM, Fields DA. Impact of maternal body mass index on neonate birthweight and body composition. Am J Obstet Gynecol. 2008;198:416 e1–6. doi: 10.1016/j.ajog.2007.10.796. [DOI] [PubMed] [Google Scholar]
- Johnson J, Clifton RG, Roberts JM, Myatt L, Hauth JC, Spong CY, Varner MW, Wapner RJ, Thorp JM, Jr, Mercer BM. Pregnancy Outcomes With Weight Gain Above or Below the 2009 Institute of Medicine Guidelines. Obstetrics & Gynecology. 2013;121:969–975. doi: 10.1097/AOG.0b013e31828aea03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nature Reviews Genetics. 2012 doi: 10.1038/nrg3230. [DOI] [PubMed] [Google Scholar]
- Karakosta P, Chatzi L, Plana E, Margioris A, Castanas E, Kogevinas M. Leptin levels in cord blood and anthropometric measures at birth: a systematic review and meta-analysis. Paediatr Perinat Epidemiol. 2011;25:150–63. doi: 10.1111/j.1365-3016.2010.01163.x. [DOI] [PubMed] [Google Scholar]
- Lawlor DA, Relton C, Sattar N, Nelson SM. Maternal adiposity--a determinant of perinatal and offspring outcomes? Nat Rev Endocrinol. 2012;8:679–88. doi: 10.1038/nrendo.2012.176. [DOI] [PubMed] [Google Scholar]
- Lepercq J, Challier JC, Guerre-Millo M, Cauzac M, Vidal H, Hauguel-de Mouzon S. Prenatal leptin production: evidence that fetal adipose tissue produces leptin. Journal of Clinical Endocrinology & Metabolism. 2001;86:2409–2413. doi: 10.1210/jcem.86.6.7529. [DOI] [PubMed] [Google Scholar]
- Marchi M, Lisi S, Curcio M, Barbuti S, Piaggi P, Ceccarini G, Nannipieri M, Anselmino M, Di Salvo C, Vitti P. Human leptin tissue distribution, but not weight loss-dependent change in expression, is associated with methylation of its promoter. Epigenetics. 2011;6:1198–1206. doi: 10.4161/epi.6.10.16600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsit CJ, Maccani MA, Padbury JF, Lester BM. Placental 11-beta hydroxysteroid dehydrogenase methylation is associated with newborn growth and a measure of neurobehavioral outcome. PloS one. 2012;7:e33794. doi: 10.1371/journal.pone.0033794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Cordero C, Amador-Licona N, Guizar-Mendoza JM, Hernandez-Mendez J, Ruelas-Orozco G. Body fat at birth and cord blood levels of insulin, adiponectin, leptin, and insulin-like growth factor-I in small-for-gestational-age infants. Arch Med Res. 2006;37:490–4. doi: 10.1016/j.arcmed.2005.11.004. [DOI] [PubMed] [Google Scholar]
- Mason MM, He Y, Chen H, Quon MJ, Reitman M. Regulation of leptin promoter function by Sp1, C/EBP, and a novel factor. Endocrinology. 1998;139:1013. doi: 10.1210/endo.139.3.5792. [DOI] [PubMed] [Google Scholar]
- Masuzaki H, Ogawa Y, Sagawa N, Hosoda K, Matsumoto T, Mise H, Nishimura H, Yoshimasa Y, Tanaka I, Mori T. Nonadipose tissue production of leptin: leptin as a novel placenta-derived hormone in humans. Nature medicine. 1997;3:1029–1033. doi: 10.1038/nm0997-1029. [DOI] [PubMed] [Google Scholar]
- Maymo JL, Pérez Pérez A, Gambino Y, Calvo JC, Sánchez-Margalet V, Varone CL. Review: Leptin gene expression in the placenta–Regulation of a key hormone in trophoblast proliferation and survival. Placenta. 2011;32:S146–S153. doi: 10.1016/j.placenta.2011.01.004. [DOI] [PubMed] [Google Scholar]
- Mehta SH, Kruger M, Sokol RJ. Being too large for gestational age precedes childhood obesity in African Americans. American journal of obstetrics and gynecology. 2011;204:265e1–265e5. doi: 10.1016/j.ajog.2010.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melzner I, Scott V, Dorsch K, Fischer P, Wabitsch M, Bruderlein S, Hasel C, Moller P. Leptin gene expression in human preadipocytes is switched on by maturation-induced demethylation of distinct CpGs in its proximal promoter. J Biol Chem. 2002;277:45420–7. doi: 10.1074/jbc.M208511200. [DOI] [PubMed] [Google Scholar]
- Michels KB, Waterland RA. The Role of Epigenetics in the Developmental Origins of Health and Disease. Epigenetic Epidemiology. 2012:105–116. [Google Scholar]
- Misra VK, Trudeau S. The influence of overweight and obesity on longitudinal trends in maternal serum leptin levels during pregnancy. Obesity (Silver Spring) 2011;19:416–21. doi: 10.1038/oby.2010.172. [DOI] [PubMed] [Google Scholar]
- Noer A, Sorensen AL, Boquest AC, Collas P. Stable CpG hypomethylation of adipogenic promoters in freshly isolated, cultured, and differentiated mesenchymal stem cells from adipose tissue. Mol Biol Cell. 2006;17:3543–56. doi: 10.1091/mbc.E06-04-0322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novakovic B, Saffery R. The ever growing complexity of placental epigenetics - role in adverse pregnancy outcomes and fetal programming. Placenta. 2012;33:959–70. doi: 10.1016/j.placenta.2012.10.003. [DOI] [PubMed] [Google Scholar]
- Oken E. Maternal and child obesity: the causal link. Obstetrics and Gynecology Clinics. 2009;36 doi: 10.1016/j.ogc.2009.03.007. [DOI] [PubMed] [Google Scholar]
- Poston L, Harthoorn LF, Van Der Beek EM. Obesity in pregnancy: implications for the mother and lifelong health of the child. A consensus statement. Pediatric research. 2011;69:175–180. doi: 10.1203/PDR.0b013e3182055ede. [DOI] [PubMed] [Google Scholar]
- Rasmussen KM, Abrams B, Bodnar LM, Butte NF, Catalano PM, Siega-Riz AM. Recommendations for weight gain during pregnancy in the context of the obesity epidemic. Obstetrics & Gynecology. 2010;116:1191–1195. doi: 10.1097/AOG.0b013e3181f60da7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravelli GP, Stein ZA, Susser MW. Obesity in young men after famine exposure in utero and early infancy. New England Journal of Medicine. 1976;295:349–353. doi: 10.1056/NEJM197608122950701. [DOI] [PubMed] [Google Scholar]
- Ren RX, Shen Y. A meta-analysis of relationship between birth weight and cord blood leptin levels in newborns. World J Pediatr. 2010;6:311–6. doi: 10.1007/s12519-010-0216-x. [DOI] [PubMed] [Google Scholar]
- Rinaudo P, Wang E. Fetal programming and metabolic syndrome. Annu Rev Physiol. 2012;74:107–30. doi: 10.1146/annurev-physiol-020911-153245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roseboom T, de Rooij S, Painter R. The Dutch famine and its long-term consequences for adult health. Early Hum Dev. 2006;82:485–91. doi: 10.1016/j.earlhumdev.2006.07.001. [DOI] [PubMed] [Google Scholar]
- Saenger P, Czernichow P, Hughes I, Reiter EO. Small for gestational age: short stature and beyond. Endocr Rev. 2007;28:219–51. doi: 10.1210/er.2006-0039. [DOI] [PubMed] [Google Scholar]
- Schalkwyk LC, Meaburn EL, Smith R, Dempster EL, Jeffries AR, Davies MN, Plomin R, Mill J. Allelic skewing of DNA methylation is widespread across the genome. Am J Hum Genet. 2010;86:196–212. doi: 10.1016/j.ajhg.2010.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubring C, Kiess W, Englaro P, Rascher W, Dötsch J, Hanitsch S, Attanasio A, Blum WF. Levels of leptin in maternal serum, amniotic fluid, and arterial and venous cord blood: relation to neonatal and placental weight. Journal of Clinical Endocrinology & Metabolism. 1997;82:1480–1483. doi: 10.1210/jcem.82.5.3935. [DOI] [PubMed] [Google Scholar]
- Stöger R. In vivo methylation patterns of the leptin promoter in human and mouse. Epigenetics. 2006;1:155–162. doi: 10.4161/epi.1.4.3400. [DOI] [PubMed] [Google Scholar]
- Swinburn BA, Sacks G, Hall KD, McPherson K, Finegood DT, Moodie ML, Gortmaker SL. The global obesity pandemic: shaped by global drivers and local environments. The Lancet. 2011;378:804–814. doi: 10.1016/S0140-6736(11)60813-1. [DOI] [PubMed] [Google Scholar]
- Tobi EW, Lumey LH, Talens RP, Kremer D, Putter H, Stein AD, Slagboom PE, Heijmans BT. DNA methylation differences after exposure to prenatal famine are common and timing- and sex-specific. Hum Mol Genet. 2009;18:4046–53. doi: 10.1093/hmg/ddp353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vickers MH, Sloboda DM. Leptin as mediator of the effects of developmental programming. Best Pract Res Clin Endocrinol Metab. 2012;26:677–87. doi: 10.1016/j.beem.2012.03.005. [DOI] [PubMed] [Google Scholar]
- Yokomori N, Tawata M, Onaya T. DNA demethylation modulates mouse leptin promoter activity during the differentiation of 3T3-L1 cells. Diabetologia. 2002;45:140–148. doi: 10.1007/s125-002-8255-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.