

Abstract
Intraepithelial γδ T lymphocytes (IEL) play important roles in repair of tissue damage at epithelial sites such as skin and intestine. Molecules that orchestrate these γδ T cell functions are not well defined. Recently, interaction of the semaphorin CD100 on skin γδ T cells with plexin B2 on keratinocytes was shown to be important for effective γδ T cell function in the epidermis, which raised the possibility that CD100 may exert similar functions in the intestinal tract. In this study, we find that CD100 is expressed on all IEL, and plexin B2 is present on all epithelial cells of the mouse colon. Using the dextran sulfate sodium (DSS) mouse model of colitis, disease severity is significantly exacerbated in CD100 deficient (CD100−/−) mice, with increased colon ulceration and mucosal infiltration with inflammatory cells. The severe colitis in CD100−/− mice is attributable to the failure of the colon epithelium to mount a proliferative response to damage. Unlike wild type γδ IEL, γδ IEL from CD100−/− mice fail to produce keratinocyte growth factor-1 (KGF-1) in response to DSS treatment. Administration of recombinant KGF-1 to CD100−/− animals ameliorates disease and reverses colitis susceptibility. These results demonstrate that CD100 mediated signals are critical for effective activation of γδ IEL to produce growth factors, including KGF-1, that are required for healing of the colon epithelium during colitis.
INTRODUCTION
Inflammatory bowel disease (IBD) is characterized by chronic inflammation of the intestine and is often accompanied by extensive epithelial ulcerations. Inflammation is thought to be driven by dysregulated cell mediated immune responses to antigens derived from commensal bacteria in the intestinal lumen (1). Resolution of disease depends on reestablishment of the epithelial cell barrier to separate luminal antigens from the underlying immune cells in the inflamed tissue (2,3). Important in the healing process are intestinal intraepithelial lymphocytes (IEL) bearing the γδ form of the T cell antigen receptor (4). These γδ IEL are in intimate contact with epithelial cells and respond to intestinal damage by secreting a number of cytokines, chemokines, and growth factors such as keratinocyte growth factor-1 (KGF-1) that promote tissue repair (4,5). Mice lacking γδ T cells (TCRδ−/−) have defects in enterocyte homeostasis and develop severe disease in different models of intestinal inflammation including the dextran sulfate sodium (DSS) mouse model of colitis (4). This severe colitis is attributed to a lack of KGF-1 production by γδIEL, a potent mitogen for epithelial cells which can stimulate proliferation and promote wound healing in the intestine (4,6). Exogenous KGF-1 ameliorates colitis when administered prior to DSS treatment (7). Conversely, mice unable to produce KGF-1 (KGF-1−/− mice) develop a severe form of DSS induced colitis (4). These lines of evidence demonstrate that γδ IEL-promoted healing of the colon epithelium by production of KGF-1 is critical to recovery from colitis.
While activation of γδ IEL is necessary for their production of KGF-1 (8), the molecular events involved in this activation are poorly understood. By analogy with the skin (9,10), numerous interactions, in addition to those through the T cell receptor (TCR), are likely to play a crucial role. The semaphorin CD100, which is expressed on the surface of skin γδ T cells, was shown recently to interact with plexin B2 on keratinocytes and to be necessary for the effector response of epidermal γδ T cells to stressed epithelial cells (10). Semaphorins are a large family of membrane bound and soluble proteins that are grouped into eight classes based on sequence similarity and shared structural domains (11). Semaphorins are mostly known for their ability to give directional cues to developing neurons by signaling through plexin family members, but recent findings indicate they have a broader physiological role in organogenesis, angiogenesis, and in metastasis of cancer cells (11). In addition, several semaphorins, including CD100, have roles in immune regulation (12,13). CD100 is a group IV transmembrane semaphorin (SEMA4D) that is expressed as a homodimer in a broad range of tissues including cells of the immune system. CD100 has higher levels of expression on T cells compared to B cells, and expression is significantly enhanced on both cell types with cellular activation (14). Originally, a costimulatory role for CD100 was proposed for human T cells, because anti-human CD100 antibodies increased T cell proliferation in the presence of sub-optimal amounts of PMA, or anti-CD2 and anti-CD3 antibodies (14). However, CD100 can also transduce signals through other proteins that act as CD100 receptors with the best characterized receptor being plexin B1. When bound by CD100, plexin B1 acts as a GTPase activating protein for r-Ras leading to inhibition of the phosphoinositide 3-kinase signaling pathway (15,16). This, and additional interactions of plexin B1 with other small GTP bound proteins, leads to modifications of the cytoskeleton and subsequent changes in cell adhesion and migration (17,18). In the immune system, plexin B1 is expressed on subsets of dendritic cells and B cells (19,20). Interaction with CD100 inhibits migration and promotes survival of these immune cells (19,20). Dendritic cells and B cells also express CD72, a low affinity receptor for CD100 (21,22). CD100 ligation results in dissociation of the tyrosine phosphatase SHP-1 from the intracellular domain of CD72 and turns off the inhibitory signals that CD72 delivers to B cells and dendritic cells (21,22).
Recently, another member of the plexin B family, plexin B2, was described as having a high affinity interaction with CD100 (10,23). The CD100/plexin B2 interaction was shown to be important for γδ T cell responses to stressed keratinocytes in the skin (10). These responses mediated by the semaphorin/plexin interaction, including secretion of growth factors and morphologic changes of the γδ T cell from a resting, dendritic shape to an activated, rounded shape, were defective in the absence of CD100 (10). Taken together, current data indicate that CD100 ligation can mediate activation and function of a wide range of immune cells. Analysis of CD100 deficient mice (CD100−/−) further support this notion (10,24,25). Despite a wide distribution of CD100 in the central nervous system, the only known defects of CD100−/− mice are associated with immune cells. In addition to the defects in epidermal γδT cell function (10), these mice have deficient antigen specific T cell responses, reduced B cell responses to T cell dependent antigens, and decreased numbers of B-1 B cells (25). In vitro assays using cells isolated from the CD100−/− mice reveal that expression of CD100 on T cells, but not dendritic cells, is essential for the T cell response to antigen (24). The hyporesponsiveness of the immune system in CD100−/− mice leads to protection against experimental autoimmune encephalomyelitis and immune complex glomerulonephritis (24,26).
If CD100 mediated signals are important for effective activation of γδ IEL, the function of these cells during colitis may be impaired in CD100−/− mice. Therefore, we hypothesized that DSS induced colitis would be more severe in CD100−/− animals due to defects in γδ IEL mediated healing of colon epithelium. In this study, we demonstrate that all IEL express CD100 and all colon epithelial cells express plexin B2 in wild-type (WT) mice. Upon induction of colitis, CD100−/− mice develop a much more severe form of disease with defects in proliferation of the colon epithelium. The γδ IEL from CD100−/− mice also have defects in proliferation and are unable to produce KGF-1, a mechanism which explains the protective role of CD100 during colitis. These results demonstrate the importance of the CD100/plexin B2 interaction in proper maintenance and repair of the colon epithelium by γδ IEL.
RESULTS
Plexin B2 is expressed on epithelial cells and CD100 on IEL in the colon
To determine whether plexin B2 is expressed in the normal murine colon, immunostaining studies were conducted with a monoclonal antibody specific for plexin B2. Strong constitutive expression of plexin B2 was found in the epithelium of the colon, while there was no expression by cells in the underlying lamina propria (Figure 1a,b). Flow cytometric analysis of isolated intestinal cells confirmed that epithelial cells expressed high levels of plexin B2 on their cell surface, while αβ and γδ IEL displayed little or no staining for plexin B2 (Figure 1c). Plexin B1 is also known to have a high affinity interaction with CD100. Real time PCR was used to compare levels of colonic expression of plexin B1 and B2 mRNA. While low levels of plexin B1 mRNA were detectable, expression of plexin B2 was markedly (>10-fold) higher in RNA isolated from colon homogenates (Figure 1d). To examine expression of CD100, a receptor for plexin B1 and B2, we developed a monoclonal antibody that recognizes the extracellular domain of CD100. CD100 was expressed on the surface of both αβ and γδ IEL but not on colon epithelial cells (Figure 1e). No expression of the low affinity binding partner of CD100, CD72, was observed by flow cytometry or RT-PCR of the colon epithelium (data not shown).
Figure 1.

CD100 is expressed on IEL and plexin B2 on mouse colon epithelium. (a) Immunohistochemical analysis of plexin B2 expression in colon of normal mice with the isotype control shown in (b) (Bar = 10 μm). (c) Epithelial cells were isolated from mouse colon and stained with anti-αβ TCR, anti-TCR-γδ, and isotype control (shaded) or anti-plexin B2 (solid line) antibodies and analyzed by flow cytometry. (d) Real time PCR analysis for plexin B1 and plexin B2 in colon homogenates and colon epithelium (mean + SD of three experiments, *= p < 0.05 compared to plexin B1). (e) Colon IEL were stained with anti-CD100 antibody and analyzed by flow cytometry.
Constitutive expression of CD100 and plexin B2 in the normal colon supported the possibility that interactions between these molecules may mediate γδ IEL functions in the colon. To address this, expression of CD100 and its ligands was examined following treatment of mice with DSS to induce acute colitis. When 2.5% DSS is added to the drinking water for five days, followed by three days recovery on normal drinking water, C57Bl/6 mice develop acute colon inflammation that is characterized by crypt loss and mucosal infiltration with inflammatory cells (4). Plexin B2 expression was detected on epithelial cells adjacent to inflammatory lesions but was not evident on immune cells found within the inflammatory infiltrate (Figure 2a,b). Colonic plexin B1 mRNA expression was not induced in colitis (Figure 2c), nor was expression of the low affinity receptor to CD100, CD72, detectable by either flow cytometry or RT-PCR in colon epithelial cells under these conditions (data not shown). This suggests that plexin B2 is the most likely binding partner for CD100 in the colon epithelium. A role for plexin B2 in the DSS-induced colitis model is suggested by in vitro blocking of plexin B2 in isolated colon tissue following in vivo DSS treatment. In vitro culture of colon segments in the presence of blocking anti-plexin B2 antibodies reduced the IFNγ response of the colon to DSS-induced colitis (Supplementary Figure S1). As IFNγ is an important mediator in the pathogenesis of IBD (27), this suggests interactions between plexin B2and CD100 may be involved in the regulation of disease progression and/or tissue repair, similar to what has been seen for this receptor-ligand pair following epithelial damage in the skin (10).
Figure 2.

CD100 and plexin B2 expression in DSS induced colitis. Colitis was induced in female mice by providing water containing DSS for five days followed by normal drinking water for three days. (a) Immunohistochemical analysis of colon from DSS-treated WT mice with plexin B2 antibody or (b) isotype control (Bar = 50 μm). Inset shows higher magnification of area represented by * (Bar = 5μm). (c) Real time PCR analysis of plexin B1 and plexin B2 in untreated and DSS treated colon (mean + SD of three experiments).
CD100−/− mice display increased susceptibility to colitis
Extensive histological and flow cytometric characterization of untreated WT and CD100−/− mice revealed normal architecture and cellular composition of the colon (Figure 3a,b), including normal numbers and localization of both αβ and γδ T cells (Supplementary Figure S2a and data not shown), suggesting that CD100 was not critical for maintaining mucosal structure or playing a major role in the development of the intestinal tract. In addition, mucin secretion (Supplementary Figure S2b) and antimicrobial peptide secretion (data not shown) in CD100−/− animals were similar to WT levels, suggesting that functional responses of the intestinal epithelium were intact in CD100−/− animals.
Figure 3.

Severe DSS induced colitis in CD100−/− mice. WT and CD100−/− mice were administered DSS-containing water for five days, and returned to normal drinking water for 3 or 14 days. (a–f) Photomicrographs (100x) of hematoxylin and eosin stained (H/E) paraffin sections of distal colon from WT mice (a, c, e) or CD100−/− mice (b, d, f). Representative sections of H/E-stained distal colon from untreated WT mice (a) or CD100−/− mice (b) reveal no differences (results representative of 3 for each group). Three days after removal of DSS, WT mice (c) display characteristic signs of moderate colitis while CD100−/− (d) mice display signs of severe colitis including ulceration of the epithelial layer (results representative of 14 for each group). (e) Two weeks after removal of DSS treated water, colitis is resolved in WT mice (e) while CD100−/− (f) mice still display signs of colitis (results representative of 6 per group) (Bar = 10 μm). (g) Daily weight measurements of mice treated with DSS (n=14 per group, mean + SEM, *= p < 0.05, **= p < 0.01). (h) Photomicrographs (4x) of H/E stained paraffin sections of colon from WT mice or CD100−/− mice. Lines represent areas of ulceration. (i) Total length of ulcers in the epithelial layer of mice 3 days after DSS treatment. (n=14 per group, bar = mean, ** = p < 0.01, rank sum test).
To elucidate the function of CD100 during acute inflammation of the colon, colitis was induced in CD100−/− mice through administration of DSS for 5 days followed by a return to normal drinking water for either 3 days or 14 days to allow for recovery. Three days after removal of DSS from the drinking water, focal areas of the colon mucosa in WT mice had the characteristic appearance of colitis including crypt dropout, mucosal infiltration with mononuclear cells, submucosal edema, and hyperproliferation and Goblet cell loss in the epithelium (Figure 3c). By comparison, CD100−/− mice displayed much more severe signs of colitis, including large areas of crypt loss and inflammation, and extensive ulcers in all animals examined (Figure 3d). It was previously shown that 14 days after removal of DSS, the epithelium is almost completely healed in C57Bl/6 WT mice, but defects persist in TCRδ−/− and KGF-1−/− mice (4), indicating a role for γδ IEL and KGF-1 in the recovery process. 14 days after removal of DSS, ulcers and inflammation were still evident in one third of the CD100−/− mice while the colon of WT mice had returned to a normal appearance (Figure 3e,f). These results suggest that CD100 plays a protective or healing role in DSS induced colitis.
Assessment of body weight, a sensitive clinical parameter of disease progression, revealed that the CD100−/− mice lost significantly more weight than WT mice in the course of the colitis (Figure 3g). Furthermore, using a previously described histological scoring system (summarized in Supplementary Figure S3a) based on epithelial damage and cellular infiltration in the mucosa, submucosa, and the muscularis (28), we found that histological scores were significantly higher in the CD100−/− mice three days after removal of DSS (Supplementary Figure S3b). Ulcers were present in all of the CD100−/− mice, but only 36% of the WT mice at that time. Histological scores two weeks after discontinuation of DSS treatment indicated that CD100−/− mice continued to have more severe disease than their WT controls (Supplementary Figure S3c). An assessment of epithelial damage using a second approach, measuring the total length of ulcers in colons three days after removal of DSS treated water, revealed striking differences between WT and CD100−/− animals. Lesion lengths in WT mice ranged from none (64% of the mice) to 0.84 mm in length in the entire colon, with a mean of 0.20 mm (Figure 3h,i). CD100−/− mice had on average 10-fold greater ulcerations than WT controls, with lesions ranging from 0.42 mm to 8.70 mm in length (Figure 3h,i). Thus, by all parameters measured, CD100−/− animals developed a substantially more severe DSS-induced colitis than their WT counterparts.
Altered IEL response in CD100−/− mice
In response to epithelial damage, γδ IEL in the colon are activated and produce cytokines and growth factors that are critical for proper healing of the mucosal layer (4,6,8). As CD100 has been shown to play an important role in γδ T cell activation in the skin (10), the increased colon epithelial damage observed in the CD100−/− mice could be the result of improper activation or function of γδ IEL. IEL activation was assessed by cytokine production, proliferation, and growth factor production. A population of γδ IEL isolated from the colons of both WT and CD100−/− animals produced TNFα at day 5+2 following DSS treatment (Supplementary Figure S4) indicating that early cytokine production by γδ IEL is unaffected by a deficiency in CD100-plexin B2 interactions. An analysis of proliferation, as assessed by in vivo BrdU incorporation showed that γδ IEL from CD100−/− mice were defective in their proliferative response to colitis (Figure 4a). While γδ IEL from WT animals increased their proliferation following DSS treatment, no increase in proliferation was seen in γδ IEL isolated from CD100−/− animals (Figure 4a). Although γδ IEL from CD100−/− animals had a somewhat higher baseline proliferation than their WT counterparts (Supplementary Figure S5a,b), this was not reflected in a general activation of these cells under homeostatic conditions (Supplementary Figure S6). Similar results were observed for αβ IEL from CD100−/− mice (Supplementary Figure S5c,d), whereas other responses, such as myeloperoxidase activity (Supplementary Figure S7a) appeared comparable in WT and CD100−/− animals both early and late in the response, suggesting a selective defect in the T cell response.. These results indicate that IEL from CD100−/− mice do not show a normal response to colitis induction. This is most likely due to an impaired response of the γδ T cells to epithelial damage resulting in deficient healing of the colon epithelium in colitis.
Figure 4.
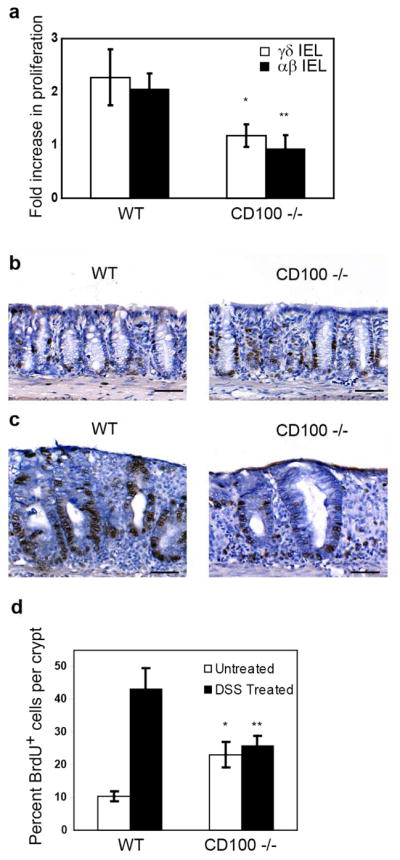
Defective epithelial proliferation and γδ IEL function in CD100−/− mice. Proliferation of cells in WT mice and CD100−/− mice was measured by in vivo BrdU incorporation. (a) Fold induction of proliferation rates of WT and CD100−/− IEL in response to DSS treatment (n= 3, * = p < 0.05 compared to WT γδ IEL, ** = p < 0.05 compared to WT αβ IEL). Immunohistochemical analysis with anti-BrdU antibody of colon sections from untreated (b) or DSS treated (c) WT and CD100−/− mice. Data are representative of six mice per group. (d) The percentage of BrdU+ epithelial cells from immunohistochemical analysis (n=6, * = p < 0.05 compared to WT untreated, ** = p < 0.05 compared to WT treated).
γδ IEL promote healing of the colon epithelium by secreting the potent epithelial mitogen KGF-1 (4). In the absence of γδ T cells, KGF-1 production is greatly reduced (4), resulting in reduced proliferation of the colon epithelium in response to colitis and exacerbation of disease. To assess the baseline proliferative rates of colon epithelial cells in WT and CD100−/− mice, in vivo BrdU incorporation was determined by immunohistochemistry on colon sections. The number of BrdU-positive epithelial cells per crypt was counted under a microscope and IEL were excluded based on cell morphology. Similar to the findings with IEL, CD100−/− mice had increased baseline proliferation of epithelial cells (Figure 4b,d). BrdU incorporation was then measured in mice 3 days after discontinuation of DSS treatment. Epithelial cells from WT mice exhibited greatly increased proliferative rates in response to colitis (Figure 4c and d), similar to previously published results (4). In contrast, there was no significant increase in proliferation of colon epithelial cells in CD100−/− mice in response to DSS treatment (Figure 4c and d). No difference in epithelial apoptosis was found between WT and CD100−/− animals either under homeostatic conditions or upon colitis induction (Supplementary Figure S7b).
The defective proliferation of colon epithelial cells in TCRδ−/− animals has been attributed to a lack of production of KGF-1 (4,6). To assess the ability of CD100−/− IEL to produce KGF-1 in response to epithelial damage, real time PCR was performed on cDNA from isolated αβ and γδ IEL. As has been described previously (4,6), γδ IEL isolated from the colon of WT mice produced KGF-1 in response to DSS treatment (Figure 5a). Production of KGF-1 is a specialized function of γδ IEL, as αβ IEL do not produce this factor (4,6). Strikingly, KGF-1 mRNA was undetectable in γδ IEL isolated from DSS-treated CD100−/− animals (Figure 5a). WT and CD100−/− IEL expressed similar levels of β-actin and GAPDH mRNA (data not shown), indicating a specific defect in KGF-1 expression by CD100−/− γδ IEL, as opposed to decreased viability of the CD100−/− IEL. We were unable to directly induce KGF-1 production via CD100 cross-linking in vitro (data not shown), suggesting that other molecular interactions, in concert with CD100 and plexin B2, are required for production of this growth factor crucial to the epithelial repair process.
Figure 5.

Defective KGF-1 production in CD100−/− mice. (a) Real time PCR analysis of KGF-1 expression by IEL. Positive control= mRNA from 3T3 cells, negative control = mRNA from the spleen of KGF−/− mice (n=3, mean + SD, * = p < 0.01 compared to WT γδ IEL treated with DSS). (b–c) Photomicrographs of H/E stained colon from CD100−/− mice that had been treated with (b) PBS or (c) recombinant KGF-1. Lines represent areas of ulceration. (d) Total length of ulcers in the epithelial layer of PBS- and KGF-1-treated mice (n=4 per group, bar = mean)
In order to demonstrate directly that lack of KGF-1 was responsible for the severe colitis in CD100−/− animals, mice were administered recombinant KGF-1, or PBS as a control, daily for 3 days, starting at day 5 of DSS treatment when animals were returned to normal drinking water. Disease was assessed by measuring total ulceration in the entire colon. PBS-treated animals (Figure 5b,d) showed the same severity of colitis as seen in untreated animals (Figure 3h). In contrast, no lesions were apparent in the colon of CD100−/− mice treated with recombinant KGF-1 (Figure 5c,d) suggesting that exogenous KGF-1 could indeed bypass the requirement for CD100-mediated signals in γδ IEL.
DISCUSSION
The semaphorin CD100 has a central role in lymphocyte function (24,25) and has recently been described to play a fundamental role in the γδ T cell response to keratinocyte damage in the skin through interaction with a plexin ligand, plexin B2 (10). Here we show defective γδ IEL activation in the colon of mice lacking CD100. The absence of signals through CD100 abolished KGF-1 production by γδ IEL, resulting in extensive damage to, and delayed repair of, the colon epithelium. These results demonstrate a central role for CD100-plexin B2 interactions in the γδ IEL response to DSS-induced damage of the colon epithelium and provide a molecular mechanism for KGF-1 production by γδ IEL.
The vital role played by γδ IEL in the repair of the colon epithelium has been known for a number of years (4). In response to DSS-induced damage, large numbers of γδ IEL localize to sites of epithelial cell damage where they express mRNA for KGF-1 (4). In TCRδ−/− animals, the absence of γδ T cells results in more severe mucosal injury and slower repair of the damaged epithelium than seen in WT animals (4). However, little is known about the molecular interactions between γδ IEL and the neighboring epithelia that mediate the critical responses of γδ IEL. It is likely that multiple interactions, in concert with those through the TCR, are important for effective γδ IEL activation, as has recently been found for γδ T cells resident in the epidermal layer of the skin (9,10). The observation here that CD100−/− mice develop a severe form of colitis upon DSS treatment similar to the colitis seen in TCRδ−/− mice led us to speculate that CD100 plays a vital role in the function of γδ IEL. This notion is in agreement with several lines of evidence pointing to CD100 being critical in B cell and αβ T cell function (24,25) as well as skin γδ T cell responses to tissue damage (10).
Although the intestinal epithelium appeared functionally intact in untreated CD100−/− animals, increased homeostatic proliferation of the colon epithelium was evident. While epithelial γδT cells are required for maintenance of epithelial homeostasis, the high baseline proliferation rate in CD100−/− mice seemed unlikely to be a result of defective γδ IEL function as mice lacking γδ T cells have decreased, rather than increased, rates of epithelial homeostatic proliferation (4,29). Instead it may well be that dysregulated WNT signaling accounts for the increased epithelial proliferation. WNT signaling is critical in maintaining the proper epithelial homeostasis of the gut (30). CD100 signaling through another plexin ligand, plexin B1, has been shown to increase levels of activated glycogen synthase kinase-3β, a potent inhibitor of the canonical WNT signaling pathway (15–17). As signals through plexin B1 and plexin B2 likely use similar pathways, the absence of CD100-plexin B2 signals may also result in altered regulation of WNT signaling and subsequent altered epithelial homeostasis.
In contrast to the high baseline proliferation, the colon epithelium of the CD100−/− mice failed to increase proliferation rates in response to DSS treatment. In wild-type animals, γδ IEL derived KGF-1 stimulates epithelial cell proliferation, a vital component of the epithelial repair process. The crucial role of KGF-1 in effective epithelial repair during colitis is evident in both rodents and humans. Increased levels of KGF-1 are observed in IBD patients, with expression corresponding to sites of inflammation (31,32). In mice, it has been demonstrated that defective proliferation of colon epithelial cells in TCRδ−/− mice is due to a lack of production of KGF-1 (4,6). Here we found CD100-deficiency resulted in ablation of KGF-1 production by γδ IEL. The lack of KGF-1 in CD100−/− animals thus likely accounts for the reduced epithelial cell proliferation seen in these animals in response to DSS. This notion is further supported by the observed restoration of epithelial repair to CD100−/− animals upon administration of exogenous KGF-1. Together these findings highlight the importance of CD100-mediated signals in γδ IEL functional integrity. Despite its importance, the signaling pathways involved in KGF-1 production by γδ IEL are poorly understood. Here we provide a link between cell surface receptor signaling, namely through CD100-plexin B2 interactions, and production of KGF-1. Although we were unable to directly induce KGF-1 through CD100 ligation in isolated cells, the identification of CD100 as a critical component for downstream KGF-1 production by γδ IEL could lead to therapeutic strategies to increase localized production of this protective factor at the site of epithelial damage.
Through interaction with neighboring epithelial cells, γδ IEL produce innate antimicrobial factors that protect against invasion of intestinal tissues by resident bacteria (33). In the DSS colitis model, a blunted γδ IEL response to damaged epithelium leading to defects in healing would likely increase the bacterial exposure of cells in the underlying lamina propria and thereby exacerbate the inflammatory response. It should be noted that CD100−/− animals do have reduced numbers of B1 B cells, and subsequently reduced IgA levels, which may also alter the mucosal immune system. However, this is unlikely to be responsible for the increased severity of colitis in CD100−/− mice as IgA deficiency does not result in a more severe course of DSS-induced colitis (34). In addition, while the lack of CD100 on αβ IEL may alter their function, a perturbed αβ IEL response is not likely to affect healing of the colon epithelium following DSS treatment, as αβ IEL are not known to regulate intestinal epithelial proliferation during homeostasis, nor to produce KGF-1 in response to colon mucosal damage (4,6,29). In addition, mice lacking αβ T cells have no significant differences from WT mice in disease severity or epithelial repair in the DSS-induced colitis model (4). Although a role for αβ IEL cannot be completely ruled out, restoration of colon epithelial integrity after injury clearly relies on a functional γδ IEL population.
In conclusion, the interaction between CD100 on γδ IEL and plexin B2 on colon epithelial cells appears to represent a vital component of the γδ IEL activation pathway and subsequent function of these IEL in mediating healing of the colon epithelium during colitis. The absence of CD100 on intestinal γδ IEL leads to an impaired growth factor response to damaged epithelium. With plexin B2 having a broad tissue distribution (10), this novel interaction may represent a general mechanism by which γδ T cells regulate inflammation and wound healing.
MATERIAL AND METHODS
Antibodies
Anti-TCR γδ (PE-GL3), anti-TCR αβ (FITC- or APC-H57–597) anti-BrdU (3D4) were purchased from BD Biosciences (San Diego, CA). Anti-mouse plexin B2 (3E7) (10) and anti-mouse CD100 (8G5) were produced as described (10) and see below). Cy5-conjugated goat anti-Armenian hamster IgG and biotinylated donkey anti-mouse IgG, as well as isotype controls, were purchased from Jackson Immunoresearch Laboratories (West Grove, PA).
Mice and colitis induction
8–12 week old female C57Bl/6 WT, CD100−/− and KGF−/− mice on the C57Bl/6 background were bred at the Scripps Research Institute (La Jolla, CA) and housed under specific pathogen-free conditions. To induce colitis, mice were given drinking water containing 2.5% (w/v) DSS ad libitum. Drinking bottles were weighed before and after administration to measure consumption. After 5 days of treatment, mice were returned to normal drinking water for an additional 1 to 14 days. Mice were weighed daily. In some experiments, mice were administered recombinant KGF-1 i.p. at 3mg/kg daily for 3 days, beginning at day 5 of DSS-treatment. In other experiments, colons were removed 5 days after DSS treatment and small segments cultured in vitro in complete DMEM+10% FCS for 48hr, as described (35), in the presence or absence of 20μg/ml anti-plexin B2 mAb, 3E7. 4–6 mice were included per group and experiments were repeated three times. All animal studies were approved by The Scripps Research Institute Institutional Animal Care and Use Committee.
Histological grading of colitis
Paraffin embedded sections of the colon were prepared and stained with hematoxylin/eosin as described previously (28). To assess the severity of DSS induced colitis, the most severe lesion present in each colon section was graded blindly by two independent observers using a published scoring scheme (28) that is summarized in Figure S2. Epithelial ulcerations were measured at 100x magnification using Axiovision AC Software with a digital camera mounted on a Nikon Eclipse E800 microscope.
Monoclonal antibody production for CD100
A female Lewis Rat was immunized once with CD100 transfected CHOK1 cells (CHOK1-CD100) in complete Freund’s adjuvant, four times with CHOK1-CD100 in Ribi adjuvant, and two times with CD100-Fc fusion protein in Ribi adjuvant and once with CD100-Fc in PBS. The spleen from the immunized rat was fused with SP3 myeloma cells. Following selection, clones were screened by flow cytometry for binding to CHOK1-CD100 cells but not to CHOK1 cells. A single positive clone (8G5) was found and subcloned twice. 8G5 was subsequently purified on Protein G sepharose beads.
Flow Cytometry
Colon epithelial cells were isolated essentially as described (4). Expression of surface markers was examined by staining cells with 0.5 μg/ml Ab in PBS containing 2% FCS and 0.2% NaN3 (FACS buffer) for 20 min at 4°C. Cells were washed with FACS buffer, and then secondary antibodies were added at 0.5 μg/ml. Cells were washed in FACS buffer and data was collected on a FACS Calibur flow cytometer using CellQuest software (BD Biosciences) and analyzed using FlowJo software (Treestar, Ashland, OR). In some experiments, IEL were pooled from 3–4 mice and then were stained and sorted based on TCR expression on a FACSVantage DiVa instrument (BD Biosciences). For intracellular flow cytometry, isolated colon cells were incubated for 4hr in 5μg/ml Brefeldin A, stained for surface marker expression followed by intracellular staining for cytokines, using the Cytofix/Cytoperm kit (BD Biosciences) as per manufacturer’s instructions. For analysis of apoptosis, colon epithelial cells were stained with Annexin V and PI using an Apoptosis Detection kit according to manufacturer’s instructions (BD Biosciences). IEL were identified by expression of the γδ or αβ TCR and epithelial cells identified by EpCAM staining.
Immunohistochemistry
Biotinylated antibody to plexin B2 (10 μg/ml in PBS containing 2% FBS) was deposited onto acetone-fixed, avidin-biotin blocked (Vector Laboratories, Burlingame, CA) frozen colon sections essentially as described (4), with the exception that 0.3% H2O2 in cold (4°C) methanol was used to block endogenous peroxidase. Specificity of the antibody was confirmed by a lack of staining with a species-matched negative control antibody.
RNA Extraction and Real Time RT-PCR Analysis
Total RNA from tissue or from a confluent flask of 3T3 fibroblast cells was isolated using Trizol reagent (Invitrogen Life Technologies, Grand Island, NY), treated with RQ1 DNAse (Promega Corp., Madison, WI) and cDNA derived from 2 μg of RNA using Superscript Reverse Transcriptase III and oligo dT primer (Invitrogen). RNA from sorted IEL was isolated and DNAse treated using the RNAqueous Microisolation Kit (Ambion Life Technologies) and cDNA generated using the Quantitect Reverse Transcription Kit (Qiagen Inc., Valencia, CA). Real time PCR was performed using the Quantitect SYBR Green PCR kit (Qaigen) on an ABI PRISM 7900 Sequence Detection System. cDNA was incubated at 95°C for 15 minutes, and then cycled (45 times) at 95°C for 15 s, 59°C for 30 s, and 72°C for 30 s. Relative mRNA levels were determined by the comparative threshold cycle (2−ΔΔCT) method and data is presented as relative to β-actin values for each sample. RNA from 3T3 cells served as a positive control and RNA from the spleen of a KGF−/− mouse was used as a negative control. All data shown result from at least three independent experiments. Sequences of all primers used in this study are the following:
plexin B1 forward, GGACCAAGGTTGCTGACATT;
plexin B1 reverse, ACCCCTGGTAGCTCCAGAAT;
plexin B2 forward, AGTCCTGATGCCAGCCTAAA;
plexin B2 reverse, TAGGCCCGTGAGACCTAGAA;
KGF-1 forward, CGGAGCAAACGGCTACGA;
KGF-1 reverse, CCCTCCTTCCATGTAGTCATAACTTC;
IFN-γ forward, CCTTTGGACCCTC;
IFN-γ reverse, AGCCAAGATGCAG;
β-Actin forward, TCTGGCTCCTAGCACCATGAAGA;
β-Actin reverse, GGGACTCATCGTACTCCTGCTTG.
Cellular Proliferation Analysis
Cells undergoing DNA replication were labeled in vivo with BrdU (55 mg/kg of body weight) from a stock solution (5 mg/ml) dissolved in PBS. BrdU was administered by i.p. injection. 24 hours later, mice were sacrificed and the colon isolated. For FACS analysis, the BD Pharmingen FITC BrdU Flow Kit (BD Biosciences) was used according to the manufacturer’s instructions. For immunohistochemistry, sections (5–8 μm) from paraffin embedded tissue were air dried, fixed with 70% ethanol, treated sequentially with 2N HCl for 20 min and 0.1 M Borax for 2 min, followed by washes in PBS, 1% Tween-20 in PBS, and 2% FBS in PBS. Slides were then incubated for 1 h with anti-BrdU antibody followed by further incubations with biotinylated monoclonal donkey anti-mouse IgG antibody peroxidase conjugated streptavidin and metal enhanced diaminobenzidine (Pierce, Rockford, IL). For untreated mice, the total number of cells and the number of BrdU positive cells present in 10 crypts were counted for each animal. For mice treated with DSS, only crypts adjacent to sites of inflammation were counted for each animal.
Myeloperoxidase Activity Analysis
Mice were either untreated or treated with DSS for 5 days and then returned to normal drinking water for 0, 7 or 21 days. Colons were isolated and homogenized in 0.05% Hexadecyltrimethylammonium bromide in phosphate buffer (HTAB) and centrifuged at 18,000g for 30min at 4°C. Supernatants were transferred to fresh tubes and 10μl added in triplicate to wells of a 96 well flat bottom plate. 90μl o-dianisidine dihydrochloride in HTAB (DDC) was added to the supernatants. 100μl of 0.0005% H2O2 in DCC was added to each well and myeloperoxidase activity measured by absorbance at 450nm.
Data Analysis
Statistical analysis was performed using Student’s t test, except for analysis of ulcer sizes where the Wilcoxon Rank Sum test was used. A probability level of P < 0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
We would like to thank Kelly Martin and Courtney St. Clair for technical help and Charlie Surh for critical reading of the manuscript. Supported by NIH grants AI52257(WLH), AI36964(WLH), T32AI07244(TFM), RR17030(LE), DK35108 (LE), DK80506 (LE), a NSF fellowship (HKK), and a fellowship from the Crohn’s and Colitis Foundation of America (TFM). This is manuscript number 18979 from the Scripps Research Institute.
Abbreviations used
- CD100−/−
CD100 deficient mice
- DSS
dextran sulfate sodium
- IBD
inflammatory bowel disease
- IEL
intestinal intraepithelial lymphocytes
- TCRδ−/−
γδ T cell deficient mice
- KGF-1
keratinocyte growth factor-1
- KGF-1−/−
keratinocyte growth factor-1 deficient mice
Footnotes
DISCLOSURE
The authors declare no conflict of interest.
References
- 1.Vora P, Shih DQ, McGovern DP, Targan SR. Current concepts on the immunopathogenesis of inflammatory bowel disease. Front Biosci. 2012;4:1451–1477. doi: 10.2741/473. [DOI] [PubMed] [Google Scholar]
- 2.Dignass AU. Mechanisms and modulation of intestinal epithelial repair. Inflamm Bowel Dis. 2001;7:68–77. doi: 10.1097/00054725-200102000-00014. [DOI] [PubMed] [Google Scholar]
- 3.Sturm A, Dignass AU. Epithelial restitution and wound healing in inflammatory bowel disease. World J Gastroenterol. 2008;14:348–353. doi: 10.3748/wjg.14.348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen Y, Chou K, Fuchs E, Havran WL, Boismenu R. Protection of the intestinal mucosa by intraepithelial γδ T cells. Proc Natl Acad Sci U S A. 2002;99:14338–14343. doi: 10.1073/pnas.212290499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boismenu R, Feng L, Xia YY, Chang JC, Havran WL. Chemokine expression by intraepithelial γδ T cells. Implications for the recruitment of inflammatory cells to damaged epithelia. J Immunol. 1996;157:985–992. [PubMed] [Google Scholar]
- 6.Yang H, Antony PA, Wildhaber BE, Teitelbaum DH. Intestinal intraepithelial lymphocyte γδ-T cell-derived keratinocyte growth factor modulates epithelial growth in the mouse. J Immunol. 2004;172:4151–4158. doi: 10.4049/jimmunol.172.7.4151. [DOI] [PubMed] [Google Scholar]
- 7.Egger B, Procaccino F, Sarosi I, Tolmos J, Buchler MW, Eysselein VE. Keratinocyte growth factor ameliorates dextran sodium sulfate colitis in mice. Dig Dis Sci. 1999;44:836–844. doi: 10.1023/a:1026642715764. [DOI] [PubMed] [Google Scholar]
- 8.Boismenu R, Havran WL. Modulation of epithelial cell growth by intraepithelial γδ T cells. Science. 1994;266:1253–1255. doi: 10.1126/science.7973709. [DOI] [PubMed] [Google Scholar]
- 9.Witherden DA, Verdino P, Rieder SE, Garijo O, Mills RE, Teyton L, et al. The junctional adhesion molecule JAML is a costimulatory receptor for epithelial γδ T cell activation. Science. 2010;329:1205–1210. doi: 10.1126/science.1192698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Witherden DA, Watanabe M, Garijo O, Rieder SE, Sarkisyan G, Cronin SJF, et al. The CD100 receptor interacts with its plexin B2 ligand to regulate epidermal γδ T cell function. Immunity. 2012;37:314–325. doi: 10.1016/j.immuni.2012.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yazdani U, Terman JR. The semaphorins. Genome Biol. 2006;7:211. doi: 10.1186/gb-2006-7-3-211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Takamatsu H, Kumanogoh A. Diverse roles for semaphorin-plexin signaling in the immune system. Trends Immunol. 2012;33:127–135. doi: 10.1016/j.it.2012.01.008. [DOI] [PubMed] [Google Scholar]
- 13.Takegahara N, Kumanogoh A, Kikutani H. Semaphorins: a new class of immunoregulatory molecules. Philos Trans R Soc Lond B Biol Sci. 2005;360:1673–1680. doi: 10.1098/rstb.2005.1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Herold C, Bismuth G, Bensussan A, Boumsell L. Activation signals are delivered through two distinct epitopes of CD100, a unique 150 kDa human lymphocyte surface structure previously defined by BB18 mAb. Int Immunol. 1995;7:1–8. doi: 10.1093/intimm/7.1.1. [DOI] [PubMed] [Google Scholar]
- 15.Ito Y, Oinuma I, Katoh H, Kaibuchi K, Negishi M. Sema4D/plexin-B1 activates GSK-3β through R-Ras GAP activity, inducing growth cone collapse. EMBO Rep. 2006;7:704–709. doi: 10.1038/sj.embor.7400737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oinuma I, Ishikawa Y, Katoh H, Negishi M. The Semaphorin 4D receptor Plexin-B1 is a GTPase activating protein for R-Ras. Science. 2004;305:862–865. doi: 10.1126/science.1097545. [DOI] [PubMed] [Google Scholar]
- 17.Kruger RP, Aurandt J, Guan KL. Semaphorins command cells to move. Nat Rev Mol Cell Biol. 2005;6:789–800. doi: 10.1038/nrm1740. [DOI] [PubMed] [Google Scholar]
- 18.Oinuma I, Katoh H, Negishi M. Semaphorin 4D/Plexin-B1-mediated R-Ras GAP activity inhibits cell migration by regulating β1 integrin activity. J Cell Biol. 2006;173:601–613. doi: 10.1083/jcb.200508204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chabbert-de Ponnat I, Marie-Cardine A, Pasterkamp RJ, Schiavon V, Tamagnone L, Thomasset N, et al. Soluble CD100 functions on human monocytes and immature dendritic cells require plexin C1 and plexin B1, respectively. Int Immunol. 2005;17:439–447. doi: 10.1093/intimm/dxh224. [DOI] [PubMed] [Google Scholar]
- 20.Granziero L, Circosta P, Scielzo C, Frisaldi E, Stella S, Geuna M, et al. CD100/Plexin-B1 interactions sustain proliferation and survival of normal and leukemic CD5+ B lymphocytes. Blood. 2003;101:1962–1969. doi: 10.1182/blood-2002-05-1339. [DOI] [PubMed] [Google Scholar]
- 21.Ishida I, Kumanogoh A, Suzuki K, Akahani S, Noda K, Kikutani H. Involvement of CD100, a lymphocyte semaphorin, in the activation of the human immune system via CD72: implications for the regulation of immune and inflammatory responses. Int Immunol. 2003;15:1027–1034. doi: 10.1093/intimm/dxg098. [DOI] [PubMed] [Google Scholar]
- 22.Kumanogoh A, Watanabe C, Lee I, Wang X, Shi W, Araki H, et al. Identification of CD72 as a lymphocyte receptor for the class IV semaphorin CD100: a novel mechanism for regulating B cell signaling. Immunity. 2000;13:621–631. doi: 10.1016/s1074-7613(00)00062-5. [DOI] [PubMed] [Google Scholar]
- 23.Masuda K, Furuyama T, Takahara M, Fujioka S, Kurinami H, Inagaki S. Sema4D stimulates axonal outgrowth of embryonic DRG sensory neurones. Genes Cells. 2004;9:821–829. doi: 10.1111/j.1365-2443.2004.00766.x. [DOI] [PubMed] [Google Scholar]
- 24.Kumanogoh A, Suzuki K, Ch’ng E, Watanabe C, Marukawa S, Takegahara N, et al. Requirement for the lymphocyte semaphorin, CD100, in the induction of antigen-specific T cells and the maturation of dendritic cells. J Immunol. 2002;169:1175–1181. doi: 10.4049/jimmunol.169.3.1175. [DOI] [PubMed] [Google Scholar]
- 25.Shi W, Kumanogoh A, Watanabe C, Uchida J, Wang X, Yasui T, et al. The class IV semaphorin CD100 plays nonredundant roles in the immune system: defective B and T cell activation in CD100-deficient mice. Immunity. 2000;13:633–642. doi: 10.1016/s1074-7613(00)00063-7. [DOI] [PubMed] [Google Scholar]
- 26.Li M, O’Sullivan KM, Jones LK, Semple T, Kumanogoh A, Kikutani H, et al. CD100 enhances dendritic cell and CD4+ cell activation leading to pathogenetic humoral responses and immune complex glomerulonephritis. J Immunol. 2006;177:3406–3412. doi: 10.4049/jimmunol.177.5.3406. [DOI] [PubMed] [Google Scholar]
- 27.Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature. 2007;448:427–434. doi: 10.1038/nature06005. [DOI] [PubMed] [Google Scholar]
- 28.Katakura K, Lee J, Rachmilewitz D, Li G, Eckmann L, Raz E. Toll-like receptor 9-induced type I IFN protects mice from experimental colitis. J Clin Invest. 2005;115:695–702. doi: 10.1172/JCI22996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Komano H, Fujiura Y, Kawaguchi M, Matsumoto S, Hashimoto Y, Obana S, et al. Homeostatic regulation of intestinal epithelia by intraepithelial γδ T cells. Proc Natl Acad Sci U S A. 1995;92:6147–6151. doi: 10.1073/pnas.92.13.6147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pinto D, Clevers H. Wnt control of stem cells and differentiation in the intestinal epithelium. Exp Cell Res. 2005;306:357–363. doi: 10.1016/j.yexcr.2005.02.022. [DOI] [PubMed] [Google Scholar]
- 31.Brauchle M, Madlener M, Wagner AD, Angermeyer K, Lauer U, Hofschneider PH, et al. Keratinocyte growth factor is highly overexpressed in inflammatory bowel disease. Am J Pathol. 1996;149:521–529. [PMC free article] [PubMed] [Google Scholar]
- 32.Finch PW, Pricolo V, Wu A, Finkelstein SD. Increased expression of keratinocyte growth factor messenger RNA associated with inflammatory bowel disease. Gastroenterology. 1996;110:441–451. doi: 10.1053/gast.1996.v110.pm8566591. [DOI] [PubMed] [Google Scholar]
- 33.Ismail AS, Severson KM, Vaishnava S, Behrendt CL, Yu X, Benjamin JL, et al. γδ intraepithelial lymphocytes are essential mediators of host-microbial homeostasis at the intestinal mucosal surface. Proc Natl Acad Sci U S A. 2011;108:8743–8748. doi: 10.1073/pnas.1019574108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Murthy AK, Dubose CN, Banas JA, Coalson JJ, Arulanandam BP. Contribution of polymeric immunoglobulin receptor to regulation of intestinal inflammation in dextran sulfate sodium-induced colitis. J Gastroenterol Hepatol. 2006;21:1372–1380. doi: 10.1111/j.1440-1746.2006.04312.x. [DOI] [PubMed] [Google Scholar]
- 35.Shui JW, Larange A, Kim G, Vela JL, Zahner S, Cheroutre H, et al. HVEM signalling at mucosal barriers provides host defence against pathogenic bacteria. Nature. 2012;488:222–225. doi: 10.1038/nature11242. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.