

Abstract
Controversy exists regarding the feasibility of preventive clinical trials in prodromal Huntington disease (HD). A primary limitation is a lack of outcome measures for persons with the gene mutation who have not yet been diagnosed with HD. Many longitudinal studies of cognitive decline in prodromal HD have not stratified samples based on disease progression, thereby obscuring differences between symptomatic and nonsymptomatic individuals. Prodromal participants from PREDICT-HD were stratified by disease progression into one of three groups: those having a High, Medium, or Low probability of motor manifestation within the next five years. Data from a total of N = 1299 participants with up to 5950 data points were subjected to linear mixed effects regression on 29 longitudinal cognitive variables, controlling for age, education, depression, and gender. Performance of the three prodromal HD groups was characterized by insidious and significant cognitive decline over time. Twenty-one variables from 19 distinct cognitive tasks revealed evidence of a disease progression gradient, meaning that the rate of deterioration varied as a function of progression level, with faster deterioration associated with greater disease progression. Nineteen measures showed significant longitudinal change in the High group, nine showed significant change in the Medium group and four showed significant cognitive decline in the Low group. Results indicate that clinical trials may be conducted in prodromal HD using the outcome measures and methods specified. The findings may help inform interventions in HD as well as other neurodegenerative disorders.
Introduction
Autosomal dominant neurodegenerative disorders such as Huntington disease (HD) provide researchers with the opportunity to track the development of cognitive impairment from the earliest measurable changes to eventual dementia. Although a wealth of research findings have established that decline is evident decades before clinical diagnosis[1], controversy exists regarding the feasibility of measuring interventions to delay or slow disease progression. We searched PubMed and Medline for articles reporting repeated cognitive assessments in persons with the HD gene expansion who were not yet diagnosed with disease. Search terms included prodromal, presymptomatic, preclinical, prediagnosed, asymptomatic, premotor, preHD, and HD. Table 1 gives a summary of sixteen papers (comprising 8 independent studies) of longitudinal cognitive change in HD before diagnosis. Four of these studies reported no significant cognitive change[2 3];[4 5] whereas the remaining twelve papers documented significant cognitive change over time[6-16]. One paper concluded that, “for premanifest HD, rates of progression of these cognitive outcomes appear to be too slow to detect with a reasonable sample size in a time period reasonable for a clinical trial” [5]. The weight of this statement is great for a field burgeoning with efforts to intervene at the earliest time possible. For well over a decade, clinical trials to slow the progression of neurodegenerative disorders [17 18] or simply to improve static impairment [19 20] have provided incentive for drug development and treatment for cognitive impairments. The purpose of the current study is to provide the largest ever longitudinal study of cognitive outcomes in prodromal HD and to compare findings with all available literature. Such a study is critical to advance efforts to design preventive clinical trials for HD. The aims of this study are to 1) investigate rates of cognitive decline between groups with different prodromal staging of disease, 2) compute standardized annual rates of cognitive change for clinical trial development in prodromal HD, and 3) compute estimated sample sizes for clinical trials designed to detect an amelioration or delay of progression prior to motor diagnosis.
Table 1.
Literature Review of Cognitive change in Prodromal HD
| First author | Year | Length of follow up (years) |
Sample(s) | # Cognitive measures |
Cognitive change findings |
|---|---|---|---|---|---|
| Giordani | 1995 | 4 | 8 + 8 − |
29 | ns |
| Campodonico | 1996 | 2 | 22 + 37− |
20 | ns |
| Kirkwood | 1999 | 3.7 | 12+ 31− |
6 | WAIS-R Digit Symbol Coding significant change in + vs − |
| Paulsen | 2001 | 2 | 260 AR 70 converted to HD |
5 | Change rate greatest in Stroop reading, then SDMT |
| Lemiere | 2002 | 1 | 21 HD 12+ 11− |
29 | Block span significant change in + vs − |
| Snowden | 2002 | 5 | 51 +; 24 converted to HD |
10 | Change rate greatest in Object memory, then Card sorting, Stroop reading, Stroop color |
| Lemiere | 2004 | 2.5 | 19 HD 12 + 11 − |
29 | SDMT, Block span, and HVLT significant change in + vs − |
| Witjes-Ane | 2007 | 3 | 33 + 73 − |
17 | ns |
| Solomon | 2008 | 10 | 43 + 112 − 21 converted to HD |
9 | significant change on 8 of 9 cog tasks in + vs − converters more rapid decline in reaction time, movement time, button tapping speed, and WAIS-R Digit Symbol Coding |
| Brandt | 2008 | 3 | 49 + 134 − 21 converted to HD |
10 | Significant change in Wisconsin Card Sorting Test performances in converters |
| Rupp | 2010 | 2.5 | 38+ 68− |
28 | Significant decline over time on 6/7 tests for “near” group and 1/7 for “far” group; memory guided latencies and errors, alternate button tapping most sensitive |
| Tabrizi | 2011 | 1 | 114 HD 118 + 117 − |
9 | Circle tracing significant change in + vs − |
| Hart | 2011 | 7 | 29 + 43 − |
15 | WMS concentration, memory and visual reproduction scales significant change in + vs − WMS concentration decline remained significant when converters removed |
| Maroof | 2011 | 21 | 110 + 138 − 19 converted to HD |
9 | 7/9 cognitive measures showed significant worsening over time; authors describe gradual declines “long before clinical onset” with abrupt worsening 5 years prior to motor diagnosis |
| Tabrizi | 2012 | 2 | 116 HD 117+ 116− |
5 | Emotion recognition and speeded tapping different in prodromal group closest to diagnosis |
| Stout | 2012 | 2 | 116 HD 117+ 116− |
12 | ns |
Note: HD = participants diagnosed with manifest HD, + = gene expansion positive, − = gene expansion negative, AR = at risk, CALT = Conditional Associative Learning Test, HVLT = Hopkins Verbal Learning Test, SDMT = Symbol Digit Modalities Test, WAIS-R = Wechsler Adult Intelligence Scale- Revised, WMS = Wechsler Memory Scale.
Methods
Participants
The data analyzed in this study were collected from September 2002 to August 2012 from N = 1299 PREDICT-HD participants (1002 with prodromal HD and 297 controls) at 32 sites. All participants had completed genetic testing for HD prior to (and independent from) study enrollment. HD is one of several trinucleotide repeat disorders which are caused by an expanded sequence of three DNA bases, cytosine, adenine, guanine (CAG). CAG is the genetic code for the amino acid glutamine. Research to date has shown that people with fewer than 36 repeated glutamines in the polyglutamine tract do not typically develop HD whereas CAG repeat lengths at or above 36 results in manifestation of disease. The number of CAG repeats is related to the age of disease onset with longer repeats showing an earlier onset, accounting for up to 60% of the variance in onset age.[21] Gene expansion status and CAG repeat length were confirmed at the initial study visit. Prodromal HD participants had CAG ≥ 36, and the gene mutation negative comparisons (controls) had CAG < 36. At study enrollment, participants were required to be 18 years of age or older and could not yet be diagnosed with manifest HD according to traditional motor criteria. Motor diagnosis is defined as the highest rating on the Diagnostic Confidence Level (DCL) of the Unified Huntington’s Disease Rating Scale (UHDRS; Huntington Study Group), which is scored after a brief motor examination. The highest rating indicates a certified examiner is at least 99% confident that the individual shows unequivocal signs of HD.[22-24] All participants provided informed consent (reviewed and approved by the Institutional Review Board at their respective sites) and were treated in accordance with the ethical standards of the American Psychological and Medical Associations. Studywide exclusion criteria included history of a significant developmental cognitive disorder, other CNS disease or injury, evidence of an unstable medical or psychiatric illness (including substance abuse), a pacemaker or metallic implants, or having taken prescribed antipsychotic medication in the last 6 months or phenothiazine derivative antiemetic medication in the 3 months prior to enrollment. Progression groups were defined based on the CAG-Age Product or CAP score[25], which is a formula derived from an accelerated failure-time (AFT) model, computed as CAP = (Age at entry) × (CAG – 33.66). CAP scores can be converted to a scaled CAP score (CAPS) based on a 5-year probability of motor diagnosis and cutoffs were derived to stratify by estimated years to diagnosis. CAP was derived by Zhang et al. (2011) as a new index of disease progression at study entry and was based on actual prospectively diagnosed prodromal PREDICT-HD participants. All previous models published were based on retrospective, self-report data using patient- and family-reported dates of disease onset age. The CAP formula was derived from the best fitting AFT model and the subgroups (Low, Medium, High) were optimal according to the criterion of the algorithm used by Zhang et al. based an earlier sample of PREDICT-HD participants.[25] Table 2 shows descriptive statistics for the prodromal HD and control groups.
Table 2.
Descriptive statistics for Prodromal HD and Control groups.
| CAP Group | ||||
|---|---|---|---|---|
| Variable | Control | Low | Medium | High |
| Probability of motor diagnosis within 5 yrs |
NA | <.67 | .67 - .85 | >.85 |
| Estimated years to motor diagnosis |
NA | >12.8 | 12.8 –7.6 | <7.6 |
| Sample Size (N) | 297 | 278 | 354 | 370 |
| Data Points (N*) | 1298 | 1229 | 1612 | 1811 |
| Prop. of Males | 0.36 | 0.35 | 0.34 | 0.43 |
| Age (Yr) | 44.28 (11.32) | 35.01 (7.87) | 41.66 (9.59) | 44.92 (10.10) |
| Education (Yr) | 14.89 (2.55) | 14.57 (2.43) | 14.56 (2.61) | 14.34 (2.76) |
| CAG Expansion | 20.27 (3.52) | 40.91 (1.62) | 42.02 (2.05) | 43.58 (2.74) |
| SCL90-Depression Baseline |
48.75 (10.35) | 52.40 (13.94) | 53.18 (14.79) | 53.96 (15.65) |
CAP=CAG by AGE probability of motor manifestation within 5 years, NA=not applicable, N=sample size, Yr=year, SCL=Symptom Checklist
Procedure
Participants completed a comprehensive cognitive assessment as part of their annual visit in the PREDICT study. Strict quality control protocols were applied to all cognitive data; details are provided in the appendix. The frequency of test administration differed over the course of the study, with a core battery administered yearly and other tasks administered every other year in order to manage participant burden while maximizing task consideration for future clinical trials. We have indicated the number of data points collected and participants in each analysis by task in Table 3. Ten neuropsychological tests administered were Symbol Digit Modalities Test (SDMT), Stroop Color and Word Test, University of Pennsylvania Smell Identification Test, Trail Making Test (TMT), Controlled Oral Word Association Test, Benton Facial Recognition Test, Matrix Reasoning and Vocabulary subtests of the Wechsler Abbreviated Scale of Intelligence (WASI), Wechsler Adult Intelligence Scale-III Letter-Number Sequencing (WAIS-III:), and Hopkins Verbal Learning Test-Revised (HVLT-R). Nine computerized tasks adapted from cognitive science paradigms were administered as well: Paced Tapping, Emotion recognition, Cued movement sequencing, Category Learning, 3-disk and 4-disk Tower of Hanoi tasks, Verbal working memory using n-back, serial learning, simple speeded tapping, simple reaction time and choice reaction time. A description of each test and task administered as well as the key test variable(s) included in the analyses is provided for each measure in the appendix. From the 19 tasks, which each yield several variables, we identified a set of 51 variables to examine baseline, cross-sectional differences from the controls. We then reduced the group to 29 variables based on the following: a) one measure from each conceptually distinct task component in the cognitive battery was retained; and b) when more than one variable was available for a specific cognitive task, we chose those variables with the greatest effect size of disease-specific difference from the controls. The final analysis involved 29 key variables derived from the 19 measures administered for longitudinal decline in the current study.
Table 3. Linear mixed effects regression results of cognitive change over time in three prodromal HD groups and normal controls. Shading indicates |Z| > 2.
| Slope | Difference with Control | Z-value of Difference | Global | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Variable | Control | Low | Medium | High | Low | Medium | High | Low | Medium | High | ΔAIC | N* | N |
| SDMT# | 0.0447 | 0.0061 | −0.0334 | −0.0958 | − 0.0386 |
−0.0780 | − 0.1405 |
−2.69 | −5.96 | −11.03 | 119.79 | 5950 | 1299 |
| Stroop Color# | −0.0063 | −0.0201 | −0.0591 | −0.1230 | − 0.0138 |
−0.0528 | − 0.1167 |
−0.96 | −4.05 | −9.20 | 91.87 | 5943 | 1298 |
| Stroop Word# | 0.0133 | −0.0123 | −0.0293 | −0.0977 | − 0.0257 |
−0.0427 | − 0.1110 |
−1.94 | −3.54 | −9.48 | 90.80 | 5952 | 1298 |
| Speeded Tappingx | 0.2536 | 0.2891 | 0.3785 | 0.5694 | 0.0355 | 0.1249 | 0.3157 | 0.76 | 2.97 | 7.74 | 72.82 | 2510 | 1028 |
| Stroop Interference# | 0.1428 | 0.1148 | 0.1021 | 0.0401 | − 0.0280 |
−0.0407 | − 0.1027 |
−2.09 | −3.33 | −8.60 | 72.47 | 5944 | 1298 |
| Smell ID# | −0.0422 | −0.0826 | −0.1182 | −0.1999 | − 0.0404 |
−0.0760 | − 0.1577 |
−1.89 | −3.89 | −8.15 | 64.63 | 4265 | 1288 |
| TMT-Ax | −0.0087 | 0.0535 | 0.0671 | 0.1873 | 0.0622 | 0.0758 | 0.1960 | 2.15 | 2.89 | 7.62 | 56.21 | 4448 | 1298 |
| TMT-Bx | −0.0588 | 0.0083 | 0.0122 | 0.1283 | 0.0671 | 0.0710 | 0.1871 | 2.42 | 2.81 | 7.59 | 55.41 | 4420 | 1298 |
| Paced Tapping# | −0.0012 | −0.0373 | −0.0796 | −0.1135 | − 0.0361 |
−0.0784 | − 0.1123 |
−1.62 | −3.83 | −5.65 | 28.70 | 2501 | 1021 |
| Emotions (static)# | 0.0300 | 0.0280 | 0.0290 | −0.0671 | − 0.0020 |
−0.0010 | − 0.0971 |
−0.07 | −0.04 | −3.69 | 20.14 | 2534 | 1029 |
| Cued Movement Sequencing: Low Cue Levelx |
0.0572 | 0.0581 | 0.1174 | 0.1881 | 0.0009 | 0.0602 | 0.1310 | 0.02 | 1.77 | 3.96 | 17.63 | 2526 | 1031 |
| Two-choice Response Timex |
0.0389 | 0.0239 | 0.0305 | 0.1254 | − 0.0150 |
−0.0084 | 0.0865 | −0.47 | −0.29 | 3.04 | 16.47 | 2459 | 1004 |
| COWAT# | 0.0757 | 0.1029 | 0.0724 | 0.0289 | 0.0271 | −0.0033 | − 0.0469 |
1.40 | −0.19 | −2.74 | 16.20 | 4056 | 1039 |
| Cued Movement Sequencing: High Cue Levelx |
0.1014 | 0.1223 | 0.1472 | 0.2416 | 0.0210 | 0.0458 | 0.1402 | 0.52 | 1.23 | 3.87 | 14.46 | 2445 | 1020 |
| Benton Facial Recognition# |
0.0749 | 0.0388 | 0.0050 | −0.0598 | − 0.0361 |
−0.0700 | − 0.1348 |
−0.87 | −1.85 | −3.67 | 10.26 | 2024 | 998 |
| Simple Response Timex |
0.0929 | 0.1118 | 0.1458 | 0.1948 | 0.0189 | 0.0529 | 0.1019 | 0.57 | 1.76 | 3.48 | 9.49 | 2495 | 1010 |
| Emotions (dynamic) # |
0.0076 | 0.0211 | −0.0146 | −0.0650 | 0.0135 | −0.0222 | − 0.0726 |
0.43 | −0.77 | −2.61 | 7.38 | 1990 | 993 |
| Cued Movement Sequencing: Medium Cue Levelx |
−0.0855 | −0.0765 | −0.0352 | 0.0280 | 0.0090 | 0.0503 | 0.1134 | 0.17 | 1.07 | 2.49 | 2.70 | 2095 | 1011 |
| Matrix Reasoning# | 0.1031 | 0.1051 | 0.0598 | −0.0224 | 0.0019 | −0.0433 | − 0.1256 |
0.03 | −0.74 | −2.21 | 1.55 | 1412 | 879 |
| Towers Four Disk Conditionx |
0.0239 | −0.0217 | 0.0280 | 0.0734 | − 0.0455 |
0.0041 | 0.0495 | −1.07 | 0.11 | 1.32 | 1.39 | 1989 | 993 |
| Letter Number Sequencing# |
−0.0377 | −0.0149 | −0.0167 | −0.0674 | 0.0227 | 0.0209 | − 0.0298 |
0.72 | 0.73 | −1.07 | 0.21 | 2017 | 996 |
Note: The first column lists the variable name. The next set of columns (Slope) show the standardized slopes for the groups, that is, annual change in SD units. The next series of columns (Difference with controls) shows the standardized difference between each CAP group and the control group in SD units. The next few columns (Z-value of Difference) show the Z-ratio for each difference depicted in the previous columns. The Z-ratio is the effect size for a specific effect, and the shading indicates |Z| > 2, which is a cutoff adopted to provide a benchmark (see previous comments). Finally, the right hand columns show global information, including the ΔAIC global effect size measure, total number of data points (N*), and total sample size (N). The cognitive outcomes are sorted in descending order by ΔAIC. Higher values indicate better performance.
= Higher values indicate worse performance. ΔAIC = Difference in AIC of full and reduced models
= total number of data points, N = sample size, SD = Standard deviation, SDMT = Symbol Digit Modalities Test, TMT-A = Trail Making Test Part A, TMT-B = Trail Making Test Part B, COWAT = Controlled Oral Word Association Test
Data Analytic Strategy
Each of the 29 variables of interest was analyzed separately using linear mixed effects regression (LMER)[26]. Details of the models are provided in the appendix. Site-to-site variability was accounted for by using a three level model with repeated measures nested within participants nested within sites. Preliminary analysis showed evidence that linear curves were adequate for the modeling of change over time. The time metric for the analysis was duration, defined as the current age minus the age at study entry. Each model had intercept and slope effects for the covariates of gender, age at entry, years of education, and depression. Two models were estimated for each outcome variable, a reduced model that had CAP group intercept differences, and a full model that added CAP group slope differences. The LMER models were estimated using maximum likelihood (ML) methods, which yield unbiased estimates under the assumption that the missing data mechanism is ignorable [27]. Due to the large number of estimated models and the large sample size, we report first on a global, followed by a specific effect size. The global effect size indexes any type of difference among the CAP groups (i.e., differing slopes among any groups). The specific effect size indexes a distinct effect between any two groups (e.g. between the High and control groups). Criteria for significance for the global effect size was based on the difference between Akaike’s Information Criterion (AIC) [28] and the specific effect size was defined as the slope difference divided by the standard error of the difference, which we denote as the Z-ratio. To facilitate comparisons among variables with different scales of measurement, the estimated slopes from the LMER analysis were expressed in SD units scaled using the control group mean and SD.
Required sample size for a hypothetical randomized clinical trial of a cognitively enhancing/protective treatment was estimated for the outcome variable with the strongest global effect size. The power analysis used values typical for clinical trials with neurodegenerative diseases[29]. Statistical power for the slope difference Z-test was considered at four levels of hypothetical treatment-placebo group differences (40% - 70%), with sampling from the High CAP group. As a point of reference for these effect sizes, the largest magnitude represents the effect required for a hypothetical treatment to reduce the most severe progression group to the next most severe (on average). The trial durations were 24 and 36 months with measurements every 6 months, consistent with the longer follow-up periods seen in other HD drug trials (e.g. 2CARE and CREST-E). The power level was 80% and the two-tailed false positive error rate (α) was 5% and 10%. The effect of the treatment was assumed to be immediate with a constant effect over time.
Results
CAP Group Comparison
Table 3 shows results for the 21 outcome variables that showed statistically reliable group slope differences. Only 8 of 29 variables considered failed to show significant decline over time in the prodromal HD group when compared with the controls. Of the 19 tests administered, 15 showed decline. The tests/tasks which failed to showed significant change over time in prodromal HD (thus, not listed in Table 3) were the Vocabulary subtest of the WAIS-III, the HVLT-R, Category learning, serial learning and the n-back test. The outcome variable with the strongest global effect was the SDMT which had a value 30 points higher than the next strongest variables, Stroop Color and Stroop Word. This was followed by a drop of 18 points for the next variables, speeded tapping and Stroop Interference, and so forth, down to the bottom of Table 3 indicating the variables with the smallest effects.
The standardized slopes shown in Table 3 indicates a disease progression gradient in which the control group performed the best over time and performance deteriorated as HD progression group increased. For example, the first row shows that the mean SDMT scores increased over time for the control group at a rate of +0.05 SD per year. However, the rate of change steadily decreased by group with the High group showing deterioration over time at a rate of −0.10 SD loss per year. Graphical representations of some effects are shown in Figure 1. The disease progression gradient was evident in the differences between CAP groups and the control group, as indexed by the Z-values. In all cases, the Z-value of the High group difference was the largest in absolute value. Nine variables (SDMT, Stroop Color, Stroop Word, Speeded Tapping, Stroop Interference, Smell ID, TMT-A, TMT-B, and Paced Tapping) had a Z-value for the Medium group difference that was large (defined as |Z| > 2), and four variables (SDMT, Stroop Interference, TMT-A, TMT-B) had a Low group difference that was large.
Figure 1.
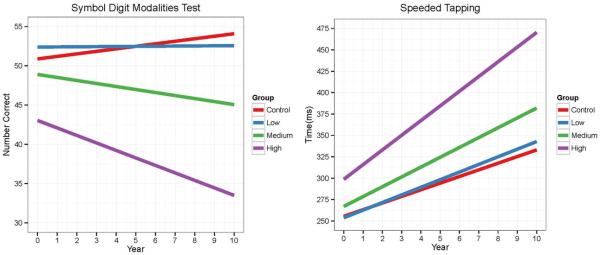
CAP group fitted curves over time for SDMT (left) and speeded tapping (right).
The left-hand graph shows the fitted curves (based on the LMER fitted model) of SDMT in its original scale by year for the CAP groups. The right-hand graph shows the fitted curves for speeded tapping.
Sample sizes for a hypothetical randomized clinical trial of a cognitively enhancing/protective treatment was estimated for the variable with the largest global effect size (i.e., SDMT). The estimated value of a single arm is shown in Table 4 as a function of study duration (24 and 36 months), two-tailed α (.05 and .10), and effects size (40% to 70%). The last row of Table 4 depicts the case of a 36-month trial with the largest slope difference and most liberal α level. The estimated sample size in this case is N = 27 per study group.
Table 4. Required single-arm sample size for a hypothetical randomized clinical trial.
| Month | α | Effect (%) |
Single N |
|---|---|---|---|
| 24 | 0.05 | 40 | 247 |
|
| |||
| 24 | 0.05 | 50 | 158 |
| 24 | 0.05 | 60 | 110 |
| 24 | 0.05 | 70 | 81 |
|
| |||
| 24 | 0.10 | 40 | 195 |
|
| |||
| 24 | 0.10 | 50 | 125 |
| 24 | 0.10 | 60 | 86 |
| 24 | 0.10 | 70 | 64 |
|
| |||
| 36 | 0.05 | 40 | 105 |
|
| |||
| 36 | 0.05 | 50 | 67 |
| 36 | 0.05 | 60 | 47 |
| 36 | 0.05 | 70 | 34 |
|
| |||
| 36 | 0.10 | 40 | 83 |
|
| |||
| 36 | 0.10 | 50 | 53 |
| 36 | 0.10 | 60 | 37 |
| 36 | 0.10 | 70 | 27 |
Discussion
In the largest sample of individuals with prodromal HD reported, findings identify numerous cognitive tasks sensitive to longitudinal decline. For all variables examined, a disease progression gradient was evident with participants who were less affected showing a smaller amount of decline compared to those nearing motor diagnosis. Sensitive tasks included tests of information processing speed, basic attention, tapping speed, inhibition, odor recognition, psychomotor response time, set switching and maintenance, motor timing, emotion recognition, verbal fluency, facial recognition, nonverbal problem-solving, planning, and sequencing. Although substantial effects (ΔAIC > 10) were found for all of these cognitive areas, it is important to highlight that efforts were made to rank order the effect sizes of the various cognitive abilities so that findings could be relevant to clinical research planning initiatives. The largest effect size was found for the SDMT, a task requiring coordination of visual scanning, working memory, fine motor speed, and concentration. These findings replicate those reported in the literature from at least six different studies[6 7 9 11 15 30] and suggest that this task is highly sensitive and consistent for tracking decline in prodromal HD. Longitudinal change for every prodromal group was substantially different from the normal control group, suggesting the SDMT might serve as a sufficient outcome measure to detect alleviation of decline over time from an effective compound or intervention. It is striking that the average rate of change for normals was slight improvement, likely reflecting practice effects or improved performances secondary to task familiarity. Longitudinal performances for every prodromal group showed decline over time, suggesting that the SDMT may be sensitive enough to warrant intervention in the prodromal group furthest from motor manifestation. With regards to neural specificity, the SDMT has long been considered a sensitive but nonspecific cognitive task, reflecting its multidimensional demands and widespread activation on functional imaging. Given the widespread brain circuitry impacted by HD, it is not surprising that one of the most sensitive tasks to disease progression is one that requires overall brain health and complex associations among various brain regions.
Many of the tasks demonstrating robust effect sizes for detection of early HD and for measuring change over time involve a timed component. Six of the studies reviewed (see Table 1) showed significant changes over time using the Stroop Color Word Test[6 10 14-16]; the Trail Making Test[6 14 16], or other measures of reaction or movement time[11 14], all of which were found to have substantial effects in the current study. Some have interpreted this finding as reason to emphasize motor and psychomotor measures of HD as superior to other potential assessed outcomes[13]. The observation that timed tasks appear most sensitive in prodromal HD has been offered as evidence that tests measuring automatic cognitive and motor routines may be more sensitive to early disease than more traditional “clinical” tests which may lack sensitivity for prodromal disease[13 31]. In many basal ganglia diseases, the impact of psychomotor and motor impairments on cognitive functioning is to be expected, although distinct methodology is required to extricate components. For instance, Aron and his colleagues showed that pure motor-based outcome measures in their study did not vary between HD patients and controls, whereas cognitive-dependent motor measures did. [32] Similarly, Lawrence et al., conducted research to disentangle cognitive components from psychomotor speed and showed separable components of speed and primary cognition, in this study visual-spatial function.[33] Finally, using a very large sample of individuals with prodromal HD, O’Rourke et al. examined scores from the Trail Making Test and demonstrated that tests of perceptual processing, visual scanning, and attention were primarily associated with performances on Part A of the test whereas executive functions (i.e., inhibition and set-shifting), processing speed, and working memory were most highly associated with Part B outcomes.[34] Careful research such as these three studies described above are needed to continue to parse out the critical cognitive, motor, and timed components of detriment secondary to diseases of the basal ganglia. It has long been observed that measures most sensitive to HD progression often depend upon speed and/or timing systems. Although the majority of the research has emphasized using a timed outcome variable similar to the studies described above, another large area of work has examined specific circuitry dependent upon internal and external timing systems in the brain. Several authors have documented a specific internal timing deficit in HD[30 35] and recent advances in neuroanatomy have provided a basis for these impairments[36]. Functional imaging research using task-activated fMRI has advanced our understanding of the importance of basal ganglia connections in the timing of movements as well as the temporal encoding and decision processes underlying action sequences [37-40].
Our findings show decline on several additional cognitive tasks not considered primarily motor or speed-dependent. For instance, primary sensory detection and recognition showed decline as captured by the smell and emotion identification tests. Stroop interference requires inhibition whereas Trails B depends upon efficient response shifting; both showed substantial effects for all three prodromal HD groups. These findings are consistent with those in the literature suggesting that measures of working memory[9 41], response shifting and concentration[6 15] showed change over time in prodromal HD. All findings in this publication can be understood in terms of neural circuitry believed to underlie HD. It is accepted that basal ganglia output targets the primary motor cortex, the prefrontal cortex involved in many cognitive and limbic functions[42], as well as discrete multisynaptic loops connecting the cerebellum and the basal ganglia with multiple areas of the cerebral cortex, further elucidating the complex array of outcome measures that prove sensitive to progression of basal ganglia disorders[43]. These findings are consistent with our cross-sectional findings showing that sensory-perceptual processing and motor planning speed best predicted time to diagnosis, after controlling for CAP scores and motor symptoms (Harrington et al., 2012).[39] Future research should illustrate specific cognitive outcomes with functional and structural imaging dysfunction in HD.
The current findings can only be interpreted within the context of the current diagnostic criteria for HD (i.e., motor abnormalities). Many authors have noted limitations of the HD diagnosis and the determination of “conversion” as a clinically subjective judgment susceptible to error[13 44]. Some have suggested that diagnostic criteria for HD be reconsidered to include behavioral, psychiatric and cognitive features[24 45] and that subgroups of prodromal HD could benefit from more sophisticated characterization and diagnosis. Five of the 16 studies reviewed followed persons with gene mutation through to “conversion”, or the point in time when a motor diagnosis was considered appropriate. Upon review, there does not appear to be any difference in the findings noted by these studies versus those that provided longitudinal data without consideration of diagnosis. Most research to date has shown that progression of HD follows a linear insidious course until a point at which time an accelerated decline occurs (documented at 8 to 15 years prior to motor diagnosis[13 14 46]). It is likely that further delineation of progression during the prodrome will be critical to best characterize the natural course of disease. Future work is needed to determine the relative sensitivity of various motor, psychomotor, and cognitive measures across the entire HD spectrum of disease.
In one of the few studies failing to document significant cognitive change, Stout et al. [5] reported “very little evidence of measurable deterioration in the premanifest group relative to controls over either 12 or 24 months.” This apparent discrepancy with our findings may be due to differences in how each study defines prodromal HD. PREDICT-HD includes all eligible participants who have positive gene expansion results but do not meet criteria for a motor diagnosis of HD at baseline (participants may obtain a motor diagnosis during the duration of the study). However, as highlighted by Paulsen and Long [47], due to the TRACK-HD entry requirements of a total motor score ≤ 5 for the premanifest group, a gap may exist between the TRACK-HD premanifest and manifest groups precisely at the state of progression for which cognitive decline accelerates. The TRACK-HD premanifest group may be comparable to the Low group defined in this study, but there is no comparable Medium and High group individuals in that study. Our data suggest that future clinical trials aimed at ameliorating cognitive decline should target participants in the High or Medium CAP group in order to balance the sometimes competing aims of intervening early and ensuring that a treatment effect can be detected.
We caution that the results cannot necessarily be taken as a definitive ordering of the importance of the cognitive variables. Cognition is not a unitary construct and the measures considered in our analysis represent a wide variety of cognitive domains. Due to the multifactorial nature of human cognition, as noted by Harrington et al. [39] in an earlier, cross-sectional analysis, composite indices may be more sensitive to the worsening of cognitive functioning in prodromal HD. Furthermore, as drug advances in other degenerative dementias have demonstrated, novel compounds may act on neural pathways associated with specific cognitive networks and selection of cognitive measures for randomized clinical trials should be carefully considered with the target compound in mind. Other aspects of cognitive task selection, such as ease of administration and reliability across sites must also be considered for clinical trial selection. However, with these caveats in mind, our data demonstrate that several cognitive measures have effect sizes suitable for use in preventive clinical trials.
In conclusion, our results demonstrate that cognitive measures are indeed sensitive to decline over time in prodromal HD. These measures may be used effectively to track cognitive functioning over time. They may also be used as clinical trial outcome measures. We provide effect sizes and sample size calculations for randomized clinical trial design. Regarding future directions for research in cognitive decline, investigations to follow include determining the relationship between cognitive trajectories and declines in functional ability and/or neuropathological changes measured by neuroimaging. Most critical to current progress is that appropriate clinical trial outcome measures are available for preventive trials in prodromal HD.
Acknowledgements
This work was supported by the National Institutes for Health, National Institute of Neurological Disorders and Stroke grant number NS40068 and CHDI Foundation, Inc. We thank the PREDICT-HD sites, the study participants, and the National Research Roster for Huntington Disease Patients and Families. Data from this investigation were presented in part at the annual meeting of the Huntington Study Group, Indianapolis, Indiana, November 2011. Drs. Paulsen, Smith, and Long substantialy contributed to conception and design, acquisition of data, or analysis and interpretation of data; 2) drafting the article or revising it critically for important intellectual content; and 3) final approval of the version to be published.
This work was supported by The National Institutes for Health, National Institute of Neurological Disorders and Stroke grant number NS40068 and CHDI Foundation, Inc. The authors declare no other competing financial interests.
References
- 1.Paulsen JS. Early detection of Huntington’s disease. Future Neurology. 2010;5(1):85–104. doi: 10.2217/fnl.09.78. doi: 10.2217/fnl.09.78[published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Campodonico JR, Codori AM, Brandt J. Neuropsychological stability over two years in asymptomatic carriers of the Huntington’s disease mutation. J Neurol Neurosurg Psychiatry. 1996;61(6):621–4. doi: 10.1136/jnnp.61.6.621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Campodonico JR, Codori AM, Brandt J. Neuropsychological stability over two years in asymptomatic carriers of the Huntington’s disease mutation. Journal of Neurology, Neurosurgery, and Psychiatry. 1996;61:621–24. doi: 10.1136/jnnp.61.6.621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Witjes-Ane M-NW, Mertens B, van Vugt JPP, et al. Longitudinal Evaluation of “Presymptomatic” Carriers of Huntington’s Disease. J Neuropsychiatry Clin Neurosci. 2007;19(3):310–17. doi: 10.1176/jnp.2007.19.3.310. doi: 10.1176/appi.neuropsych.19.3.310[published Online First: Epub Date]|. [DOI] [PubMed] [Google Scholar]
- 5.Stout JC, Jones R, Labuschagne I, et al. Evaluation of longitudinal 12 and 24 month cognitive outcomes in premanifest and early Huntington’s disease. Journal of Neurology, Neurosurgery & Psychiatry. 2012;83(7):687–94. doi: 10.1136/jnnp-2011-301940. doi: 10.1136/jnnp-2011-301940[published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hart E, Middelkoop H, Jurgens CK, et al. Seven-year clinical follow-up of premanifest carriers of Huntington’s disease. PLoS Curr. 2011;3 doi: 10.1371/currents.RRN1288. RRN1288 doi: 10.1371/currents.RRN1288[published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kirkwood SC, Siemers E, Hodes ME, et al. Subtle changes among presymptomatic carriers of the Huntington’s disease gene. J Neurol Neurosurg Psychiatry. 2000;69(6):773–9. doi: 10.1136/jnnp.69.6.773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lemiere C. Non-invasive assessment of airway inflammation in occupational lung diseases. Current opinion in allergy and clinical immunology. 2002;2(2):109–14. doi: 10.1097/00130832-200204000-00005. [DOI] [PubMed] [Google Scholar]
- 9.Lemiere J, Decruyenaere M, Evers-Kiebooms G, et al. Cognitive changes in patients with Huntington’s disease (HD) and asymptomatic carriers of the HD mutation--a longitudinal follow-up study. Journal of neurology. 2004;251(8):935–42. doi: 10.1007/s00415-004-0461-9. doi: 10.1007/s00415-004-0461-9[published Online First: Epub Date]|. [DOI] [PubMed] [Google Scholar]
- 10.Paulsen JS, Ready RE, Hamilton JM, et al. Neuropsychiatric aspects of Huntington’s disease. J Neurol Neurosurg Psychiatry. 2001;71(3):310–4. doi: 10.1136/jnnp.71.3.310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Solomon AC, Stout JC, Weaver M, et al. Ten-year rate of longitudinal change in neurocognitive and motor function in prediagnosis Huntington disease. Mov Disord. 2008;23(13):1830–6. doi: 10.1002/mds.22097. doi: 10.1002/mds.22097[published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tabrizi SJ, Scahill RI, Durr A, et al. Biological and clinical changes in premanifest and early stage Huntington’s disease in the TRACK-HD study: the 12-month longitudinal analysis. Lancet neurology. 2011;10(1):31–42. doi: 10.1016/S1474-4422(10)70276-3. doi: 10.1016/S1474-4422(10)70276-3[published Online First: Epub Date]|. [DOI] [PubMed] [Google Scholar]
- 13.Brandt J, Inscore AB, Ward J, et al. Neuropsychological deficits in Huntington’s disease gene carriers and correlates of early “conversion”. J Neuropsychiatry Clin Neurosci. 2008;20(4):466–72. doi: 10.1176/appi.neuropsych.20.4.466. doi: 10.1176/appi.neuropsych.20.4.466[published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rupp J, Blekher T, Jackson J, et al. Progression in prediagnostic Huntington disease. J Neurol Neurosurg Psychiatry. 2010;81(4):379–84. doi: 10.1136/jnnp.2009.176982. doi: 10.1136/jnnp.2009.176982[published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Snowden JS, Craufurd D, Thompson J, et al. Psychomotor, Executive, and Memory Function in Preclinical Huntington’s Disease. Journal of clinical and experimental neuropsychology. 2002;24(2):133–45. doi: 10.1076/jcen.24.2.133.998. doi: 10.1076/jcen.24.2.133.998[published Online First: Epub Date]|. [DOI] [PubMed] [Google Scholar]
- 16.Maroof DA, Gross AL, Brandt J. Modeling longitudinal change in motor and cognitive processing speed in presymptomatic Huntington’s disease. Journal of clinical and experimental neuropsychology. 2011;33(8):901–9. doi: 10.1080/13803395.2011.574606. doi: 10.1080/13803395.2011.574606[published Online First: Epub Date]|. [DOI] [PubMed] [Google Scholar]
- 17.Rogers SL, Farlow MR, Doody RS, et al. Donepezil Study Group A 24-week, double-blind, placebo-controlled trial of donepezil in patients with Alzheimer’s disease. Neurology. 1998;50(1):136–45. doi: 10.1212/wnl.50.1.136. [DOI] [PubMed] [Google Scholar]
- 18.Reisberg B, Doody R, Stoffler A, et al. Memantine in moderate-to-severe Alzheimer’s disease. N Engl J Med. 2003;348(14):1333–41. doi: 10.1056/NEJMoa013128. doi: 10.1056/NEJMoa013128[published Online First: Epub Date]|. [DOI] [PubMed] [Google Scholar]
- 19.Keefe RS, Bilder RM, Davis SM, et al. Neurocognitive effects of antipsychotic medications in patients with chronic schizophrenia in the CATIE Trial. Arch Gen Psychiatry. 2007;64(6):633–47. doi: 10.1001/archpsyc.64.6.633. doi: 10.1001/archpsyc.64.6.633[published Online First: Epub Date]|. [DOI] [PubMed] [Google Scholar]
- 20.Buchanan RW, Freedman R, Javitt DC, et al. Recent advances in the development of novel pharmacological agents for the treatment of cognitive impairments in schizophrenia. Schizophr Bull. 2007;33(5):1120–30. doi: 10.1093/schbul/sbm083. doi: 10.1093/schbul/sbm083[published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Walker FO. Huntington’s disease. Lancet. 2007;369(9557):218–28. doi: 10.1016/S0140-6736(07)60111-1. doi: 10.1016/S0140-6736(07)60111-1[published Online First: Epub Date]|. [DOI] [PubMed] [Google Scholar]
- 22.Huntington Study Group Unified Huntington’s Disease Rating Scale: reliability and consistency. Mov Disord. 1996;11(2):136–42. doi: 10.1002/mds.870110204. doi: 10.1002/mds.870110204[published Online First: Epub Date]|. [DOI] [PubMed] [Google Scholar]
- 23.Biglan KM, Ross CA, Langbehn DR, et al. Motor abnormalities in premanifest persons with Huntington’s disease: the PREDICT-HD study. Movement Disorders. 2009;24:1763–72. doi: 10.1002/mds.22601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Biglan KM, Zhang Y, Long JD, et al. Refining the diagnosis of Huntington disease: the PREDICT-HD study. Front Aging Neurosci. 2013;5:12. doi: 10.3389/fnagi.2013.00012. doi: 10.3389/fnagi.2013.00012[published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang Y, Long JD, Mills JA, et al. Indexing disease progression at study entry with individuals at-risk for Huntington Disease. American Journal of Medical Genetics Part B: Neuropsychiatric Genetics. 2011;156:751–63. doi: 10.1002/ajmg.b.31232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Verbeke G, Molenberghs G. Linear Mixed Models for Longitudinal Data. Springer; New York: 2000. [Google Scholar]
- 27.Little RJA, Rubin DB. Statistical analysis with missing data. 2nd ed. Wiley; New York: 2002. [Google Scholar]
- 28.Akaike H. Information theory and an extension of the maximum likelihood principle. In: Petrov BN, Csaki F, editors. 2nd International Symposium on Information Theory. Akademiai Kiado; Budapest: 1973. pp. 267–81. [Google Scholar]
- 29.Altmann DR, Jasperse B, Barkhof F, et al. Sample sizes for brain atrophy outcomes in trials for secondary progressive multiple sclerosis. Neurology. 2009;72(7):595–601. doi: 10.1212/01.wnl.0000335765.55346.fc. doi: 10.1212/01.wnl.0000335765.55346.fc[published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Paulsen JS, Zimbelman JL, Hinton SC, et al. fMRI biomarker of early neuronal dysfunction in presymptomatic Huntington’s Disease. AJNR. American journal of neuroradiology. 2004;25(10):1715–21. [PMC free article] [PubMed] [Google Scholar]
- 31.Thompson JC, Poliakoff E, Sollom AC, et al. Automaticity and attention in Huntington’s disease: when two hands are not better than one. Neuropsychologia. 2010;48(1):171–8. doi: 10.1016/j.neuropsychologia.2009.09.002. doi: 10.1016/j.neuropsychologia.2009.09.002[published Online First: Epub Date]|. [DOI] [PubMed] [Google Scholar]
- 32.Aron AR, Watkins L, Sahakian BJ, et al. Task-set switching deficits in early-stage Huntington’s disease: implications for basal ganglia function. Journal of cognitive neuroscience. 2003;15(5) doi: 10.1162/089892903322307357. doi: 10.1162/089892903322307357[published Online First: Epub Date]|. [DOI] [PubMed] [Google Scholar]
- 33.Lawrence AD, Sahakian BJ, Hodges JR, et al. Executive and mnemonic functions in early Huntington’s disease. Brain. 1996;119(Pt 5):1633–45. doi: 10.1093/brain/119.5.1633. [DOI] [PubMed] [Google Scholar]
- 34.O’Rourke JJ, Beglinger LJ, Smith MM, et al. The Trail Making Test in prodromal Huntington disease: contributions of disease progression to test performance. Journal of clinical and experimental neuropsychology. 2011;33(5):567–79. doi: 10.1080/13803395.2010.541228. doi: 10.1080/13803395.2010.541228[published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zimbelman JL, Paulsen JS, Mikos A, et al. fMRI detection of early neural dysfunction in preclinical Huntington’s disease. Journal of the International Neuropsychological Society: JINS. 2007;13(5):758–69. doi: 10.1017/S1355617707071214. doi: 10.1017/S1355617707071214[published Online First: Epub Date]|. [DOI] [PubMed] [Google Scholar]
- 36.Bostan AC, Strick PL. The cerebellum and basal ganglia are interconnected. Neuropsychology review. 2010;20(3):261–70. doi: 10.1007/s11065-010-9143-9. doi: 10.1007/s11065-010-9143-9[published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Harrington DL, Zimbelman JL, Hinton SC, et al. Neural Modulation of Temporal Encoding, Maintenance, and Decision Processes. Cerebral Cortex. 2010;20(6):1274–85. doi: 10.1093/cercor/bhp194. doi: 10.1093/cercor/bhp194[published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Elsinger CL, Harrington DL, Rao SM. From preparation to online control: Reappraisal of neural circuitry mediating internally generated and externally guided actions. Neuroimage. 2006;31(3):1177–87. doi: 10.1016/j.neuroimage.2006.01.041. doi: 10.1016/j.neuroimage.2006.01.041[published Online First: Epub Date]|. [DOI] [PubMed] [Google Scholar]
- 39.Harrington DL, Smith MM, Zhang Y, et al. Cognitive domains that predict time to diagnosis in prodromal Huntington disease. Journal of Neurology, Neurosurgery, and Psychiatry. 2012;83(6):612–19. doi: 10.1136/jnnp-2011-301732. doi: 10.1136/jnnp-2011-301732[published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rao SM, Harrington DL, Haaland KY, et al. Distributed Neural Systems Underlying the Timing of Movements. The Journal of Neuroscience. 1997;17(14):5528–35. doi: 10.1523/JNEUROSCI.17-14-05528.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lemiere J, Decruyenaere M, Evers-Kiebooms G, et al. Longitudinal study evaluating neuropsychological changes in so-called asymptomatic carriers of the Huntington’s disease mutation after 1 year. Acta neurologica Scandinavica. 2002;106(3):131–41. doi: 10.1034/j.1600-0404.2002.01192.x. [DOI] [PubMed] [Google Scholar]
- 42.Alexander GE, DeLong MR, Strick PL. Parallel organization of functionally segregated circuits linking basal ganglia and cortex. Annual review of neuroscience. 1986;9:357–81. doi: 10.1146/annurev.ne.09.030186.002041. doi: 10.1146/annurev.ne.09.030186.002041[published Online First: Epub Date]|. [DOI] [PubMed] [Google Scholar]
- 43.Bostan AC, Dum RP, Strick PL. The basal ganglia communicate with the cerebellum. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(18):8452–6. doi: 10.1073/pnas.1000496107. doi: 10.1073/pnas.1000496107[published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Duff K, Paulsen JS, Beglinger LJ, et al. “Frontal” behaviors before the diagnosis of Huntington’s disease and their relationship to markers of disease progression: evidence of early lack of awareness. J Neuropsychiatry Clin Neurosci. 2010;22(2):196–207. doi: 10.1176/appi.neuropsych.22.2.196. doi: 10.1176/appi.neuropsych.22.2.196[published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Witjes-Ane MN, Mertens B, van Vugt JP, et al. Longitudinal evaluation of “presymptomatic” carriers of Huntington’s disease. J Neuropsychiatry Clin Neurosci. 2007;19(3):310–7. doi: 10.1176/jnp.2007.19.3.310. doi: 10.1176/appi.neuropsych.19.3.310[published Online First: Epub Date]|. [DOI] [PubMed] [Google Scholar]
- 46.Paulsen JS, Langbehn DR, Stout JC, et al. Detection of Huntington’s disease decades before diagnosis: the Predict-HD study. J Neurol Neurosurg Psychiatry. 2008;79(8):874–80. doi: 10.1136/jnnp.2007.128728. doi: 10.1136/jnnp.2007.128728[published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Paulsen JS, Long JD. Neurodegenerative disease: Establishing a clinical trial battery for Huntington disease. Nat Rev Neurol. 2012;8(5):250–51. doi: 10.1038/nrneurol.2012.67. [DOI] [PMC free article] [PubMed] [Google Scholar]